1. Introduction
Earth is a dynamic and energetic system, and terrestrial processes have long ago obscured most geochemical records of the early solar system. Therefore, we look to extraterrestrial materials that better preserve that early history. Carbonaceous asteroids are some of the most important solar system objects because they are composed of primordial materials that escaped harsh planetary differentiation processes, although some were modified by post-accretion hydrothermal processes. Thus, they provide clues about the very beginning of the formation of our solar system and our planet. The JAXA Hayabusa2 spacecraft returned samples from the dark carbonaceous near-Earth asteroid 162,173 Ryugu [
1]. Approximately 5.4 g of material was delivered to Earth in 2020; surface material (“A” series samples) was collected from the first touch-down site, while subsurface material (“C” series samples) was obtained from a second site in a crater produced by the impact of a projectile fired from the spacecraft. These unique samples provide access to terrestrially uncontaminated and unprocessed chemical components that are otherwise inaccessible in meteorites that fall on Earth. Initial analyses (e.g., [
2,
3,
4]) indicated that the Ryugu particles contain abundant organic compounds, inorganic materials, and bound water, and are mineralogically and chemically similar to CI chondrite meteorites.
The major objectives of the Hayabusa2 mission were to understand the origin and the evolutionary history of primitive, organic-rich asteroids, as well as implications for the formation of the planets. Ryugu’s parent body likely formed in the colder outer ranges of the solar system, beyond the “snow line” for H
2O and CO
2 ices, capturing an important spatial aspect of Solar System formation, and was subsequently mildly heated to mobilize fluids that interacted with and significantly altered the original mineral and organic components, forming secondary phases such as phyllosilicates, carbonates, sulfides, and magnetite [
3,
4]. However, details of this primordial chemical evolution of Ryugu are not yet fully understood. Signatures of such aqueous processes are imprinted on the grain-scale mineralogy and chemistry of Ryugu samples, the study of which can shed significant light on the formation and chemical evolution of its parent body. In particular, element-specific studies identifying microscale oxidation state and chemical speciation are key to understanding and constraining both the formation and alteration history of very early solar system materials (e.g., [
5,
6,
7]).
We were allocated two Ryugu samples by JAXA for these and related measurements. A0030 is from the surface-collected material, a 2.84 mm grain weighing 6.9 mg. C0034 is from the subsurface series, a 2.09 mm grain weighing 2.4 mg.
Synchrotron X-ray fluorescence (XRF) and X-ray absorption spectroscopy (XAS) microprobes are generally non-destructive, element-specific tools to characterize, at the micrometer to submicrometer spatial scale in heterogeneous materials, (a) the distributions and associations of most elements, and (b) their oxidation state, chemical speciation and local structure, in crystalline and non-crystalline phases. As such, these naturally complement electron microscopic techniques, providing chemical details and context, and scaling up the field of view in materials that are heterogeneous on a wide range of spatial scales. This study applies a novel combination of two less common XRF microprobe techniques to explore the chemistry within intact Ryugu samples without physical sectioning.
2. Methods
Synchrotron X-ray microprobes are most commonly used for 2-D mapping and spectroscopy of polished thin sections, due to the advantages of flat geometry and minimum thickness. Owing to the deep penetration and sampling depth of hard X-rays, sample thickness should be comparable to the probe beam footprint to optimize spatial resolution and avoid seeing overlap from grains below the surface. In order to overcome that limitation, and to preserve intact specimens, pink-beam fluorescence computed microtomography (FCT) uses XRF and absorption contrast to image the distributions and co-locations of elements (Cr and heavier) in a virtual slice through an intact sample without physically cutting it. On the other hand, “tender” energy (1–5 keV) fluorescence X-ray absorption microspectroscopy (μXRF and μXAS) probes lighter elements (Mg through Ti) [
8]. Having a shallower sampling depth, tender-energy microbeam fluorescence measurements can be made on a whole grain regardless of sample thickness. Three-dimensional spatial resolution is comparable to the focused beam size for both techniques, optimizing clarity of the results. Applied to unprocessed grains, pink-beam FCT and tender-energy microspectroscopy are complementary, covering both heavier and lighter elements. In order to better relate these different measurements, we employ two trace-element proxy pairs. Strontium is well known to be co-located with Ca in most systems. Selenium is chemically similar to S, although its use as a proxy is less extensive; in this study, we use S and Se μXAS (at tender and hard X-ray energies, respectively) to substantiate applicability of this proxy in Ryugu samples.
