
Theoretical Predictions of Structure, Mechanics, Dislocation, and Electronics Properties of FeTi Alloy at High Pressure

Abstract
:1. Introduction
2. Methodology
3. Results and Discussions
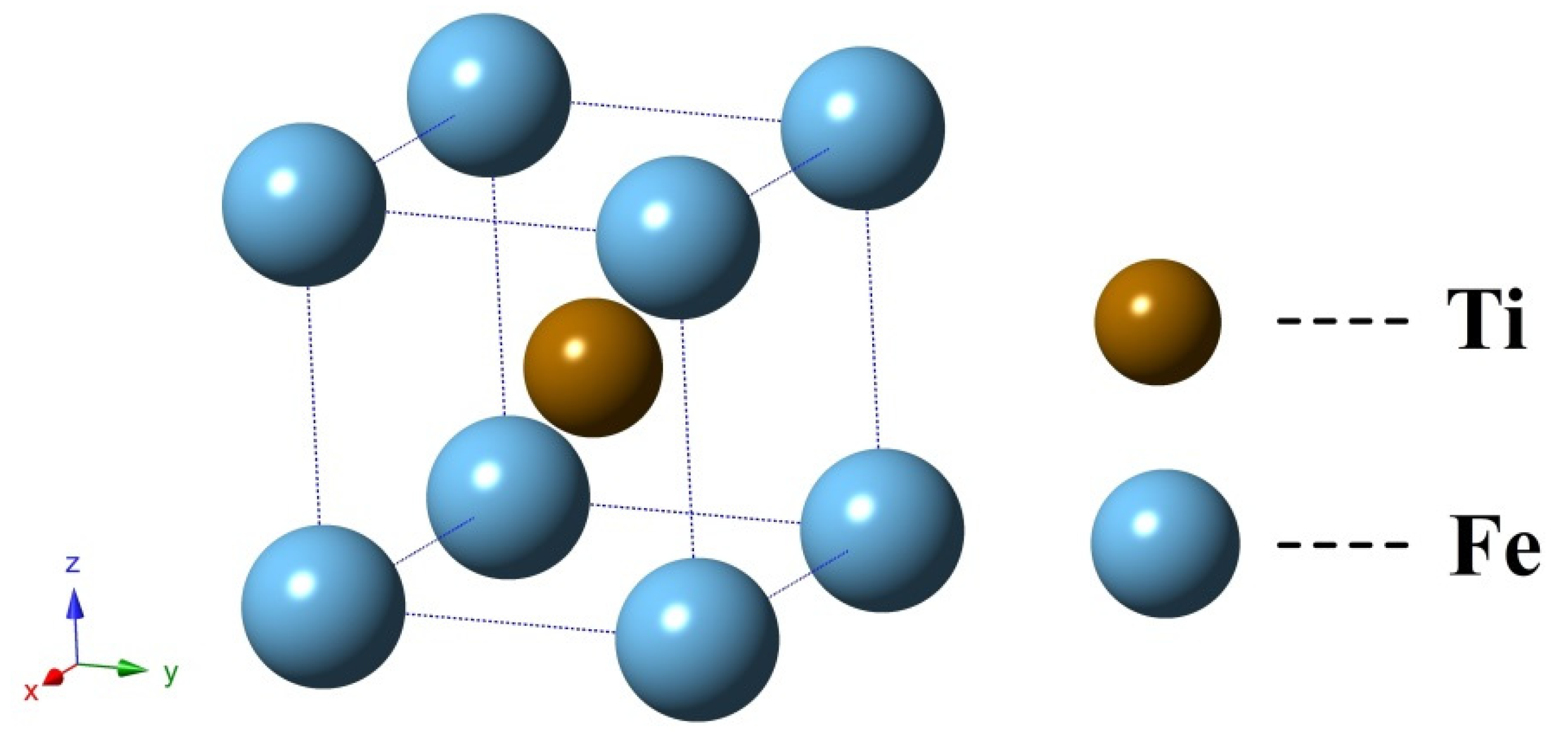
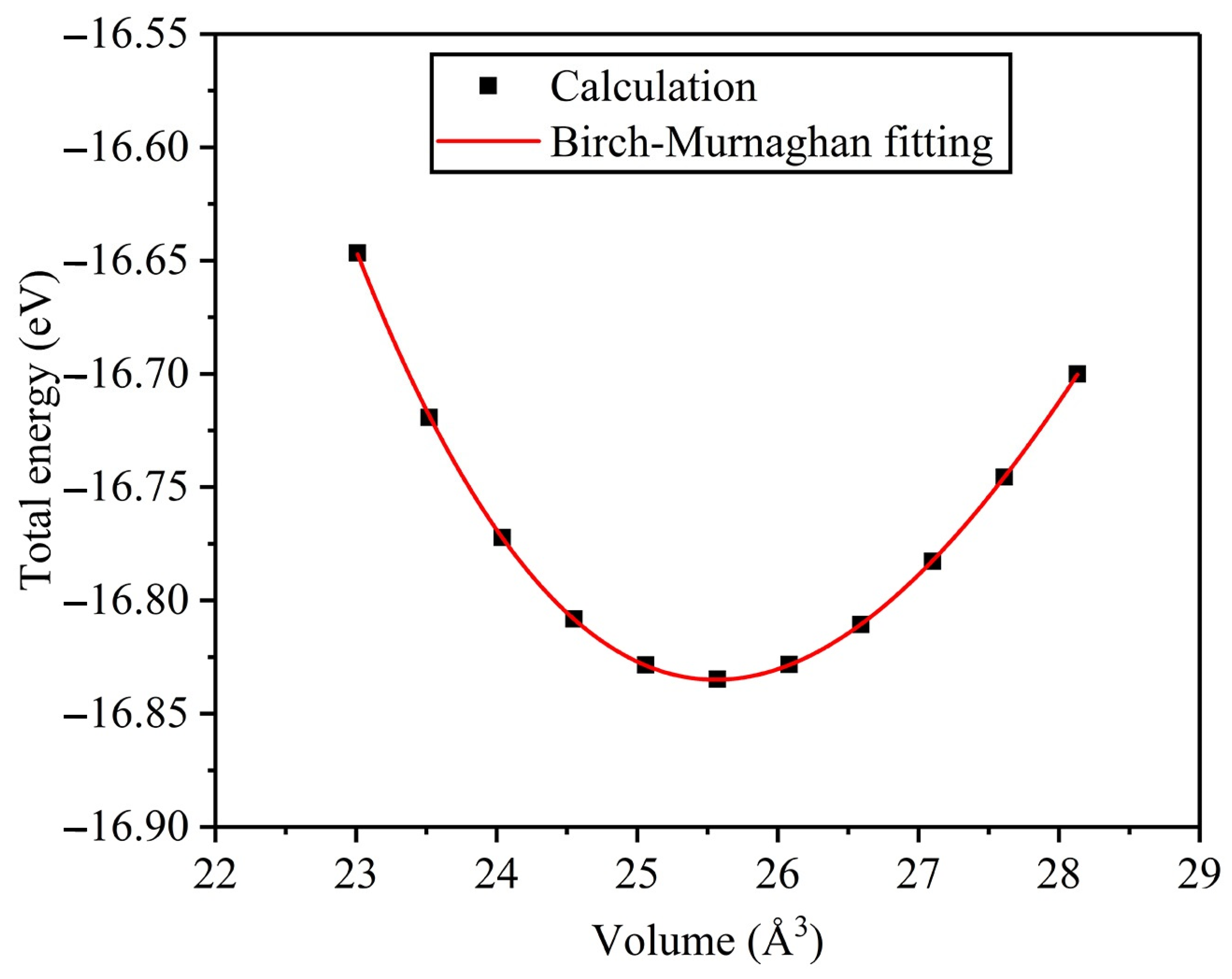
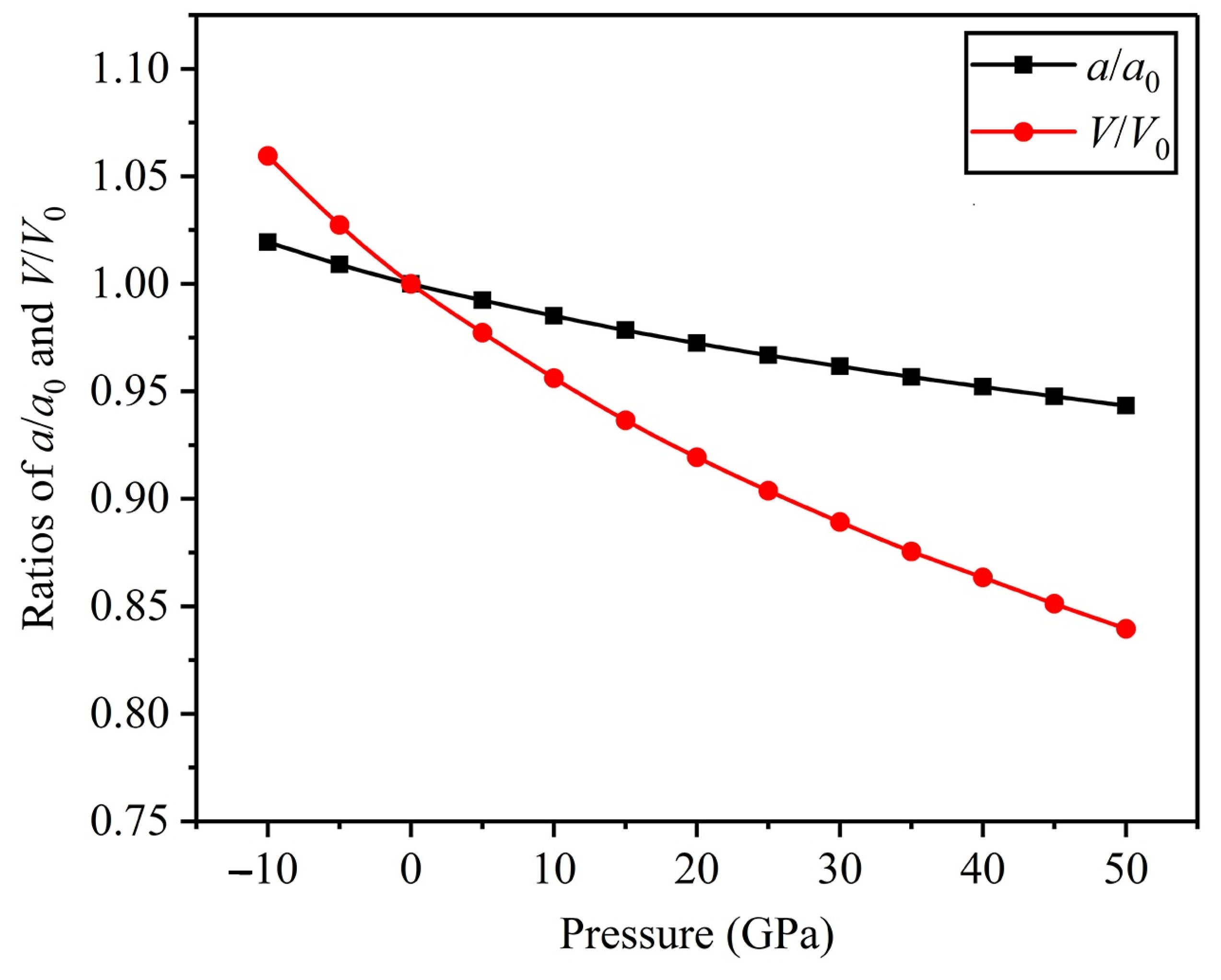
3.1. Structure Properties and Stability
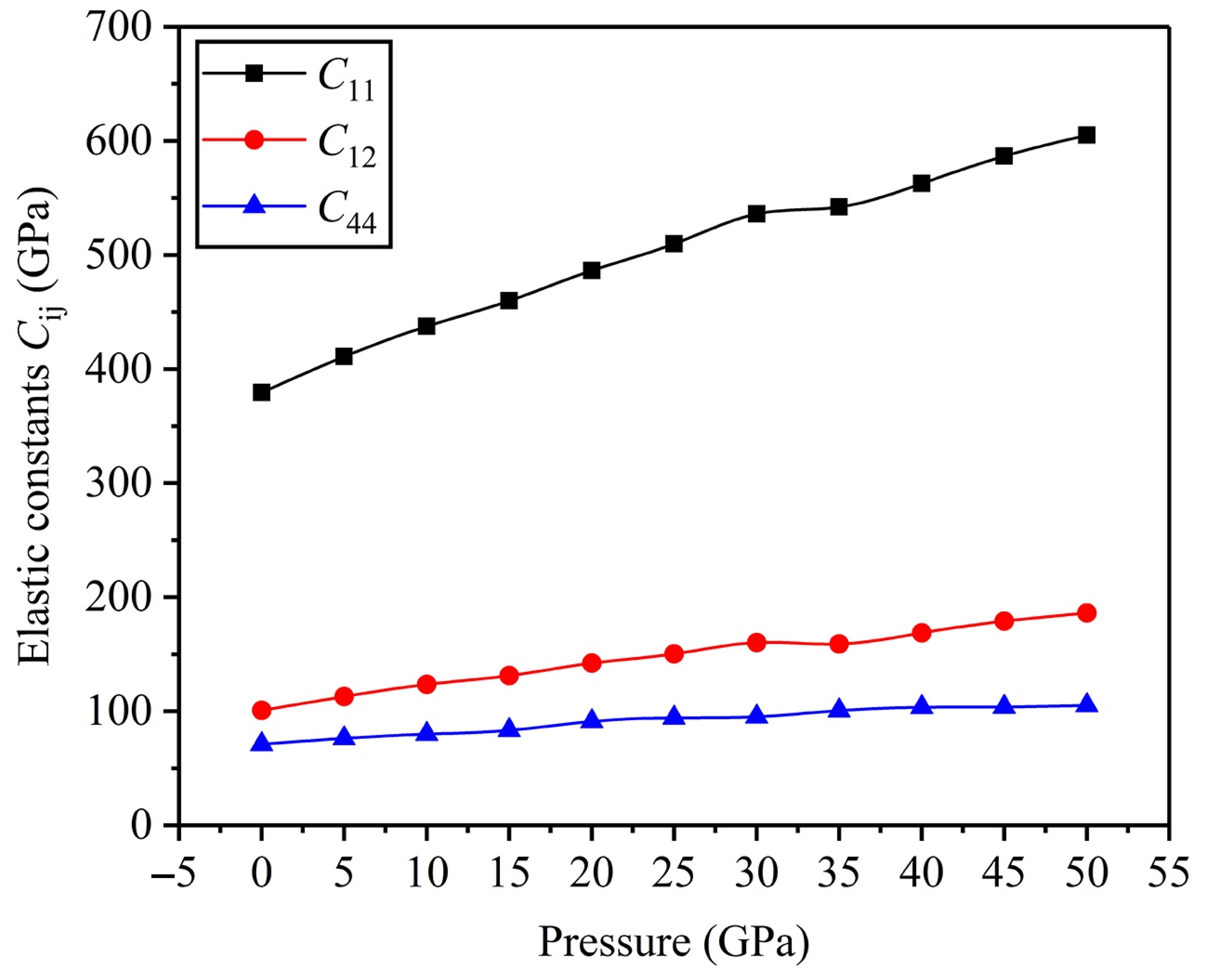
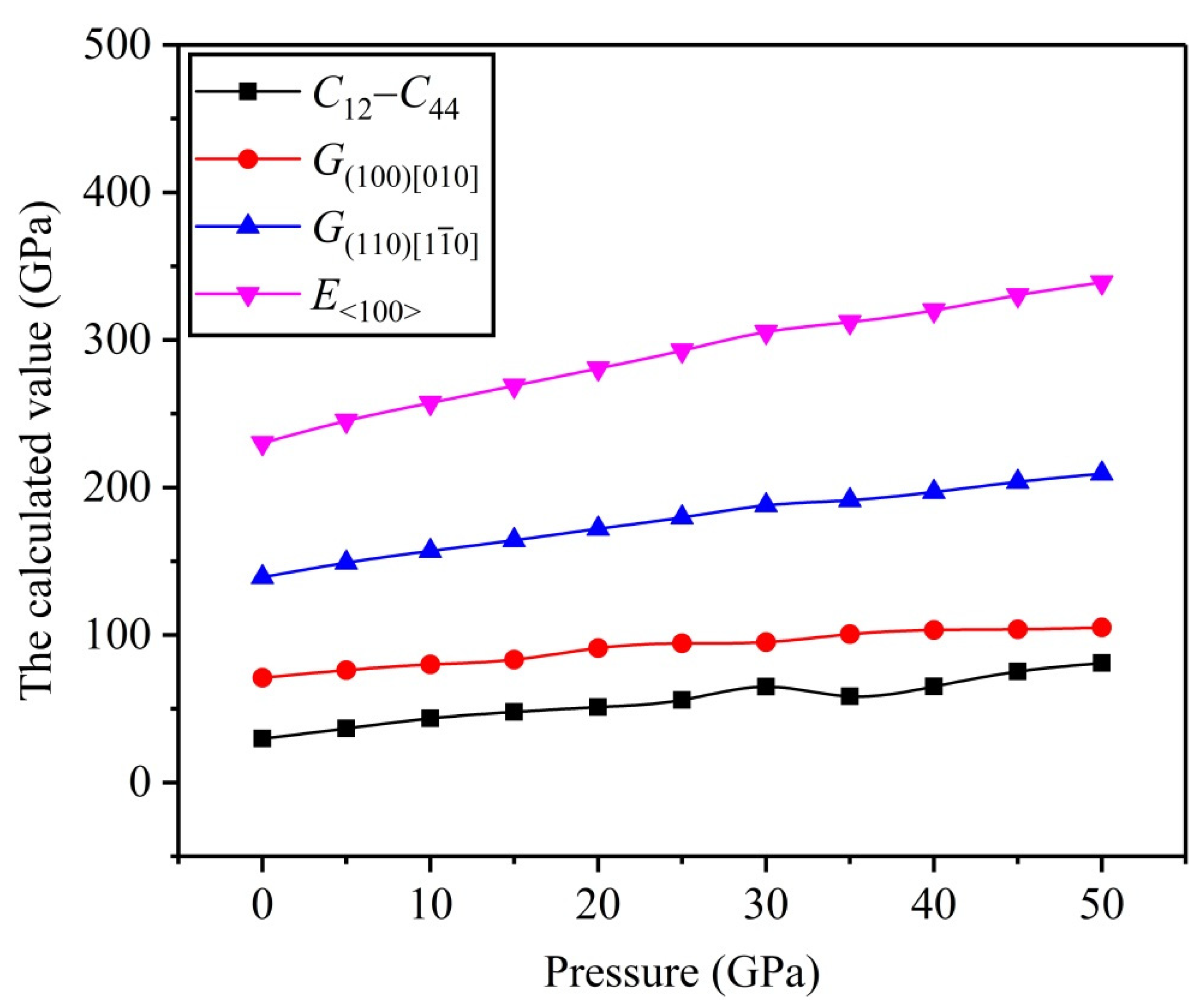
3.2. Mechanical Properties
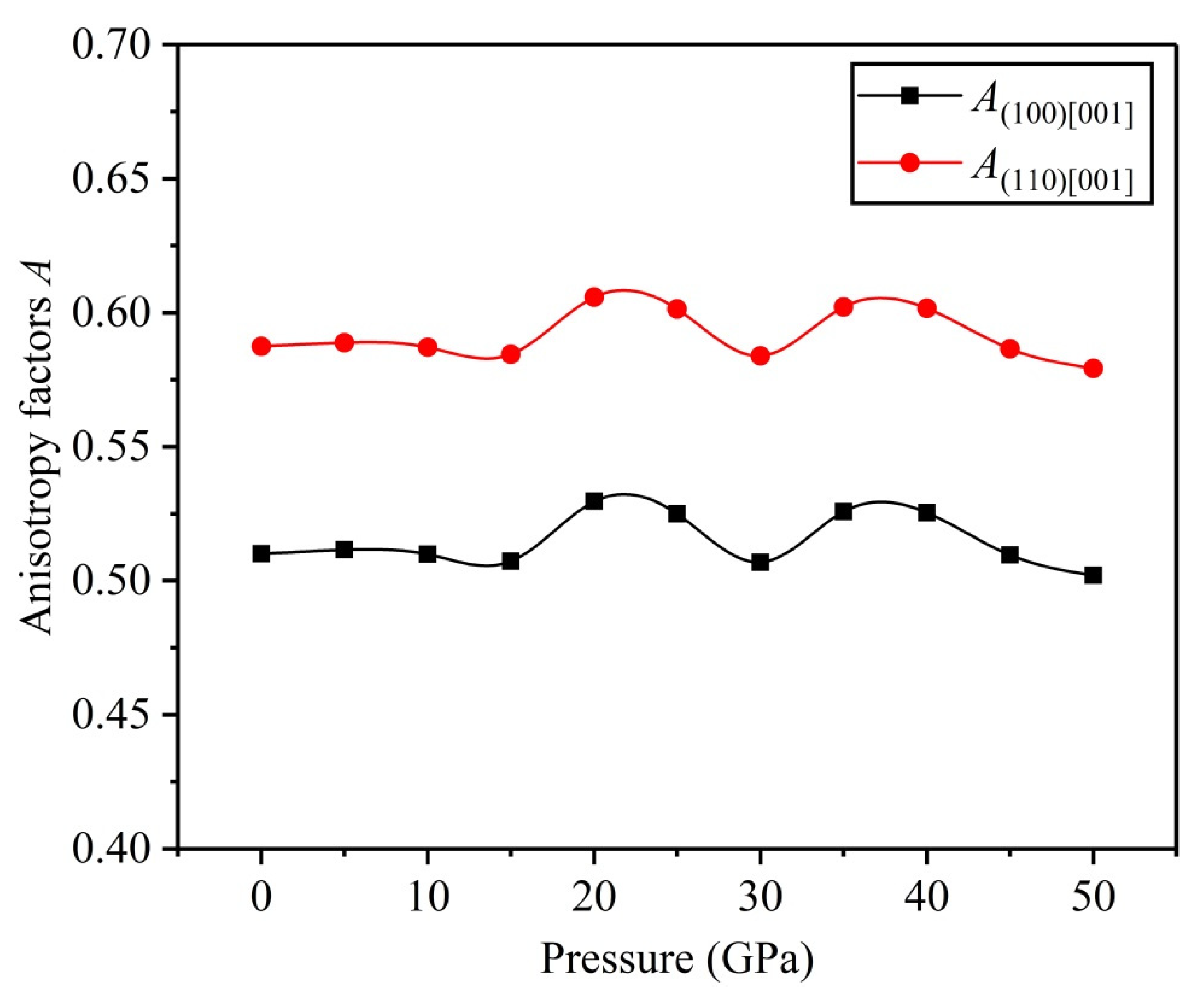
3.3. Anisotropy
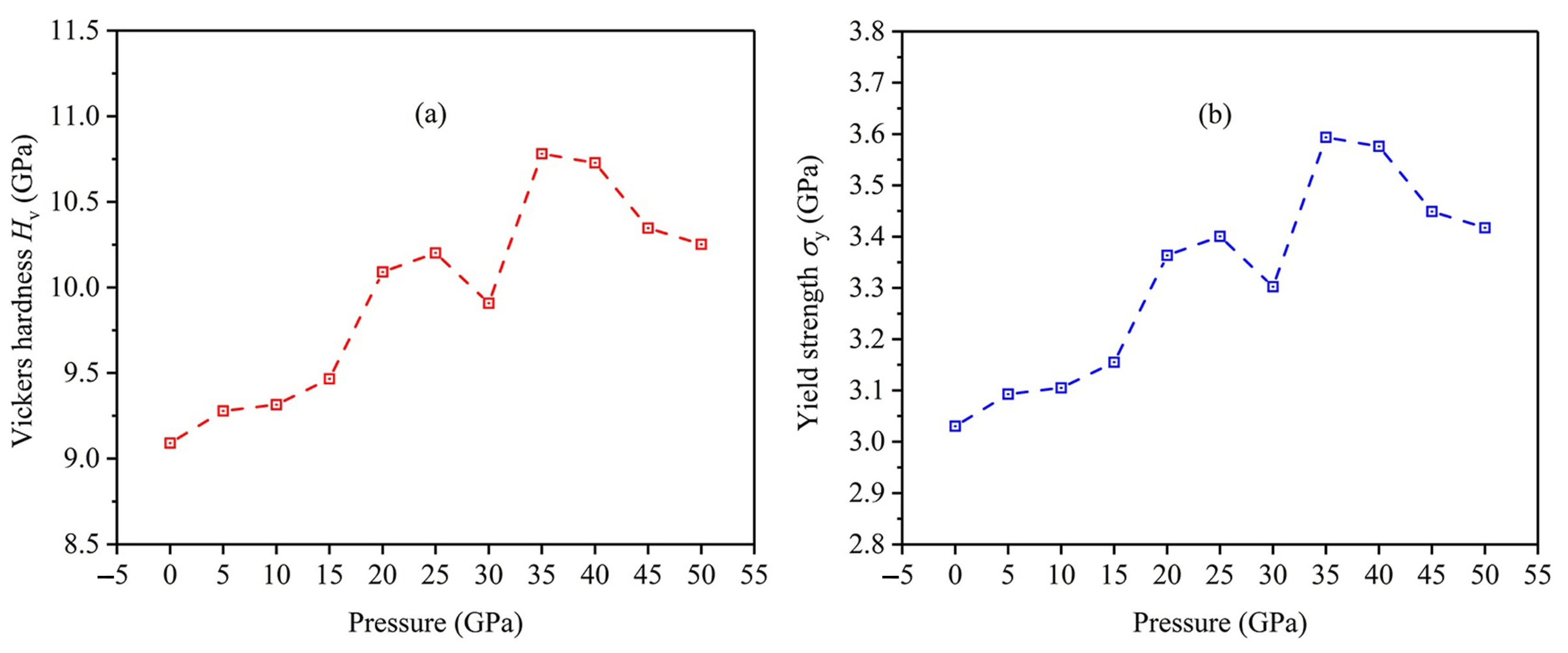
3.4. Hardness and Yield Strength
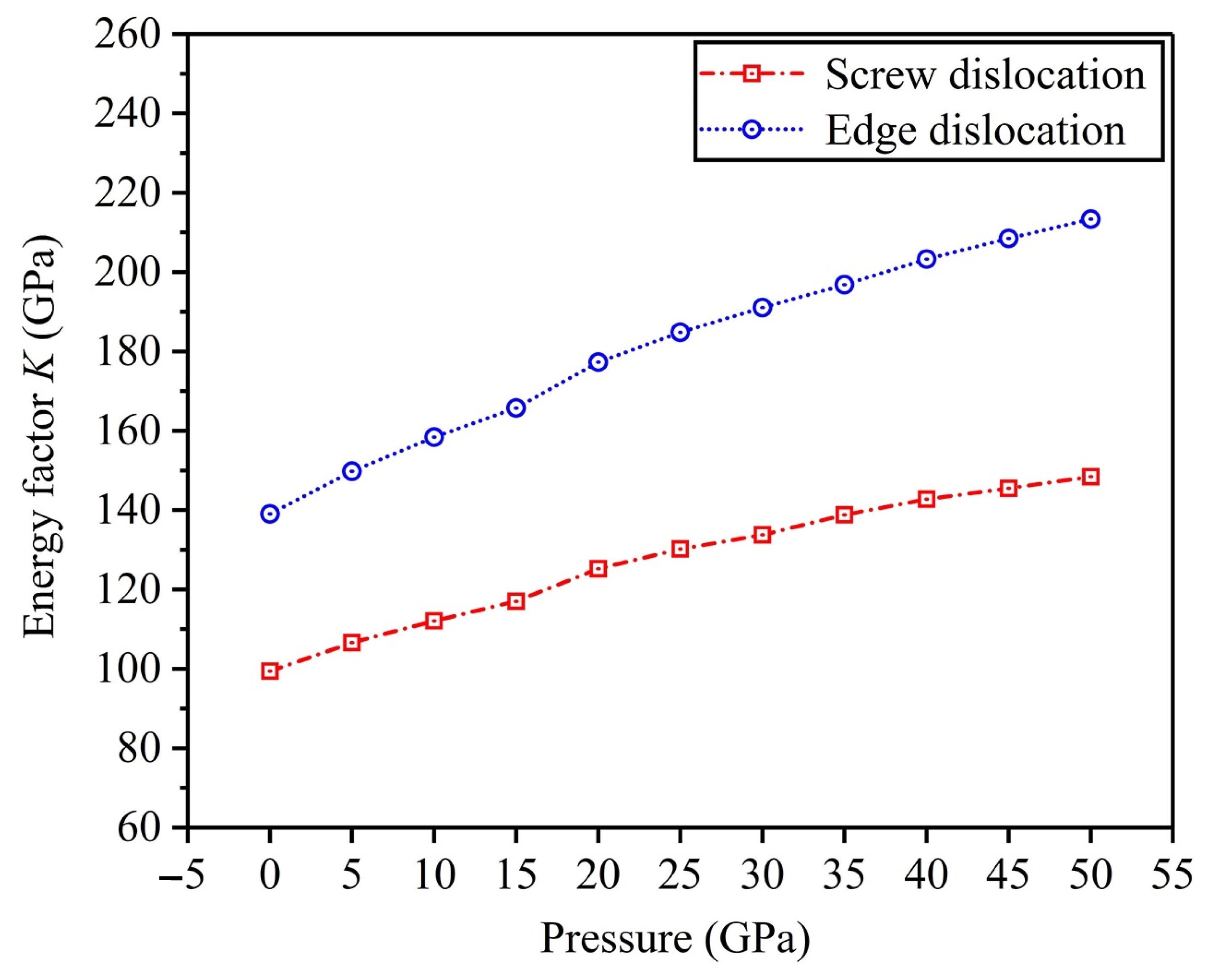
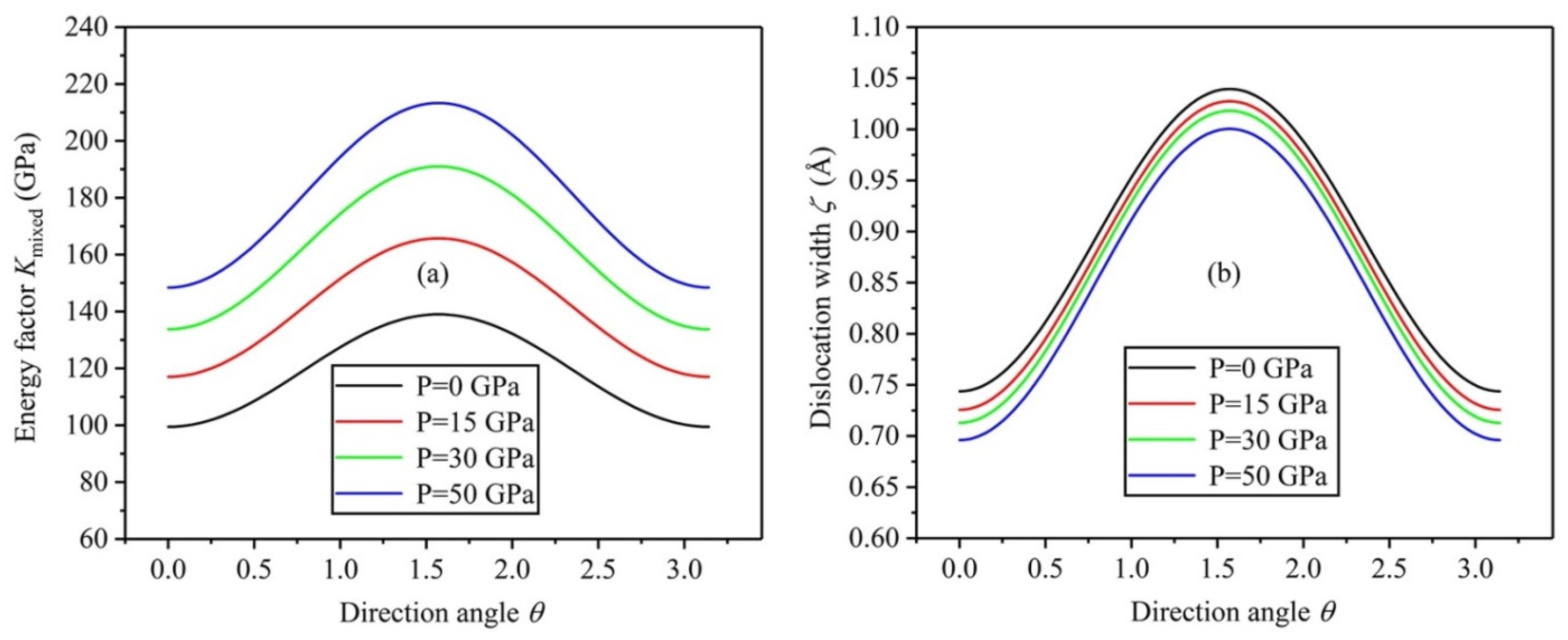
3.5. Energy Factor K
3.6. Electronic Properties
4. Conclusions
- (1)
- In the range of applied pressure (0–50 GPa), an FeTi alloy can exist stably and is not prone to producing structural phase transition.
- (2)
- With increasing pressure, the abilities to resist bulk and elastic deformations get stronger. An FeTi alloy is a kind of material with good ductility, and its ductility can be enhanced at high pressure.
- (3)
- Shear deformation in the direction is more difficult than that in the direction under the same pressure, and high pressure prevents elastic deformation in the direction. Cauchy pressure indicates that the metallic bond is predominant and gets stronger under high pressure.
- (4)
- High pressure can generally enhance the Vickers hardness and yield strength of an FeTi alloy, and the maximum values of Vickers hardness and yield strength occur at GPa.
- (5)
- High pressure inhibits dislocation nucleation and twinning deformation and then reduce the plasticity of FeTi hydrogen storage alloys. The edge dislocation is more difficult to nucleate with regard to screw dislocation.
