
A First-Principles Study of the Structural and Thermo-Mechanical Properties of Tungsten-Based Plasma-Facing Materials


Abstract
1. Introduction
2. Computational Methods
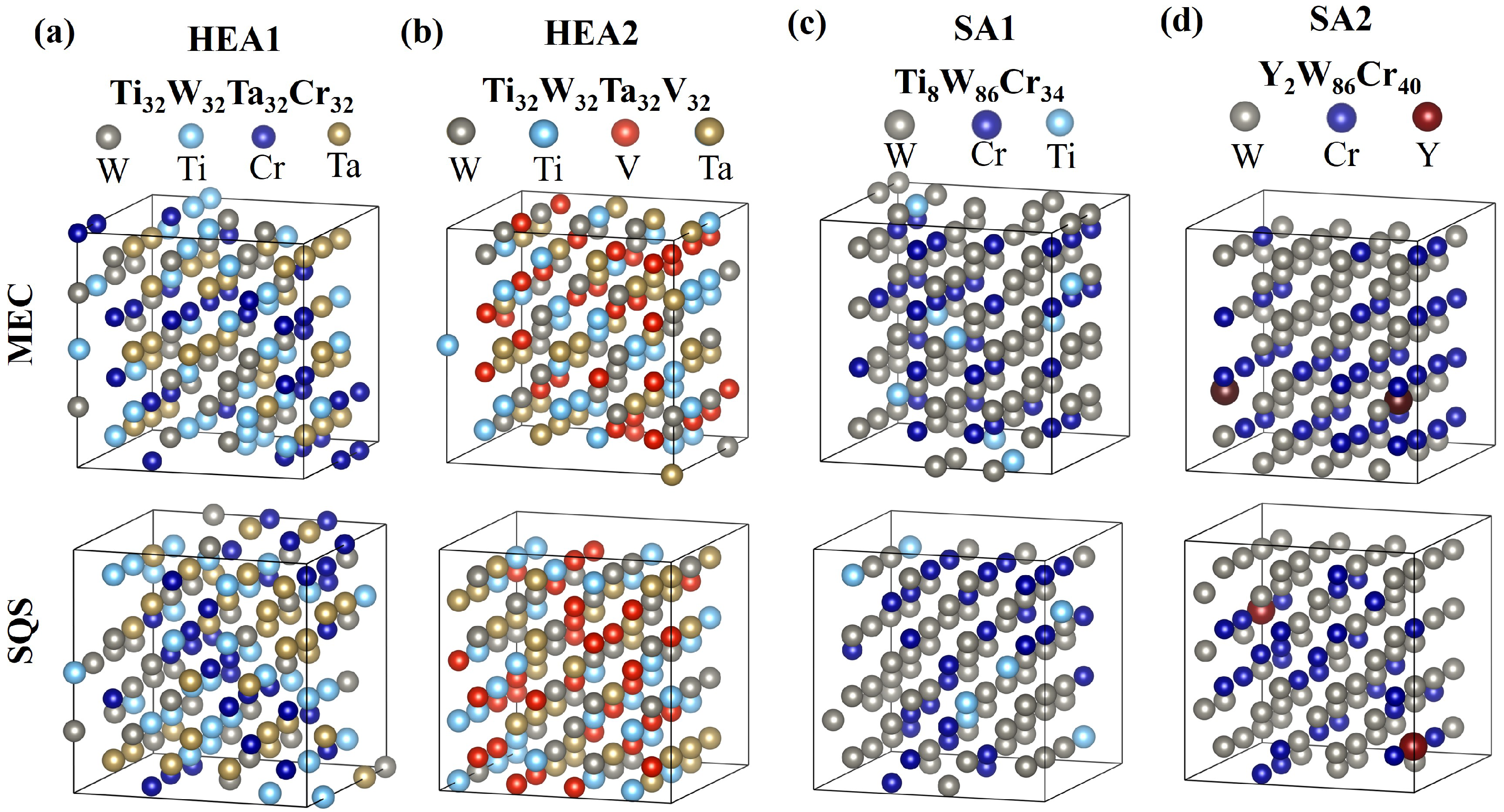
2.1. Generation of Random and Minimum-Energy Configurations
2.2. Quasiharmonic Approximation
2.3. Elastic Properties
3. Results
3.1. Structural Characteristics of the Lattice Configurations
3.2. Phonons
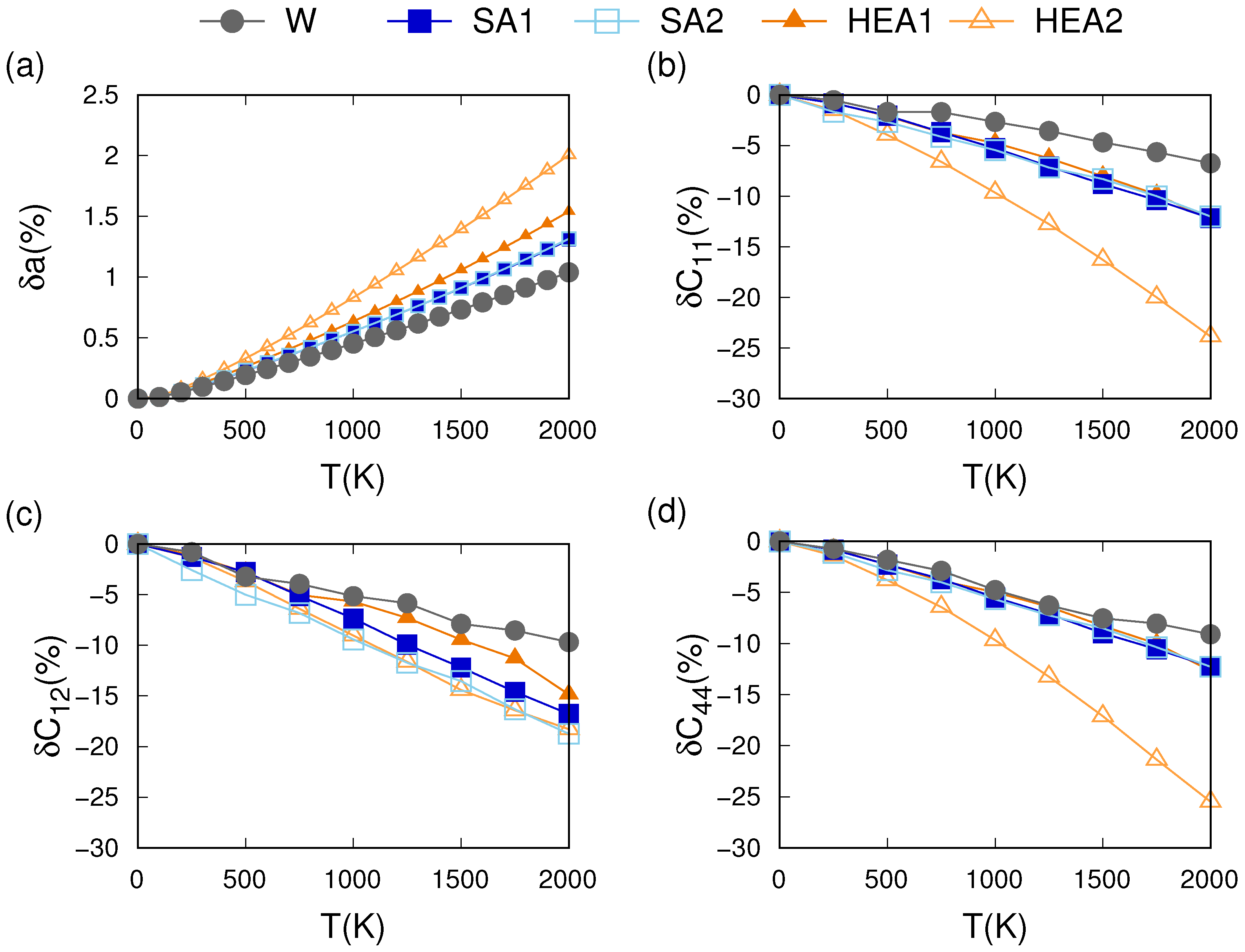
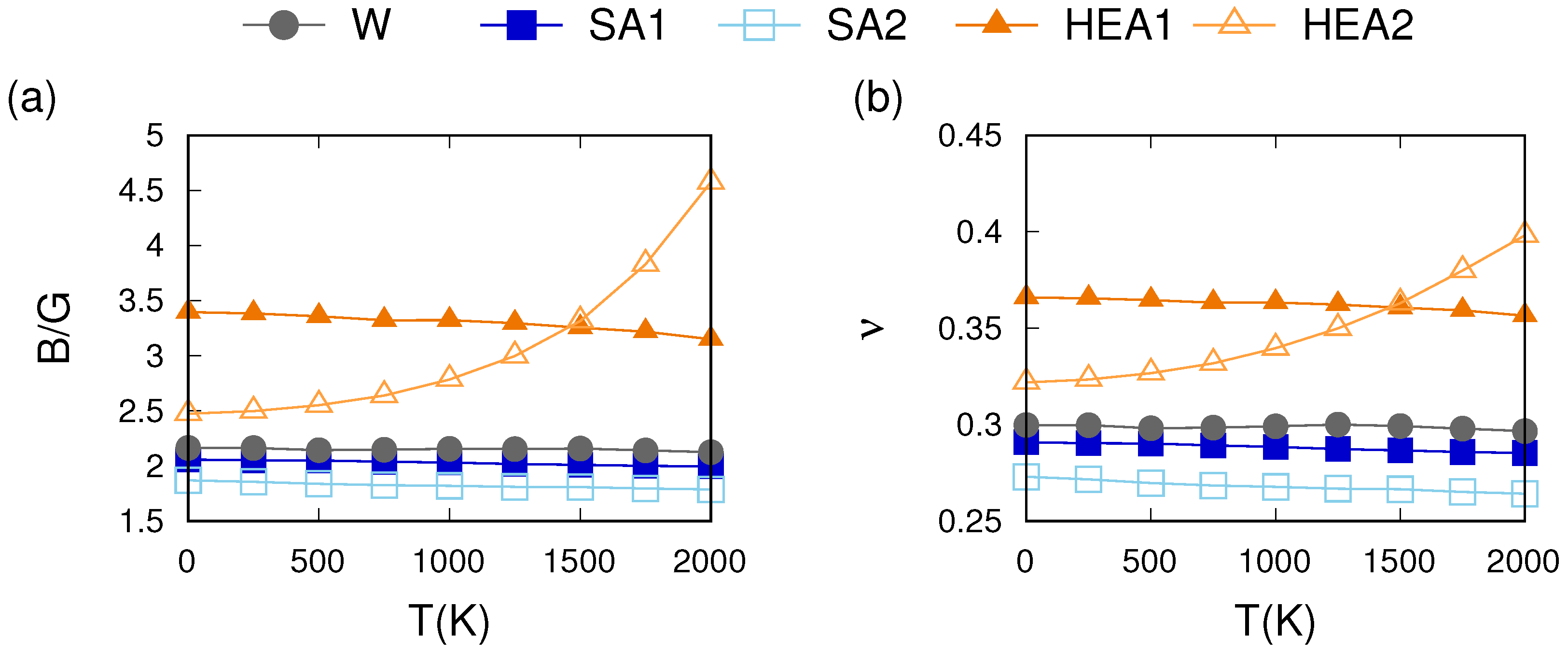
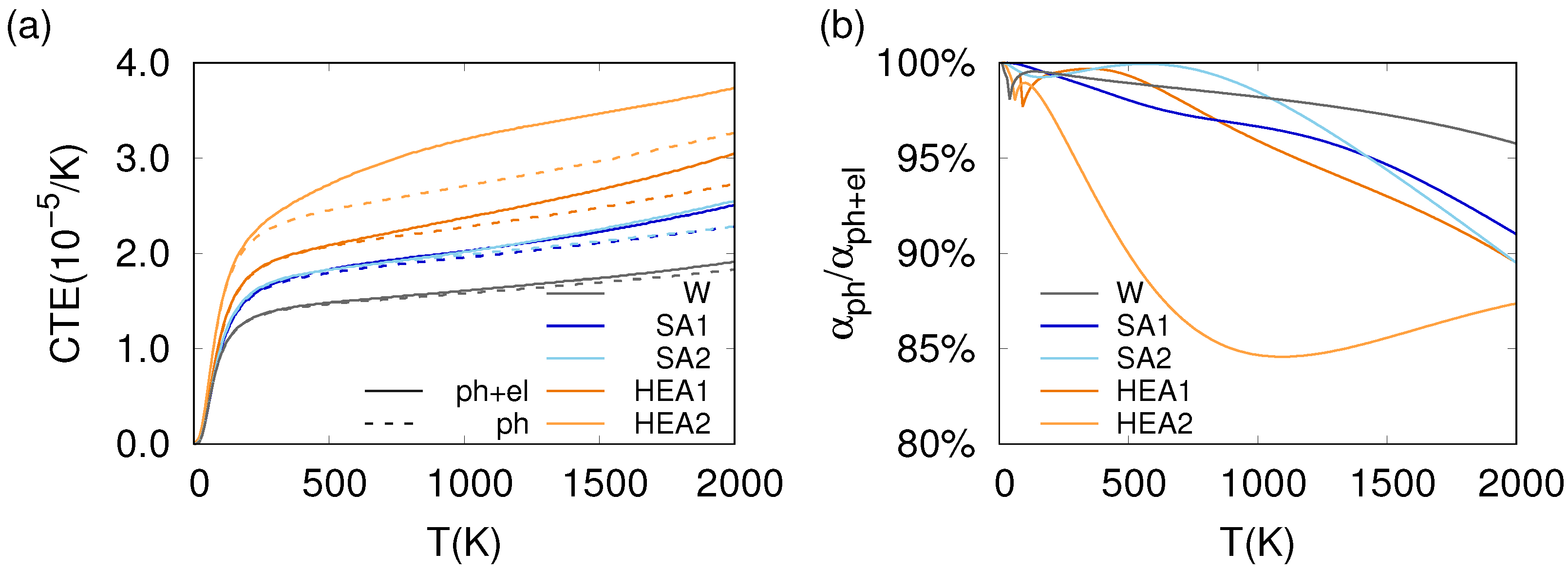
3.3. Thermo-Mechanical Behavior
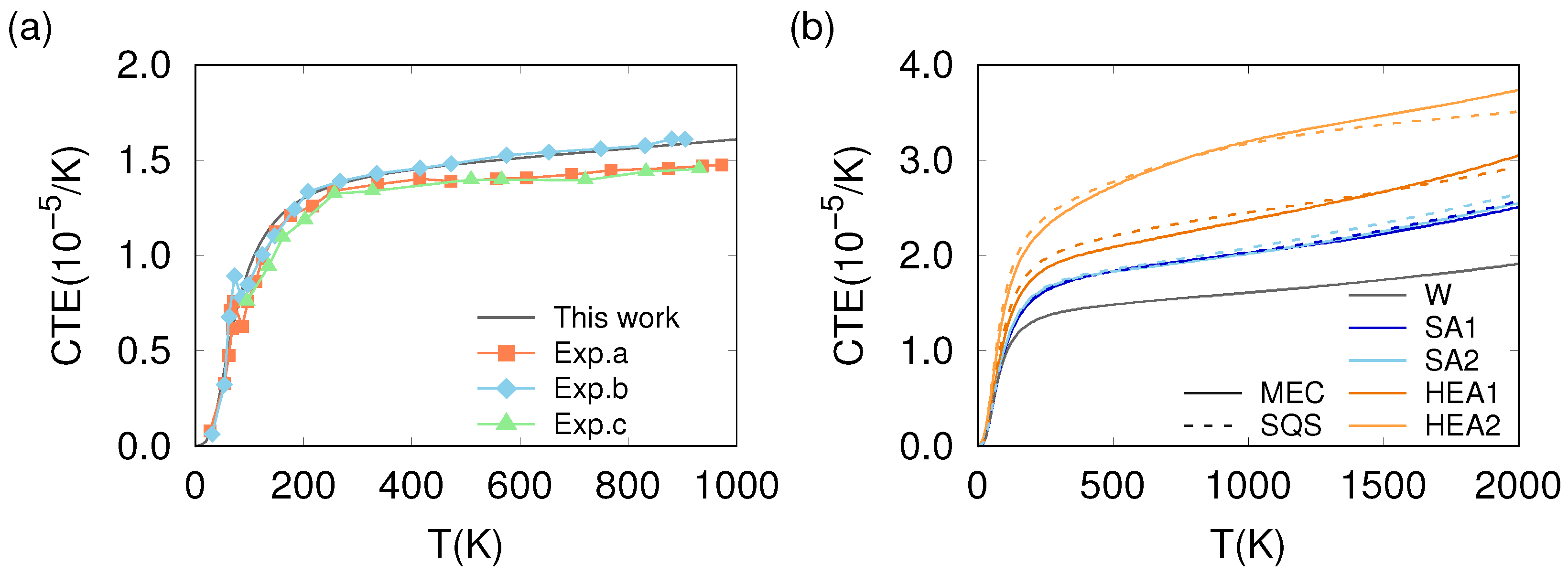
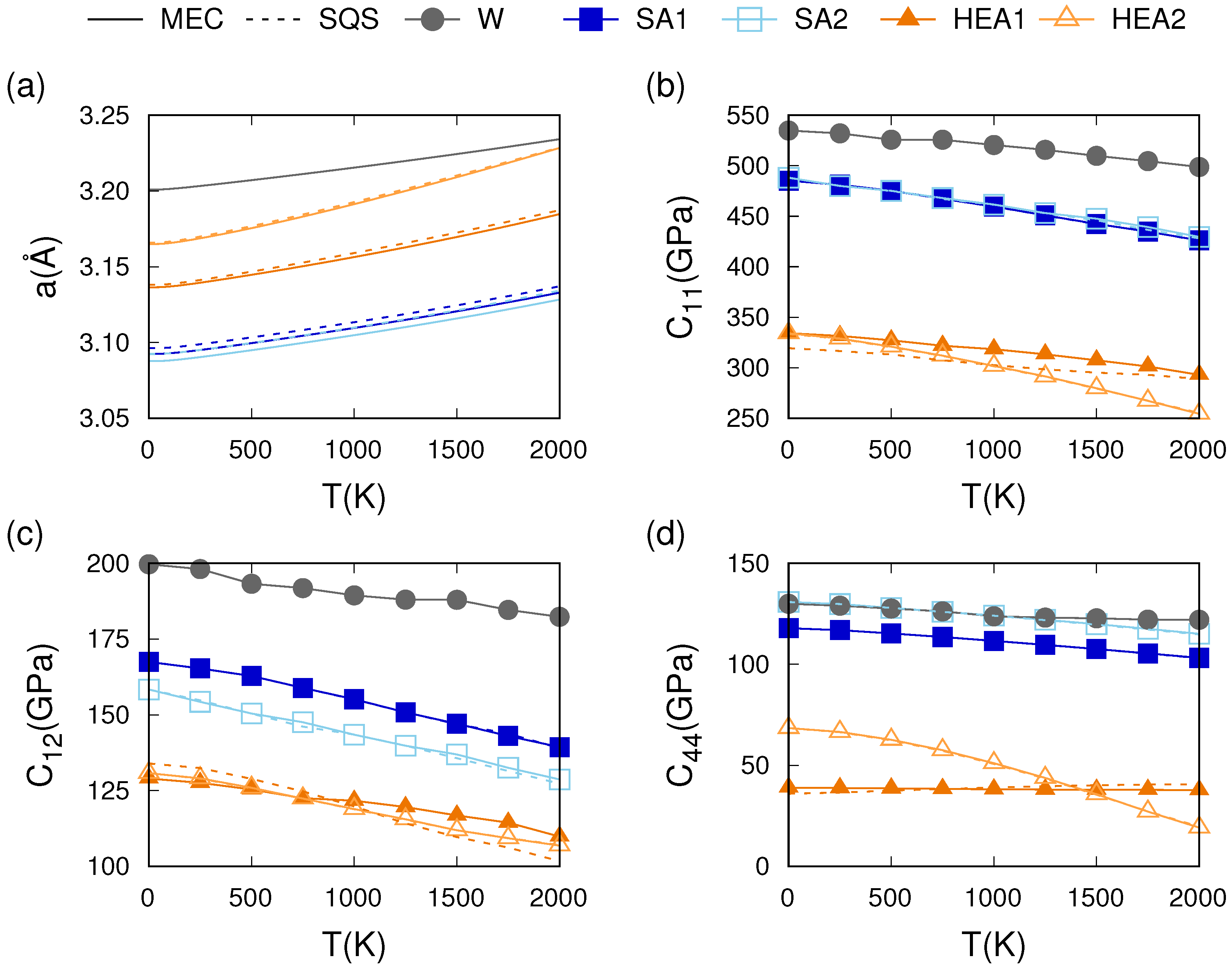
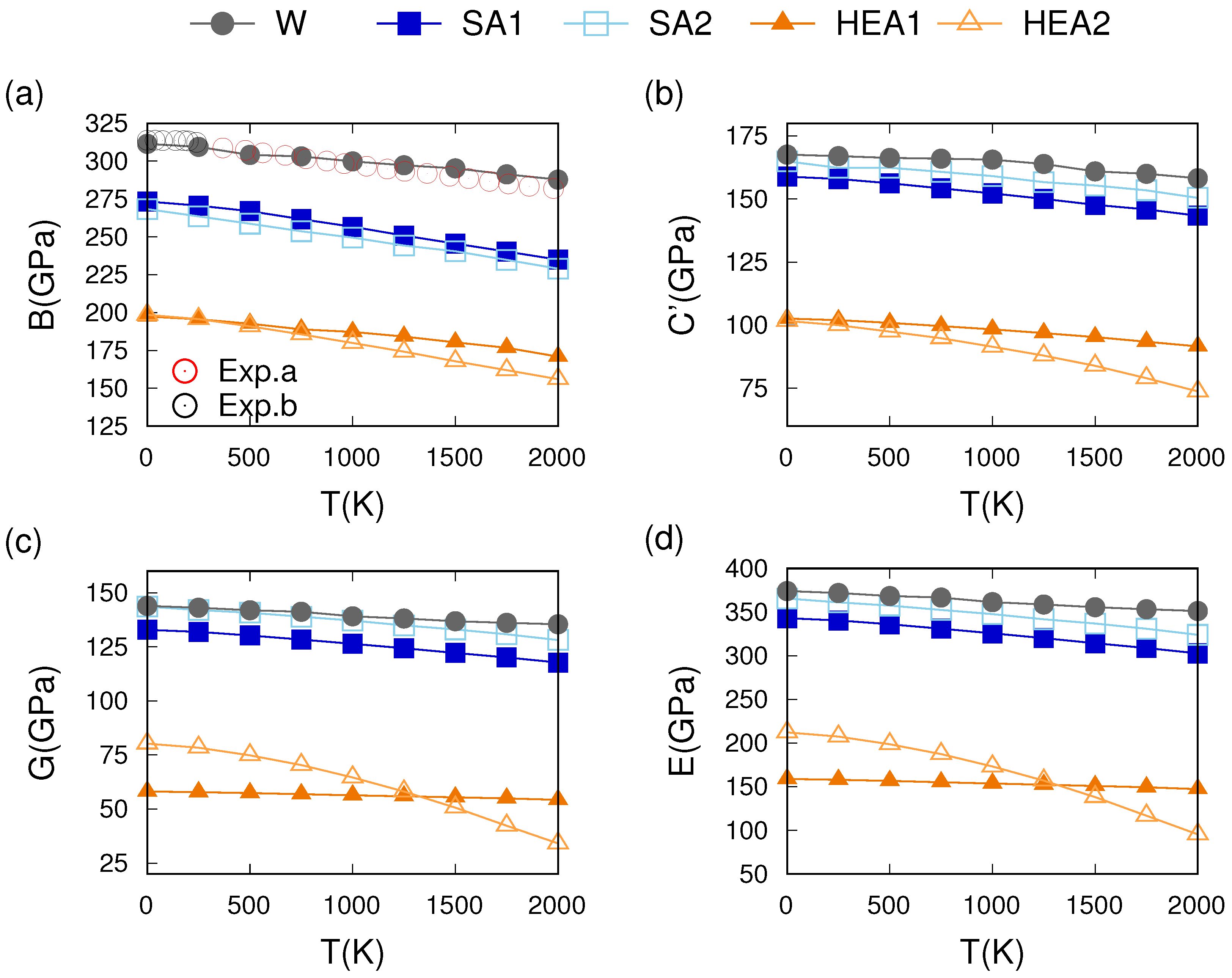
4. Discussion
4.1. Effects of the Chemical Composition
4.2. Phonon and Electron Contributions to the Thermo-Mechanical Behavior
4.3. Impact of Phonons on the Thermo-Mechanical Properties
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A. Thermo-Mechanical Response of SQS Configurations
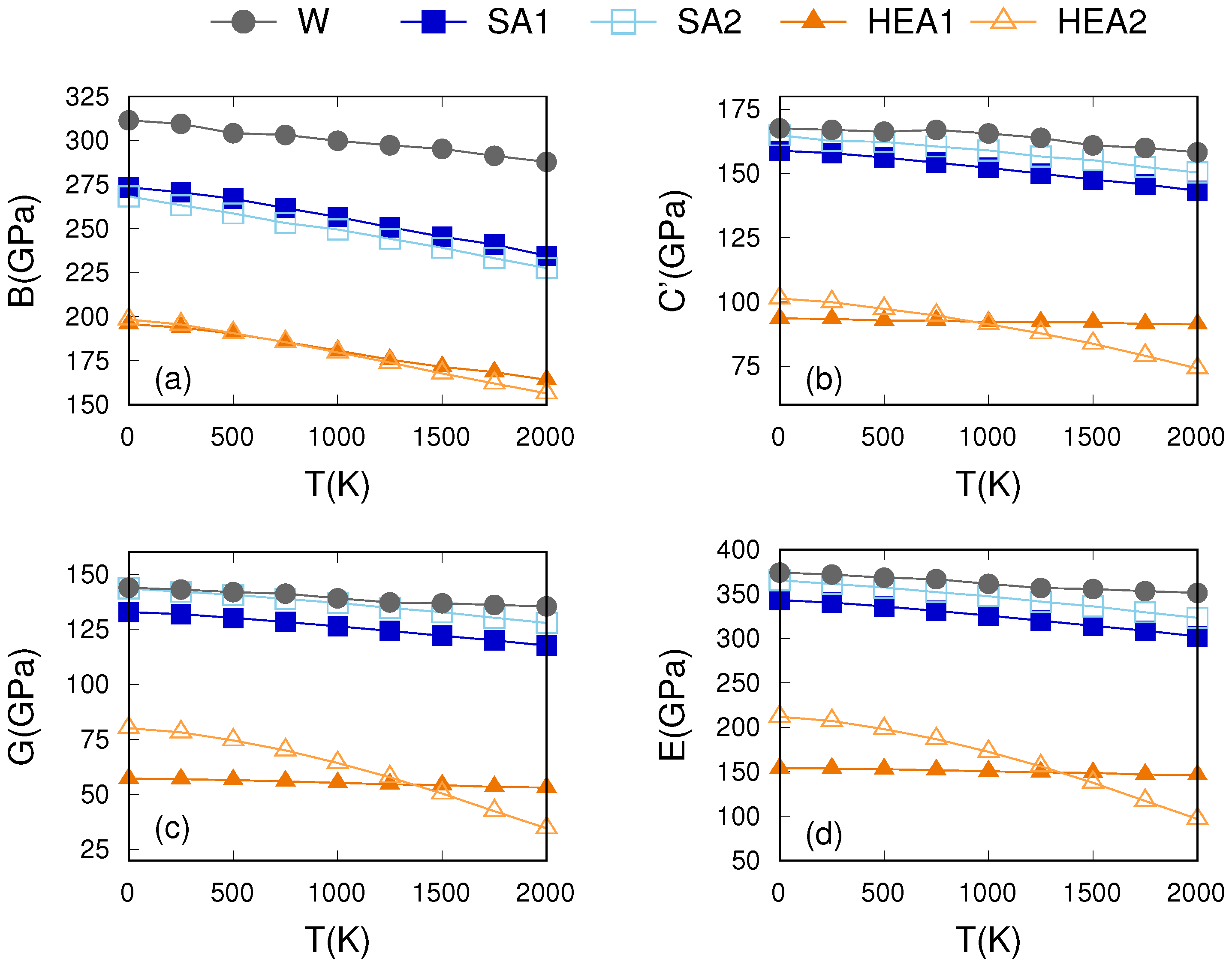
References
- Zinkle, S.J.; Ghoniem, N.M. Prospects for accelerated development of high performance structural materials. J. Nucl. Mater. 2011, 417, 2–8. [Google Scholar] [CrossRef]
- Rieth, M.; Dudarev, S.; Gonzalez de Vicente, S.; Aktaa, J.; Ahlgren, T.; Antusch, S.; Armstrong, D.; Balden, M.; Baluc, N.; Barthe, M.F.; et al. Recent progress in research on tungsten materials for nuclear fusion applications in Europe. J. Nucl. Mater. 2013, 432, 482–500. [Google Scholar] [CrossRef]
- Lassner, E.; Wolf-Dieter, S. Tungsten: Properties, Chemistry, Technology of the Element, Alloys, and Chemical Compounds; Springer: New York, NY, USA, 1999. [Google Scholar]
- Bolt, H.; Barabash, V.; Krauss, W.; Linke, J.; Neu, R.; Suzuki, S.; Yoshida, N.; Team, A.U. Materials for the plasma-facing components of fusion reactors. J. Nucl. Mater. 2004, 329, 66–73. [Google Scholar] [CrossRef]
- Giannattasio, A.; Roberts, S.G. Strain-rate dependence of the brittle-to-ductile transition temperature in tungsten. Philos. Mag. 2007, 87, 2589–2598. [Google Scholar] [CrossRef]
- Yeh, J.W.; Chen, S.K.; Lin, S.J.; Gan, J.Y.; Chin, T.S.; Shun, T.T.; Tsau, C.H.; Chang, S.Y. Nanostructured high-entropy alloys with multiple principal elements: Novel alloy design concepts and outcomes. Adv. Eng. Mater. 2004, 6, 299–303. [Google Scholar] [CrossRef]
- Tsai, M.H.; Yeh, J.W. High-entropy alloys: A critical review. Mater. Res. Lett. 2014, 2, 107–123. [Google Scholar] [CrossRef]
- Li, Z.; Pradeep, K.G.; Deng, Y.; Raabe, D.; Tasan, C.C. Metastable high-entropy dual-phase alloys overcome the strength–ductility trade-off. Nature 2016, 534, 227–230. [Google Scholar] [CrossRef]
- George, E.P.; Raabe, D.; Ritchie, R.O. High-entropy alloys. Nat. Rev. Mater. 2019, 4, 515–534. [Google Scholar] [CrossRef]
- Murty, B.S.; Yeh, J.W.; Ranganathan, S.; Bhattacharjee, P. High-Entropy Alloys; Elsevier: Amsterdam, The Netherlands, 2019. [Google Scholar]