Pink-beam FCT measurements utilized the X-ray Fluorescence Microprobe (XFM) beamline at NSLS-II. XFM is designed for monochromatic operation in the 2.3–23 keV range and optimized for high-quality, spatially resolved XAS in conjunction with element-specific XRF imaging and microdiffraction. It is also designed to operate in a pink-beam mode that delivers a 1 μm focus with up to 1000× more flux than monochromatic mode. The filtered “pink” beam X-rays are a subset of the full spectrum “white beam” from the synchrotron source, in this case ranging from approximately 12 to 20 keV.
Conventional 2-D hard X-ray microprobe measurements, with effective sampling depth tens to hundreds of μm, would require sample thickness comparable to the size of the beam focus. In contrast, FCT provides a virtual slice whose thickness is defined by the vertical dimension of the probe beam, in this case 1 μm. Pixel size in the plane of the slice is defined by measurement parameters and ultimately limited by beam size. This effectively provides measurement of multiple 1 μm thick virtual slices of much larger areas than could be physically obtained by mechanical ultramicrotome or FIB sectioning, particularly in such friable material as the Ryugu samples, while preserving precious samples unaltered. Here we report pink-beam FCT of a fragment of sample C0034. Details of how these measurements were made are provided in the
Supplementary Material.
These FCT measurements identify major- and trace-element distributions and co-locations, while concurrent measurement of transmission through the sample provides a map of absorption contrast to identify specific phases within a sample. However, it is important to note that there are limitations to quantitative interpretations of concentration or elemental ratios. Over large cross-sections, the virtual slices show a decrease in signal toward the center. Similar artifacts of intensity occur within large high-density grains. These absorption effects are highly dependent on an element’s fluorescence energy, and are more pronounced for lighter elements. For example, Fe shows a steeper gradient of intensity than Se. Elements lighter than Cr are only visible near the edges of the slice. There is a similar energy- and depth-dependence for elemental sensitivity. Heavier elements such as Se can be detected at trace concentrations of ~100 ppm, while lighter elements require higher concentrations up to wt%. While it is challenging to present whole-slice images at a fixed color scale, it is clearer to view smaller areas of large slices. Tender-energy μXRF and μXAS measurements of whole grains were used to probe important lighter elements. “Tender”-energy X-rays are ~1–5 keV in energy, between more typical “hard” and “soft” X-rays. This work used a monochromatic beam tuned for the K absorption edges of, e.g., S and P, and to stimulate fluorescence from elements Mg through Ca. Since the sampling depth in this energy range is a few μm, comparable to beam focus size, these near-surface measurements can be made regardless of sample thickness. Here we report tender-energy microspectroscopy of whole grains C0034 and A0030, and the fragment of C0034 used for FCT.
Measurements utilized the TES beamline at NSLS-II, which was designed and optimized for μXRF and μXAS in the tender-energy range [
8]. The monochromatic beam was focused by a KB mirror pair to a user-tunable spot size of 2 to 10 μm, and the sample was raster-scanned on-the-fly across this focus in a helium-filled sample chamber. Some tender-energy measurements were made at the XFM beamline employing an accessory helium-filled sample chamber. Although the XFM beamline is not optimized for the tender-energy range, it is capable of reaching down into that range as far as the S K-edge. Samples are interchangeable between TES and XFM.
Microbeam XAS measurements included both short and long-range scans. X-ray absorption near-edge spectroscopy (μXANES) provides element-specific information about oxidation state and chemical species, while extended X-ray absorption fine structure (μEXAFS) analysis provides details of local structure within about 10 Å. These are accessory measurements for both hard and tender energies, and are employed here for P, S, Se, Ca and Mn, to enhance the interpretation of FCT and XRF results. These spectroscopic techniques, particularly at tender energies, provide an opportunity to identify phases in samples that are too thick for effective microbeam XRD. μXANES spectra are measured by focusing the beam at a desired location and scanning the incident beam energy across an element’s absorption edge while measuring that element’s characteristic X-ray fluorescence. The absorption edge position is indicative of oxidation state, and spectral features can be used to identify chemical species relative to reference standards. These features, typically peaks and resonances, arise from potential electronic transitions that are sensitive to bond configurations (first- and second-neighbor) and crystal field effects, as well as the absorbing element’s structural and symmetry environments. Detailed spectral feature analysis deconstructs XANES spectra into these specific features that can be quantitatively associated with chemical species. μEXAFS measures the interference patterns of photoelectrons from the absorbing atom being reflected by neighboring atoms within about 10 Å. These oscillations extend up to about 1000 eV above the absorption edge, requiring longer scans. μXAS spectra were processed and normalized using the Athena software [
9]. EXAFS data were processed, Fourier transformed to radial distribution functions, and fit to model structures using the Athena and Artemis software packages [
9].
