Catalytic Antioxidants in the Kidney


Abstract
:
1. Introduction
2. Antioxidant Enzymes and Kidney Disease
2.1. Superoxide Dismutase and Kidney Disease
2.2. Catalase and Kidney Disease
2.3. Glutathione Peroxidase and Kidney Diseases
LOOH + 2GSH → GSSG + H2O + LOH
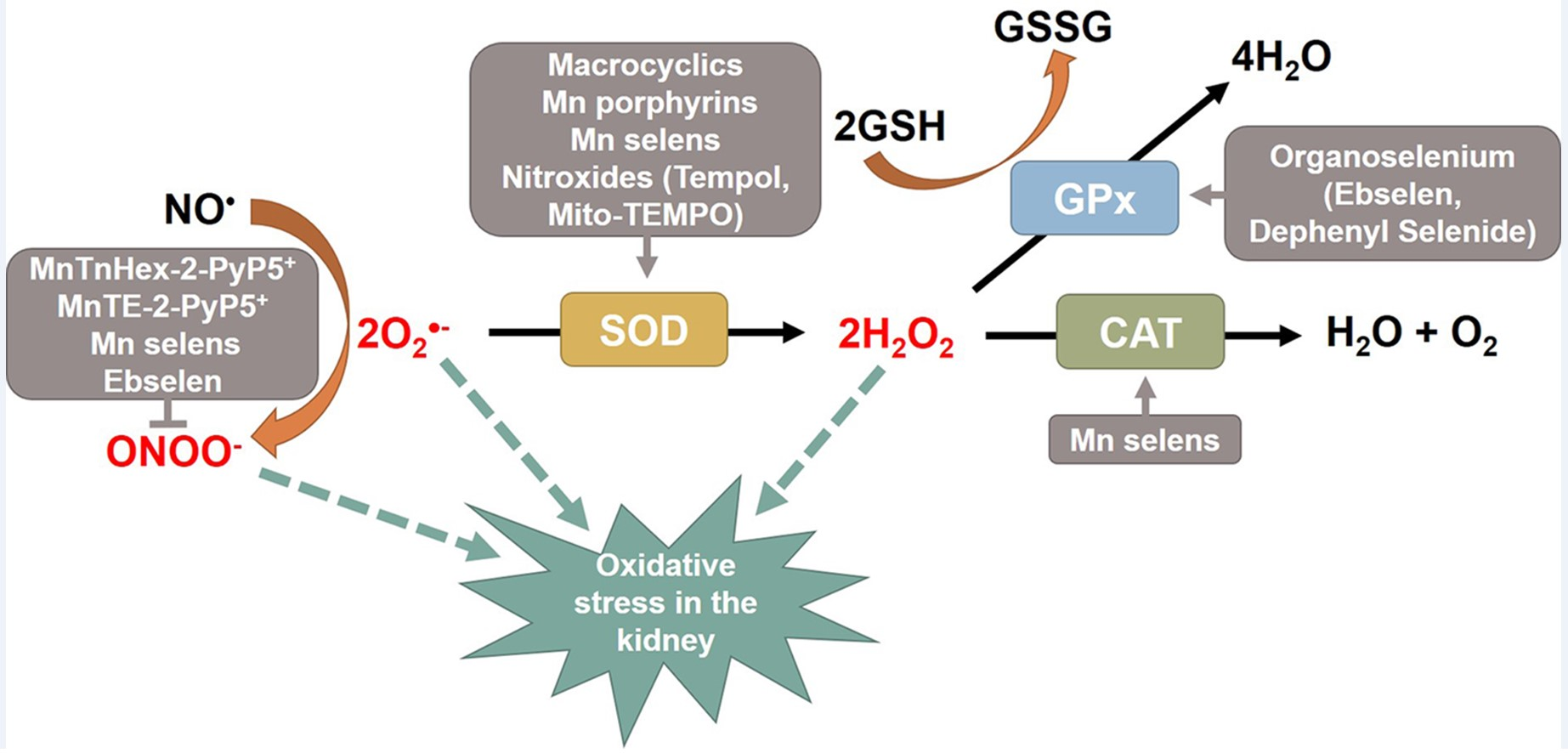
3. Catalytic Antioxidants
3.1. Catalytic Antioxidants as SOD and CAT Mimics
3.1.1. Macrocyclics
3.1.2. Mn porphyrins
3.1.3. Manganosalens
3.1.4. Nitroxides
3.2. Catalytic Antioxidants as GPx Mimics
3.2.1. Ebselen
3.2.2. Diphenyl Diselenide
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sies, H. Oxidative Stress: A Concept in Redox Biology and Medicine. Redox Biol. 2015, 4, 180–183. [Google Scholar] [CrossRef] [Green Version]
- Murphy, M.P. How Mitochondria Produce Reactive Oxygen Species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, N.; Jiang, S.; Persson, P.B.; Persson, E.A.G.; Lai, E.Y.; Patzak, A. Reactive Oxygen Species in Renal Vascular Function. Acta Physiol. 2020, 229, e13477. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Branicky, R.; Noë, A.; Hekimi, S. Superoxide Dismutases: Dual Roles in Controlling ROS Damage and Regulating ROS Signaling. J. Cell Biol. 2018, 217, 1915–1928. [Google Scholar] [CrossRef]
- Go, Y.M.; Jones, D.P. Redox Compartmentalization in Eukaryotic Cells. Biochim. Biophys. Acta 2008, 1780, 1273–1290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matés, J.M.; Pérez-Gómez, C.; Núñez de Castro, I. Antioxidant Enzymes and Human Diseases. Clin. Biochem. 1999, 32, 595–603. [Google Scholar] [CrossRef]
- Espinosa-Diez, C.; Miguel, V.; Mennerich, D.; Kietzmann, T.; Sanchez-Perez, P.; Cadenas, S.; Lamas, S. Antioxidant Responses and Cellular Adjustments to Oxidative Stress. Redox Biol. 2015, 6, 183–197. [Google Scholar] [CrossRef] [Green Version]
- Sharma, K. Obesity and Diabetic Kidney Disease: Role of Oxidant Stress and Redox Balance. Antioxid. Redox Signal. 2016, 25, 208–216. [Google Scholar] [CrossRef] [PubMed]
- Dennis, J.M.; Witting, P.K. Protective Role for Antioxidants in Acute Kidney Disease. Nutrients 2017, 9, 718. [Google Scholar] [CrossRef] [Green Version]
- Irazabal, M.V.; Torres, V.E. Reactive Oxygen Species and Redox Signaling in Chronic Kidney Disease. Cells 2020, 9, 1342. [Google Scholar] [CrossRef]
- Ratliff, B.B.; Abdulmahdi, W.; Pawar, R.; Wolin, M.S. Oxidant Mechanisms in Renal Injury and Disease. Antioxid. Redox Signal. 2016, 25, 119–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Day, B.J. Catalytic Antioxidants: A Radical Approach to New Therapeutics. Drug Discov. Today 2004, 9, 557–566. [Google Scholar] [CrossRef]
- Mirończuk-Chodakowska, I.; Witkowska, A.M.; Zujko, M.E. Endogenous Non-Enzymatic Antioxidants in the Human Body. Adv. Med. Sci. 2018, 63, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Pisoschi, A.M.; Pop, A. The Role of Antioxidants in the Chemistry of Oxidative Stress: A Review. Eur. J. Med. Chem. 2015, 97, 55–74. [Google Scholar] [CrossRef] [PubMed]
- Rouco, L.; González-Noya, A.M.; Pedrido, R.; Maneiro, M. Pursuing the Elixir of Life: In Vivo Antioxidative Effects of Manganosalen Complexes. Antioxidants 2020, 9, 727. [Google Scholar] [CrossRef] [PubMed]
- Zelko, I.N.; Mariani, T.J.; Folz, R.J. Superoxide Dismutase Multigene Family: A Comparison of the CuZn-SOD (SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3) Gene Structures, Evolution, and Expression. Free Radic. Biol. Med. 2002, 33, 337–349. [Google Scholar] [CrossRef]
- Marklund, S.L. Extracellular Superoxide Dismutase and Other Superoxide Dismutase Isoenzymes in Tissues from Nine Mammalian Species. Biochem. J. 1984, 222, 649–655. [Google Scholar] [CrossRef] [Green Version]
- Van Remmen, H.; Salvador, C.; Yang, H.; Huang, T.T.; Epstein, C.J.; Richardson, A. Characterization of the Antioxidant Status of the Heterozygous Manganese Superoxide Dismutase Knockout Mouse. Arch. Biochem. Biophys. 1999, 363, 91–97. [Google Scholar] [CrossRef]
- Schieber, M.; Chandel, N.S. ROS Function in Redox Signaling and Oxidative Stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [Green Version]
- Brzoska, K.; Sochanowicz, B.; Siomek, A.; Olinski, R.; Kruszewski, M. Alterations in the Expression of Genes Related to NF-kappaB Signaling in Liver and Kidney of CuZnSOD-Deficient Mice. Mol. Cell. Biochem. 2011, 353, 151–157. [Google Scholar] [CrossRef]
- Siomek, A.; Brzoska, K.; Sochanowicz, B.; Gackowski, D.; Rozalski, R.; Foksinski, M.; Zarakowska, E.; Szpila, A.; Guz, J.; Bartlomiejczyk, T.; et al. Cu,Zn-superoxide Dismutase Deficiency in Mice Leads to Organ-Specific Increase in Oxidatively Damaged DNA and NF-kappaB1 Protein Activity. Acta Biochim. Pol. 2010, 57, 577–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamanobe, T.; Okada, F.; Iuchi, Y.; Onuma, K.; Tomita, Y.; Fujii, J. Deterioration of Ischemia/Reperfusion-Induced Acute Renal Failure in SOD1-Deficient Mice. Free Radic. Res. 2007, 41, 200–207. [Google Scholar] [CrossRef] [PubMed]
- Yin, M.; Wheeler, M.D.; Connor, H.D.; Zhong, Z.; Bunzendahl, H.; Dikalova, A.; Samulski, R.J.; Schoonhoven, R.; Mason, R.P.; Swenberg, J.A.; et al. Cu/Zn-Superoxide Dismutase Gene Attenuates Ischemia-Reperfusion Injury in the Rat Kidney. J. Am. Soc. Nephrol. 2001, 12, 2691–2700. [Google Scholar] [PubMed]
- Carlström, M.; Brown, R.D.; Sällström, J.; Larsson, E.; Zilmer, M.; Zabihi, S.; Eriksson, U.J.; Persson, A.E. SOD1 Deficiency Causes Salt Sensitivity and Aggravates Hypertension in Hydronephrosis. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 297, R82–R92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlström, M.; Lai, E.Y.; Ma, Z.; Steege, A.; Patzak, A.; Eriksson, U.J.; Lundberg, J.O.; Wilcox, C.S.; Persson, A.E. Superoxide Dismutase 1 Limits Renal Microvascular Remodeling and Attenuates Arteriole and Blood Pressure Responses to Angiotensin II via Modulation of Nitric Oxide Bioavailability. Hypertension 2010, 56, 907–913. [Google Scholar] [CrossRef] [Green Version]
