1. Introduction
Microarray is now a mature technology in the field of molecular biology for monitoring of gene expression profiling at the genome level. Using two-color microarray technology, the biological activities of two samples are compared on an array on which thousands of specific deoxyribonucleic (DNA) sequences are printed or synthesized
in situ. Two fluorescent dyes (generally red and green) are used for labeling of the samples for hybridization. Then, after washing, scanning and quantification of images corresponding to the red and green fluorescence of the array, numerical values, or intensities are associated with each probe (gene). These values are normalized to correct for undesirable technical variations [
1,
2]. Using one-color microarray technology, each sample is hybridized on one array. One fluorescence color is used, and expression intensities are obtained for genes on the arrays used. These values should also be normalized [
3]. For a given gene, a ratio of values from two samples is used as a measurement of its expression change. Throughout this paper, we use logarithm two scale to transform the values. Hence, we use
intensities and
ratios for sample intensities and their ratios. In general,
ratios are used for two-color microarray data, while
intensities are used for one-color microarray data.
In a screening study, the goal is to select genes with differential expression from data whose characteristics are unknown. To study the performance of gene selection methods, we often use synthetic data, which can also serve in developing new methods. To properly play their role, synthetic data must resemble as closely as possible the real data they represent. This is achieved by simulating the physical phenomena through a model. The parameters of this model are approximations of the physical laws that govern the observed phenomenon, and a good knowledge of the physical laws is therefore required to obtain a good model. A complex model that takes into account several components of a phenomenon may become less flexible and may be difficult to modify in order to include new factors. In contrast, a simple model can be effective and easy to modify to take into account an unexpected situation.
One way to generate synthetic data is to use real microarray values as seeds [
4,
5,
6,
7]. In [
4], data were generated using a normal distribution and a real microarray dataset. This dataset was used to estimate some hyperparameters, and the percentage of the differentially expressed (DE) genes was fixed; see details in [
4]. The simulation data obtained in [
4] may be useful for statistical methods, but they differ from observed data, since the
-intensities obtained vary between the unrealistic bounds 16 and 30. In [
5], two samples are selected from the control and the test samples. The measurements associated with their genes are modified (exchange of values between the two samples…) in order to obtain two statistically undistinguishable samples. Finally, a given number of down- and up-regulated genes is used in this dataset. This procedure necessitates a real microarray dataset, which cannot be available. The procedure proposed in [
6] is a modular system, including all steps of the microarray technology. This includes slide layout, hybridization, scanning and image processing. Many models already available in the literature are used with new ones in [
6]. The system of [
6] results in data as close as possible to real biological data, but is quite complex, and the large number of its parameter settings may discourage its use. In [
7], two simulation methods are used. In the first, the number of DE genes and the number of levels of changes for these genes are fixed. Then, for each gene, a mean and a standard deviation are drawn from a uniform distribution. Finally, these data (mean and standard deviation) are used as normal distribution parameters to get expression values for the genes. In the second simulation method, the mean and standard deviation of test (
,
) and control (