- El-Atwani, O.; Li, N.; Li, M.; Devaraj, A.; Baldwin, J.; Schneider, M.M.; Sobieraj, D.; Wróbel, J.S.; Nguyen-Manh, D.; Maloy, S.A.; et al. Outstanding radiation resistance of tungsten-based high-entropy alloys. Sci. Adv. 2019, 5, eaav2002. [Google Scholar] [CrossRef]
- Waseem, O.A.; Ryu, H.J. Powder metallurgy processing of a WxTaTiVCr high-entropy alloy and its derivative alloys for fusion material applications. Sci. Rep. 2017, 7, 1926. [Google Scholar] [CrossRef]
- Zou, Y.; Ma, H.; Spolenak, R. Ultrastrong ductile and stable high-entropy alloys at small scales. Nat. Commun. 2015, 6, 7748. [Google Scholar] [CrossRef] [PubMed]
- Senkov, O.N.; Miracle, D.B.; Chaput, K.J.; Couzinie, J.P. Development and exploration of refractory high entropy alloys—A review. J. Mater. Res. 2018, 33, 3092–3128. [Google Scholar] [CrossRef]
- El-Atwani, O.; Alvarado, A.; Unal, K.; Fensin, S.; Hinks, J.; Greaves, G.; Baldwin, J.; Maloy, S.; Martinez, E. Helium implantation damage resistance in nanocrystalline W-Ta-V-Cr high entropy alloys. Mater. Today Energy 2021, 19, 100599. [Google Scholar] [CrossRef]
- Senkov, O.N.; Wilks, G.B.; Scott, J.M.; Miracle, D.B. Mechanical properties of Nb25Mo25Ta25W25 and V20Nb20Mo20Ta20W20 refractory high entropy alloys. Intermetallics 2011, 19, 698–706. [Google Scholar] [CrossRef]
- Litnovsky, A.; Klein, F.; Tan, X.; Ertmer, J.; Coenen, J.; Linsmeier, C.; Gonzalez-Julian, J.; Bram, M.; Povstugar, I.; Morgan, T.; et al. Advanced Self-Passivating Alloys for an Application under Extreme Conditions. Metals 2021, 11, 1255. [Google Scholar] [CrossRef]
- Sobieraj, D.; Wróbel, J.S.; Gilbert, M.R.; Litnovsky, A.; Klein, F.; Kurzydłowski, K.J.; Nguyen-Manh, D. Composition stability and Cr-rich phase formation in W-Cr-Y and W-Cr-Ti smart alloys. Metals 2021, 11, 743. [Google Scholar] [CrossRef]
- Litnovsky, A.; Wegener, T.; Klein, F.; Linsmeier, C.; Rasinski, M.; Kreter, A.; Unterberg, B.; Vogel, M.; Kraus, S.; Breuer, U.; et al. Smart alloys for a future fusion power plant: First studies under stationary plasma load and in accidental conditions. Nucl. Mater. Energy 2017, 12, 1363–1367. [Google Scholar] [CrossRef]
- Klein, F.; Gilbert, M.R.; Litnovsky, A.; Gonzalez-Julian, J.; Weckauf, S.; Wegener, T.; Schmitz, J.; Linsmeier, C.; Bram, M.; Coenen, J.W. Tungsten–chromium–yttrium alloys as first wall armor material: Yttrium concentration, oxygen content and transmutation elements. Fusion Eng. Des. 2020, 158, 111667. [Google Scholar] [CrossRef]
- Koch, F.; Bolt, H. Self passivating W-based alloys as plasma facing material for nuclear fusion. Phys. Scr. 2007, 2007, 100. [Google Scholar] [CrossRef]
- Muzyk, M.; Nguyen-Manh, D.; Kurzydłowski, K.J.; Baluc, N.L.; Dudarev, S.L. Phase stability, point defects, and elastic properties of W-V and W-Ta alloys. Phys. Rev. B 2011, 84, 104115. [Google Scholar] [CrossRef]
- Muzyk, M.; Nguyen-Manh, D.; Wróbel, J.; Kurzydłowski, K.; Baluc, N.; Dudarev, S. First-principles model for phase stability, radiation defects and elastic properties Of W–Ta and W–V alloys. J. Nucl. Mater. 2013, 442, S680–S683. [Google Scholar] [CrossRef]