- Cepas, V.; Collino, M.; Mayo, J.C.; Sainz, R.M. Redox Signaling and Advanced Glycation Endproducts (AGEs) in Diet-Related Diseases. Antioxidants 2020, 9, 142. [Google Scholar] [CrossRef] [Green Version]
- DeRubertis, F.R.; Craven, P.A.; Melhem, M.F.; Salah, E.M. Attenuation of Renal Injury in db/db Mice Overexpressing Superoxide Dismutase: Evidence for Reduced Superoxide-Nitric Oxide Interaction. Diabetes 2004, 53, 762–768. [Google Scholar] [CrossRef] [Green Version]
- Craven, P.A.; Melhem, M.F.; Phillips, S.L.; DeRubertis, F.R. Overexpression of Cu2+/Zn2+ Superoxide Dismutase Protects against Early Diabetic Glomerular Injury in Transgenic Mice. Diabetes 2001, 50, 2114–2125. [Google Scholar] [CrossRef] [Green Version]
- Kitada, M.; Xu, J.; Ogura, Y.; Monno, I.; Koya, D. Manganese Superoxide Dismutase Dysfunction and the Pathogenesis of Kidney Disease. Front. Physiol. 2020, 11, 755. [Google Scholar] [CrossRef]
- Parajuli, N.; Marine, A.; Simmons, S.; Saba, H.; Mitchell, T.; Shimizu, T.; Shirasawa, T.; Macmillan-Crow, L.A. Generation and Characterization of a Novel Kidney-Specific Manganese Superoxide Dismutase Knockout Mouse. Free Radic. Biol. Med. 2011, 51, 406–416. [Google Scholar] [CrossRef] [Green Version]
- Parajuli, N.; MacMillan-Crow, L.A. Role of Reduced Manganese Superoxide Dismutase in Ischemia-Reperfusion Injury: A Possible Trigger for Autophagy and Mitochondrial Biogenesis? Am. J. Physiol. Renal Physiol. 2013, 304, F257–F267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pisani, A.; Sabbatini, M.; Riccio, E.; Rossano, R.; Andreucci, M.; Capasso, C.; De Luca, V.; Carginale, V.; Bizzarri, M.; Borrelli, A.; et al. Effect of a Recombinant Manganese Superoxide Dismutase on Prevention of Contrast-Induced Acute Kidney Injury. Clin. Exp. Nephrol. 2014, 18, 424–431. [Google Scholar] [CrossRef] [PubMed]
- Jin, K.; Vaziri, N.D. Salt-Sensitive Hypertension in Mitochondrial Superoxide Dismutase Deficiency Is Associated with Intra-Renal Oxidative Stress and Inflammation. Clin. Exp. Nephrol. 2014, 18, 445–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez-Iturbe, B.; Sepassi, L.; Quiroz, Y.; Ni, Z.; Wallace, D.C.; Vaziri, N.D. Association of Mitochondrial SOD Deficiency with Salt-Sensitive Hypertension and Accelerated Renal Senescence. J. Appl. Physiol. 2007, 102, 255–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, C.; Huang, A.; Wu, Z.; Kaminski, P.M.; Wolin, M.S.; Hintze, T.H.; Kaley, G.; Sun, D. Increased Superoxide Leads to Decreased Flow-Induced Dilation in Resistance Arteries of Mn-SOD-Deficient Mice. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H2225–H2231. [Google Scholar] [CrossRef] [PubMed]
- Forbes, J.M.; Thorburn, D.R. Mitochondrial Dysfunction in Diabetic Kidney Disease. Nat. Rev. Nephrol. 2018, 14, 291–312. [Google Scholar] [CrossRef]
- Sharma, K. Mitochondrial Dysfunction in the Diabetic Kidney. Adv. Exp. Med. Biol. 2017, 982, 553–562. [Google Scholar]
- Li, C.; Matavelli, L.C.; Akhtar, S.; Siragy, H.M. (Pro)renin Receptor Contributes to Renal Mitochondria Dysfunction, Apoptosis and Fibrosis in Diabetic Mice. Sci. Rep. 2019, 9, 11667. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.Y.; Lim, J.H.; Youn, H.H.; Hong, Y.A.; Yang, K.S.; Park, H.S.; Chung, S.; Ko, S.H.; Shin, S.J.; Choi, B.S.; et al. Resveratrol Prevents Renal Lipotoxicity and Inhibits Mesangial Cell Glucotoxicity in a Manner Dependent on the AMPK-SIRT1-PGC1alpha Axis in db/db Mice. Diabetologia 2013, 56, 204–217. [Google Scholar] [CrossRef] [Green Version]
- De Cavanagh, E.M.; Ferder, L.; Toblli, J.E.; Piotrkowski, B.; Stella, I.; Fraga, C.G.; Inserra, F. Renal Mitochondrial Impairment Is Attenuated by AT1 Blockade in Experimental Type I Diabetes. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H456–H465. [Google Scholar] [CrossRef] [Green Version]
- Hong, Y.A.; Lim, J.H.; Kim, M.Y.; Kim, T.W.; Kim, Y.; Yang, K.S.; Park, H.S.; Choi, S.R.; Chung, S.; Kim, H.W.; et al. Fenofibrate Improves Renal Lipotoxicity Through Activation of AMPK-PGC-1alpha in db/db Mice. PLoS ONE 2014, 9, e96147. [Google Scholar]
- Fujita, H.; Fujishima, H.; Chida, S.; Takahashi, K.; Qi, Z.; Kanetsuna, Y.; Breyer, M.D.; Harris, R.C.; Yamada, Y.; Takahashi, T. Reduction of Renal Superoxide Dismutase in Progressive Diabetic Nephropathy. J. Am. Soc. Nephrol. 2009, 20, 1303–1313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dugan, L.L.; You, Y.H.; Ali, S.S.; Diamond-Stanic, M.; Miyamoto, S.; DeCleves, A.E.; Andreyev, A.; Quach, T.; Ly, S.; Shekhtman, G.; et al. AMPK Dysregulation Promotes Diabetes-Related Reduction of Superoxide and Mitochondrial Function. J. Clin. Investig. 2013, 123, 4888–4899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, O.; Marklund, S.L.; Geiger, H.; Pedrazzini, T.; Busse, R.; Brandes, R.P. Extracellular Superoxide Dismutase Is a Major Determinant of Nitric Oxide Bioavailability: In Vivo and Ex Vivo Evidence from ecSOD-Deficient Mice. Circ. Res. 2003, 93, 622–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suliman, H.B.; Ali, M.; Piantadosi, C.A. Superoxide Dismutase-3 Promotes Full Expression of the EPO Response to Hypoxia. Blood 2004, 104, 43–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, M.P.; Sullivan, J.C.; Wach, P.F.; Boesen, E.I.; Yamamoto, T.; Fukai, T.; Harrison, D.G.; Pollock, D.M.; Pollock, J.S. Protective Role of Extracellular Superoxide Dismutase in Renal Ischemia/Reperfusion Injury. Kidney Int. 2010, 78, 374–381. [Google Scholar] [CrossRef] [Green Version]
- Tan, R.J.; Zhou, D.; Xiao, L.; Zhou, L.; Li, Y.; Bastacky, S.I.; Oury, T.D.; Liu, Y. Extracellular Superoxide Dismutase Protects against Proteinuric Kidney Disease. J. Am. Soc. Nephrol. 2015, 26, 2447–2459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujita, H.; Fujishima, H.; Takahashi, K.; Sato, T.; Shimizu, T.; Morii, T.; Shimizu, T.; Shirasawa, T.; Qi, Z.; Breyer, M.D.; et al. SOD1, but Not SOD3, Deficiency Accelerates Diabetic Renal Injury in C57BL/6-Ins2(Akita) Diabetic Mice. Metabolism 2012, 61, 1714–1724. [Google Scholar] [CrossRef] [Green Version]