) samples are estimated from observed real data and are used as normal distribution parameters to get expression values for all genes. There is no flexibility in the number of the DE genes of the method in [
7]. A hierarchical model is used in [
8,
9], where the variance of each gene is simulated using two parameter settings and the
distribution. This variance is then used to generate values from a normal distribution. DE genes are finally defined using levels compared to a threshold. The procedure used in [
8,
9] allows one to generate data with parameters derived from assumptions about real data. However, the percentage of the DE genes depends on a parameter that is difficult to control
a priori. For some of the above methods, there is no distinction between the number of weakly expressed genes and those strongly expressed.
The model proposed in [
10] provides synthetic data fairly close to the characteristics of true data. In this model, the level of expression of a gene is obtained by the superposition of several components. These components allow one to define the DE genes and the overall level of variability in the data. However, it lacks flexibility in choosing the number of DE genes and the number of genes over- and under-regulated. In this paper, we are interested in generating synthetic microarray data associated with two biological conditions. These conditions correspond to comparison of wild-type samples
versus knock-out ones, treated samples
versus non-treated ones,
etc. For these situations, we use the generic terms of control
versus test samples. Two condition biological data can be obtained using either one-color or two-color microarray technology leading to
intensities or
ratios, respectively. We assume that the same reference sample was used for
ratio data.
This paper is organized as follows. In the next section, we present the model used and describe its parameter settings. In
Section 3, we present commented results obtained using our model. Conclusions are drawn in
Section 4.
3. Results and Discussion
To evaluate the performance of the proposed model, we performed simulations, in which we studied the influence of different parameter settings. Their default values are: , , , , , , , , , , , , ). We performed 100 independent simulations by changing the initialization of the generator through parameter, rseed. For each simulation, we performed a Student t-test and selected genes with a p-value less than . Using the DE information, selected genes were split into two: true and false DE genes. The p-value threshold, , leads to an expected error (false discovery rate) equal to for default settings. When studying one parameter, the others are set to their default value.
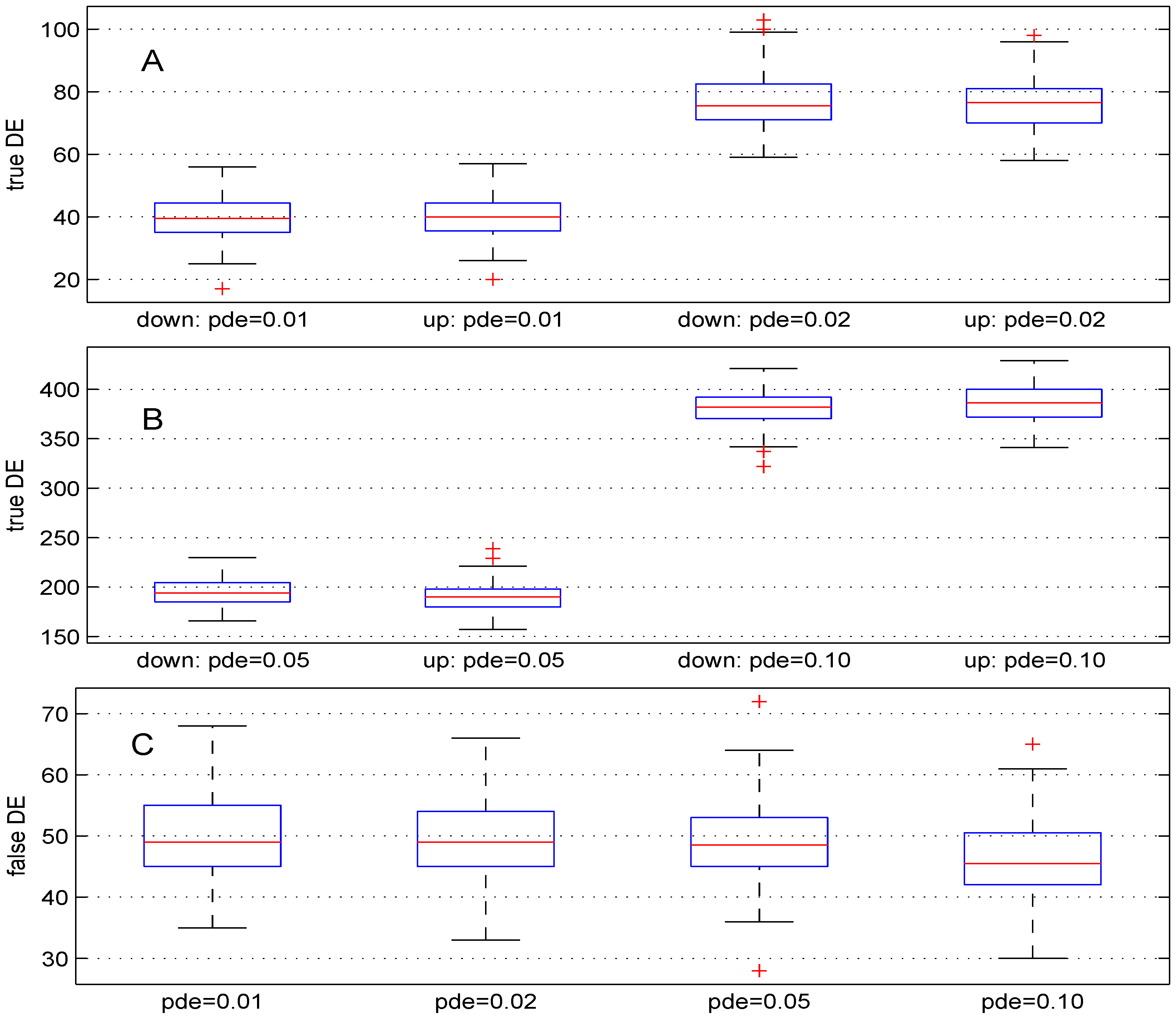
3.1. Parameter Pde
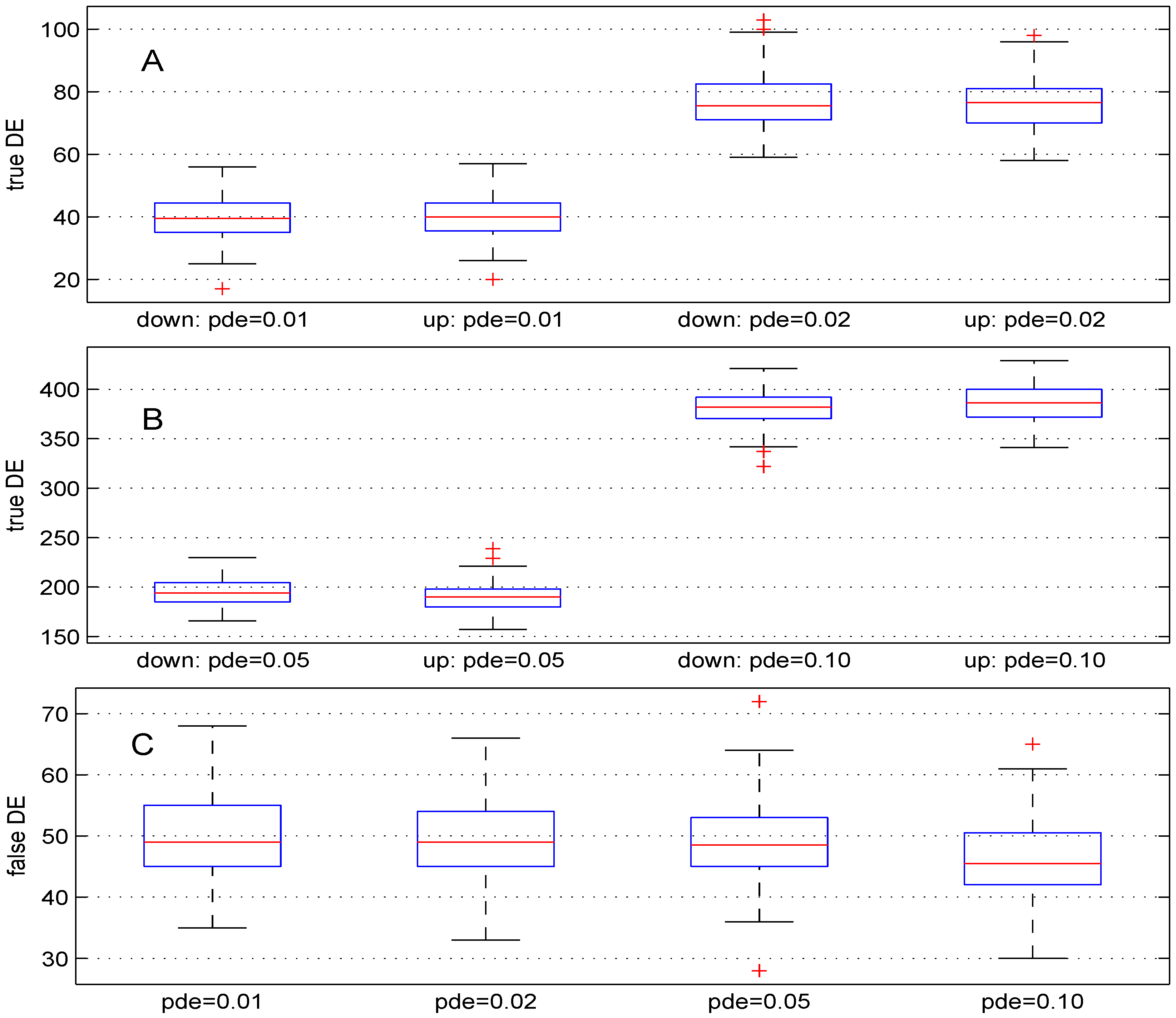
We used four different values (
,
,
and
). For these values, the theoretical numbers of DE genes are, respectively, 100, 200, 500 and
. The boxplots in
Figure 2 show the results obtained. The median numbers of true down- (up-) regulated genes are
(40),
(
), 194 (190) and 382 (
) for the above values of parameter
pde, respectively. In comparison with the expected number of DE genes, the recovery powers of the Student
t-test are
,
,
and
. Better power results can be obtained for this test by using smaller value for parameter
. Panel C of
Figure 2 shows the number of false DE genes obtained using the four values for parameter
pde. The median numbers of the false DE genes are 49, 49,
and
for the above values of parameter
pde, respectively.
Figure 2.
Boxplots of the number of down- and up-regulated genes (true DE, false DE) with four values of the parameter pde. 100 simulations were used for these results.
Figure 2.
Boxplots of the number of down- and up-regulated genes (true DE, false DE) with four values of the parameter pde. 100 simulations were used for these results.
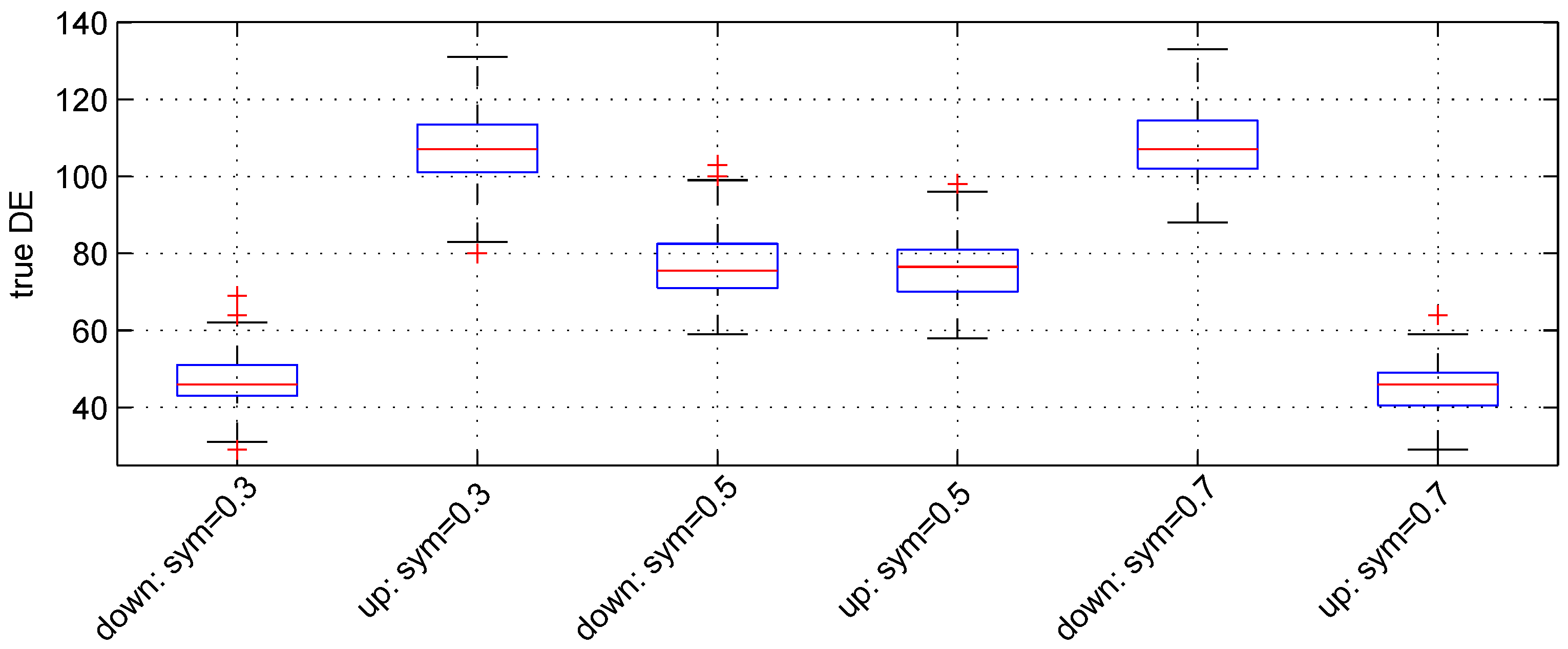
3.2. Parameter Sym
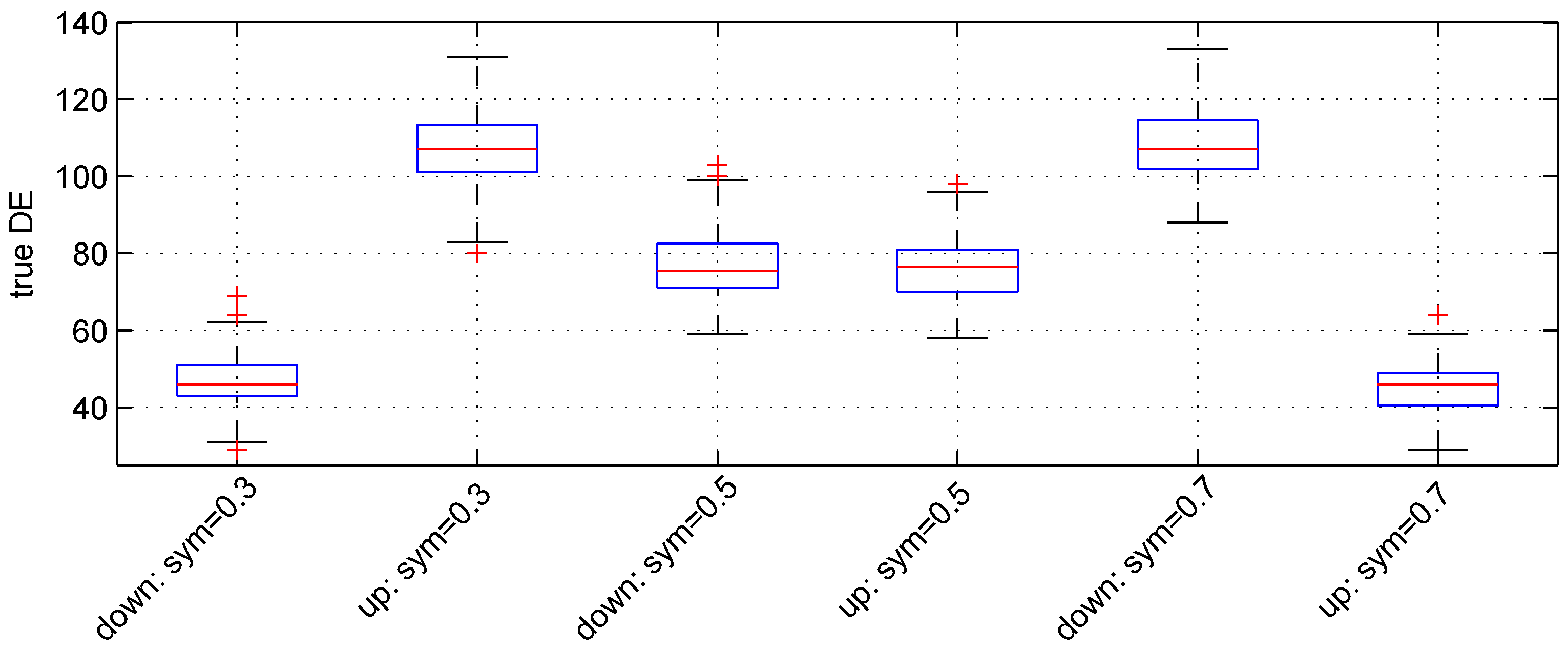
We used the following three values:
,
and
. For these values, the expected numbers for pairs of down- and up-regulated genes are, respectively, (
), (
) and (
).
