
Tunability of Martensitic Transformation with Cohesive Energies for Fe80−xMnxCo10Cr10 High-Entropy Alloys



Abstract
1. Introduction
2. Method and Simulation Details
2.1. EAM Potential Optimization
2.2. Molecular Dynamic Simulation Details
3. Results and Discussion
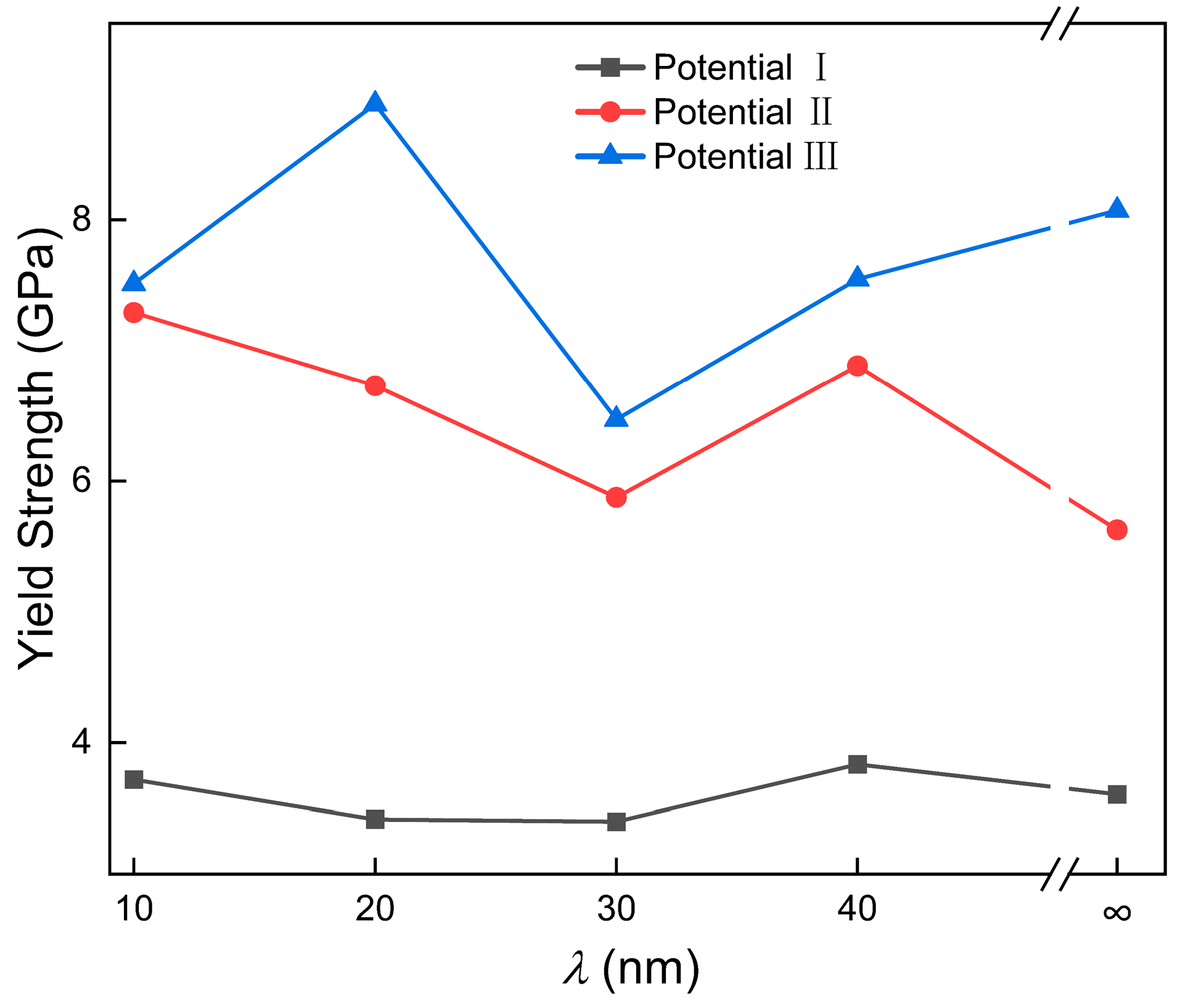
3.1. Potential Optimization
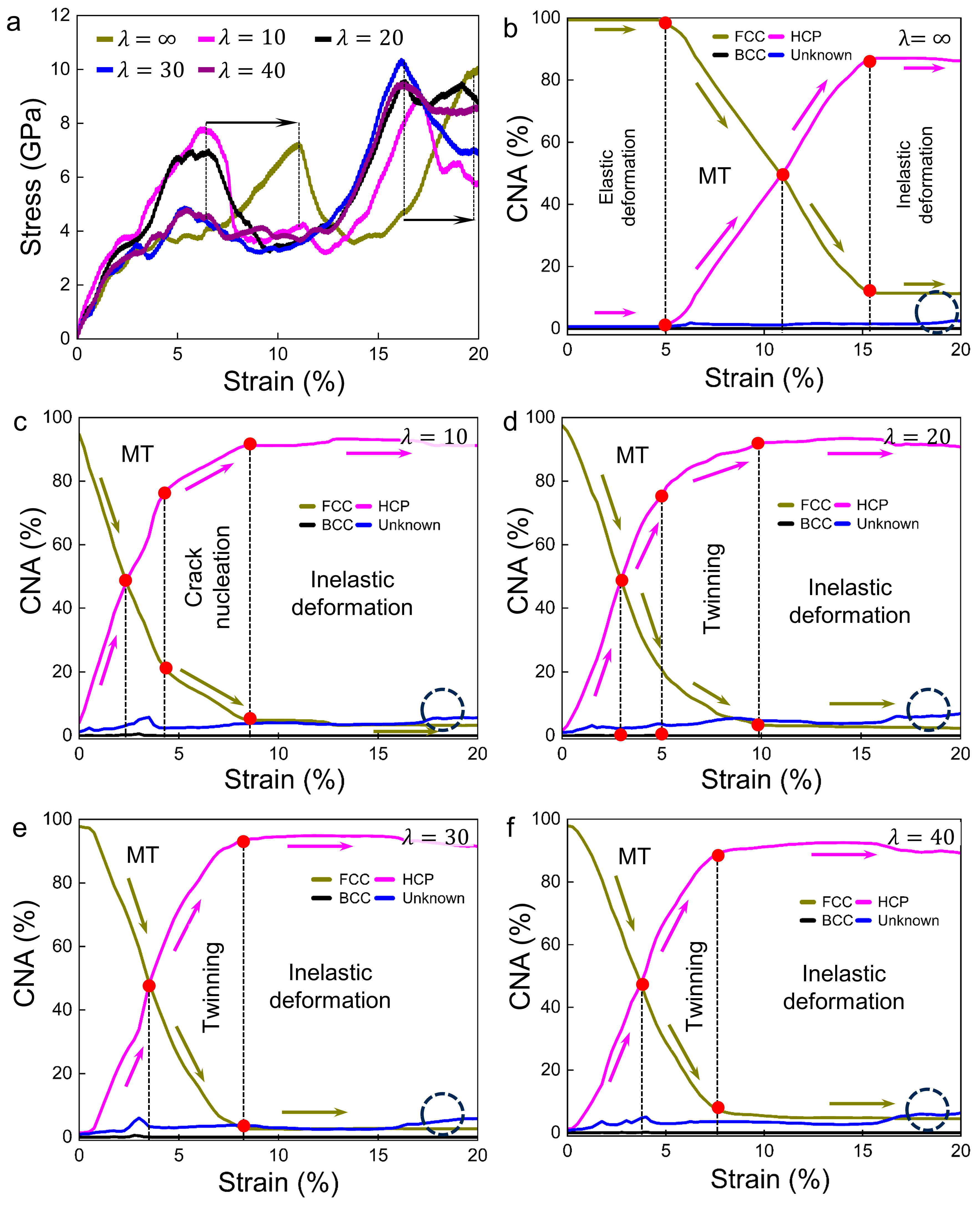
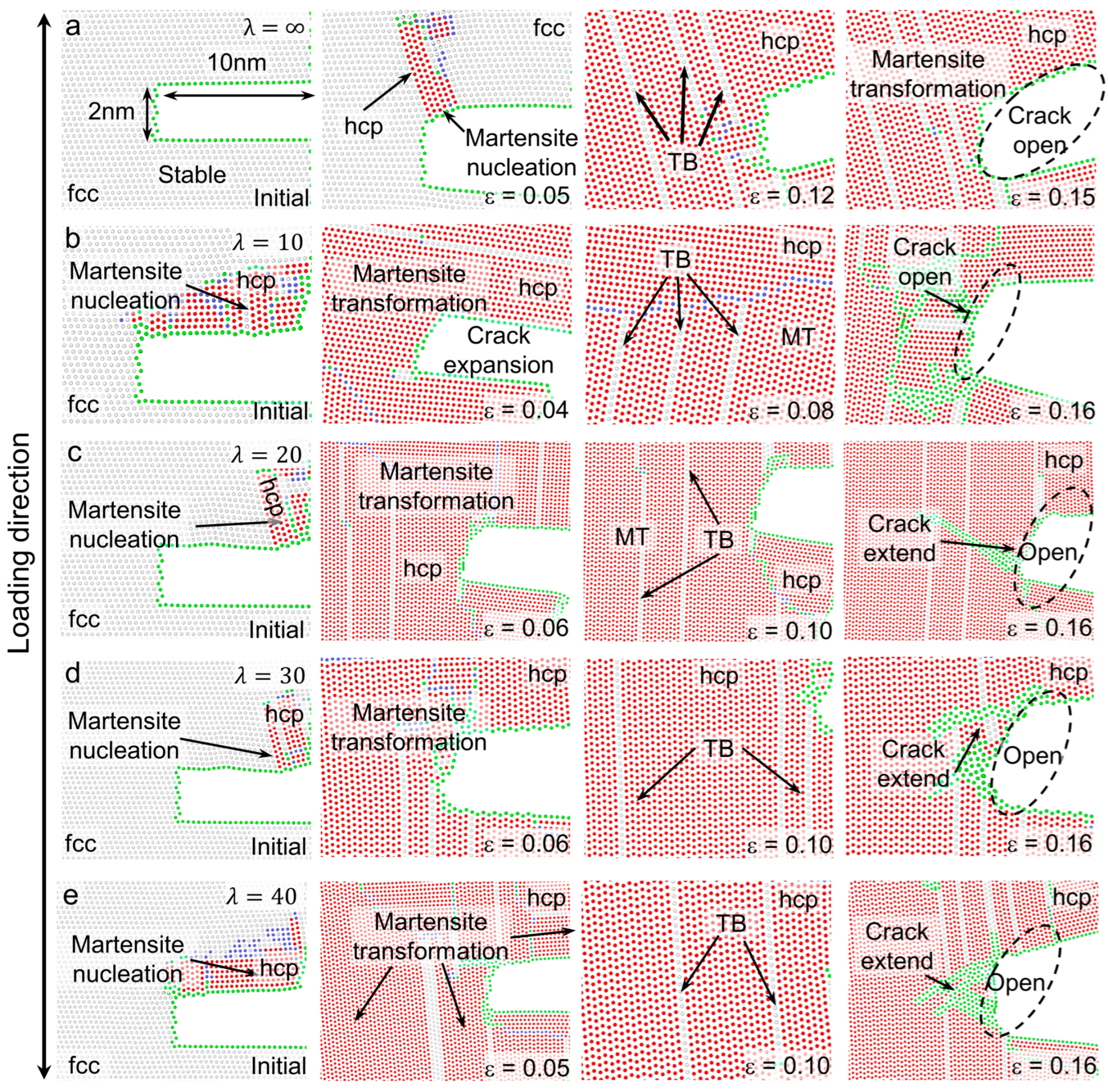
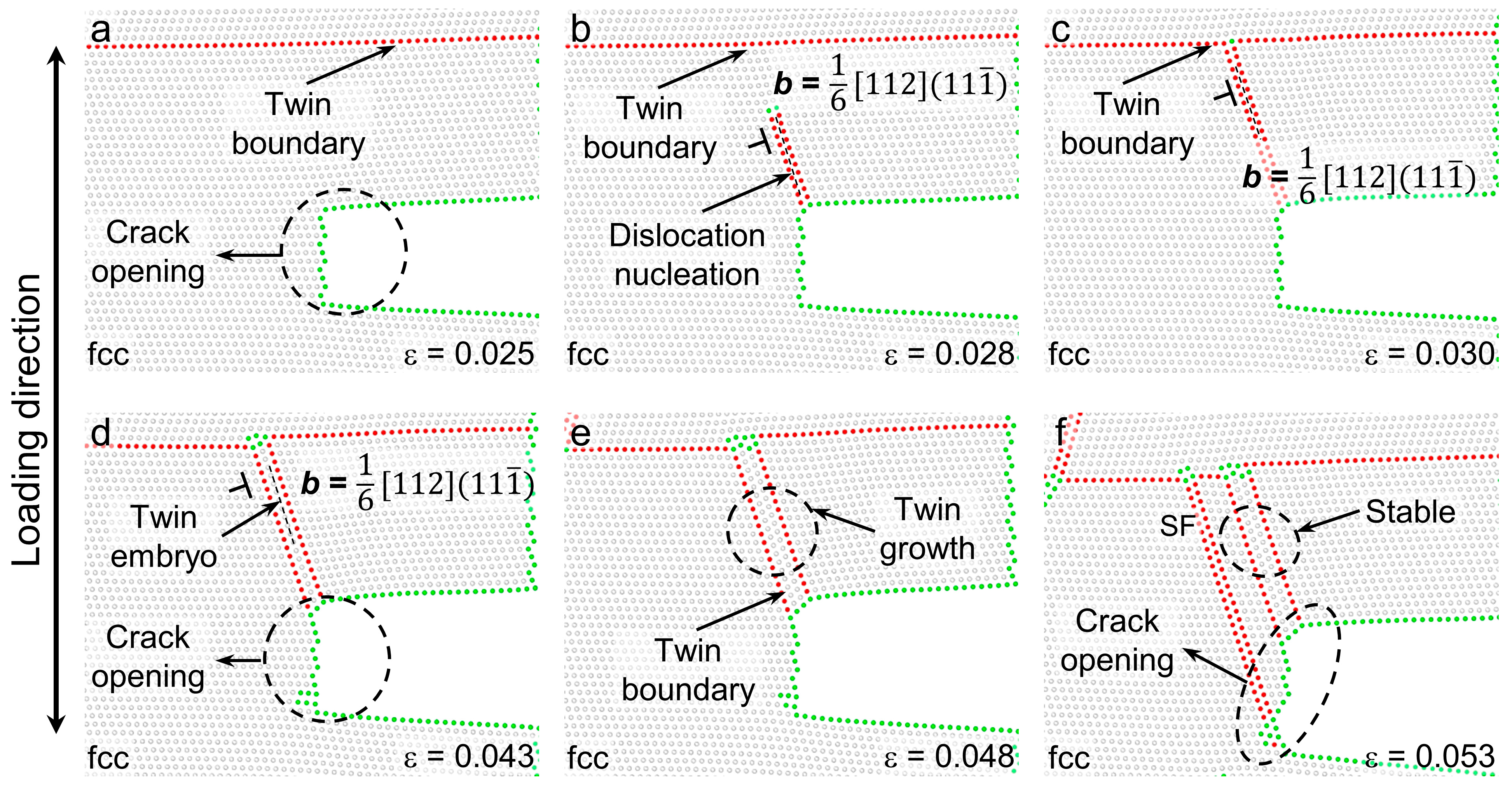
3.2. Deformation Processes for Models with of 71 meV/atom
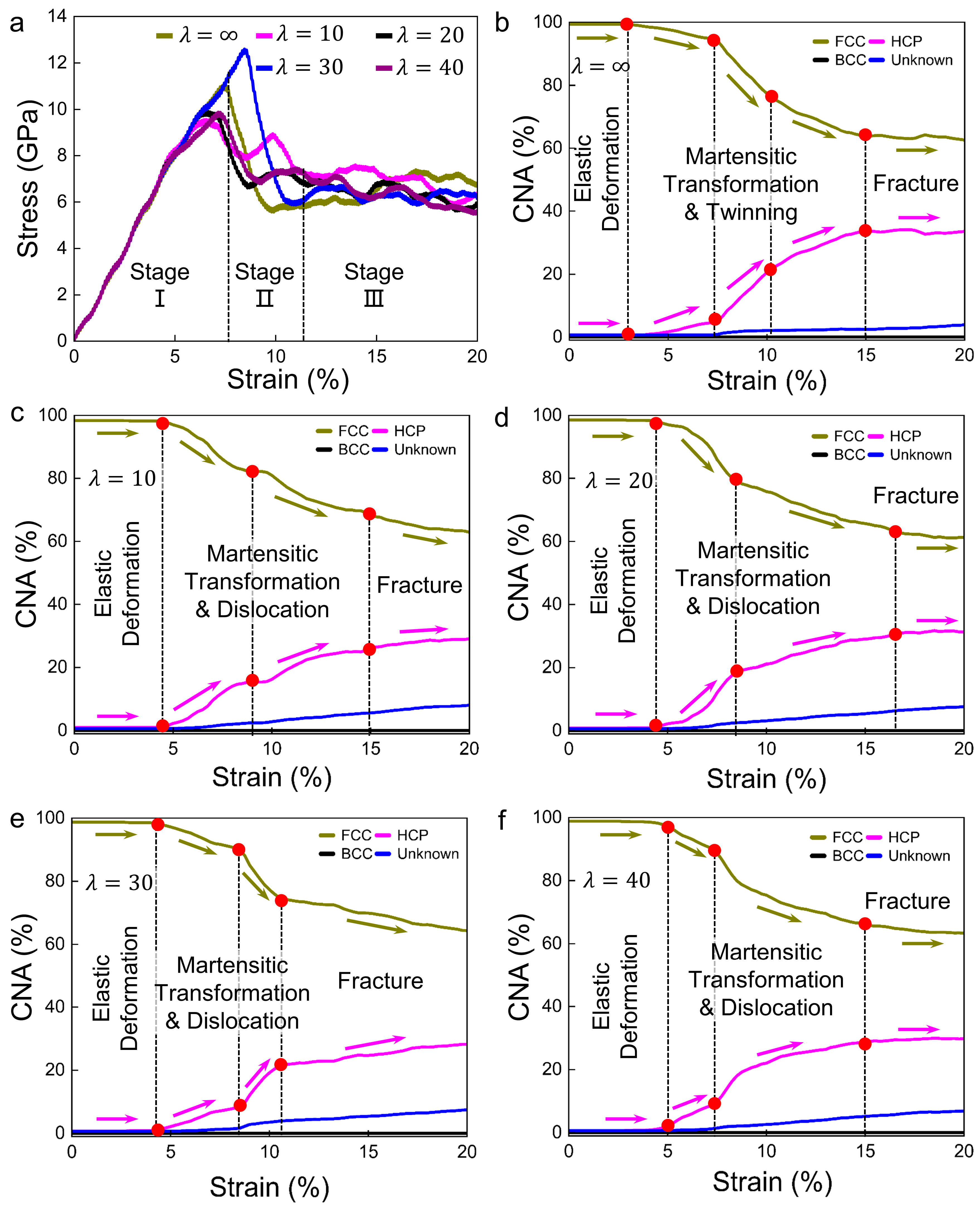
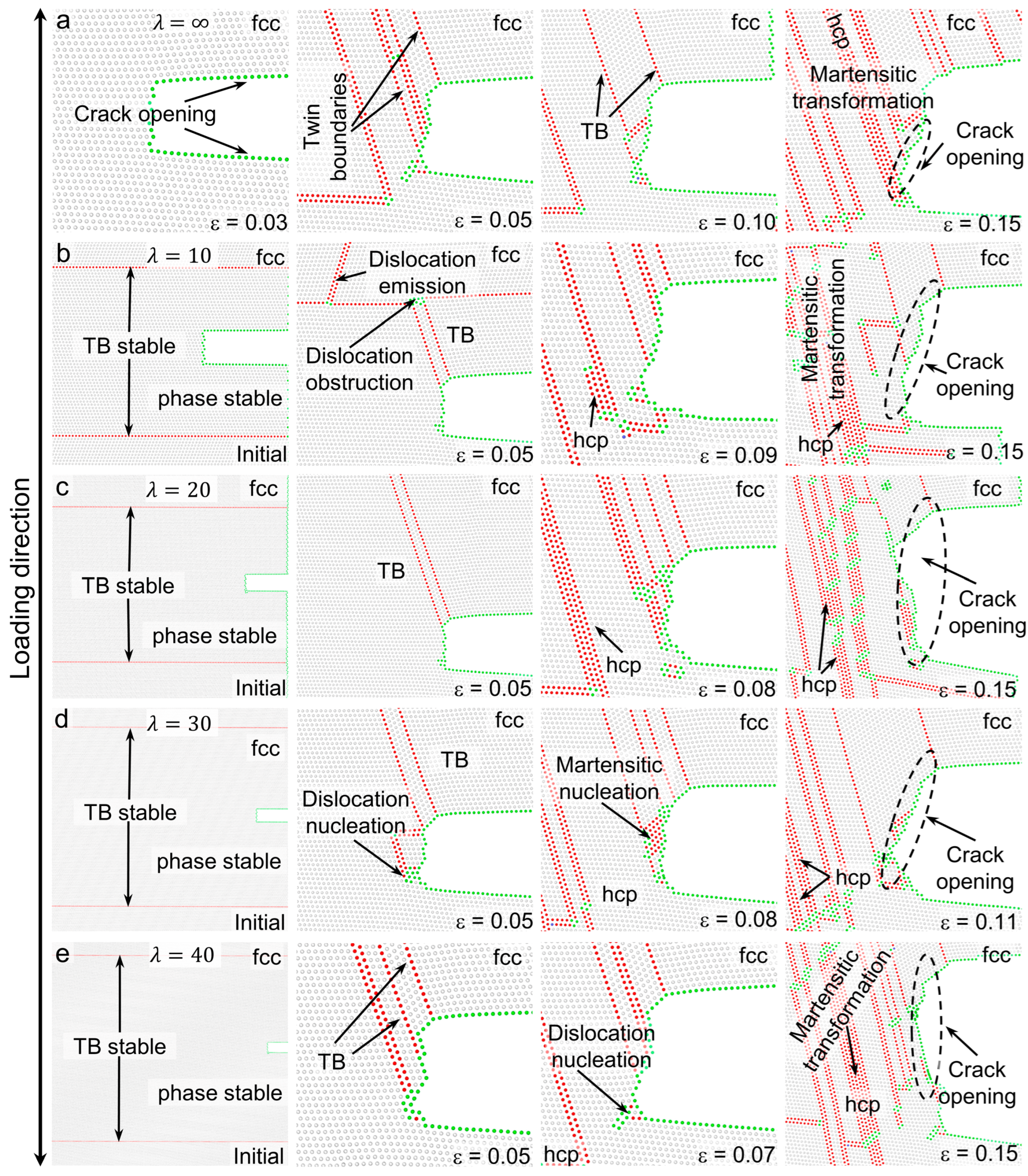
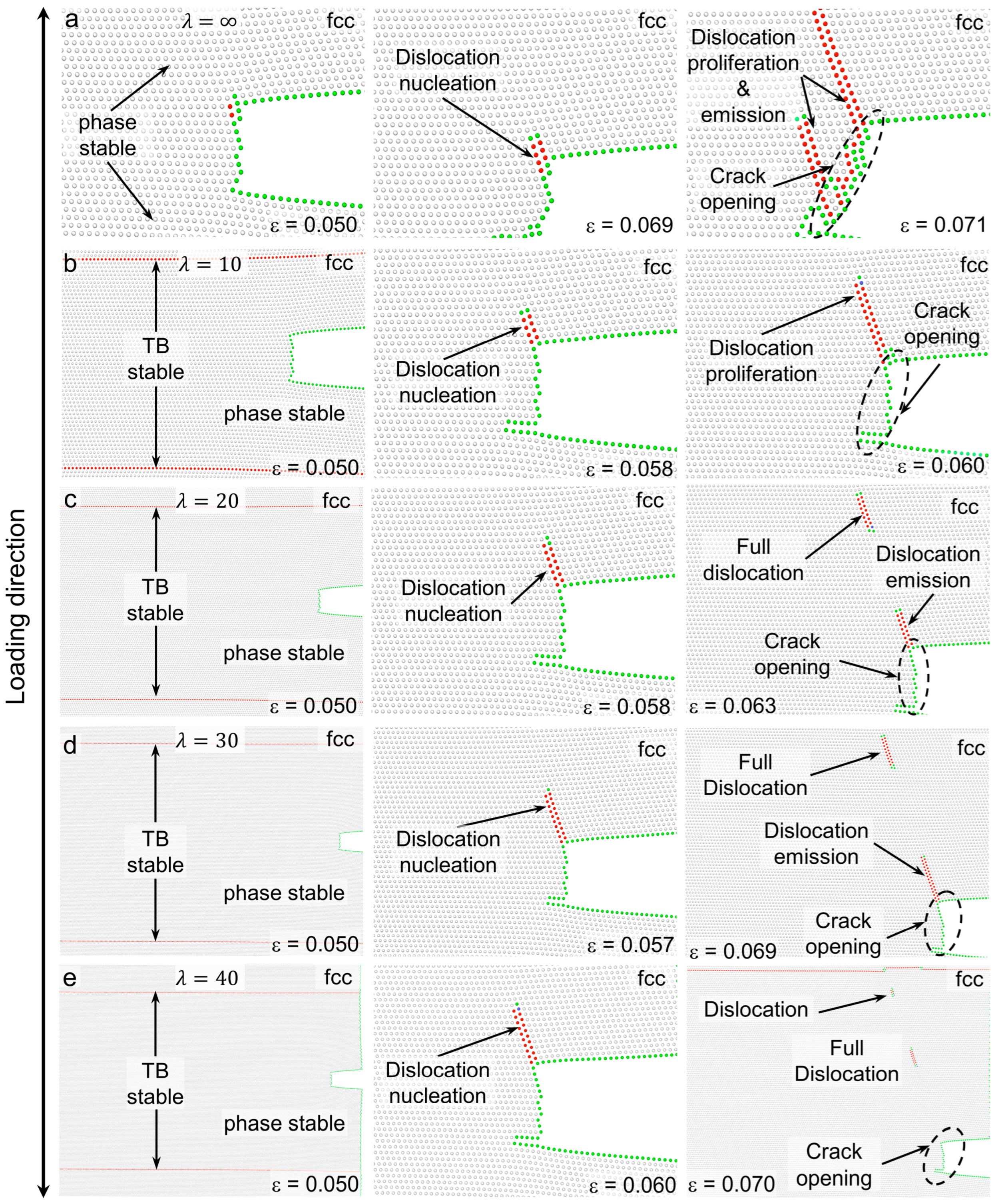
3.3. Deformation Processes for Models with of 0 meV/atom
3.4. Deformation Processes for Models with of −50 meV/atom
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Nomenclature
| HEAs | High-entropy alloys |
| TRIP-DP-HEA | Transformation-induced plasticity-assisted dual-phase alloy |
| MD | Molecular dynamic |
| MT | Martensitic transformation |
| DFT | Density functional theory |
| SFE | Stacking fault energy (mJ/m2) |
| TB | Twin boundary |
| λ | Twin boundary spacings |
| LAMMPS | Large-Scale Atom/Molecular Massively Parallel Simulator |
| CNA | Common Neighbor Analysis |
| VCA | Virtual crystal approximation |
| bcc | Body-centered cubic |
| fcc | Face-centered cubic |
| hcp | Hexagonal close-packed |
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Summary of the Fitting Coefficients The Units of Energy and Distance Are eV and Å | |||
|---|---|---|---|