XANES spectra can also guide
speciation imaging via multi-energy mapping, wherein an XRF map is repeated at incident beam energies tuned to particular XANES features characteristic of different chemical species of an element. Fluorescence intensities in these maps are then processed to isolate the contributions of each species in each pixel in order to map the relative distribution of those species (see e.g., [
10,
11]). This technique was used here for S.
3. Results
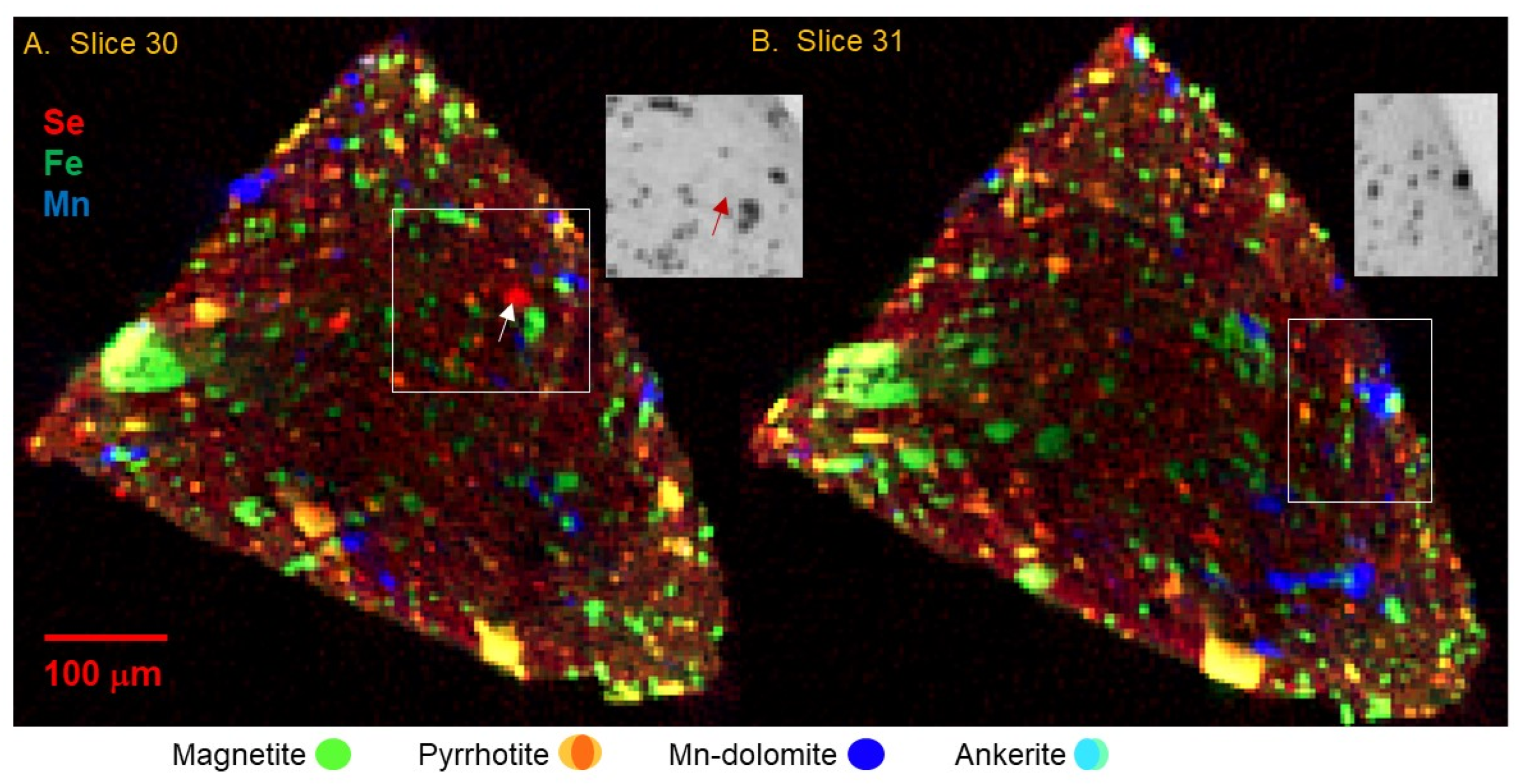
Figure 1 depicts the distribution of Se, Fe, and Mn in reconstructed tomographic slices numbered 30 and 31, at 4 μm spatial resolution, for a fragment of sample C0034.