Figure 3 shows the boxplot of results obtained. The median numbers of true down- and up-regulated genes observed are (46, 107), (
,
) and (107, 46) for the above values of parameter
, respectively. Hence, the recovery powers of the Student
t-test are
,
and
.
Figure 3.
Boxplots of the number of down- and up-regulated genes (true DE) with three values of the parameter sym. 100 simulations were used for these results.
Figure 3.
Boxplots of the number of down- and up-regulated genes (true DE) with three values of the parameter sym. 100 simulations were used for these results.
The median number of false DE genes is 49 for the three values of parameter sym.
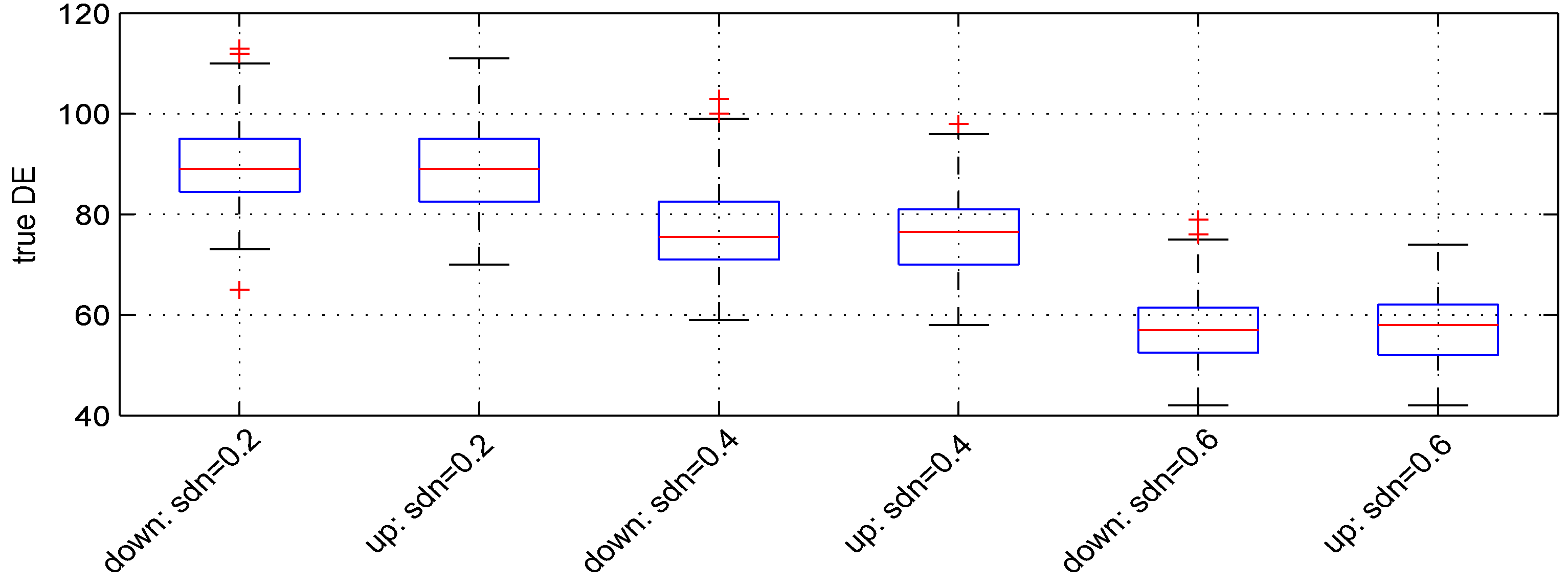
3.3. Parameter
We used three values:
,
and
.
Figure 4 shows the boxplots of the results obtained. The median numbers of down- and up-regulated genes observed are (89, 89), (