| Potential II | Potential II | ||
| r0 | 1.8 | r1 | 2.4 |
| r2 | 3.0 | r3 | 3.6 |
| r4 | 4.2 | r5 | 4.8 |
| r6 | 5.4 | r7 | 6.0 |
| a1 | −9.66770760793731 × 10−1 | a2 | −4.320220866033277 × 10−4 |
| a3 | 3.478971484000802 × 10−7 | a4 | 2.000000000000000 × 10−1 |
| a5 | 40 | ||
| b0 | 2.101375956583815 × 103 | b1 | −2.077171964986614 × 103 |
| b2 | 5.162776790386556 × 102 | ||
| c1,3 | −5.2106037336697 × 10−2 | c1,4 | 2.778188909933398 × 102 |
| c1,5 | −3.7570947633811610 × 101 | c2,3 | 4.62601873279574 × 10−1 |
| c2,4 | 5.397455863083986 × 100 | c2,5 | 1.329131989361568 × 100 |
| c3,3 | −4.344697177683853 × 100 | c3,4 | 3.444687503275756 × 100 |
| c3,5 | −2.556175142394920 × 100 | c4,3 | −1.539582959232970 × 100 |
| c4,4 | 8.16212811008731 × 10−1 | c4,5 | −4.4854759939966 × 10−2 |
| c5,3 | −9.19177586830533 × 10−1 | c5,4 | −1.264203911131165 × 100 |
| c5,5 | −9.80724404586589 × 10−1 | c6,3 | 8.201649434365 × 10−3 |
| c6,4 | −2.817017818372356 × 100 | c6,5 | 1.58226822562389 × 10−1 |
| c7,3 | 4.47354593228291 × 10−1 | c7,4 | −1.232064925519546 × 100 |
| c7,5 | 8.53120624954035 × 10−1 | ||
| Summary of the Fitting Coefficients The Units of Energy and Distance Are eV and Å | |||
|---|---|---|---|
| Potential III | Potential III | ||
| r0 | 1.8 | r1 | 2.4 |
| r2 | 3.0 | r3 | 3.6 |
| r4 | 4.2 | r5 | 4.8 |
| r6 | 5.4 | r7 | 6.0 |
| a1 | −1.081659385091810 × 100 | a2 | −1.515231941409 × 10−3 |
| a3 | 6.943744787859560 × 10−7 | a4 | 2.000000000000000 × 10−1 |
| a5 | 40 | ||
| b0 | 5.451752816213673 × 103 | b1 | −5.754856912375750 × 103 |