- Kuo, C.W.; Shen, C.J.; Tung, Y.T.; Chen, H.L.; Chen, Y.H.; Chang, W.H.; Cheng, K.C.; Yang, S.H.; Chen, C.M. Extracellular Superoxide Dismutase Ameliorates Streptozotocin-Induced Rat Diabetic Nephropathy via Inhibiting the ROS/ERK1/2 Signaling. Life Sci. 2015, 135, 77–86. [Google Scholar] [CrossRef]
- Hong, Y.A.; Lim, J.H.; Kim, M.Y.; Kim, Y.; Park, H.S.; Kim, H.W.; Choi, B.S.; Chang, Y.S.; Kim, H.W.; Kim, T.Y.; et al. Extracellular Superoxide Dismutase Attenuates Renal Oxidative Stress Through the Activation of Adenosine Monophosphate-Activated Protein Kinase in Diabetic Nephropathy. Antioxid. Redox Signal. 2018, 28, 1543–1561. [Google Scholar] [CrossRef]
- Ho, Y.S.; Xiong, Y.; Ma, W.; Spector, A.; Ho, D.S. Mice Lacking Catalase Develop Normally but Show Differential Sensitivity to Oxidant Tissue Injury. J. Biol. Chem. 2004, 279, 32804–32812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Z.; Kang, Y.J. Cellular and Subcellular Localization of Catalase in the Heart of Transgenic Mice. J. Histochem. Cytochem. 2000, 48, 585–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, I.; Lee, J.; Huh, J.Y.; Park, J.; Lee, H.B.; Ho, Y.S.; Ha, H. Catalase Deficiency Accelerates Diabetic Renal Injury Through Peroxisomal Dysfunction. Diabetes 2012, 61, 728–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sunami, R.; Sugiyama, H.; Wang, D.H.; Kobayashi, M.; Maeshima, Y.; Yamasaki, Y.; Masuoka, N.; Ogawa, N.; Kira, S.; Makino, H. Acatalasemia Sensitizes Renal Tubular Epithelial Cells to Apoptosis and Exacerbates Renal Fibrosis after Unilateral Ureteral Obstruction. Am. J. Physiol. Renal Physiol. 2004, 286, F1030–F1038. [Google Scholar] [CrossRef]
- Kobayashi, M.; Sugiyama, H.; Wang, D.H.; Toda, N.; Maeshima, Y.; Yamasaki, Y.; Masuoka, N.; Yamada, M.; Kira, S.; Makino, H. Catalase Deficiency Renders Remnant Kidneys More Susceptible to Oxidant Tissue Injury and Renal Fibrosis in Mice. Kidney Int. 2005, 68, 1018–1031. [Google Scholar] [CrossRef] [Green Version]
- Takiue, K.; Sugiyama, H.; Inoue, T.; Morinaga, H.; Kikumoto, Y.; Kitagawa, M.; Kitamura, S.; Maeshima, Y.; Wang, D.H.; Masuoka, N.; et al. Acatalasemic Mice Are Mildly Susceptible to Adriamycin Nephropathy and Exhibit Increased Albuminuria and Glomerulosclerosis. BMC Nephrol. 2012, 13, 14. [Google Scholar] [CrossRef]
- Brezniceanu, M.L.; Liu, F.; Wei, C.C.; Tran, S.; Sachetelli, S.; Zhang, S.L.; Guo, D.F.; Filep, J.G.; Ingelfinger, J.R.; Chan, J.S. Catalase Overexpression Attenuates Angiotensinogen Expression and Apoptosis in Diabetic Mice. Kidney Int. 2007, 71, 912–923. [Google Scholar] [CrossRef] [Green Version]
- Brezniceanu, M.L.; Liu, F.; Wei, C.C.; Chénier, I.; Godin, N.; Zhang, S.L.; Filep, J.G.; Ingelfinger, J.R.; Chan, J.S. Attenuation of Interstitial Fibrosis and Tubular Apoptosis in db/db Transgenic Mice Overexpressing Catalase in Renal Proximal Tubular Cells. Diabetes 2008, 57, 451–459. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Lo, C.S.; Chenier, I.; Maachi, H.; Filep, J.G.; Ingelfinger, J.R.; Zhang, S.L.; Chan, J.S. Overexpression of Catalase Prevents Hypertension and Tubulointerstitial Fibrosis and Normalization of Renal Angiotensin-Converting Enzyme-2 Expression in Akita Mice. Am. J. Physiol. Renal Physiol. 2013, 304, F1335–F1346. [Google Scholar] [CrossRef] [Green Version]
- Abdo, S.; Shi, Y.; Otoukesh, A.; Ghosh, A.; Lo, C.S.; Chenier, I.; Filep, J.G.; Ingelfinger, J.R.; Zhang, S.L.; Chan, J.S. Catalase Overexpression Prevents Nuclear Factor Erythroid 2-Related Factor 2 Stimulation of Renal Angiotensinogen Gene Expression, Hypertension, and Kidney Injury in Diabetic Mice. Diabetes 2014, 63, 3483–3496. [Google Scholar] [CrossRef] [Green Version]
- Godin, N.; Liu, F.; Lau, G.J.; Brezniceanu, M.L.; Chénier, I.; Filep, J.G.; Ingelfinger, J.R.; Zhang, S.L.; Chan, J.S. Catalase Overexpression Prevents Hypertension and Tubular Apoptosis in Angiotensinogen Transgenic Mice. Kidney Int. 2010, 77, 1086–1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flohe, L.; Günzler, W.A.; Schock, H.H. Glutathione Peroxidase: A Selenoenzyme. FEBS Lett. 1973, 32, 132–134. [Google Scholar] [CrossRef] [Green Version]
- Schafer, F.Q.; Buettner, G.R. Redox Environment of the Cell as Viewed Through the Redox State of the Glutathione Disulfide/Glutathione Couple. Free Radic. Biol. Med. 2001, 30, 1191–1212. [Google Scholar] [CrossRef]
- Lei, X.G.; Cheng, W.H. New Roles for an Old Selenoenzyme: Evidence from Glutathione Peroxidase-1 Null and Overexpressing Mice. J. Nutr. 2005, 135, 2295–2298. [Google Scholar] [CrossRef] [Green Version]
- Day, B.J. Catalase and Glutathione Peroxidase Mimics. Biochem. Pharmacol. 2009, 77, 285–296. [Google Scholar] [CrossRef] [Green Version]
- Behne, D.; Kyriakopoulos, A. Mammalian Selenium-Containing Proteins. Annu. Rev. Nutr. 2001, 21, 453–473. [Google Scholar] [CrossRef]
- Muse, K.E.; Oberley, T.D.; Sempf, J.M.; Oberley, L.W. Immunolocalization of Antioxidant Enzymes in Adult Hamster Kidney. Histochem. J. 1994, 26, 734–753. [Google Scholar] [CrossRef]
- Wiedenmann, T.; Dietrich, N.; Fleming, T.; Altamura, S.; Deelman, L.E.; Henning, R.H.; Muckenthaler, M.U.; Nawroth, P.P.; Hammes, H.P.; Wagner, A.H.; et al. Modulation of Glutathione Peroxidase Activity by Age-Dependent Carbonylation in Glomeruli of Diabetic Mice. J. Diabetes Complicat. 2018, 32, 130–138. [Google Scholar] [CrossRef]
- Olson, G.E.; Whitin, J.C.; Hill, K.E.; Winfrey, V.P.; Motley, A.K.; Austin, L.M.; Deal, J.; Cohen, H.J.; Burk, R.F. Extracellular Glutathione Peroxidase (Gpx3) Binds Specifically to Basement Membranes of Mouse Renal Cortex Tubule Cells. Am. J. Physiol. Renal Physiol. 2010, 298, F1244–F1253. [Google Scholar] [CrossRef] [Green Version]