Figure 2 and
Figure 3 show the same elements in slices 32 and 33, measured and reconstructed at finer 2 μm spatial resolution. Consecutive numbered slices were measured at 10 μm increments in z.
Figure 4,
Figure 5 and
Figure 6 highlight closer views of these and other elements in selected areas of interest.
Figure 7 and
Figure 8 show tender-energy XRF maps of the near-surface distributions of lighter elements P, S, and Si in whole-particle A0030 and C0034, respectively.
Figure 9 shows maps of lighter elements from Mg to Mn in the same fragment of C0034 as measured by FCT.
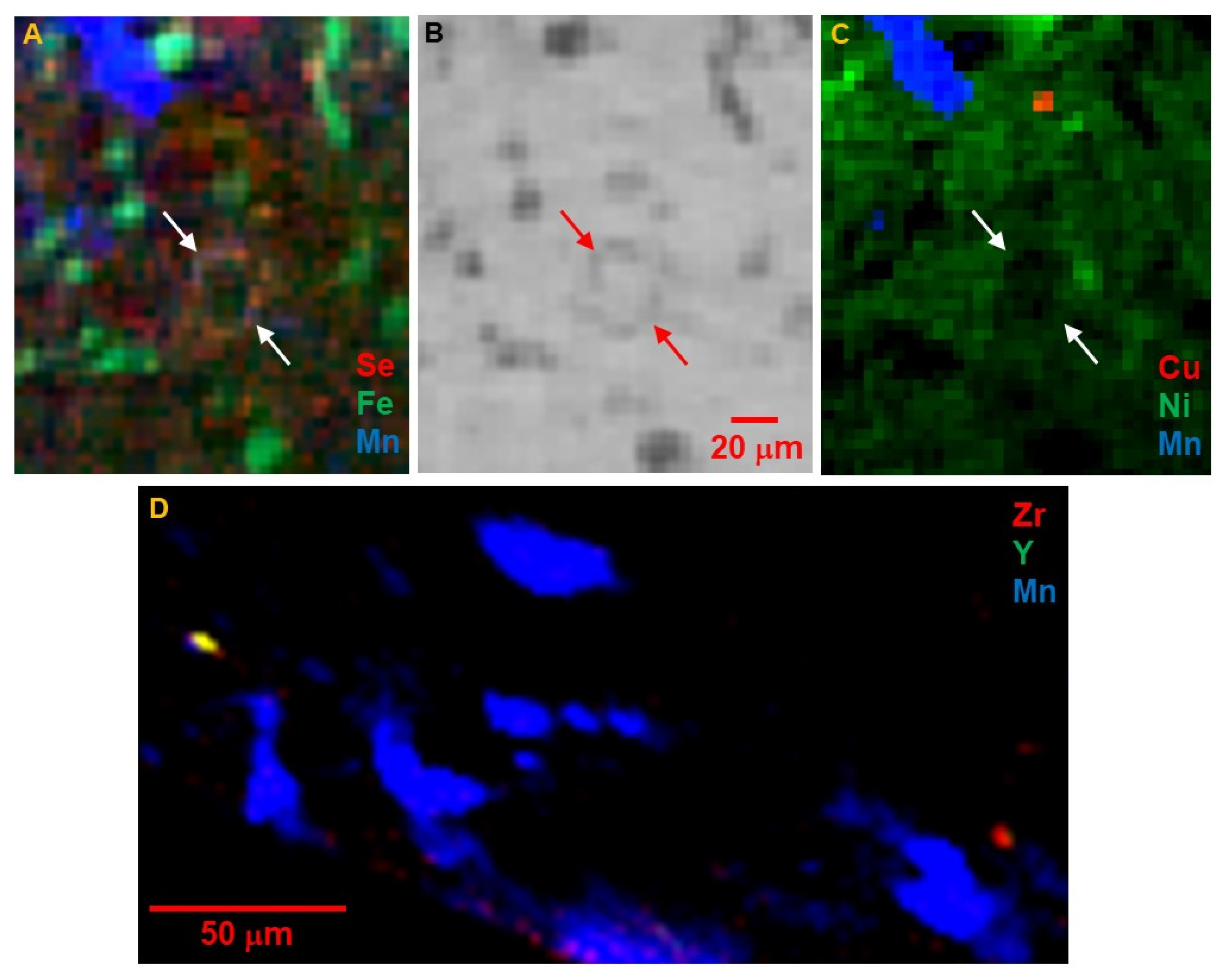
The tomograms show a complex texture that includes larger mineral grains up to >50 μm across, in a finer (<4 μm grains) matrix. The larger grains are mostly rounded in shape, although there are notably more euhedral rod-like or platy shapes as in
Figure 4C,D. A subtle fabric is apparent, particularly in the close-up images in
Figure 5, in which chains of like particles are oriented in slightly curved striae from upper left to lower right, while rod-like shapes (platy crystals intersecting the tomographic slice) are oriented at 90 degrees to that texture.