,
) and (57, 58), leading to detection powers of
,
and
, respectively. The
t-test detection power decreases when
increases.
Figure 4.
Boxplots of the number of down- and up-regulated genes (true DE) with three values of the parameter . 100 simulations were used for these results.
Figure 4.
Boxplots of the number of down- and up-regulated genes (true DE) with three values of the parameter . 100 simulations were used for these results.
The median numbers of false DE genes are 54, 49 and 49 for the 3 values of parameter , respectively.
3.4. Parameters , , ,
and
We performed simulations to examine the influence of these parameters. For each parameter, 100 simulations were used, and the results obtained are summarized in
Table 1. Increasing the parameter
setting introduces more change for the DE genes, while its decrease leads to the opposite effect. Parameter
acts as noise. The effect of the modification of some parameters is investigated further in the MA plot representations described in the following paragraph.
Table 1.
Number of down- and up-regulated detected genes using the Student t-test and various parameter settings.
Table 1.
Number of down- and up-regulated detected genes using the Student t-test and various parameter settings.
| Parameters | (down, up) | power |
|---|
| (100, 100) | |
| (41, 40) | |
| (, 90) | |
| (82, 83) | |
| (69, ) | |
| (76, 6) | |
| (81, 81) | |
| (89, 88) | |
| (92, 92) | |
| (66, 66) | |
| (92, 91) | |
| (, ) | |
| (75, 76) | |
| (76, ) | |
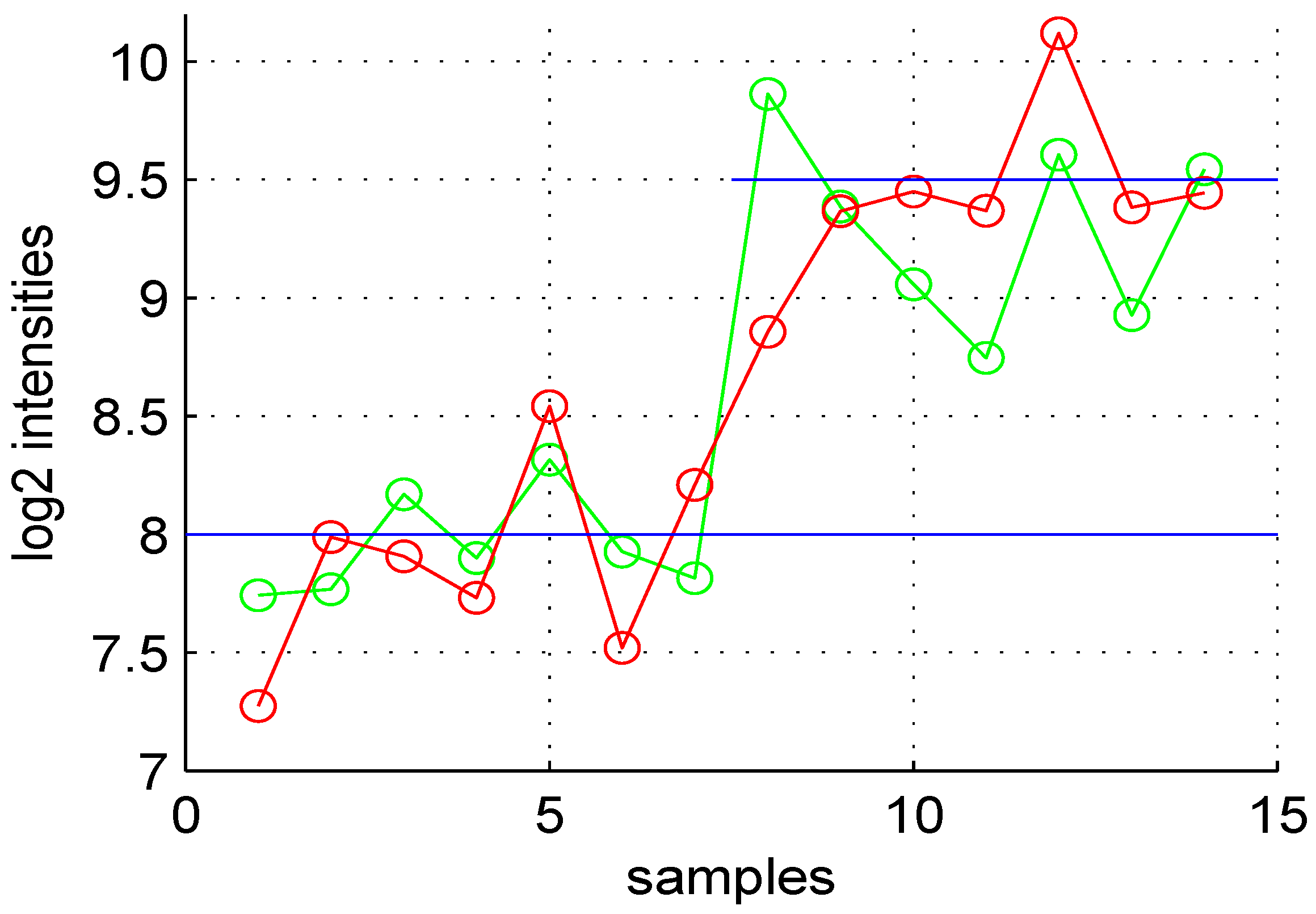
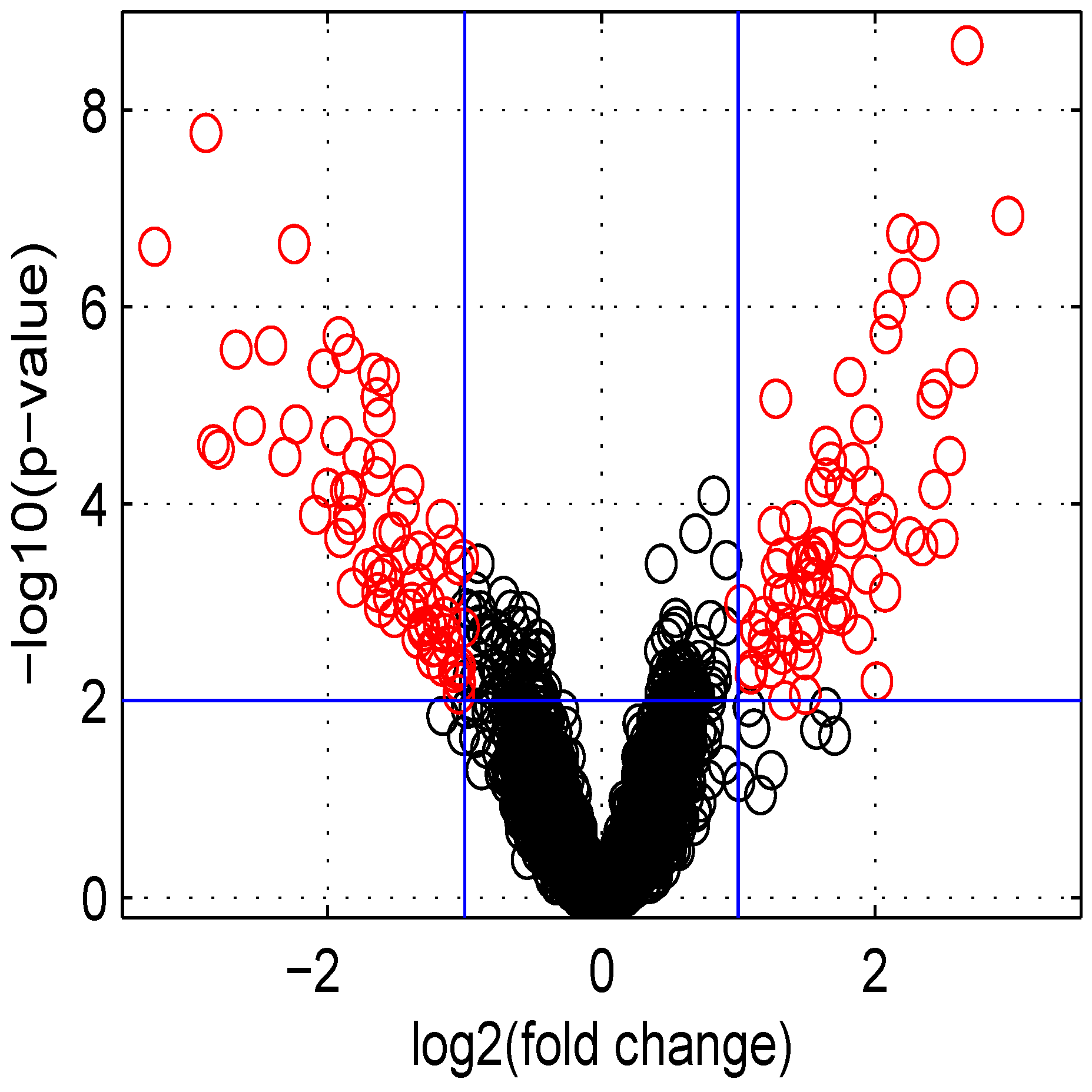
3.5. Volcano and MA Plots
Using default settings, we performed one simulation. Then we computed the Student
t-test
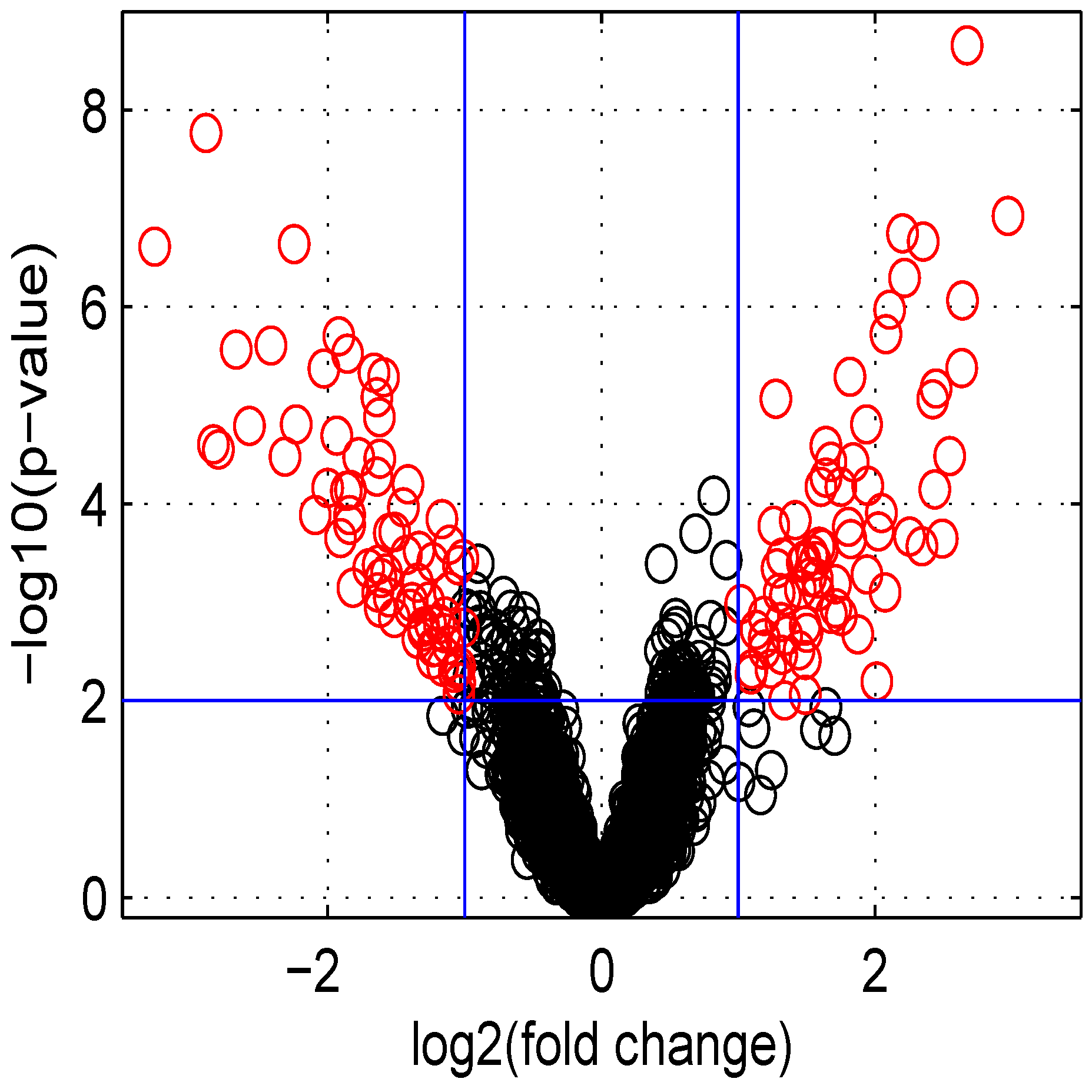