- De Haan, J.B.; Bladier, C.; Griffiths, P.; Kelner, M.; O’Shea, R.D.; Cheung, N.S.; Bronson, R.T.; Silvestro, M.J.; Wild, S.; Zheng, S.S.; et al. Mice with a Homozygous Null Mutation for the Most Abundant Glutathione Peroxidase, Gpx1, Show Increased Susceptibility to the Oxidative Stress-Inducing Agents Paraquat and Hydrogen Peroxide. J. Biol. Chem. 1998, 273, 22528–22536. [Google Scholar] [CrossRef] [Green Version]
- De Haan, J.B.; Stefanovic, N.; Nikolic-Paterson, D.; Scurr, L.L.; Croft, K.D.; Mori, T.A.; Hertzog, P.; Kola, I.; Atkins, R.C.; Tesch, G.H. Kidney Expression of Glutathione Peroxidase-1 Is Not Protective against Streptozotocin-Induced Diabetic Nephropathy. Am. J. Physiol. Renal Physiol. 2005, 289, F544–F551. [Google Scholar] [CrossRef] [PubMed]
- Esposito, L.A.; Kokoszka, J.E.; Waymire, K.G.; Cottrell, B.; MacGregor, G.R.; Wallace, D.C. Mitochondrial Oxidative Stress in Mice Lacking the Glutathione Peroxidase-1 Gene. Free Radic. Biol. Med. 2000, 28, 754–766. [Google Scholar] [CrossRef] [Green Version]
- Mai, H.N.; Chung, Y.H.; Shin, E.J.; Kim, D.J.; Jeong, J.H.; Nguyen, T.T.; Nam, Y.; Lee, Y.J.; Nah, S.Y.; Yu, D.Y.; et al. Genetic Depletion of Glutathione Peroxidase-1 Potentiates Nephrotoxicity Induced by Multiple Doses of Cocaine via Activation of Angiotensin II AT1 Receptor. Free Radic. Res. 2016, 50, 467–483. [Google Scholar] [CrossRef] [PubMed]
- Chu, Y.; Lan, R.S.; Huang, R.; Feng, H.; Kumar, R.; Dayal, S.; Chan, K.S.; Dai, D.F. Glutathione Peroxidase-1 Overexpression Reduces Oxidative Stress, and Improves Pathology and Proteome Remodeling in the Kidneys of Old Mice. Aging Cell 2020, 19, e13154. [Google Scholar] [CrossRef] [PubMed]
- Chiu, Y.W.; Kuo, M.C.; Kuo, H.T.; Chang, J.M.; Guh, J.Y.; Lai, Y.H.; Chen, H.C. Alterations of Glomerular and Extracellular Levels of Glutathione Peroxidase in Patients and Experimental Rats with Diabetic Nephropathy. J. Lab. Clin. Med. 2005, 145, 181–186. [Google Scholar] [CrossRef]
- Chew, P.; Yuen, D.Y.; Stefanovic, N.; Pete, J.; Coughlan, M.T.; Jandeleit-Dahm, K.A.; Thomas, M.C.; Rosenfeldt, F.; Cooper, M.E.; de Haan, J.B. Antiatherosclerotic and Renoprotective Effects of Ebselen in the Diabetic Apolipoprotein E/GPx1-Double Knockout Mouse. Diabetes 2010, 59, 3198–3207. [Google Scholar] [CrossRef] [Green Version]
- Ottaviano, F.G.; Tang, S.S.; Handy, D.E.; Loscalzo, J. Regulation of the Extracellular Antioxidant Selenoprotein Plasma Glutathione Peroxidase (GPx-3) in Mammalian Cells. Mol. Cell. Biochem. 2009, 327, 111–126. [Google Scholar] [CrossRef] [Green Version]
- Burk, R.F.; Olson, G.E.; Winfrey, V.P.; Hill, K.E.; Yin, D. Glutathione Peroxidase-3 Produced by the Kidney Binds to a Population of Basement Membranes in the Gastrointestinal Tract and in Other Tissues. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 301, G32–G38. [Google Scholar] [CrossRef] [Green Version]
- Pang, P.; Abbott, M.; Abdi, M.; Fucci, Q.A.; Chauhan, N.; Mistri, M.; Proctor, B.; Chin, M.; Wang, B.; Yin, W.; et al. Pre-Clinical Model of Severe Glutathione Peroxidase-3 Deficiency and Chronic Kidney Disease Results in Coronary Artery Thrombosis and Depressed Left Ventricular Function. Nephrol. Dial. Transplant. 2018, 33, 923–934. [Google Scholar] [CrossRef]
- Martin-Sanchez, D.; Fontecha-Barriuso, M.; Martinez-Moreno, J.M.; Ramos, A.M.; Sanchez-Niño, M.D.; Guerrero-Hue, M.; Moreno, J.A.; Ortiz, A.; Sanz, A.B. Ferroptosis and Kidney Disease. Nefrologia 2020, 40, 384–394. [Google Scholar] [CrossRef]
- Hu, Z.; Zhang, H.; Yang, S.K.; Wu, X.; He, D.; Cao, K.; Zhang, W. Emerging Role of Ferroptosis in Acute Kidney Injury. Oxid. Med. Cell. Longev. 2019, 2019, 8010614. [Google Scholar] [CrossRef] [PubMed]
- Belavgeni, A.; Meyer, C.; Stumpf, J.; Hugo, C.; Linkermann, A. Ferroptosis and Necroptosis in the Kidney. Cell Chem. Biol. 2020, 27, 448–462. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of Ferroptotic Cancer Cell Death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedmann Angeli, J.P.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the Ferroptosis Regulator Gpx4 Triggers Acute Renal Failure in Mice. Nat. Cell Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Bi, R.; Quan, F.; Cao, Q.; Lin, Y.; Yue, C.; Cui, X.; Yang, H.; Gao, X.; Zhang, D. Ferroptosis Involves in Renal Tubular Cell Death in Diabetic Nephropathy. Eur. J. Pharmacol. 2020, 888, 173574. [Google Scholar] [CrossRef]
- Giustarini, D.; Dalle-Donne, I.; Tsikas, D.; Rossi, R. Oxidative Stress and Human Diseases: Origin, Link, Measurement, Mechanisms, and Biomarkers. Crit. Rev. Clin. Lab. Sci. 2009, 46, 241–281. [Google Scholar] [CrossRef]
- Kurutas, E.B. The Importance of Antioxidants Which Play the Role in Cellular Response against Oxidative/Nitrosative Stress: Current State. Nutr. J. 2016, 15, 71. [Google Scholar] [CrossRef] [Green Version]
- Haber, A.; Gross, Z. Catalytic Antioxidant Therapy by Metallodrugs: Lessons from Metallocorroles. Chem. Commun. 2015, 51, 5812–5827. [Google Scholar] [CrossRef]
- Patel, M.; Day, B.J. Metalloporphyrin Class of Therapeutic Catalytic Antioxidants. Trends Pharmacol. Sci. 1999, 20, 359–364. [Google Scholar] [CrossRef]
- Pasternack, R.F.; Halliwell, B. Superoxide Dismutase Activities of an Iron Porphyrin and Other Iron Complexes. J. Am. Chem. Soc. 1979, 101, 1026–1031. [Google Scholar] [CrossRef]
- Batinić-Haberle, I.; Spasojević, I.; Hambright, P.; Benov, L.; Crumbliss, A.L.; Fridovich, I. Relationship among Redox Potentials, Proton Dissociation Constants of Pyrrolic Nitrogens, and In Vivo and In Vitro Superoxide Dismutating Activities of Manganese(III) and Iron(III) Water-Soluble Porphyrins. Inorg. Chem. 1999, 38, 4011–4022. [Google Scholar] [CrossRef]