- Wei, N.; Jia, T.; Zhang, X.; Liu, T.; Zeng, Z.; Yang, X. First-principles study of the phase stability and the mechanical properties of W-Ta and W-Re alloys. AIP Adv. 2014, 4, 057103. [Google Scholar] [CrossRef]
- Yang, C.; Qi, L. Ab initio calculations of ideal strength and lattice instability in W-Ta and W-Re alloys. Phys. Rev. B 2018, 97, 014107. [Google Scholar] [CrossRef]
- Huang, C.H.; Gharaee, L.; Zhao, Y.; Erhart, P.; Marian, J. Mechanism of nucleation and incipient growth of Re clusters in irradiated W-Re alloys from kinetic Monte Carlo simulations. Phys. Rev. B 2017, 96, 094108. [Google Scholar] [CrossRef]
- Jiang, D.; Ouyang, C.; Liu, S. Mechanical properties of W–Ti alloys from first-principles calculations. Fusion Eng. Des. 2016, 106, 34–39. [Google Scholar]
- Li, X.; Schönecker, S.; Li, R.; Li, X.; Wang, Y.; Zhao, J.; Johansson, B.; Vitos, L. Ab initio calculations of mechanical properties of bcc W–Re–Os random alloys: Effects of transmutation of W. J. Phys. Condens. Matter 2016, 28, 295501. [Google Scholar] [CrossRef]
- Hu, Y.J.; Shang, S.L.; Wang, Y.; Darling, K.A.; Butler, B.G.; Kecskes, L.J.; Liu, Z.K. Effects of alloying elements and temperature on the elastic properties of W-based alloys by first-principles calculations. J. Alloys Compd. 2016, 671, 267–275. [Google Scholar] [CrossRef]
- Giusepponi, S.; Celino, M. The ideal tensile strength of tungsten and tungsten alloys by first-principles calculations. J. Nucl. Mater. 2013, 435, 52–55. [Google Scholar] [CrossRef]
- Qian, J.; Wu, C.; Fan, J.; Gong, H. Effect of alloying elements on stacking fault energy and ductility of tungsten. J. Alloys Compd. 2018, 737, 372–376. [Google Scholar] [CrossRef]
- Nguyen-Manh, D.; Wróbel, J.S.; Klimenkov, M.; Lloyd, M.J.; Messina, L.; Dudarev, S.L. First-principles model for voids decorated by transmutation solutes: Short-range order effects and application to neutron irradiated tungsten. Phys. Rev. Mater. 2021, 5, 065401. [Google Scholar] [CrossRef]
- Suzudo, T.; Yamaguchi, M.; Hasegawa, A. Stability and mobility of rhenium and osmium in tungsten: First principles study. Model. Simul. Mater. Sci. Eng. 2014, 22, 075006. [Google Scholar] [CrossRef]
- Linke, J.; Du, J.; Loewenhoff, T.; Pintsuk, G.; Spilker, B.; Steudel, I.; Wirtz, M. Challenges for plasma-facing components in nuclear fusion. Matter Radiat. Extrem. 2019, 4. [Google Scholar] [CrossRef]
- Hossain, M.; Marian, J. Stress-dependent solute energetics in W–Re alloys from first-principles calculations. Acta Mater. 2014, 80, 107–117. [Google Scholar] [CrossRef]
- Gharaee, L.; Marian, J.; Erhart, P. The role of interstitial binding in radiation induced segregation in W-Re alloys. J. Appl. Phys. 2016, 120, 025901. [Google Scholar] [CrossRef]
- Giusepponi, S.; Celino, M. The effects of vacancies in the mechanical properties of tungsten: A first-principles study. Nucl. Instruments Methods Phys. Res. Sect. B Beam Interact. Mater. Atoms 2015, 342, 70–75. [Google Scholar] [CrossRef]