| b2 | 1.522035944735593 × 103 | ||
| c1,3 | 5.510424603160274 × 102 | c1,4 | −2.207729708000522 × 103 |
| c1,5 | 2.474846739550133 × 103 | c2,3 | −3.628422779567440 × 100 |
| c2,4 | 9.789322689368860 × 100 | c2,5 | −5.955323422751416 × 100 |
| c3,3 | 1.933180399702355 × 100 | c3,4 | 1.1043734351838088 × 101 |
| c3,5 | 3.966275516567340 × 100 | c4,3 | −1.139122907374148 × 100 |
| c4,4 | 8.174054355850220 × 100 | c4,5 | −3.837897146665342 × 100 |
| c5,3 | −1.261352893543118 × 100 | c5,4 | 1.897898187922895 × 100 |
| c5,5 | −7.74660867853492 × 10−1 | c6,3 | −1.720878011359318 × 100 |
| c6,4 | −1.30970194552900 × 10−1 | c6,5 | −1.060029898052776 × 100 |
| c7,3 | 3.66636529045938 × 10−1 | c7,4 | −8.86829309275372 × 10−1 |
| c7,5 | 6.95113617224733 × 10−1 | ||
References
- Moschetti, M.; Perrière, L.; Couzinié, J.; Kruzic, J.; Gludovatz, B. A novel strategy for the design of compositionally complex alloys for advanced nuclear applications. Appl. Mater. Today 2024, 38, 102164. [Google Scholar] [CrossRef]
- Li, Z.; Pradeep, K.G.; Deng, Y.; Raabe, D.; Tasan, C.C. Metastable high-entropy dual-phase alloys overcome the strength–ductility trade-off. Nature 2016, 534, 227–230. [Google Scholar] [CrossRef]
- Miracle, D.B.; Senkov, O.N. A critical review of high entropy alloys and related concepts. Acta Mater. 2017, 122, 448–511. [Google Scholar] [CrossRef]
- Otto, F.; Dlouhý, A.; Somsen, C.; Bei, H.; Eggeler, G.; George, E.P. The influences of temperature and microstructure on the tensile properties of a CoCrFeMnNi high-entropy alloy. Acta Mater. 2013, 61, 5743–5755. [Google Scholar] [CrossRef]