- Batinić-Haberle, I.; Rebouças, J.S.; Spasojević, I. Superoxide Dismutase Mimics: Chemistry, Pharmacology, and Therapeutic Potential. Antioxid. Redox Signal. 2010, 13, 877–918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonetta, R. Potential Therapeutic Applications of MnSODs and SOD-Mimetics. Chemistry 2018, 24, 5032–5041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aston, K.; Rath, N.; Naik, A.; Slomczynska, U.; Schall, O.F.; Riley, D.P. Computer-Aided Design (CAD) of Mn(II) Complexes: Superoxide Dismutase Mimetics with Catalytic Activity Exceeding the Native Enzyme. Inorg. Chem. 2001, 40, 1779–1789. [Google Scholar] [CrossRef] [PubMed]
- Riley, D.P.; Lennon, P.J.; Neumann, W.L.; Weiss, R.H. Toward the Rational Design of Superoxide Dismutase Mimics: Mechanistic Studies for the Elucidation of Substituent Effects on the Catalytic Activity of Macrocyclic Manganese(II) Complexes. J. Am. Chem. Soc. 1997, 119, 6522–6528. [Google Scholar] [CrossRef]
- Golden, T.R.; Patel, M. Catalytic Antioxidants and Neurodegeneration. Antioxid. Redox Signal. 2009, 11, 555–570. [Google Scholar] [CrossRef] [Green Version]
- Cuzzocrea, S.; Mazzon, E.; Dugo, L.; Serraino, I.; Di Paola, R.; Britti, D.; De Sarro, A.; Pierpaoli, S.; Caputi, A.; Masini, E.; et al. A Role for Superoxide in Gentamicin-Mediated Nephropathy in Rats. Eur. J. Pharmacol. 2002, 450, 67–76. [Google Scholar] [CrossRef]
- Miriyala, S.; Spasojevic, I.; Tovmasyan, A.; Salvemini, D.; Vujaskovic, Z.; St Clair, D.; Batinic-Haberle, I. Manganese Superoxide Dismutase, MnSOD and Its Mimics. Biochim. Biophys. Acta 2012, 1822, 794–814. [Google Scholar] [CrossRef] [Green Version]
- Azadmanesh, J.; Borgstahl, G.E.O. A Review of the Catalytic Mechanism of Human Manganese Superoxide Dismutase. Antioxidants 2018, 7, 25. [Google Scholar] [CrossRef] [Green Version]
- Soldevila-Barreda, J.J.; Sadler, P.J. Approaches to the Design of Catalytic Metallodrugs. Curr. Opin. Chem. Biol. 2015, 25, 172–183. [Google Scholar] [CrossRef] [Green Version]
- Batinic-Haberle, I.; Rajic, Z.; Tovmasyan, A.; Reboucas, J.S.; Ye, X.; Leong, K.W.; Dewhirst, M.W.; Vujaskovic, Z.; Benov, L.; Spasojevic, I. Diverse Functions of Cationic Mn(III) N-Substituted Pyridylporphyrins, Recognized as SOD Mimics. Free Radic. Biol. Med. 2011, 51, 1035–1053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tovmasyan, A.; Sheng, H.; Weitner, T.; Arulpragasam, A.; Lu, M.; Warner, D.S.; Vujaskovic, Z.; Spasojevic, I.; Batinic-Haberle, I. Design, Mechanism of Action, Bioavailability and Therapeutic Effects of mn Porphyrin-Based Redox Modulators. Med. Princ. Pract. 2013, 22, 103–130. [Google Scholar] [CrossRef] [PubMed]
- Batinic-Haberle, I.; Tovmasyan, A.; Spasojevic, I. An Educational Overview of the Chemistry, Biochemistry and Therapeutic Aspects of Mn Porphyrins—From Superoxide Dismutation to H2O2-Driven Pathways. Redox Biol. 2015, 5, 43–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leu, D.; Spasojevic, I.; Nguyen, H.; Deng, B.; Tovmasyan, A.; Weitner, T.; Sampaio, R.S.; Batinic-Haberle, I.; Huang, T.T. CNS Bioavailability and Radiation Protection of Normal Hippocampal Neurogenesis by a Lipophilic Mn Porphyrin-Based Superoxide Dismutase Mimic, MnTnBuOE-2-PyP(5). Redox Biol. 2017, 12, 864–871. [Google Scholar] [CrossRef]
- Faulkner, K.M.; Liochev, S.I.; Fridovich, I. Stable Mn(III) Porphyrins Mimic Superoxide Dismutase In Vitro and Substitute for It In Vivo. J. Biol. Chem. 1994, 269, 23471–23476. [Google Scholar]
- Spasojević, I.; Chen, Y.; Noel, T.J.; Fan, P.; Zhang, L.; Rebouças, J.S.; St Clair, D.K.; Batinić-Haberle, I. Pharmacokinetics of the Potent Redox-Modulating Manganese Porphyrin, MnTE-2-PyP(5+), in Plasma and Major Organs of B6C3F1 Mice. Free Radic. Biol. Med. 2008, 45, 943–949. [Google Scholar] [CrossRef] [Green Version]
- Batinić-Haberle, I. Manganese Porphyrins and Related Compounds as Mimics of Superoxide Dismutase. Methods Enzymol. 2002, 349, 223–233. [Google Scholar]
- Tovmasyan, A.; Reboucas, J.S.; Benov, L. Simple Biological Systems for Assessing the Activity of Superoxide Dismutase Mimics. Antioxid. Redox Signal. 2014, 20, 2416–2436. [Google Scholar] [CrossRef] [Green Version]
- Gad, S.C.; Sullivan, D.W., Jr.; Spasojevic, I.; Mujer, C.V.; Spainhour, C.B.; Crapo, J.D. Nonclinical Safety and Toxicokinetics of MnTnBuOE-2-PyP5+ (BMX-001). Int. J. Toxicol. 2016, 35, 438–453. [Google Scholar] [CrossRef]
- Saba, H.; Batinic-Haberle, I.; Munusamy, S.; Mitchell, T.; Lichti, C.; Megyesi, J.; MacMillan-Crow, L.A. Manganese Porphyrin Reduces Renal Injury and Mitochondrial Damage during Ischemia/Reperfusion. Free Radic. Biol. Med. 2007, 42, 1571–1578. [Google Scholar] [CrossRef] [Green Version]
- Batinic-Haberle, I.; Tome, M.E. Thiol Regulation by Mn Porphyrins, Commonly Known as SOD Mimics. Redox Biol. 2019, 25, 101139. [Google Scholar] [CrossRef] [PubMed]
- Batinic-Haberle, I.; Tovmasyan, A.; Spasojevic, I. Mn Porphyrin-Based Redox-Active Drugs: Differential Effects as Cancer Therapeutics and Protectors of Normal Tissue against Oxidative Injury. Antioxid. Redox Signal. 2018, 29, 1691–1724. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.L.; Hilton, G.; Mortensen, J.; Regner, K.; Johnson, C.P.; Nilakantan, V. MnTMPyP, a Cell-Permeant SOD Mimetic, Reduces Oxidative Stress and Apoptosis Following Renal Ischemia-Reperfusion. Am. J. Physiol. Renal Physiol. 2009, 296, F266–F276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mortensen, J.; Shames, B.; Johnson, C.P.; Nilakantan, V. MnTMPyP, a Superoxide Dismutase/Catalase Mimetic, Decreases Inflammatory Indices in Ischemic Acute Kidney Injury. Inflamm. Res. 2011, 60, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Seok, Y.M.; Jung, K.J.; Park, K.M. Reactive Oxygen Species/Oxidative Stress Contributes to Progression of Kidney Fibrosis Following Transient Ischemic Injury in Mice. Am. J. Physiol. Renal Physiol. 2009, 297, F461–F470. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.I.; Kim, J.; Jang, H.S.; Noh, M.R.; Lipschutz, J.H.; Park, K.M. Reduction of Oxidative Stress during Recovery Accelerates Normalization of Primary Cilia Length that Is Altered after Ischemic Injury in Murine Kidneys. Am. J. Physiol. Renal Physiol. 2013, 304, F1283–F1294. [Google Scholar] [CrossRef]