p-value and fold change for all genes. These values were used in the volcano plot of
Figure 5. Red circles represent genes having a
p-value less than
and a fold change greater than two or less than
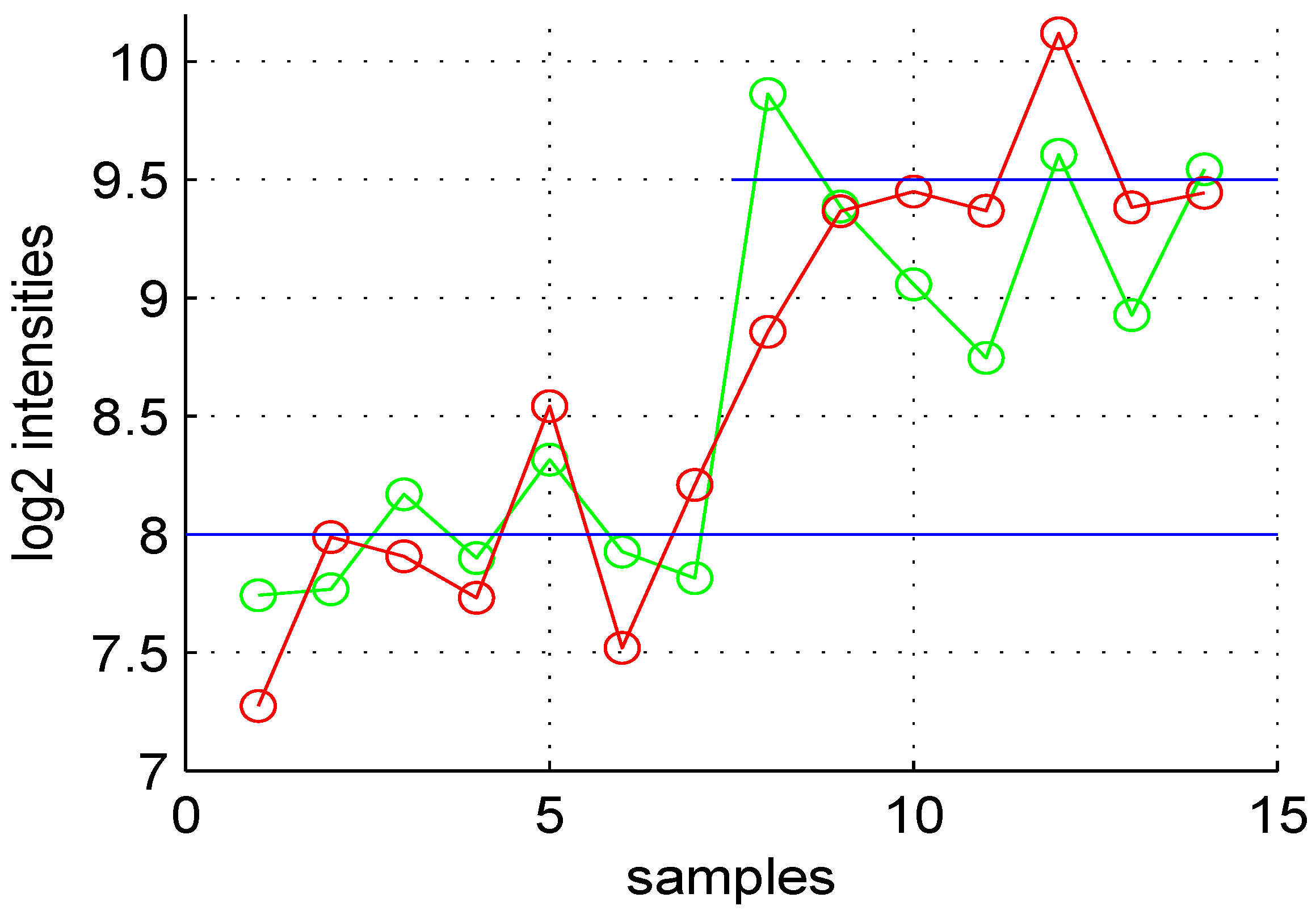
. Intensity measurements of two samples can be used to create two new variables:
and
, where
and
are
intensities of samples
and
, respectively. A value, one (
) for M means that the corresponding gene is up- (down-) regulated two-fold. A plot of
M (
ratio)
versus A (
intensities average) is denoted “MA plot" [
11]. The MA plots in
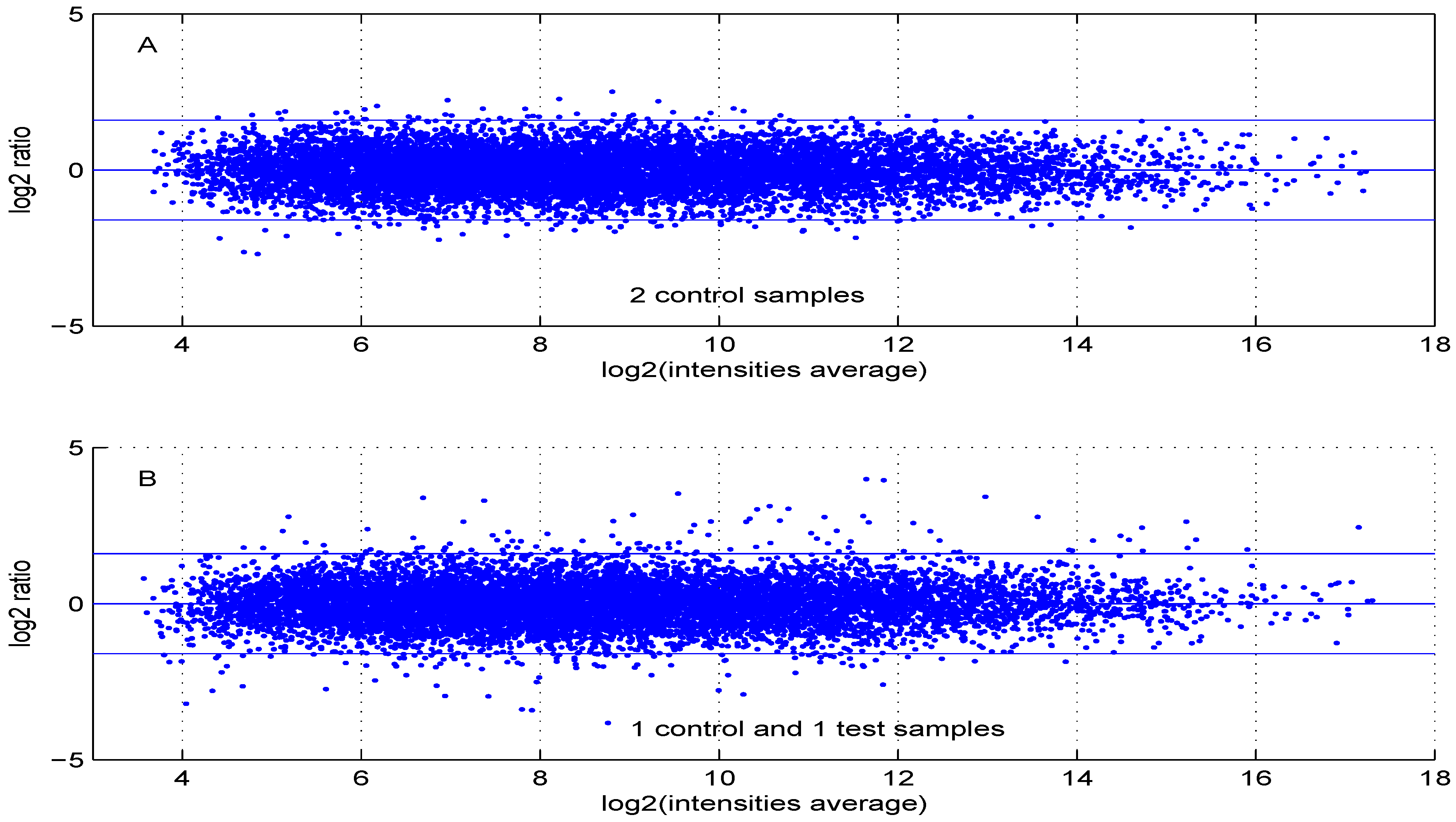
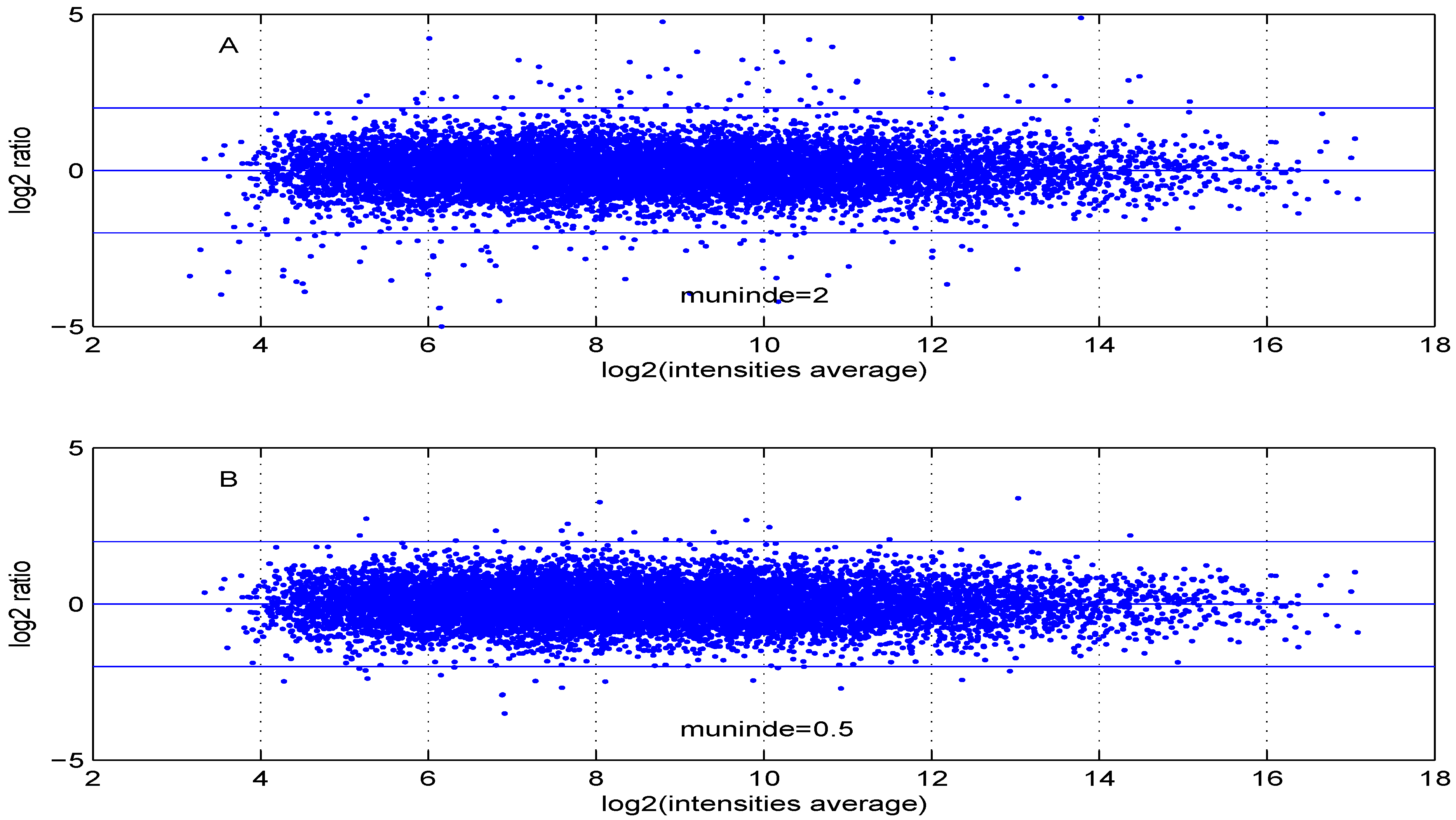
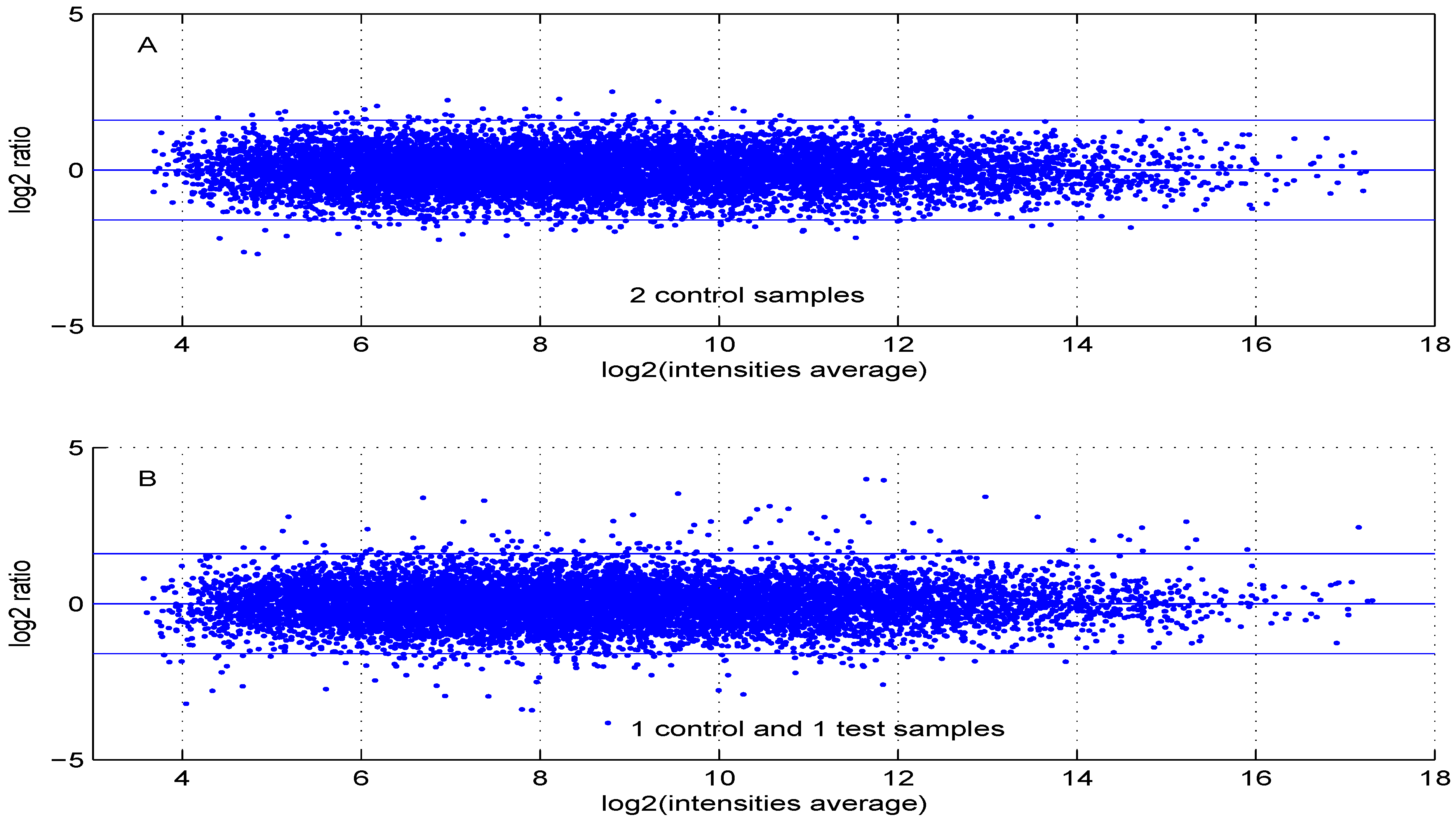
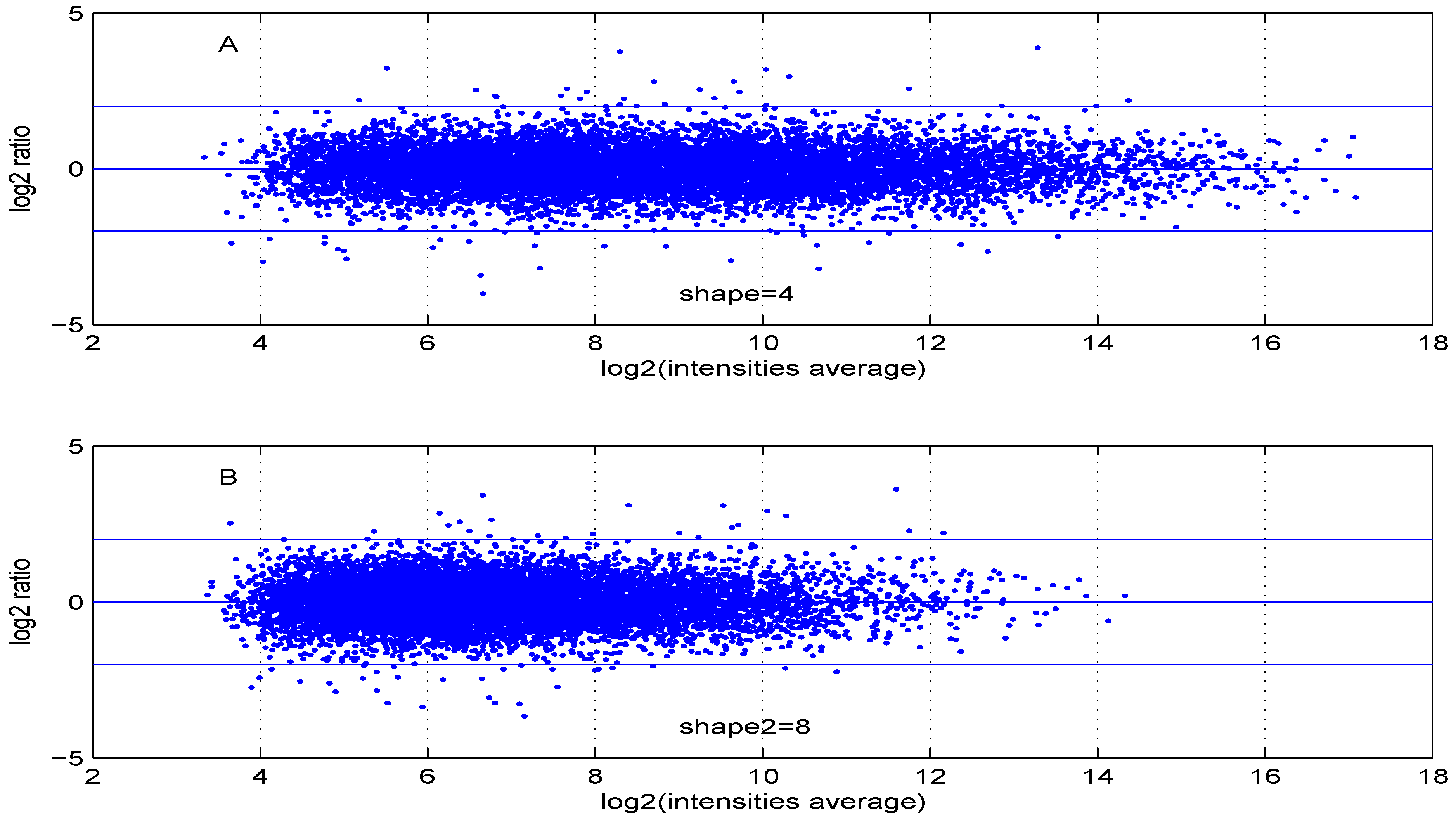
Figure 6 are obtained using either two control samples (panel A) or one control and one test sample (panel B).