- Chen, Z.; Zhu, W.; Wang, H.; He, Q.; Fang, Q.; Liu, X.; Li, J.; Yang, Y. Overcoming strength-toughness trade-off in a eutectic high entropy alloy by optimizing chemical and microstructural heterogeneities. Commun. Mater. 2024, 5, 12. [Google Scholar] [CrossRef]
- Zhang, Y.; Zuo, T.T.; Tang, Z.; Gao, M.C.; Dahmen, K.A.; Liaw, P.K.; Lu, Z.P. Microstructures and properties of high-entropy alloys. Prog. Mater. Sci. 2014, 61, 1–93. [Google Scholar] [CrossRef]
- Zhang, C.; Zhang, F.; Jin, K.; Bei, H.; Chen, S.; Cao, W.; Zhu, J.; Lv, D. Understanding of the Elemental Diffusion Behavior in Concentrated Solid Solution Alloys. J. Phase Equilibria Diffus. 2017, 38, 434–444. [Google Scholar] [CrossRef]
- Li, Z.; Tasan, C.C.; Springer, H.; Gault, B.; Raabe, D. Interstitial atoms enable joint twinning and transformation induced plasticity in strong and ductile high-entropy alloys. Sci. Rep. 2017, 7, 40704. [Google Scholar] [CrossRef]
- Li, Z.; Raabe, D. Influence of compositional inhomogeneity on mechanical behavior of an interstitial dual-phase high-entropy alloy. Mater. Chem. Phys. 2018, 210, 29–36. [Google Scholar] [CrossRef]
- Huang, H.; Wu, Y.; He, J.; Wang, H.; Liu, X.; An, K.; Wu, W.; Lu, Z. Phase-Transformation Ductilization of Brittle High-Entropy Alloys via Metastability Engineering. Adv. Mater. 2017, 29, 1701678. [Google Scholar] [CrossRef]