- Han, S.J.; Jang, H.S.; Kim, J.I.; Lipschutz, J.H.; Park, K.M. Unilateral Nephrectomy Elongates Primary Cilia in the Remaining Kidney via Reactive Oxygen Species. Sci. Rep. 2016, 6, 22281. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Holthoff, J.H.; Seely, K.A.; Pathak, E.; Spencer, H.J., III; Gokden, N.; Mayeux, P.R. Development of Oxidative Stress in the Peritubular Capillary Microenvironment Mediates Sepsis-Induced Renal Microcirculatory Failure and Acute Kidney Injury. Am. J. Pathol. 2012, 180, 505–516. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Jittikanont, S.; Falk, S.A.; Li, P.; Feng, L.; Gengaro, P.E.; Poole, B.D.; Bowler, R.P.; Day, B.J.; Crapo, J.D.; et al. Interaction among Nitric Oxide, Reactive Oxygen Species, and Antioxidants during Endotoxemia-Related Acute Renal Failure. Am. J. Physiol. Renal Physiol. 2003, 284, F532–F537. [Google Scholar] [CrossRef] [Green Version]
- Khan, I.; Batinic-Haberle, I.; Benov, L.T. Effect of Potent Redox-Modulating Manganese Porphyrin, MnTM-2-PyP, on the Na(+)/H(+) Exchangers NHE-1 and NHE-3 in the Diabetic Rat. Redox Rep. 2009, 14, 236–242. [Google Scholar] [CrossRef]
- Ali, D.K.; Oriowo, M.; Tovmasyan, A.; Batinic-Haberle, I.; Benov, L. Late Administration of Mn Porphyrin-Based SOD Mimic Enhances Diabetic Complications. Redox Biol. 2013, 1, 457–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Day, B.J.; Shawen, S.; Liochev, S.I.; Crapo, J.D. A Metalloporphyrin Superoxide Dismutase Mimetic Protects against Paraquat-Induced Endothelial Cell Injury, In Vitro. J. Pharmacol. Exp. Ther. 1995, 275, 1227–1232. [Google Scholar] [PubMed]
- Tovmasyan, A.; Maia, C.G.; Weitner, T.; Carballal, S.; Sampaio, R.S.; Lieb, D.; Ghazaryan, R.; Ivanovic-Burmazovic, I.; Ferrer-Sueta, G.; Radi, R.; et al. A Comprehensive Evaluation of Catalase-Like Activity of Different Classes of Redox-Active Therapeutics. Free Radic. Biol. Med. 2015, 86, 308–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rebouças, J.S.; Spasojević, I.; Batinić-Haberle, I. Pure Manganese(III) 5,10,15,20-tetrakis(4-benzoic acid)porphyrin (MnTBAP) Is Not a Superoxide Dismutase Mimic in Aqueous Systems: A Case of Structure-Activity Relationship as a Watchdog Mechanism in Experimental Therapeutics and Biology. J. Biol. Inorg. Chem. 2008, 13, 289–302. [Google Scholar] [CrossRef]
- Batinić-Haberle, I.; Cuzzocrea, S.; Rebouças, J.S.; Ferrer-Sueta, G.; Mazzon, E.; Di Paola, R.; Radi, R.; Spasojević, I.; Benov, L.; Salvemini, D. Pure MnTBAP Selectively Scavenges Peroxynitrite over Superoxide: Comparison of Pure and Commercial MnTBAP Samples to MnTE-2-PyP in Two Models of Oxidative Stress Injury, an SOD-Specific Escherichia coli Model and Carrageenan-Induced Pleurisy. Free Radic. Biol. Med. 2009, 46, 192–201. [Google Scholar] [CrossRef] [Green Version]
- Zahmatkesh, M.; Kadkhodaee, M.; Moosavi, S.M.; Jorjani, M.; Kajbafzadeh, A.; Golestani, A.; Ghaznavi, R. Beneficial Effects of MnTBAP, a Broad-Spectrum Reactive Species Scavenger, in Rat Renal Ischemia/Reperfusion Injury. Clin. Exp. Nephrol. 2005, 9, 212–218. [Google Scholar] [CrossRef]
- Zahmatkesh, M.; Kadkhodaee, M.; Arab, H.A.; Shams, S. Effects of Co-Administration of an iNOS Inhibitor with a Broad-Spectrum Reactive Species Scavenger in Rat Renal Ischemia/Reperfusion Injury. Nephron. Exp. Nephrol. 2006, 103, e119–e125. [Google Scholar] [CrossRef]
- Pan, H.; Shen, K.; Wang, X.; Meng, H.; Wang, C.; Jin, B. Protective Effect of Metalloporphyrins against Cisplatin-Induced Kidney Injury in Mice. PLoS ONE 2014, 9, e86057. [Google Scholar] [CrossRef]
- Zhuang, Y.; Yasinta, M.; Hu, C.; Zhao, M.; Ding, G.; Bai, M.; Yang, L.; Ni, J.; Wang, R.; Jia, Z.; et al. Mitochondrial Dysfunction Confers Albumin-Induced NLRP3 Inflammasome Activation and Renal Tubular Injury. Am. J. Physiol. Renal Physiol. 2015, 308, F857–F866. [Google Scholar] [CrossRef]
- Bi, X.; Wang, J.; Liu, Y.; Wang, Y.; Ding, W. MnTBAP Treatment Ameliorates Aldosterone-Induced Renal Injury by Regulating Mitochondrial Dysfunction and NLRP3 Inflammasome Signalling. Am. J. Transl. Res. 2018, 10, 3504–3513. [Google Scholar]
- Yu, J.; Mao, S.; Zhang, Y.; Gong, W.; Jia, Z.; Huang, S.; Zhang, A. MnTBAP Therapy Attenuates Renal Fibrosis in Mice with 5/6 Nephrectomy. Oxid. Med. Cell. Longev. 2016, 2016, 7496930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baudry, M.; Etienne, S.; Bruce, A.; Palucki, M.; Jacobsen, E.; Malfroy, B. Salen-Manganese Complexes Are Superoxide Dismutase-Mimics. Biochem. Biophys. Res. Commun. 1993, 192, 964–968. [Google Scholar] [CrossRef]
- Doctrow, S.R.; Huffman, K.; Marcus, C.B.; Musleh, W.; Bruce, A.; Baudry, M.; Malfroy, B. Salen-Manganese Complexes: Combined Superoxide Dismutase/Catalase Mimics with Broad Pharmacological Efficacy. Adv. Pharmacol. 1997, 38, 247–269. [Google Scholar]
- Gianello, P.; Saliez, A.; Bufkens, X.; Pettinger, R.; Misseleyn, D.; Hori, S.; Malfroy, B. EUK-134, a Synthetic Superoxide Dismutase and Catalase Mimetic, Protects Rat Kidneys from Ischemia-Reperfusion-Induced Damage. Transplantation 1996, 62, 1664–1666. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, P.K.; Patel, N.S.; Kvale, E.O.; Brown, P.A.; Stewart, K.N.; Mota-Filipe, H.; Sharpe, M.A.; Di Paola, R.; Cuzzocrea, S.; Thiemermann, C. EUK-134 Reduces Renal Dysfunction and Injury Caused by Oxidative and Nitrosative Stress of the Kidney. Am. J. Nephrol. 2004, 24, 165–177. [Google Scholar] [CrossRef] [PubMed]
- Samai, M.; Sharpe, M.A.; Gard, P.R.; Chatterjee, P.K. Comparison of the Effects of the Superoxide Dismutase Mimetics EUK-134 and Tempol on Paraquat-Induced Nephrotoxicity. Free Radic. Biol. Med. 2007, 43, 528–534. [Google Scholar] [CrossRef]
- McDonald, M.C.; d’Emmanuele di Villa Bianca, R.; Wayman, N.S.; Pinto, A.; Sharpe, M.A.; Cuzzocrea, S.; Chatterjee, P.K.; Thiemermann, C. A Superoxide Dismutase Mimetic with Catalase Activity (EUK-8) Reduces the Organ Injury in Endotoxic Shock. Eur. J. Pharmacol. 2003, 466, 181–189. [Google Scholar] [CrossRef]