The dominant Fe phases are expected to be oxides and sulfides, with lesser amounts in silicates (primarily serpentine and ferruginous saponite) and carbonate.
Figure 1,
Figure 2,
Figure 3,
Figure 4,
Figure 5 and
Figure 6 show that Fe dominates the larger particles, and that these have the highest absorption contrast consistent with high Fe concentration as found in oxides and sulfides. The presence of Se with Fe distinguishes the sulfides. Selenium was chosen as a trace-element proxy for S to locate sulfides and other S-bearing phases. A clear Se signal is present in ~30% of the >5 μm Fe-bearing grains, but in ~80% of the fine-grained Fe-bearing particles in the matrix. Some of the large Fe-bearing grains, particularly as shown in
Figure 4C, are an intergrowth of sulfide and oxide; the importance of this is discussed below. There are also several instances, such as those shown in
Figure 4A,B and
Figure 6A,B, of small Fe-sulfide grains forming a rim on a round structure of Fe-poor low-absorption material. This is similar to the necklace-like string of Fe sulfide particles surrounding a phyllosilicate nodule as described in
Figure 6 of [
4].
Manganese is negatively correlated with most high-Fe phases and is present in distinct 5 to 50 μm rounded grains. The low absorption and presence of Sr (
Figure 5D) indicate these are carbonates. As shown in
Figure 10, Mn μXANES of several different spots on the whole grain are very similar; all show that Mn is in the 2+ oxidation state, and spectral structure indicates it is incorporated into a rhombohedral, bilayered carbonate such as dolomite MgCa(CO
3)
2. Concentration of Mn is consistent across the Mn-bearing carbonates. Although much lower in Fe than the Fe oxide and sulfide phases, there is still significant Fe in the carbonates, ~1 wt% of both Fe and Mn. These carbonates are also evident as Ca hot spots correlated with Mn in the tender-energy whole-particle surface maps (
Figure 9) and limited information from FCT (
Figure 5E). A small number of grains with similar Mn content but higher absorption contrast and ~10× higher Fe (turquoise-colored grains in
Figure 1,
Figure 2,
Figure 3 and
Figure 5), high Ca and Sr, and inferred lower Mg, are ankerite FeCa(CO
3)
2. Ankerite grains are locally associated with or overgrown by dolomite. Calcium μXANES (
Figure 11) of several spots on dolomite and ankerite grains confirm each of these elements.
Figure 5D indicates that Sr concentration in the dolomite is zoned with slightly higher Sr in grain centers.
Copper is found as small <10 μm distinct hot spots. Most of these spots also have high Se (as illustrated in
Figure 5C and
Figure 6C), but low Fe, indicating a Cu sulfide, as previously reported for Ryugu [
3]. Among other elements, Ni is dilutely distributed throughout the matrix, rather than in hot spots (
Figure 6C), but is notably absent from the large Fe oxide and sulfide grains, the carbonates, and the low-absorption interiors of the round features rimmed by small Fe sulfides. Zinc is sparsely distributed as small fine-grained areas of moderate concentrations (
Figure 5C). Zirconium and Y are rare, but concentrated in very small <5 μm hot spots, often together (
Figure 6D). Grains with only Zr may be zircon or baddeleyite.
The distribution of P, shown in
Figure 6,
Figure 7 and
Figure 8, is in sparse hot spots of up to ~25 μm. Most, but not all, of these also contain Ca, indicating that they are likely apatite Ca
5(PO
4)
3(F, OH, Cl). In the tomograms, most of the small Sr hot spots that are not carbonates are likely apatite. K-edge μXANES of P matches apatite for most high-P spots measured (
Figure 11C). Notably, however, there are a few spots having P without Ca, and the P μXANES spectra for these are distinctly different from apatite. These are phosphates but may be inorganic or organic species; a more detailed μXAS study of P speciation in additional Ryugu samples is underway.
Sulfur is present in numerous grains up to ~40 μm as well as in much finer grains distributed through the matrix, matching the distribution of Se seen in the tomograms. Initial S speciation mapping (
Figure 12B) reveals the distribution of different oxidation states. Most of the large S-bearing grains are sulfide, but there are a number of separate spots containing sulfate. These are lower in total S concentration, but are not spatially associated with the sulfides. There were also a very few areas of “intermediate” S species. This species may be a disulfide such as pyrite [
13], elemental S [
13,
14], or an organic S species such as a thiol [
15], thiophene or sulfoxide [
16].