- Wang, P.; Song, Z.; Lin, Y.; Li, Q.; Wang, H. The nucleation mechanism of martensite and its interaction with dislocation dipoles in dual-phase high-entropy alloys. J. Alloys Compd. 2022, 909, 164685. [Google Scholar] [CrossRef]
- Fang, Q.; Chen, Y.; Li, J.; Jiang, C.; Liu, B.; Liu, Y.; Liaw, P.K. Probing the phase transformation and dislocation evolution in dual-phase high-entropy alloys. Int. J. Plast. 2019, 114, 161–173. [Google Scholar] [CrossRef]
- Qi, Y.; Zhao, M.; Feng, M. Molecular simulation of microstructure evolution and plastic deformation of nanocrystalline CoCrFeMnNi high-entropy alloy under tension and compression. J. Alloys Compd. 2021, 851, 156923. [Google Scholar] [CrossRef]
- Jiao, Y.; Xu, L.; Dan, W.; Xu, Y.; Zhang, W. Atomic-scale study of the mechanical properties of dual-phase fcc/bcc crystallites: Influences of alloying elements and phase boundaries. J. Mater. Sci. 2022, 57, 11111–11131. [Google Scholar] [CrossRef]
- Kim, Y.-M.; Shin, Y.-H.; Lee, B.-J. Modified embedded-atom method interatomic potentials for pure Mn and the Fe–Mn system. Acta Mater. 2009, 57, 474–482. [Google Scholar] [CrossRef]
- Lee, B.-J. A modified embedded-atom method interatomic potential for the Fe–C system. Acta Mater. 2006, 54, 701–711. [Google Scholar] [CrossRef]
- Wang, P.; Lin, Y.; Cao, Y.; Zhao, H.; Li, Q.; Wang, H. Atomistic simulations of martensitic transformation processes for metastable FeMnCoCr high-entropy alloy. Sci. China Technol. Sci. 2023, 66, 998–1006. [Google Scholar] [CrossRef]
- Chen, Y. Kinetic Monte Carlo Modeling of Martensitic Phase Transformation Dynamics. In Handbook of Materials Modeling; Springer: Berlin/Heidelberg, Germany, 2018; pp. 1–21. [Google Scholar] [CrossRef]