- Magder, S.; Parthenis, D.G.; Ghouleh, I.A. Preservation of Renal Blood Flow by the Antioxidant EUK-134 in LPS-Treated Pigs. Int. J. Mol. Sci. 2015, 16, 6801–6817. [Google Scholar] [CrossRef] [Green Version]
- Vera, M.; Torramade-Moix, S.; Martin-Rodriguez, S.; Cases, A.; Cruzado, J.M.; Rivera, J.; Escolar, G.; Palomo, M.; Diaz-Ricart, M. Antioxidant and Anti-Inflammatory Strategies Based on the Potentiation of Glutathione Peroxidase Activity Prevent Endothelial Dysfunction in Chronic Kidney Disease. Cell. Physiol. Biochem. 2018, 51, 1287–1300. [Google Scholar] [CrossRef]
- Krishna, M.C.; Grahame, D.A.; Samuni, A.; Mitchell, J.B.; Russo, A. Oxoammonium Cation Intermediate in the Nitroxide-Catalyzed Dismutation of Superoxide. Proc. Natl. Acad. Sci. USA. 1992, 89, 5537–5541. [Google Scholar] [CrossRef] [Green Version]
- Wilcox, C.S.; Pearlman, A. Chemistry and Antihypertensive Effects of Tempol and Other Nitroxides. Pharmacol. Rev. 2008, 60, 418–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatterjee, P.K.; Cuzzocrea, S.; Brown, P.A.; Zacharowski, K.; Stewart, K.N.; Mota-Filipe, H.; Thiemermann, C. Tempol, a Membrane-Permeable Radical Scavenger, Reduces Oxidant Stress-Mediated Renal Dysfunction and Injury in the Rat. Kidney Int. 2000, 58, 658–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aksu, U.; Ergin, B.; Bezemer, R.; Kandil, A.; Milstein, D.M.; Demirci-Tansel, C.; Ince, C. Scavenging Reactive Oxygen Species Using Tempol in the Acute Phase of Renal Ischemia/Reperfusion and Its Effects on Kidney Oxygenation and Nitric Oxide Levels. Intensive Care Med. Exp. 2015, 3, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leach, M.; Frank, S.; Olbrich, A.; Pfeilschifter, J.; Thiemermann, C. Decline in the Expression of Copper/Zinc Superoxide Dismutase in the Kidney of Rats with Endotoxic Shock: Effects of the Superoxide Anion Radical Scavenger, Tempol, on Organ Injury. Br. J. Pharmacol. 1998, 125, 817–825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nassar, T.; Kadery, B.; Lotan, C.; Da’as, N.; Kleinman, Y.; Haj-Yehia, A. Effects of the Superoxide Dismutase-Mimetic Compound Tempol on Endothelial Dysfunction in Streptozotocin-Induced Diabetic Rats. Eur. J. Pharmacol. 2002, 436, 111–118. [Google Scholar] [CrossRef]
- Rodriguez, F.; Lopez, B.; Perez, C.; Fenoy, F.J.; Hernandez, I.; Stec, D.E.; Li Volti, G.; Salom, M.G. Chronic Tempol Treatment Attenuates the Renal Hemodynamic Effects Induced by a Heme Oxygenase Inhibitor in Streptozotocin Diabetic Rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 301, R1540–R1548. [Google Scholar] [CrossRef] [Green Version]
- Luan, J.; Li, W.; Han, J.; Zhang, W.; Gong, H.; Ma, R. Renal Protection of In Vivo Administration of Tempol in Streptozotocin-Induced Diabetic Rats. J. Pharmacol. Sci. 2012, 119, 167–176. [Google Scholar] [CrossRef] [Green Version]
- Rafikova, O.; Salah, E.M.; Tofovic, S.P. Renal and Metabolic Effects of Tempol in Obese ZSF1 Rats—Distinct Role for Superoxide and Hydrogen Peroxide in Diabetic Renal Injury. Metabolism 2008, 57, 1434–1444. [Google Scholar] [CrossRef]
- Shokoji, T.; Nishiyama, A.; Fujisawa, Y.; Hitomi, H.; Kiyomoto, H.; Takahashi, N.; Kimura, S.; Kohno, M.; Abe, Y. Renal Sympathetic Nerve Responses to Tempol in Spontaneously Hypertensive Rats. Hypertension 2003, 41, 266–273. [Google Scholar] [CrossRef] [Green Version]
- Welch, W.J.; Mendonca, M.; Blau, J.; Karber, A.; Dennehy, K.; Patel, K.; Lao, Y.S.; José, P.A.; Wilcox, C.S. Antihypertensive Response to Prolonged Tempol in the Spontaneously Hypertensive Rat. Kidney Int. 2005, 68, 179–187. [Google Scholar] [CrossRef] [Green Version]
- Onuma, S.; Nakanishi, K. Superoxide Dismustase Mimetic Tempol Decreases Blood Pressure by Increasing Renal Medullary Blood Flow in Hyperinsulinemic-Hypertensive Rats. Metabolism 2004, 53, 1305–1308. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, A.; Yoshizumi, M.; Hitomi, H.; Kagami, S.; Kondo, S.; Miyatake, A.; Fukunaga, M.; Tamaki, T.; Kiyomoto, H.; Kohno, M.; et al. The SOD Mimetic Tempol Ameliorates Glomerular Injury and Reduces Mitogen-Activated Protein Kinase Activity in Dahl Salt-Sensitive Rats. J. Am. Soc. Nephrol. 2004, 15, 306–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Son, D.; Kojima, I.; Inagi, R.; Matsumoto, M.; Fujita, T.; Nangaku, M. Chronic Hypoxia Aggravates Renal Injury via Suppression of Cu/Zn-SOD: A Proteomic Analysis. Am. J. Physiol. Renal Physiol. 2008, 294, F62–F72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, W.; Chen, S.S.; Chen, Y.; Ahokas, R.A.; Sun, Y. Kidney Fibrosis in Hypertensive Rats: Role of Oxidative Stress. Am. J. Nephrol. 2008, 28, 548–554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaziri, N.D.; Dicus, M.; Ho, N.D.; Boroujerdi-Rad, L.; Sindhu, R.K. Oxidative Stress and Dysregulation of Superoxide Dismutase and NADPH Oxidase in Renal Insufficiency. Kidney Int. 2003, 63, 179–185. [Google Scholar] [CrossRef] [Green Version]
- Trnka, J.; Blaikie, F.H.; Smith, R.A.; Murphy, M.P. A Mitochondria-Targeted Nitroxide Is Reduced to Its Hydroxylamine by Ubiquinol in Mitochondria. Free Radic. Biol. Med. 2008, 44, 1406–1419. [Google Scholar] [CrossRef] [PubMed]
- Dikalov, S. Cross Talk between Mitochondria and NADPH Oxidases. Free Radic. Biol. Med. 2011, 51, 1289–1301. [Google Scholar] [CrossRef] [Green Version]
- Patil, N.K.; Parajuli, N.; MacMillan-Crow, L.A.; Mayeux, P.R. Inactivation of Renal Mitochondrial Respiratory Complexes and Manganese Superoxide Dismutase during Sepsis: Mitochondria-Targeted Antioxidant Mitigates Injury. Am. J. Physiol. Renal Physiol. 2014, 306, F734–F743. [Google Scholar] [CrossRef] [Green Version]