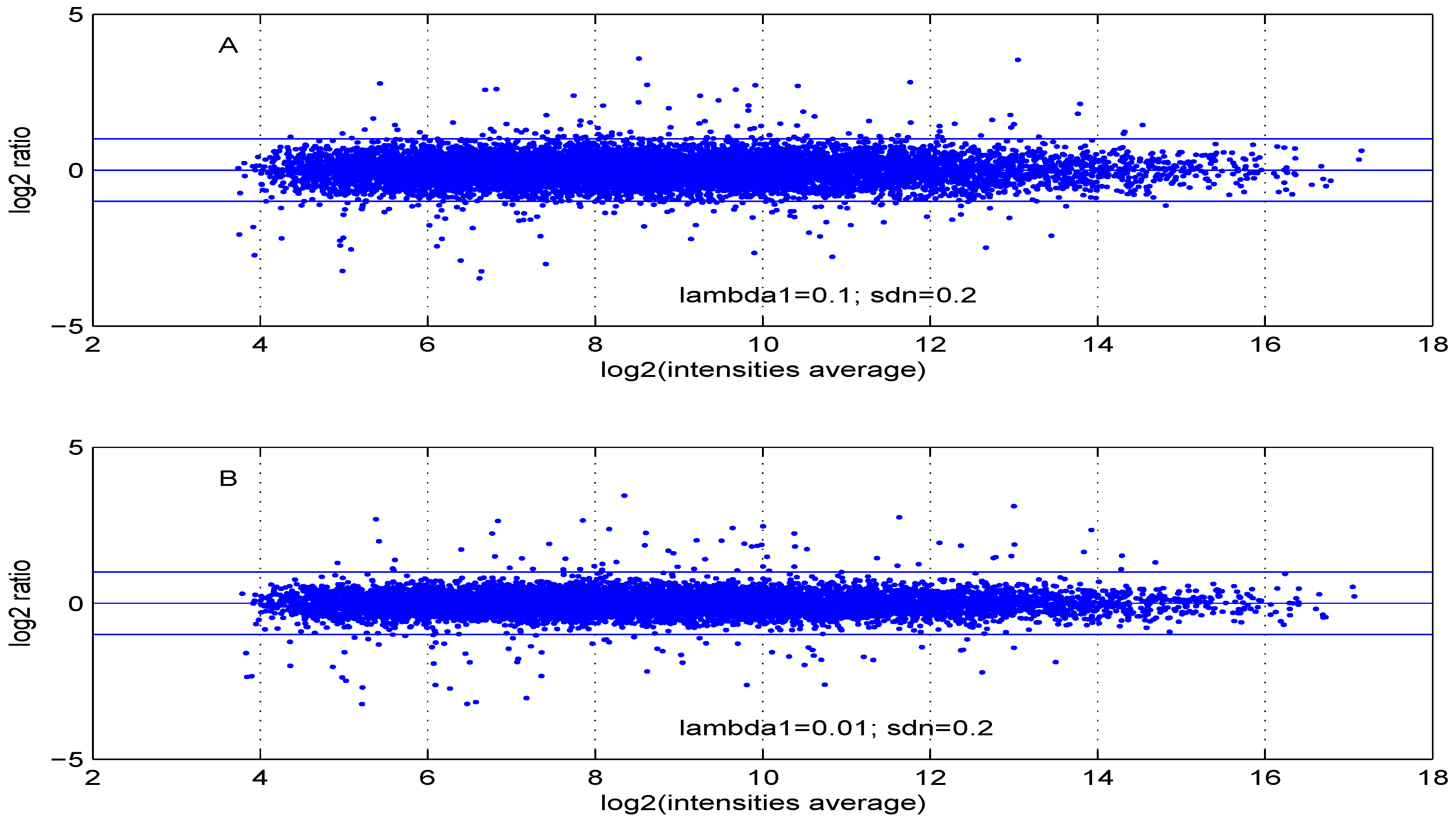
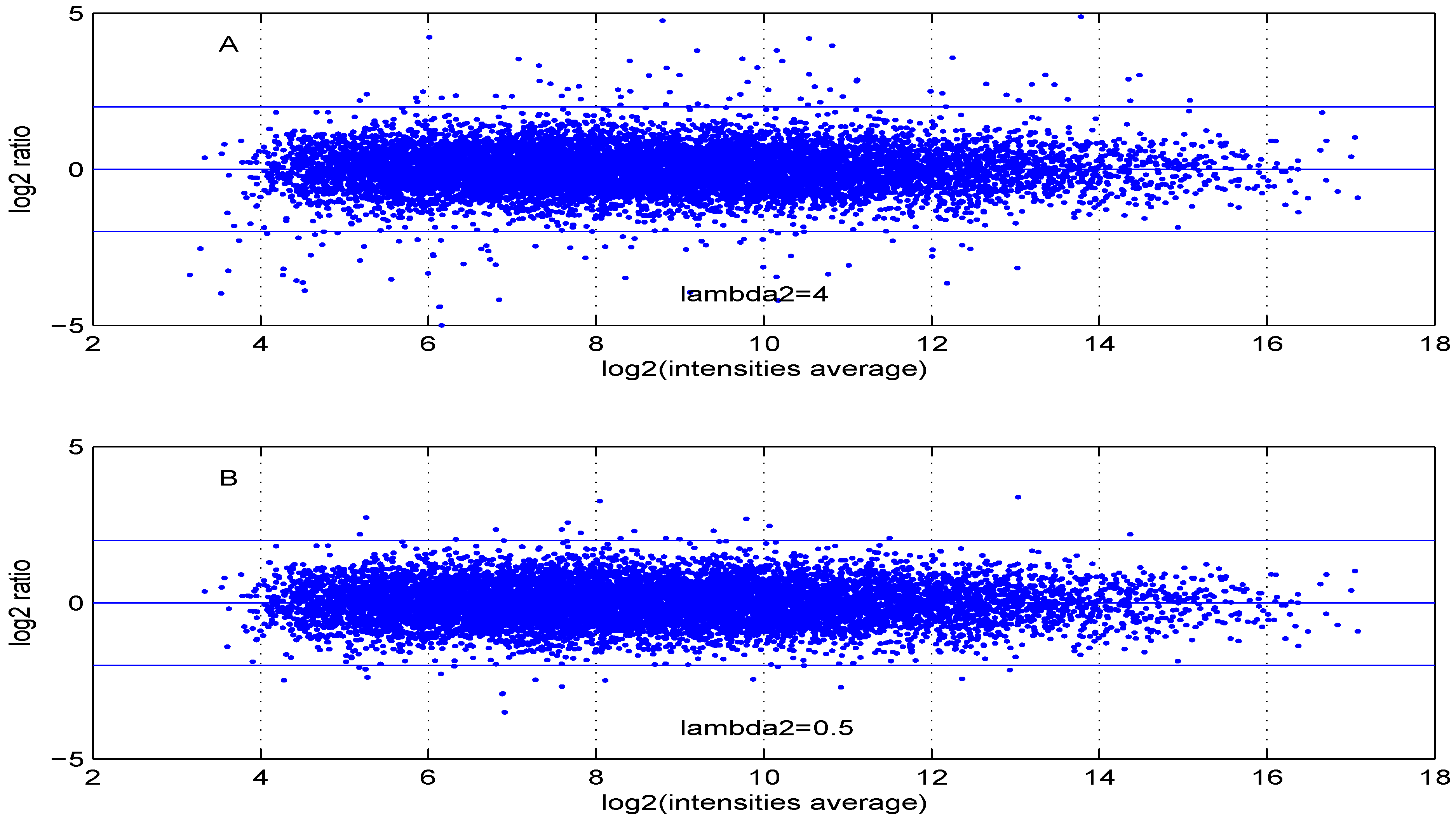
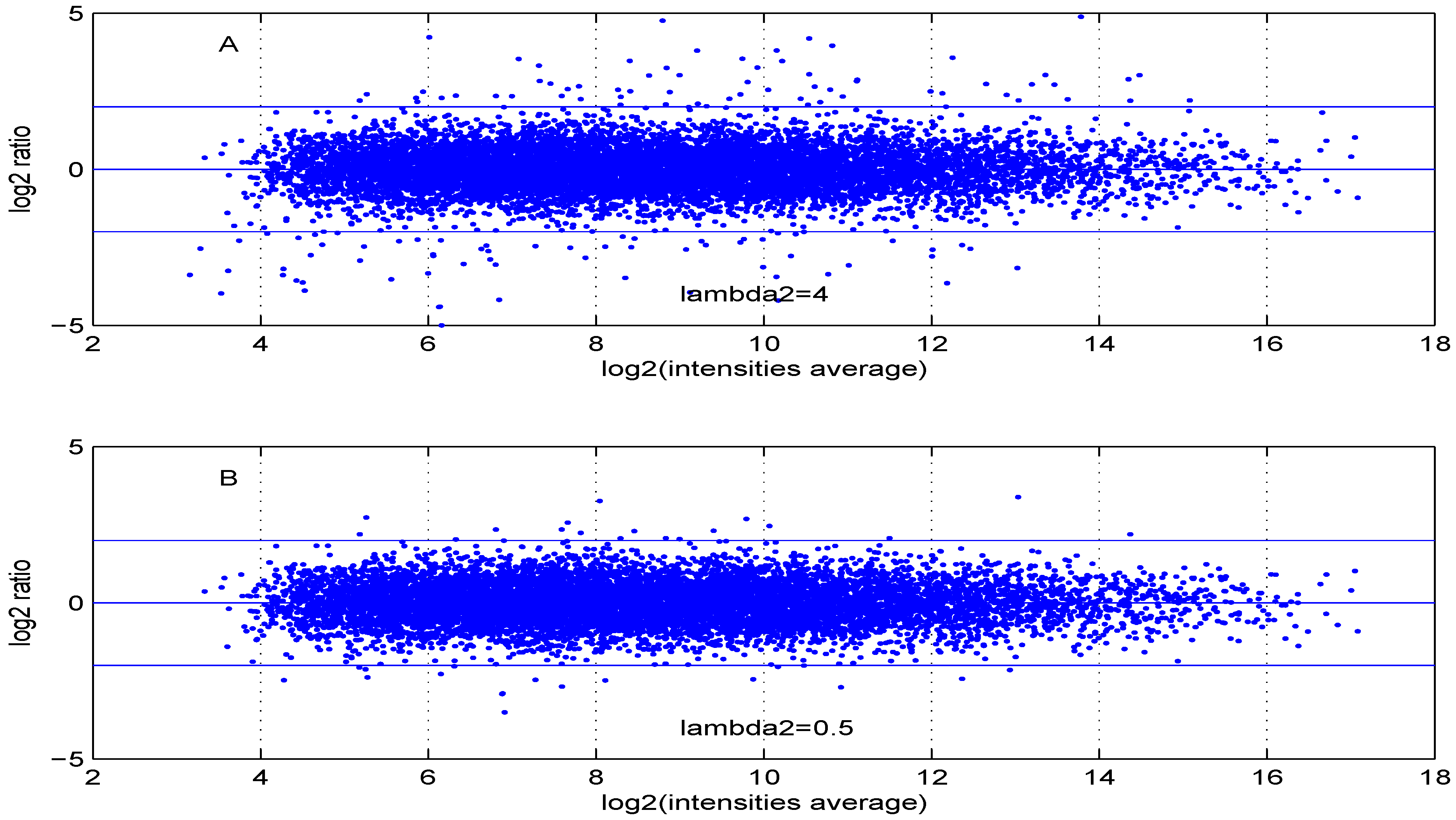
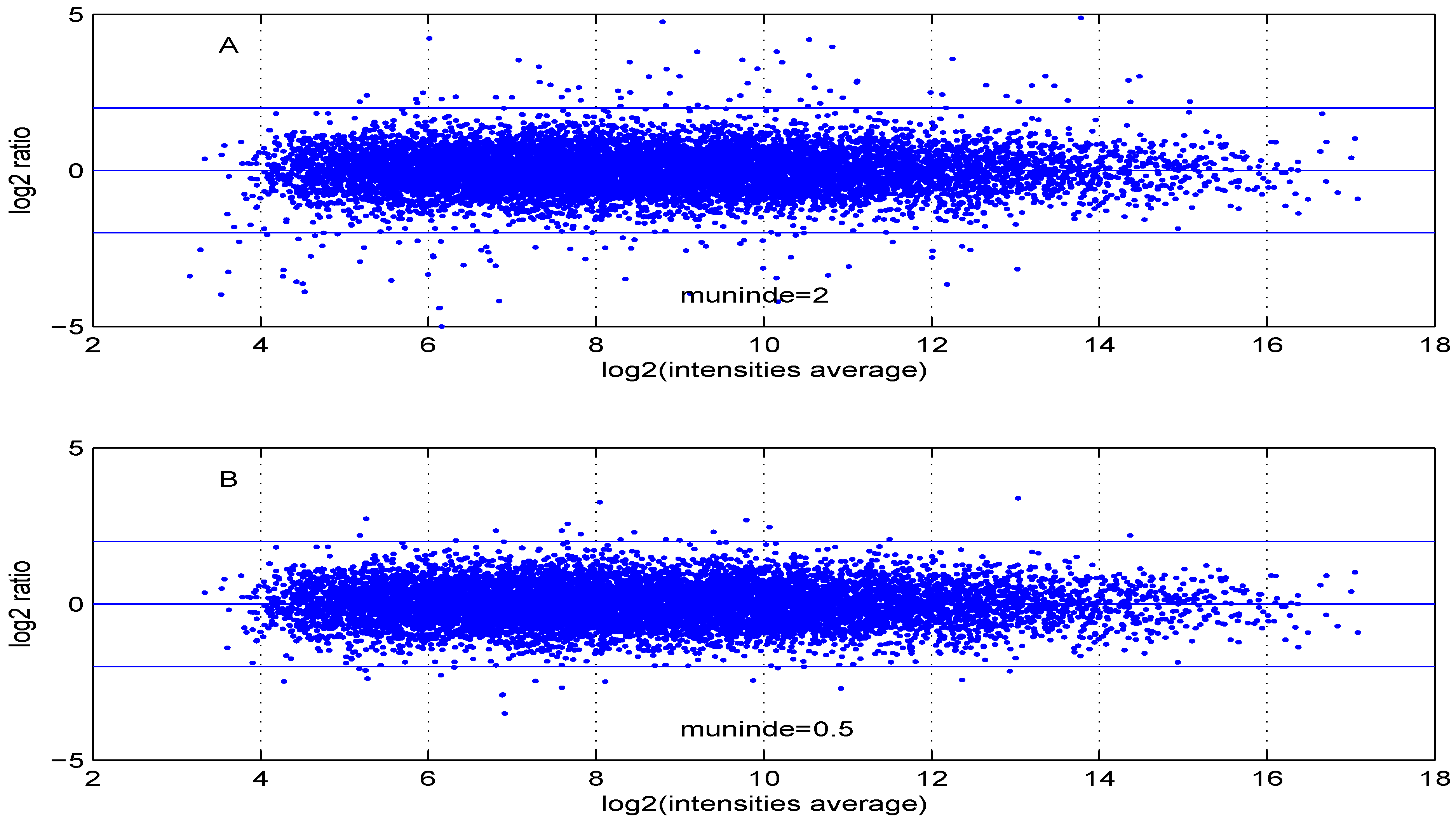
Additional MA plots in
Figure 7,
Figure 8,
Figure 9 and
Figure 10 showing the effect of some parameter settings. A small value for
leads to a less dense cloud of points. A larger change is observed for higher values of
than for smaller ones. The same applies to parameter,
. Increasing the parameter
value leads to a decrease of the dynamic range of the data.
Figure 5.
Volcano plot of data obtained.
Figure 5.
Volcano plot of data obtained.
Figure 6.
MA plot using (A) two control samples or (B) one control and one test sample data.
Figure 6.
MA plot using (A) two control samples or (B) one control and one test sample data.
Figure 7.
MA plot with one control and one test sample, using (A) or (B) .
Figure 7.
MA plot with one control and one test sample, using (A) or (B) .
Figure 8.
MA plot with one control and one test sample, using (A) or (B) .
Figure 8.
MA plot with one control and one test sample, using (A) or (B) .
Figure 9.
MA plot with one control and one test sample, using (A) or (B) .
Figure 9.
MA plot with one control and one test sample, using (A) or (B) .
Figure 10.
MA plot with one control and one test sample, using (A) or (B) .
Figure 10.
MA plot with one control and one test sample, using (A) or (B) .
3.6. Discussion
The choice of some setting parameters for the proposed model is easy and can be dictated by the experimental design. This applies to
n,
,
and
ratio. Parameters
lb and
ub and
sym and
pde concern the dynamic range of variation of the data to generate and define the DE genes. The intervals indicated for
lb and
ub are those observed for data from common platforms. Parameters,
,
and
, have no effect if real microarray data are used as a seed. The number of DE genes (
pde) and the proportion of under- and over-regulated genes (
sym) is at the discretion of the user. The parameter,