- (6)
- The TDOS reveals that high pressure is inclined to promote the structural stability for FeTi hydrogen storage alloys instead of structural transition induced by high pressure.
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Abdin, Z.; Zafaranloo, A.; Rafiee, A.; Mérida, W.; Lipiński, W.; Khalilpour, K.R. Hydrogen as an energy vector. Renew. Sustain. Energy Rev. 2020, 120, 109620. [Google Scholar] [CrossRef]
- Salmon, N.; Bañares-Alcántara, R. Green ammonia as a spatial energy vector: A review. Sustain. Energy Fuels 2021, 5, 2814–2839. [Google Scholar] [CrossRef]
- Guilbert, D.; Vitale, G. Hydrogen as a Clean and Sustainable Energy Vector for Global Transition from Fossil-Based to Zero-Carbon. Clean Technol. 2021, 3, 881–909. [Google Scholar] [CrossRef]
- Shang, Y.Y.; Liu, S.F.; Liang, Z.D.; Pyczak, F.; Lei, Z.F.; Heidenreich, T.; Schkel, A.; Kai, J.J.; Gizer, G.; Dornheim, M. Developing sustainable FeTi alloys for hydrogen storage by recycling. Commun. Mater. 2022, 3, 101. [Google Scholar] [CrossRef]
- Padhee, S.P.; Roy, A.; Pati, S. Mechanistic insights into efficient reversible hydrogen storage in ferrotitanium. Int. J. Hydrogen Energy 2020, 46, 906–921. [Google Scholar] [CrossRef]
- Niu, J.Z.; Guo, Y.H.; Li, K.; Liu, W.J.; Zhou, L. Improved mechanical, bio-corrosion properties and in vitro cell responses of Ti-Fe alloys as candidate dental implants. Mater. Sci. Eng. C 2021, 122, 111917. [Google Scholar] [CrossRef]
- Liang, L.; Wang, F.; Rong, M.H.; Wang, Z.M.; Yang, S.T.; Wang, J.; Zhou, H.Y. Recent Advances on Preparation Method of Ti-Based Hydrogen Storage Alloy. J. Mater. Sci. Chem. Eng. 2020, 8, 18–38. [Google Scholar] [CrossRef]