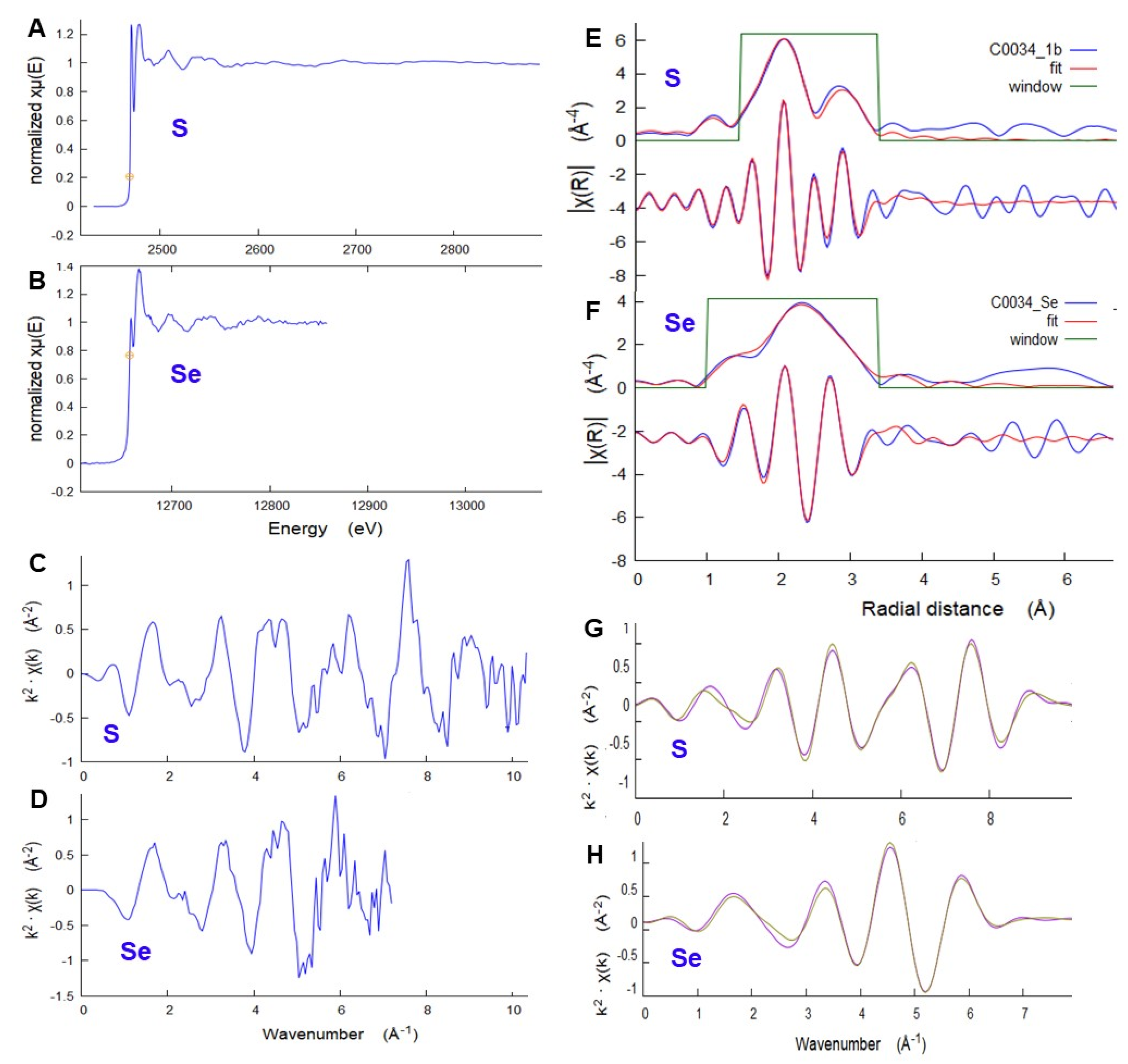
Microbeam XAS of S and Se were used to further characterize the sulfide species and confirm the applicability of Se as a proxy for S in FCT measurements of these samples. Sulfur K-edge μXANES of several 5 × 5 μm spots on the whole grains (
Figure 13A) all show the same sulfide species, pyrrhotite, some with variable minor amounts of sulfate. These included examples of both the large single grains and the finer sulfides disseminated in the matrix. Selenium μXANES, although of limited spatial resolution due to large sampling depth, shows a selenide with similar spectral shape to S in pyrrhotite, and a very minor oxidized selenate component (
Figure 13B). Detailed spectral feature analyses (
Supplementary Material) give a range of sulfate component from 0 to 18.3% of total S in different 5 × 5 μm spots, while selenate was limited to <1% of total Se. The smaller observed fraction of selenate may be the result of a different extent of oxidation for Se compared to S, but could also reflect the sampling depths for which the S analyses show more localized variations weighted toward particle surfaces. In contrast, Se is sampled to greater depth and therefore exhibits more of a bulk average including the interiors of larger sulfide particles.