- Setyawan, W.; Nandipati, G.; Kurtz, R.J. Ab initio study of interstitial cluster interaction with Re, Os, and Ta in W. J. Nucl. Mater. 2017, 484, 30–41. [Google Scholar] [CrossRef]
- Romaner, L.; Ambrosch-Draxl, C.; Pippan, R. Effect of rhenium on the dislocation core structure in tungsten. Phys. Rev. Lett. 2010, 104, 195503. [Google Scholar] [CrossRef]
- Li, H.; Wurster, S.; Motz, C.; Romaner, L.; Ambrosch-Draxl, C.; Pippan, R. Dislocation-core symmetry and slip planes in tungsten alloys: Ab initio calculations and microcantilever bending experiments. Acta Mater. 2012, 60, 748–758. [Google Scholar] [CrossRef]
- Cereceda, D.; Stukowski, A.; Gilbert, M.; Queyreau, S.; Ventelon, L.; Marinica, M.; Perlado, J.; Marian, J. Assessment of interatomic potentials for atomistic analysis of static and dynamic properties of screw dislocations in W. J. Phys. Condens. Matter 2013, 25, 085702. [Google Scholar] [CrossRef]
- Stukowski, A.; Cereceda, D.; Swinburne, T.D.; Marian, J. Thermally-activated non-Schmid glide of screw dislocations in W using atomistically-informed kinetic Monte Carlo simulations. Int. J. Plast. 2015, 65, 108–130. [Google Scholar] [CrossRef]
- Cereceda, D.; Diehl, M.; Roters, F.; Shanthraj, P.; Raabe, D.; Perlado, J.M.; Marian, J. Linking atomistic, kinetic Monte Carlo and crystal plasticity simulations of single-crystal tungsten strength. GAMM-Mitteilungen 2015, 38, 213–227. [Google Scholar] [CrossRef]
- He, S.; Overly, E.; Bulatov, V.; Marian, J.; Cereceda, D. Coupling 2D atomistic information to 3D kink-pair enthalpy models of screw dislocations in bcc metals. Phys. Rev. Mater. 2019, 3, 103603. [Google Scholar] [CrossRef]
- Wu, X.; You, Y.W.; Kong, X.S.; Chen, J.L.; Luo, G.N.; Lu, G.H.; Liu, C.; Wang, Z. First-principles determination of grain boundary strengthening in tungsten: Dependence on grain boundary structure and metallic radius of solute. Acta Mater. 2016, 120, 315–326. [Google Scholar] [CrossRef]
- Qian, Y.; Gilbert, M.R.; Dezerald, L.; Cereceda, D. Using first-principles calculations to predict the mechanical properties of transmuting tungsten under first wall fusion power-plant conditions. J. Phys. Condens. Matter 2021, 33, 345901. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.; Gilbert, M.R.; Dezerald, L.; Nguyen-Manh, D.; Cereceda, D. Ab initio study of tungsten-based alloys under fusion power-plant conditions. J. Nucl. Mater. 2023, 581, 154422. [Google Scholar] [CrossRef]
- Vesti, A.; Music, D.; Olsson, P.A. First-principles study on thermal expansion of W-Re sigma and chi phases. Nucl. Mater. Energy 2024, 39, 101684. [Google Scholar] [CrossRef]
- Gong, X.; Dal Corso, A. Pressure and temperature dependent ab-initio quasi-harmonic thermoelastic properties of tungsten. J. Phys. Condens. Matter 2024, 36, 285702. [Google Scholar] [CrossRef]
- Van Der Giessen, E.; Schultz, P.A.; Bertin, N.; Bulatov, V.V.; Cai, W.; Csányi, G.; Foiles, S.M.; Geers, M.G.; González, C.; Hütter, M.; et al. Roadmap on multiscale materials modeling. Model. Simul. Mater. Sci. Eng. 2020, 28, 043001. [Google Scholar] [CrossRef]