rseed, allows one to produce the same data at different times using the same computer. This parameter is also useful for generating test data for different analysis algorithms. Settings,
,
,
,
and
, control the global behavior of the data generated. More precisely,
allows one to make gene changes dependent on the average level of expression.
introduces variation in the expression changes for DE genes. These changes are defined by
and
. Parameter,
, also acts as noise. The parameter,
, allows one to perturb the data generated. The example of
Figure 4 shows that the data obtained can differ from those expected for large values of
. The signal to noise ratio (defined as the ratio of standard deviations) for default settings is
. This rises to
for
.
An interesting microarray study was reported in [
12]. In that study, the same RNA samples were processed by many laboratories using three leading microarray platforms: Affymetrix (five labs), two-color cDNA (three labs) and two-color oligonucleotides (two labs). The results presented show a good agreement across-platforms in contrast to some results previously reported in the literature, see, for instance, references cited in [
12]. The microarray data generated for the study described in [
12] are available from the Gene Expression Omnibus [
13] under accession number GSE2521. These data can be used as seed for our model, which can then be integrated into user friendly data analysis software, such as Partek Genomics Suite, GeneSpring GX,
etc., for demonstration and/or teaching purposes.
3.7. R Code
For immediate use of the proposed model, we provide an R code function
madsim.R (MicroArray Data Simulation Model), which is deposed as a package on the Comprehensible R Archive Network (CRAN) server for download [
14]. The outputs of this function are the data generated (
xdata) and the indexes of DE genes (
xid). Real data can be used as seed for each gene. An example of such data is available in the data folder of the package. Further explanations are available from the package’s help function.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}