The detailed local structure around S and Se in a selected C0034 sulfide grain were studied using μEXAFS (
Figure 14). Data for both elements closely match the specific distribution of interatomic distances in a model pyrrhotite structure [
17], including first-neighbor S-Fe (Se-Fe) and second-neighbor S-S (Se-S) distances, adjusting only for uniform overall expansion. The resulting first-neighbor average distance is 2.42(1) Å for S and 2.54(1) Å for Se. Local expansion of the structure to accommodate the larger Se is 0.12 Å, comparable to the 0.14 Å difference in ionic radius [
18] between S
2− and Se
2−. This local distortion relaxes to 0.02 Å for the second-neighbor distances. Details of EXAFS analyses are provided in
Supplementary Materials.
Silicon is broadly distributed, mainly as phyllosilicates in the fine matrix, but there is no evidence of large silicate grains. Aluminum (
Figure 9D) is concentrated in very few distinct grains up to 30 μm in size. Magnesium displays a similar distribution to Si but is also present in carbonates, spots without any Si.
Whole-grain measurements of elements heavier than Ca become less informative as z increases, due to the increasing effective sampling depth leading to overlapping signals from grains buried below the surface. Titanium is present as discrete small grains consistent with ilmenite, while Cr is dilutely distributed throughout the matrix as well as in small hot spots (
Figure 9E).
4. Discussion and Conclusions
The novel combination of non-invasive synchrotron microprobe techniques applied here sheds light on the chemistry and evolution of the fluids that interacted with primitive material in the Ryugu parent body. Evidence supports multiple fluid episodes, or significantly evolving fluid character, recorded in grain-scale chemistry.
Sulfur is a major player in fluid chemistry and redox processes, and is closely involved with Fe chemistry. Yet, sulfur chemistry, reactions, and the formation of sulfur-based compounds (especially S-bearing organic molecules) on carbonaceous asteroids are not well-constrained [
19]. The varying abundance of sulfur in the “A” and “C” Ryugu samples (~3.3 ± 0.7 wt% and ~5.5 ± 0.7 wt%, respectively), indicates a heterogeneous distribution of sulfides [
20]. XAS at the 5 μm scale identified the dominant sulfide as pyrrhotite, both in large grains and finer particles in the matrix. This is in contrast to observed microscale heterogeneity of sulfide speciation in samples from carbonaceous meteorites such as Tarda, Murchison, Cold Bokkeveld, Aguas Zarcas, Northwest Africa 12748, and Tagish Lake [
10,
14,
16,
21].
Equilibrium condensation modeling indicates that S condenses from a cooling gas of solar composition as an Fe-sulfide [
22], and the characterization of sulfides in primitive, anhydrous chondritic porous interplanetary dust particles, believed to be the least altered samples of nebula condensate available for laboratory study [
23], indicates the dominant sulfide is pyrrhotite [
24]. As a result, any sulfate would be an alteration phase. Sulfates are usually considered to be weathering products from terrestrial environments, as discussed in, e.g., [
25] for CI chondrites. Our observation of oxidized S with sulfide may result from oxidation by atmospheric exposure during sample handling, or from beam-induced oxidation during XAS measurement (e.g., [
26]), but we can discount these possibilities on the basis that (a) not all sulfide spots showed the presence of sulfate, there were no obvious μm-scale reaction rims at the scale of this study, and the amount of sulfate was not correlated to particle size as would be the case for surface oxidation; (b) particle C0034 had significantly more sulfate with the sulfides as compared to A0030, but both were handled similarly and equally exposed to atmosphere; and (c) there was no evidence of increasing sulfate with beam exposure when scans were repeated on the same spot. The formation of sulfate post-collection reaction products has been observed in Ryugu samples by electron microscopy [
2], but these are small enough to be volumetrically insignificant compared to the scale of our measurements (tens of μm
3).