- Wang, P.; Xu, S.; Liu, J.; Li, X.; Wei, Y.; Wang, H.; Gao, H.; Yang, W. Atomistic simulation for deforming complex alloys with application toward TWIP steel and associated physical insights. J. Mech. Phys. Solids 2017, 98, 290–308. [Google Scholar] [CrossRef]
- Wang, P.; Wang, H. Meta-Atom Molecular Dynamics for Studying Material Property Dependent Deformation Mechanisms of Alloys. J. Appl. Mech. 2017, 84, 111002. [Google Scholar] [CrossRef]
- Wang, P.; Wu, Y.; Liu, J.; Wang, H. Impacts of atomic scale lattice distortion on dislocation activity in high-entropy alloys. Extrem. Mech. Lett. 2017, 17, 38–42. [Google Scholar] [CrossRef]
- Evans, D.J.; Holian, B.L. The Nose–Hoover thermostat. J. Chem. Phys. 1985, 83, 4069–4074. [Google Scholar] [CrossRef]
- Plimpton, S. Fast Parallel Algorithms for Short-Range Molecular Dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef]
- Stukowski, A. Visualization and analysis of atomistic simulation data with OVITO–the Open Visualization Tool. Model. Simul. Mater. Sci. Eng. 2010, 18, 015012. [Google Scholar] [CrossRef]
- Faken, D.; Jónsson, H. Systematic analysis of local atomic structure combined with 3D computer graphics. Comput. Mater. Sci. 1994, 2, 279–286. [Google Scholar] [CrossRef]
- Bu, Y.; Li, Z.; Liu, J.; Wang, H.; Raabe, D.; Yang, W. Nonbasal Slip Systems Enable a Strong and Ductile Hexagonal-Close-Packed High-Entropy Phase. Phys. Rev. Lett. 2019, 122, 075502. [Google Scholar] [CrossRef]