- Dobson, J.F.; Vignale, G.; Das, M.P. Electronic Density Functional Theory: Recent Progress and New Directions; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2013. [Google Scholar]
- Featherston, F.H.; Neighbours, J. Elastic constants of tantalum, tungsten, and molybdenum. Phys. Rev. 1963, 130, 1324. [Google Scholar] [CrossRef]
- Sutton, P.M. The variation of the elastic constants of crystalline aluminum with temperature between 63 K and 773 K. Phys. Rev. 1953, 91, 816. [Google Scholar] [CrossRef]
- Kamm, G.; Alers, G. Low-temperature elastic moduli of aluminum. J. Appl. Phys. 1964, 35, 327–330. [Google Scholar] [CrossRef]
- Walker, E.; Bujard, P. Anomalous temperature behaviour of the shear elastic constant C44 in tantalum. Solid State Commun. 1980, 34, 691–693. [Google Scholar] [CrossRef]
- Gerlich, D.; Fisher, E. The high temperature elastic moduli of aluminum. J. Phys. Chem. Solids 1969, 30, 1197–1205. [Google Scholar] [CrossRef]
- Eschrig, H. T> 0 ensemble-state density functional theory via Legendre transform. Phys. Rev. B—Condensed Matter Mater. Phys. 2010, 82, 205120. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, J.; Zhang, H.; Manga, V.; Shang, S.; Chen, L.; Liu, Z. A first-principles approach to finite temperature elastic constants. J. Phys. Condens. Matter 2010, 22, 225404. [Google Scholar] [CrossRef]
- Pham, H.H.; Williams, M.E.; Mahaffey, P.; Radovic, M.; Arroyave, R.; Cagin, T. Finite-temperature elasticity of fcc Al: Atomistic simulations and ultrasonic measurements. Phys. Rev. B—Condensed Matter Mater. Phys. 2011, 84, 064101. [Google Scholar] [CrossRef]
- Shang, S.; Wang, W.; Wang, Y.; Du, Y.; Zhang, J.; Patel, A.; Liu, Z. Temperature-dependent ideal strength and stacking fault energy of fcc Ni: A first-principles study of shear deformation. J. Phys. Condens. Matter 2012, 24, 155402. [Google Scholar] [CrossRef]
- Shang, S.; Zacherl, C.; Fang, H.; Wang, Y.; Du, Y.; Liu, Z. Effects of alloying element and temperature on the stacking fault energies of dilute Ni-base superalloys. J. Phys. Condens. Matter 2012, 24, 505403. [Google Scholar] [CrossRef]
- Duong, T.C.; Singh, N.; Arróyave, R. First-principles calculations of finite-temperature elastic properties of Ti2AlX (X= C or N). Comput. Mater. Sci. 2013, 79, 296–302. [Google Scholar] [CrossRef]
- Shang, S.L.; Wang, Y.; Kim, D.; Liu, Z.K. First-principles thermodynamics from phonon and Debye model: Application to Ni and Ni3Al. Comput. Mater. Sci. 2010, 47, 1040–1048. [Google Scholar] [CrossRef]
- Sobieraj, D.; Wróbel, J.S.; Rygier, T.; Kurzydłowski, K.J.; El Atwani, O.; Devaraj, A.; Saez, E.M.; Nguyen-Manh, D. Chemical short-range order in derivative Cr–Ta–Ti–V–W high entropy alloys from the first-principles thermodynamic study. Phys. Chem. Chem. Phys. 2020, 22, 23929–23951. [Google Scholar] [CrossRef] [PubMed]
- Zunger, A.; Wei, S.H.; Ferreira, L.; Bernard, J.E. Special quasirandom structures. Phys. Rev. Lett. 1990, 65, 353. [Google Scholar] [CrossRef] [PubMed]