- Ko, W.S.; Park, K.B.; Park, H.K. Density functional theory study on the role of ternary alloying elements in TiFe-based hydrogen storage alloys. J. Mater. Sci. Chem. Eng. 2021, 33, 148–158. [Google Scholar] [CrossRef]
- Liu, H.; Zhang, J.X.; Sun, P.; Zhou, C.S.; Liu, Y. An overview of TiFe alloys for hydrogen storage: Structure, processes, properties, and applications. J. Energy Storage 2023, 68, 107772. [Google Scholar] [CrossRef]
- Dematteis, E.M.; Berti, N.; Cuevas, F.; Latroche, M.; Baricco, M. Substitutional effects in TiFe for hydrogen storage: A comprehensive review. Mater. Adv. 2021, 2, 2524–2560. [Google Scholar] [CrossRef]
- Zhou, C.S.; Zhang, J.X.; Bowman, R.C.; Fang, Z.Z. Roles of Ti-Based Catalysts on Magnesium Hydride and Its Hydrogen Storage Properties. Inorganics 2021, 9, 36. [Google Scholar] [CrossRef]
- Padhee, S.P.; Roy, A.; Pati, S. Role of Mn-substitution towards the enhanced hydrogen storage performance in FeTi. Int. J. Hydrogen Energy 2022, 47, 9357–9371. [Google Scholar] [CrossRef]
- Alvares, E.; Jerabek, P.; Shang, Y.; Santhosh, A.; Pistidda, C.; Heo, T.W.; Sundman, B.; Dornheim, M. Modeling the thermodynamics of the FeTi hydrogenation under para-equilibrium: An ab-initio and experimental study. Calphad 2022, 77, 102426. [Google Scholar] [CrossRef]
- Nong, Z.; Zhu, J.; Yang, X.; Cao, Y.; Lai, Z.; Liu, Y. First-principles study of hydrogen storage and diffusion in B2 FeTi alloy. Comput. Mater. Sci. 2014, 81, 517–523. [Google Scholar] [CrossRef]
- Benyelloul, K.; Bouhadda, Y.; Bououdina, M.; Faraoun, H.I.; Aourag, H.; Seddik, L. The effect of hydrogen on the mechanical properties of FeTi for hydrogen storage applications. Int. J. Hydrogen Energy 2014, 39, 12667–12675. [Google Scholar] [CrossRef]