- Medvedeva, N.I.; Park, M.S.; Van Aken, D.C.; Medvedeva, J.E. First-principles study of Mn, Al and C distribution and their effect on stacking fault energies in fcc Fe. J. Alloys Compd. 2014, 582, 475–482. [Google Scholar] [CrossRef]
- Mizuno, M.; Sugita, K.; Araki, H. Defect energetics for diffusion in CrMnFeCoNi high-entropy alloy from first-principles calculations. Comput. Mater. Sci. 2019, 170, 109163. [Google Scholar] [CrossRef]
- Bellaiche, L.; Vanderbilt, D. Virtual crystal approximation revisited: Application to dielectric and piezoelectric properties of perovskites. Phys. Rev. B 2000, 61, 7877–7882. [Google Scholar] [CrossRef]
- Grässel, O.; Krüger, L.; Frommeyer, G.; Meyer, L.W. High strength Fe–Mn–(Al, Si) TRIP/TWIP steels development—Properties—Application. Int. J. Plast. 2000, 16, 1391–1409. [Google Scholar] [CrossRef]
- Qian, S.U.N.; Xiong-Bo, Y.; Ya-Jun, G.A.O.; Jian-Wei, Z. Molecular Dynamics Simulation of the Deformation Behavior of Ag Nanowires with Different Twin Boundary Density under Tension Loading. Acta Phys.-Chim. Sin. 2014, 30, 2015–2023. [Google Scholar] [CrossRef]
- Suzuki, T.; Shimono, M.; Kajiwara, S. On the mechanism for martensitic transformation from fcc to bcc. Mater. Sci. Eng. A 2001, 312, 104–108. [Google Scholar] [CrossRef]




| Properties | Target | Potential Ⅰ [17] | Potential Ⅱ | Potential Ⅲ |
|---|---|---|---|---|
| a (fcc) (Å) | 3.608 [26] | 3.609 | 3.610 | 3.608 |
| Ec_fcc (eV/atom) | −4.679 [*] | −4.679 | −4.729 | −4.729 |
| C11 (GPa) | 265 [12] | 332 | 329 | 317 |