- van de Walle, A.; Asta, M.D.; Ceder, G. The Alloy Theoretic Automated Toolkit: A User Guide. Calphad 2002, 26, 539–553. [Google Scholar] [CrossRef]
- van de Walle, A. Multicomponent multisublattice alloys, nonconfigurational entropy and other additions to the Alloy Theoretic Automated Toolkit. Calphad 2009, 33, 266–278. [Google Scholar] [CrossRef]
- Methfessel, M.; Paxton, A. High-precision sampling for Brillouin-zone integration in metals. Phys. Rev. B 1989, 40, 3616. [Google Scholar] [CrossRef]
- Tamm, A.; Aabloo, A.; Klintenberg, M.; Stocks, M.; Caro, A. Atomic-scale properties of Ni-based FCC ternary, and quaternary alloys. Acta Mater. 2015, 99, 307–312. [Google Scholar] [CrossRef]
- Hastings, W.K. Monte Carlo sampling methods using Markov chains and their applications. Biometrika 1970. [Google Scholar] [CrossRef]
- Kang, S.; Tamm, A. Density functional study of atomic arrangements in CrMnFeCoNi high-entropy alloy and their impact on vacancy formation energy and segregation. Comput. Mater. Sci. 2023, 230, 112456. [Google Scholar] [CrossRef]
- Zhang, F.; Zhao, S.; Jin, K.; Xue, H.; Velisa, G.; Bei, H.; Huang, R.; Ko, J.; Pagan, D.; Neuefeind, J.; et al. Local structure and short-range order in a NiCoCr solid solution alloy. Phys. Rev. Lett. 2017, 118, 205501. [Google Scholar] [CrossRef]
- Zhang, R.; Zhao, S.; Ding, J.; Chong, Y.; Jia, T.; Ophus, C.; Asta, M.; Ritchie, R.O.; Minor, A.M. Short-range order and its impact on the CrCoNi medium-entropy alloy. Nature 2020, 581, 283–287. [Google Scholar] [CrossRef]
- Ding, J.; Yu, Q.; Asta, M.; Ritchie, R.O. Tunable stacking fault energies by tailoring local chemical order in CrCoNi medium-entropy alloys. Proc. Natl. Acad. Sci. USA 2018, 115, 8919–8924. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.; Gilbert, M.R.; Dezerald, L.; Nguyen-Manh, D.; Cereceda, D. First-principles study of the energetics and the local chemical ordering of tungsten-based alloys. arXiv 2024, arXiv:2410.03998. [Google Scholar]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef]
- Langreth, D.C.; Perdew, J.P. Theory of nonuniform electronic systems. I. Analysis of the gradient approximation and a generalization that works. Phys. Rev. B 1980, 21, 5469. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188. [Google Scholar] [CrossRef]
- Togo, A.; Chaput, L.; Tanaka, I.; Hug, G. First-principles phonon calculations of thermal expansion in Ti3SiC2, Ti3AlC2, and Ti3GeC2. Phys. Rev. B 2010, 81, 174301. [Google Scholar] [CrossRef]
- Mounet, N.; Marzari, N. First-principles determination of the structural, vibrational and thermodynamic properties of diamond, graphite, and derivatives. Phys. Rev. B 2005, 71, 205214. [Google Scholar] [CrossRef]
- Peng, J.; Najmaei, S.; Dubey, M.; Chung, P.W. Dominant ZA phonons and thermal carriers in HfS2. J. Appl. Phys. 2019, 126. [Google Scholar] [CrossRef]
- Wolverton, C.; Ozoliņš, V.; Zunger, A. First-principles theory of short-range order in size-mismatched metal alloys: Cu-Au, Cu-Ag, and Ni-Au. Phys. Rev. B 1998, 57, 4332. [Google Scholar] [CrossRef]