- Zhu, L.F.; Friák, M.; Udyansky, A.; Ma, D.; Neugebauer, J. Ab initio based study of finite-temperature structural, elastic and thermodynamic properties of FeTi. Intermetallics 2014, 45, 11–17. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projected augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Pack, J.D.; Monkhorst, H.J. Special Points for Brillonin-Zone Integrations. Phys. Rev. B 1976, 13, 5188. [Google Scholar]
- Birch, F. Finite elastic strain of cubic crystals. Phys. Rev. 1947, 71, 809. [Google Scholar] [CrossRef]
- Lv, P.; Peng, C.; Liu, Q.Y.; Zhong, C.L.; Huang, D.F.; Liu, Z.C.; Zhou, Q.B.; Zhao, R.X. Effect of regulating the different proportions of Zr to Mn elements on the hydrogen storage properties of titanium–iron–manganese–hydrogen storage alloys. RSC Adv. 2023, 13, 10157–10167. [Google Scholar] [CrossRef] [PubMed]
- Park, K.B.; Fadonougbo, J.O.; Park, C.S.; Lee, J.H.; Na, T.K.; Kang, H.S.; Ko, W.S.; Park, H.K. Effect of Fe substitution on first hydrogenation kinetics of TiFe-based hydrogen storage alloys after air exposure. Int. J. Hydrogen Energy 2021, 46, 30780–30789. [Google Scholar] [CrossRef]
- Nong, Z.; Zhu, J.; Yu, H.; Lai, Z. First principles calculation of intermetallic compounds in FeTiCoNiVCrMnCuAl system high entropy alloy. Trans. Nonferrous Met. Soc. 2012, 22, 1437–1444. [Google Scholar] [CrossRef]
- Patil, S.K.R.; Khare, S.V.; Tuttle, B.R.; Bording, J.K.; Kodambaka, S. Mechanical stability of possible structures of PtN investigated using first-principles calculations. Phys. Rev. B 2006, 73, 104118. [Google Scholar] [CrossRef]
- Wang, J.; Yip, S.; Phillpot, S.R.; Wolf, D. Crystal instabilities at finite strain. Phys. Rev. Lett. 1993, 71, 4182. [Google Scholar] [CrossRef]
- Wallace, D.C. Thermodynamics of Crystals. Am. J. Phys. 1972, 40, 1718. [Google Scholar] [CrossRef]