| C12 (GPa) | 184 [12] | 133 | 138 | 154 |
| C44 (GPa) | 113 [12] | 51 | 45 | 40 |
| γ111 (mJ/m2) | 2367 [17] | 2457 | 2443 | 1471 |
| γ110 (mJ/m2) | 2718 [17] | 2580 | 2626 | 1800 |
| γ100 (mJ/m2) | 2783 [17] | 2481 | 2446 | 1479 |
| γsf (mJ/m2) | - | −385 | 16 | 301 |
| γusf (mJ/m2) | 200 [27] | 199 | 241 | 460 |
| Evac (eV) | 2 [28] | 2 | 2.2 | 2.17 |
| a (hcp) (Å) | 2.544 [26] | 2.547 | 2.544 | 2.544 |
| c (hcp) (Å) | 4.110 [26] | 4.098 | 4.152 | 4.110 |
| Ec_hcp (eV/atom) | −4.750 [*] | −4.750 | −4.729 | −4.679 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cao, Y.; Zhang, X.; Zhou, D.; Wang, P.; Pan, D.; Wang, H. Tunability of Martensitic Transformation with Cohesive Energies for Fe80−xMnxCo10Cr10 High-Entropy Alloys. Metals 2024, 14, 728. https://doi.org/10.3390/met14060728
Cao Y, Zhang X, Zhou D, Wang P, Pan D, Wang H. Tunability of Martensitic Transformation with Cohesive Energies for Fe80−xMnxCo10Cr10 High-Entropy Alloys. Metals. 2024; 14(6):728. https://doi.org/10.3390/met14060728
Chicago/Turabian StyleCao, Yu, Xiaoliang Zhang, Daoxuan Zhou, Peng Wang, Deng Pan, and Hongtao Wang. 2024. "Tunability of Martensitic Transformation with Cohesive Energies for Fe80−xMnxCo10Cr10 High-Entropy Alloys" Metals 14, no. 6: 728. https://doi.org/10.3390/met14060728
APA StyleCao, Y., Zhang, X., Zhou, D., Wang, P., Pan, D., & Wang, H. (2024). Tunability of Martensitic Transformation with Cohesive Energies for Fe80−xMnxCo10Cr10 High-Entropy Alloys. Metals, 14(6), 728. https://doi.org/10.3390/met14060728