- Eriksson, O.; Wills, J.; Wallace, D. Electronic, quasiharmonic, and anharmonic entropies of transition metals. Phys. Rev. B 1992, 46, 5221. [Google Scholar] [CrossRef] [PubMed]
- Le Page, Y.; Saxe, P. Symmetry-general least-squares extraction of elastic data for strained materials from ab initio calculations of stress. Phys. Rev. B 2002, 65, 104104. [Google Scholar] [CrossRef]
- Hill, R. The elastic behaviour of a crystalline aggregate. Proc. Phys. Society. Sect. A 1952, 65, 349. [Google Scholar] [CrossRef]
- Ye, X.; He, Z.; Gao, F.; Pan, B. Electronic excitation induced non-thermal phase transition of tungsten. J. Alloys Compd. 2023, 952, 170087. [Google Scholar] [CrossRef]
- Boda, A.; Sk, M.A.; Shenoy, K.; Mohan, S. Diffusion, permeation and solubility of hydrogen, deuterium and tritium in crystalline tungsten: First principles DFT simulations. Int. J. Hydrogen Energy 2020, 45, 29095–29109. [Google Scholar] [CrossRef]
- Ye, X.; Pan, B. A First-principles analysis of Structural, Electronic, Elastic, and vacant properties of BCC, FCC and HCP tungsten at different electronic temperatures. Nucl. Mater. Energy 2023, 35, 101447. [Google Scholar] [CrossRef]
- Lowrie, R.; Gonas, A. Single-crystal elastic properties of tungsten from 24 to 1800 C. J. Appl. Phys. 1967, 38, 4505–4509. [Google Scholar] [CrossRef]
- Togo, A.; Chaput, L.; Tadano, T.; Tanaka, I. Implementation strategies in phonopy and phono3py. J. Phys. Condens. Matter 2023, 35, 353001. [Google Scholar] [CrossRef]
- Debernardi, A.; Alouani, M.; Dreyssé, H. Ab initio thermodynamics of metals: Al and W. Phys. Rev. B 2001, 63, 064305. [Google Scholar] [CrossRef]
- Dorogokupets, P.; Ponomarev, E.; Melekhova, E. Optimization of experimental data on the heat capacity, volume, and bulk moduli of minerals. Petrology C/C Petrologiia 1999, 7, 574–591. [Google Scholar]
- Novikova, S.I. Thermal Expansion of Solids; Izdatel Nauka: Moscow, Russia, 1974. [Google Scholar]
- Pugh, S. XCII. Relations between the elastic moduli and the plastic properties of polycrystalline pure metals. Lond. Edinb. Dublin Philos. Mag. J. Sci. 1954, 45, 823–843. [Google Scholar] [CrossRef]
- Greaves, G.N.; Greer, A.L.; Lakes, R.S.; Rouxel, T. Poisson’s ratio and modern materials. Nat. Mater. 2011, 10, 823–837. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Cai, N.; Chen, T.; Wang, S.; Li, B. Experimental and theoretical studies on the elasticity of tungsten to 13 GPa. J. Appl. Phys. 2018, 124. [Google Scholar] [CrossRef]
- Koči, L.; Ma, Y.; Oganov, A.; Souvatzis, P.; Ahuja, R. Elasticity of the superconducting metals V, Nb, Ta, Mo, and W at high pressure. Phys. Rev. B—Condensed Matter Mater. Phys. 2008, 77, 214101. [Google Scholar] [CrossRef]
- Geantă, V.; Voiculescu, I.; Stefănoiu, R.; Chereches, T.; Zecheru, T.; Matache, L.; Rotariu, A. Dynamic impact behaviour of high entropy alloys used in the military domain. IOP Conf. Ser. Mater. Sci. Eng. 2018, 374, 012041. [Google Scholar] [CrossRef]