- Buchenau, U.; Schober, H.R.; Welter, J.M.; Arnold, G.; Wagner, R. Lattice dynamics of Fe0.5Ti0.5. Phys. Rev. B 1983, 27, 955–962. [Google Scholar] [CrossRef]
- Liebertz, J.; Stähr, S.; Haussühl, S. Growth and properties of single crystals of FeTi. Krist. Tech. 1980, 15, 1257–1260. [Google Scholar] [CrossRef]
- Cheng, D.; Zhao, S.; Wang, S.; Ye, H. First-principles study of the elastic properties and electronic structure of NiTi, CoTi and FeTi. Philos. Mag. A 2001, 81, 1625–1632. [Google Scholar] [CrossRef]
- Souissi, M.; Numakura, H. Elastic properties of Fe–C and Fe–N martensites. ISIJ Int. 2015, 55, 1512–1521. [Google Scholar] [CrossRef]
- Shang, S.L.; Wang, Y.; Liu, Z.K.; Ye, H. First-principles elastic constants of α- and θ-Al2O3. Appl. Phys. Lett. 2007, 90, 101909. [Google Scholar] [CrossRef]
- Ganeshan, S.; Shang, S.L.; Zhang, H.; Wang, Y.; Mantina, M.; Liu, Z.K. Elastic constants of binary Mg compounds from first-principles calculations. Intermetallics 2009, 17, 313–318. [Google Scholar] [CrossRef]
- Boucetta, S. Theoretical study of elastic, mechanical and thermodynamic properties of MgRh intermetallic compound. J. Magnes. Alloy 2014, 2, 59–63. [Google Scholar] [CrossRef]
- Yang, X.W.; Zhu, J.C.; Lai, Z.H.; Liu, Y.; He, D.; Nong, Z.S. Finite element analysis of quenching temperature field, residual stress and distortion in A357 aluminum alloy large complicated thin-wall workpieces. Trans. Nonferrous Met. Soc. 2013, 23, 1751–1760. [Google Scholar] [CrossRef]
- Lu, Y.; Yang, Y.; Zheng, F.; Wang, B.T.; Zhang, P. Electronic, mechanical, and thermodynamic properties of americium dioxide. J. Nucl. Mater. 2013, 441, 411–420. [Google Scholar] [CrossRef]
- Hill, R. The elastic behavior of crystalline aggregate. Proc. Phys. Soc. A 1952, 65, 349–354. [Google Scholar] [CrossRef]