The partial oxidation of sulfide observed in μXANES is therefore interpreted as original to the material, indicating an incomplete but significant oxidation process occurred on the parent body. This is consistent with the absence of Fe metal and formation of magnetite of various morphologies [
27], as well as a study of isotopic compositions of Ryugu carbonates [
28] which concluded that dolomite precipitated later in the alteration process in a more oxygen-rich CO
2-dominated environment. It is also corroborated by the textural evidence of partial replacement of pyrrhotite by magnetite. The presence of sulfates spatially separated from sulfides in C0034 further implies that some resulting sulfate was transported by fluids. While it is possible that there were some more reactive sulfide species present, and these oxidized to sulfate on exposure to Earth’s atmosphere, such reactive species would likely have reacted during aqueous alteration by the somewhat oxidizing fluids that precipitated, e.g., magnetite. Observations of a few examples of intermediate S species in a layered geometry with sulfide and sulfate (
Figure 12) raise interesting questions about formation of such a structure. Bose et al. observed this intermediate S in another Ryugu sample [
15] and potentially related organic S species in Murchison [
16]. Beyond the scope of the measurements reported here, the relationship of organic S species to other organic and inorganic phases will provide important information on the evolution of organic molecules in the early solar system.
Iron is present mainly as oxides and sulfides, the former being dominant among larger grains and the latter being prevalent in the fine-grained matrix. There is a notable absence of large Fe-bearing silicate grains. Several of the rod-like or platy Fe-rich grains, and some of the rounded Fe-rich grains, are composed of intergrown oxide and sulfide in an apparent partial-replacement alteration texture. Electron microscopy studies of Ryugu samples showing framboidal magnetite suggest that it was formed from a fluid (e.g., [
3,
4]). The large, often euhedral sulfide grains imply recrystallization by a fluid that was O-poor but did not have access to much of the fine-grained sulfides, so it must not have been the same fluid responsible for the alteration that formed the hydrated silicates that now dominate the matrix. Preservation of fine-scale structures such as sulfide rims on round features indicate that silicates were altered in situ without mobilization of silica, and the fluid responsible for silicate alteration did not significantly react with these sulfides, but likely mobilized Mg that later formed dolomite. Likewise, if the large sulfide grains are early fluid-mediated recrystallization products, then a later more O-rich fluid was responsible for altering some of the sulfide to magnetite but this fluid also did not have access to the smaller sulfides embedded in the silicate matrix. Alternatively, a single fluid may have evolved significantly over time, at some point becoming more O-rich.
This temporal separation between silicate alteration and formation of magnetite and dolomite is further supported by the observed distribution of Cr and Ni. If those elements were initially minor components of silicate phases or silicate-dominated assemblages, and alteration of the silicates occurred in situ and preserved textures like sulfide rims, then Cr and Ni would remain dispersed throughout the phyllosilicates either in the phyllosilicates themselves or in particles finer than the resolution of this study. Importantly, Ni is absent from the phyllosilicate nodule rimmed by sulfide grains, indicating that the original core of that structure was a low-Ni silicate, and the silicate-altering fluid did not transport Ni.
Carbonates are important in Ryugu and form large grains [
29], but are generally limited in Ca and Fe content (i.e., no calcite or siderite). Most that we observed are Mn-rich dolomite, evidenced by the Ca and Mn XANES, the presence of Ca (and Sr), and the low absorbance indicating a significant Mg component; this agrees with [
28], who found that dolomite was the most abundant carbonate in the Ryugu samples they analyzed. The dolomite we observed has significant minor concentrations of Fe and Mn substituting for Ca, and is the primary host for Mn in the sample. It is also consistent with remote spectroscopic observations interpreted as ferroan magnesite (sometimes called “breunnerite”) on Bennu [
30], with similar indications for Ryugu [
31]. The Mn, Fe, Sr, and Ca contents of the carbonates help constrain the composition of the fluid involved in their formation. While Fe and Mn content is quite uniform across the dolomite grains, the observed zoning of Sr indicates that the fluid became depleted in Sr during carbonate formation. Although Fe is abundant overall in the sample, and has undergone significant chemical processing, its low concentration in the dolomite implies that there was limited available Fe
2+ in the carbonate-forming fluid relative to the greater availability of Mn
2+ and Mg
2+. Magnesium would have been mobilized during silicate alteration, perhaps linking that process to dolomite formation. The few ankerites we found crystallized first, as suggested by their spatial association with dolomite, and may have depleted Fe in the fluid. Alternatively, this may be the result of timing, such that the availability of carbonate did not coincide with conditions when more Fe was mobilized.



 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}