- Li, Z.; Zhao, S.; Ritchie, R.O.; Meyers, M.A. Mechanical properties of high-entropy alloys with emphasis on face-centered cubic alloys. Prog. Mater. Sci. 2019, 102, 296–345. [Google Scholar] [CrossRef]
- Luo, D.M.; Ma, L.; Yang, J.; Ding, N.; Liu, S.Y.; Tang, B.Y. Temperature-dependent elastic properties of high entropy ceramic (ZrTaNbTi) C from self-consistent quasi-harmonic approximation. Solid State Commun. 2021, 336, 114432. [Google Scholar] [CrossRef]
- Körmann, F.; Ikeda, Y.; Grabowski, B.; Sluiter, M.H. Phonon broadening in high entropy alloys. NPJ Comput. Mater. 2017, 3, 36. [Google Scholar] [CrossRef]
- Turner, S.R.; Pailhès, S.; Bourdarot, F.; Ollivier, J.; Sidis, Y.; Castellan, J.P.; Zanotti, J.M.; Berrod, Q.; Porcher, F.; Bosak, A.; et al. Phonon behavior in a random solid solution: A lattice dynamics study on the high-entropy alloy FeCoCrMnNi. Nat. Commun. 2022, 13, 7509. [Google Scholar] [CrossRef]
- Dsouza, R.; Poul, M.; Huber, L.; Swinburne, T.D.; Neugebauer, J. Sampling-free computation of finite temperature material properties in isochoric and isobaric ensembles using the mean-field anharmonic bond model. Phys. Rev. B 2024, 109, 064108. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chemical Composition (at %) [No. Atoms in the Supercell] | |||||||
|---|---|---|---|---|---|---|---|
| W | Cr | Ti | Y | Ta | V | ||
| W | 100 [128] | - | - | - | - | - | |
| W-HEA1 | [64] and this work | 25 [32] | 25 [32] | 25 [32] | - | 25 [32] | - |
| W-HEA2 | [64] and this work | 25 [32] | - | 25 [32] | - | 25 [32] | 25 [32] |
| W-SA1 | [19] | 67.16 | 26.98 | 5.86 | - | - | - |
| This work | 67.19 [86] | 26.56 [34] | 6.26 [8] | - | - | - | |
| W-SA2 | [20] | 67.93 | 31.11 | - | 0.958 | - | - |
| This work | 67.19 [86] | 31.25 [40] | - | 1.56 [2] | - | - | |
| HEA1 | HEA2 | SA1 | SA2 | |
|---|---|---|---|---|
| (Å) | 3.140 | 3.165 | 3.095 | 3.093 |
| (Å) | 3.138 | 3.173 | 3.090 | 3.043 |
| (Å) | 3.135 | 3.160 | 3.088 | 3.083 |
| (Å) | 0.01 | 0.16 | 0.12 | 0.15 |
| (GPa) | −0.01 | 0.001 | 0.02 | 0.008 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peng, J.; Qian, Y.; Cereceda, D. A First-Principles Study of the Structural and Thermo-Mechanical Properties of Tungsten-Based Plasma-Facing Materials. Metals 2024, 14, 1197. https://doi.org/10.3390/met14101197
Peng J, Qian Y, Cereceda D. A First-Principles Study of the Structural and Thermo-Mechanical Properties of Tungsten-Based Plasma-Facing Materials. Metals. 2024; 14(10):1197. https://doi.org/10.3390/met14101197
Chicago/Turabian StylePeng, Jie, Yichen Qian, and David Cereceda. 2024. "A First-Principles Study of the Structural and Thermo-Mechanical Properties of Tungsten-Based Plasma-Facing Materials" Metals 14, no. 10: 1197. https://doi.org/10.3390/met14101197
APA StylePeng, J., Qian, Y., & Cereceda, D. (2024). A First-Principles Study of the Structural and Thermo-Mechanical Properties of Tungsten-Based Plasma-Facing Materials. Metals, 14(10), 1197. https://doi.org/10.3390/met14101197