- Iotova, D.; Kioussis, N.; Lim, S.P. Electronic structure and elastic properties of the Ni3X (X=Mn, Al, Ga, Si, Ge) intermetallics. Phys. Rev. B 1996, 54, 14413–14422. [Google Scholar] [CrossRef] [PubMed]
- Cottrel, A.H. Advanced Structural Materials. In Proceedings of the European Conference on Advanced Materials and Processes, Cambridge, UK, 22–24 July 1991. [Google Scholar]
- Fu, H.; Li, D.; Peng, F.; Gao, T.; Cheng, X. Structural and elastic properties of γTiAl under high pressure from electronic structure calculations. J. Alloys Compd. 2009, 473, 255–261. [Google Scholar] [CrossRef]
- Mattesini, M.; Ahuja, R.; Johansson, B. Cubic Hf3N4 and Zr3N4: A class of hard materials. Phys. Rev. B 2003, 68, 184108. [Google Scholar] [CrossRef]
- Yoo, M.H. On the theory of anomalous yield behavior of Ni3Al—Effect of elastic anisotropy. Scr. Metall. 1986, 20, 915–920. [Google Scholar] [CrossRef]
- Fu, H.; Li, X.F.; Liu, W.F.; Ma, Y.; Tao, G.; Hong, X. Electronic and dynamical properties of NiAl studied from first principles. Intermetallics 2011, 19, 1959–1967. [Google Scholar] [CrossRef]
- Lau, K.; McCurdy, A.K. Elastic anisotropy factors for orthorhombic, tetragonal, and hexagonal crystals. Phys. Rev. B 1998, 58, 8980–8984. [Google Scholar] [CrossRef]
- Reed, R.P.; Clark, A.F. Materials at Low Temperatures; American Society for Metals: Russell, OH, USA, 1983. [Google Scholar]
- Friák, M.; Šob, M.; Vitek, V. Ab initio calculation of tensile strength in iron. Philos. Mag. 2003, 83, 3529–3537. [Google Scholar] [CrossRef]
- Fu, H.; Peng, W.; Gao, T. Structural and elastic properties of ZrC under high pressure. Mater. Chem. Phys. 2009, 115, 789–794. [Google Scholar] [CrossRef]
- Fu, H.; Li, D.; Peng, F.; Gao, T.; Cheng, X. Ab initio calculations of elastic constants and thermodynamic properties of NiAl under high pressures. Comput. Mater. Sci. 2008, 44, 774–778. [Google Scholar] [CrossRef]
- Pettifor, D.G. Theoretical predictions of structure and related properties of intermetallics. Mater. Sci. Tech. 1992, 8, 345–349. [Google Scholar] [CrossRef]
- Johnson, R.A. Analytic nearest-neighbor model for fcc metals. Phys. Rev. B 1988, 37, 3924–3931. [Google Scholar] [CrossRef]
- Tse, J.S. Intrinsic hardness of crystalline solids. J. Superhard Mater. 2010, 32, 177–191. [Google Scholar] [CrossRef]
- Smedskjaer, M.M.; Mauro, J.C.; Yue, Y. Prediction of glass hardness using temperature-dependent constraint theory. Phys. Rev. Lett. 2010, 105, 115503. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Wang, X.; Zhang, F.; Xue, D. Electronegativity identification of novel superhard materials. Phys. Rev. Lett. 2008, 100, 235504. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; He, J.; Wu, E.; Liu, S.; Yu, D.; Li, D.; Zhang, S.; Tian, Y. Hardness of covalent crystals. Phys. Rev. Lett. 2003, 91, 015502. [Google Scholar] [CrossRef] [PubMed]
- Šimůnek, A.; Vackář, J. Hardness of Covalent and Ionic Crystals: First-Principle Calculations. Phys. Rev. Lett. 2006, 96, 085501. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Niu, H.; Li, D.; Li, Y. Modeling hardness of polycrystalline materials and bulk metallic glasses. Intermetallics 2011, 19, 1275–1281. [Google Scholar] [CrossRef]
- Savin, M.M.; Chernov, V.M.; Strokova, A.M. Energy factor of dislocations in hexagonal crystals. Phys. Status Solidi A 1976, 35, 747–754. [Google Scholar] [CrossRef]
- Foreman, A.J.E. Dislocation energies in anisotropic crystals. Acta Metall. 1955, 3, 322–330. [Google Scholar] [CrossRef]
- Reid, C.N. Dislocation widths in anisotropic B.C.C. crystals. Acta Metall. 1966, 14, 13–16. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| FeTi | Present | Experiments | Theoretical Data |
|---|---|---|---|
| Lattice parameter | 2.945 | 2.978 [23], 2.972 [24] | 2.943 [15], 2.953 [14], 2.95 [16] 2.96 [5], 2.97 [25] |
| FeTi | Present | Experiments | Theoretical Calculations |
|---|---|---|---|
| C11 | 379.44 | 310 [30], 325 [29] | 304 [31], 351.70 [14], 384.74 [15], 385 [16] |
| C12 | 100.89 | 86.2 [30], 121 [29] | 136 [31], 86.82 [14], 102.42 [15], 95 [16] |
| C44 | 71.04 | 74.9 [30], 69 [29] | 138 [31], 73.08 [14], 66.96 [15], 72 [16] |
| B | 193.74 | 160.8 [30], 189 [29] | 175.11 [14], 196.52 [15], 191.66 [16] |
| E | 241.28 | 223 [30], 213 [29] | 236.88 [14], 235.84 [15], 244.58 [16] |
| G | 93.34 | 88 [30], 81 [29] | 92.93 [14], 90.71 [15], 95 [16] |
| σ | 0.21 | 0.26 [30], 0.31 [29] | 0.27 [14], 0.29 [15], 0.28 [16] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, L.; Chen, H.; Wang, Z.; Ma, L.; Tang, P. Theoretical Predictions of Structure, Mechanics, Dislocation, and Electronics Properties of FeTi Alloy at High Pressure. Metals 2023, 13, 1547. https://doi.org/10.3390/met13091547
Zhang L, Chen H, Wang Z, Ma L, Tang P. Theoretical Predictions of Structure, Mechanics, Dislocation, and Electronics Properties of FeTi Alloy at High Pressure. Metals. 2023; 13(9):1547. https://doi.org/10.3390/met13091547
Chicago/Turabian StyleZhang, Linkun, Hong Chen, Zhipeng Wang, Li Ma, and Pingying Tang. 2023. "Theoretical Predictions of Structure, Mechanics, Dislocation, and Electronics Properties of FeTi Alloy at High Pressure" Metals 13, no. 9: 1547. https://doi.org/10.3390/met13091547