- Ding, W.; Liu, T.; Bi, X.; Zhang, Z. Mitochondria-Targeted Antioxidant Mito-Tempo Protects against Aldosterone-Induced Renal Injury In Vivo. Cell. Physiol. Biochem. 2017, 44, 741–750. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, Y.; Ding, W.; Wang, Y. Mito-TEMPO Alleviates Renal Fibrosis by Reducing Inflammation, Mitochondrial Dysfunction, and Endoplasmic Reticulum Stress. Oxid. Med. Cell. Longev. 2018, 2018, 5828120. [Google Scholar] [CrossRef] [Green Version]
- Chu, S.; Mao, X.; Guo, H.; Wang, L.; Li, Z.; Zhang, Y.; Wang, Y.; Wang, H.; Zhang, X.; Peng, W. Indoxyl Sulfate Potentiates Endothelial Dysfunction via Reciprocal Role for Reactive Oxygen Species and RhoA/ROCK Signaling in 5/6 Nephrectomized Rats. Free Radic. Res. 2017, 51, 237–252. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Feng, Q.; Kumagai, T.; Torikai, K.; Ohigashi, H.; Osawa, T.; Noguchi, N.; Niki, E.; Uchida, K. Ebselen, a Glutathione Peroxidase Mimetic Seleno-Organic Compound, as a Multifunctional Antioxidant. Implication for Inflammation-Associated Carcinogenesis. J. Biol. Chem. 2002, 277, 2687–2694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldew, G.S.; McVie, J.G.; van der Valk, M.A.; Los, G.; de Goeij, J.J.; Vermeulen, N.P. Selective Reduction of Cis-diamminedichloroplatinum(II) Nephrotoxicity by Ebselen. Cancer Res. 1990, 50, 7031–7036. [Google Scholar] [PubMed]
- Baldew, G.S.; Boymans, A.P.; Mol, J.G.; Vermeulen, N.P. The Influence of Ebselen on the Toxicity of Cisplatin in LLC-PK1 Cells. Biochem. Pharmacol. 1992, 44, 382–387. [Google Scholar] [CrossRef]
- Husain, K.; Morris, C.; Whitworth, C.; Trammell, G.L.; Rybak, L.P.; Somani, S.M. Protection by Ebselen against Cisplatin-Induced Nephrotoxicity: Antioxidant System. Mol. Cell. Biochem. 1998, 178, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, M.; Iizuka, K.; Terada, A.; Hara, M.; Nishijima, H.; Shimada, A.; Nakada, K.; Satoh, Y.; Akama, Y. Prevention of Nephrotoxicity of Cisplatin by Repeated Oral Administration of Ebselen in Rats. Tohoku J. Exp. Med. 2000, 191, 209–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhanarajan, R.; Abraham, P.; Isaac, B. Protective Effect of Ebselen, a Selenoorganic Drug, against Gentamicin-Induced Renal Damage in Rats. Basic Clin. Pharmacol. Toxicol. 2006, 99, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Kizilgun, M.; Poyrazoglu, Y.; Oztas, Y.; Yaman, H.; Cakir, E.; Cayci, T.; Akgul, O.E.; Kurt, Y.G.; Yaren, H.; Kunak, Z.I.; et al. Beneficial Effects of N-acetylcysteine and Ebselen on Renal Ischemia/Reperfusion Injury. Ren. Fail. 2011, 33, 512–517. [Google Scholar] [CrossRef] [Green Version]
- Ozgur, T.; Tutanc, M.; Zararsiz, I.; Motor, S.; Ozturk, O.H.; Yaldiz, M.; Kurtgoz, O.Y. The Protective Effect of Ebselen on Radiocontrast-Induced Nephrotoxicity. Ren. Fail. 2012, 34, 991–997. [Google Scholar] [CrossRef] [Green Version]
- Gealekman, O.; Brodsky, S.V.; Zhang, F.; Chander, P.N.; Friedli, C.; Nasjletti, A.; Goligorsky, M.S. Endothelial Dysfunction as a Modifier of Angiogenic Response in Zucker Diabetic Fat Rat: Amelioration with Ebselen. Kidney Int. 2004, 66, 2337–2347. [Google Scholar] [CrossRef] [Green Version]
- Tan, S.M.; Sharma, A.; Yuen, D.Y.; Stefanovic, N.; Krippner, G.; Mugesh, G.; Chai, Z.; de Haan, J.B. The Modified Selenenyl Amide, M-hydroxy Ebselen, Attenuates Diabetic Nephropathy and Diabetes-Associated Atherosclerosis in ApoE/GPx1 Double Knockout Mice. PLoS ONE 2013, 8, e69193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, S.M.; Sharma, A.; Stefanovic, N.; de Haan, J.B. Late-Intervention Study with Ebselen in an Experimental Model of Type 1 Diabetic Nephropathy. Free Radic. Res. 2015, 49, 219–227. [Google Scholar] [CrossRef]
- Huang, X.; Dong, Z.; Liu, J.; Mao, S.; Xu, J.; Luo, G.; Shen, J. Selenium-Mediated Micellar Catalyst: An Efficient Enzyme Model for Glutathione Peroxidase-Like Catalysis. Langmuir 2007, 23, 1518–1522. [Google Scholar] [CrossRef] [PubMed]
- Rosa, R.M.; Sulzbacher, K.; Picada, J.N.; Roesler, R.; Saffi, J.; Brendel, M.; Henriques, J.A. Genotoxicity of Diphenyl Diselenide in Bacteria and Yeast. Mutat. Res. 2004, 563, 107–115. [Google Scholar] [CrossRef]
- Brandão, R.; Acker, C.I.; Leite, M.R.; Barbosa, N.B.; Nogueira, C.W. Diphenyl Diselenide Protects against Glycerol-Induced Renal Damage in Rats. J. Appl. Toxicol. 2009, 29, 612–618. [Google Scholar] [CrossRef] [PubMed]
- Brandão, R.; Moresco, R.N.; Bellé, L.P.; Leite, M.R.; de Freitas, M.L.; Bianchini, A.; Nogueira, C.W. Diphenyl Diselenide Potentiates Nephrotoxicity Induced by Mercuric Chloride in Mice. J. Appl. Toxicol. 2011, 31, 773–782. [Google Scholar] [CrossRef]
- Barbosa, N.B.; Rocha, J.B.; Soares, J.C.; Wondracek, D.C.; Gonçalves, J.F.; Schetinger, M.R.; Nogueira, C.W. Dietary Diphenyl Diselenide Reduces the STZ-Induced Toxicity. Food Chem. Toxicol. 2008, 46, 186–194. [Google Scholar] [CrossRef]
- Fulco, B.C.W.; Jung, J.T.K.; Chagas, P.M.; Rosa, S.G.; Prado, V.C.; Nogueira, C.W. Diphenyl Diselenide Is as Effective as Ebselen in a Juvenile Rat Model of Cisplatin-Induced Nephrotoxicity. J. Trace Elem. Med. Biol. 2020, 60, 126482. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Groups | Structures | Compounds | Diseases | Models | Species or Cells | Ref. |
|---|---|---|---|---|---|---|
| SOD mimics: metal-based | Macrocyclics | M40403 | AKI | Gentamycin | Rats | [97] |
| Mn Porphyrins | AEOL10112 (MnTM-2-PyP5+) | DKD | Streptozotocin | Rats | [120,121] | |
| AEOL10113 (MnTE-2-PyP5+) | AKI | Lipopolysaccharide | Mice | [119] | ||
| MnTM-4-PyP5+ | AKI | I/R injury | Rats, Mice | [113,114,115,116] | ||
| Cecal ligation and puncture | Mice | [118] | ||||
| CKD | Unilateral nephrectomy | Mice | [117] | |||
| MnTnHex-2-PyP5+ | AKI | I/R injury | Rats | [110] | ||
| Non-SOD mimics: metal-based | Mn Porphyrins | AEOL10201 (MnTBAP3-) | AKI | I/R injury | Rats | [126,127] |
| Cisplatin | Mice | [128] | ||||
| CKD | Albumin | Mice | [129] | |||
| Aldosterone | Mice/HK-2 cells | [130] | ||||
| 5/6 nephrectomy | Mice/mPT cells | [131] | ||||
| SOD/CAT mimics: metal-based | Mn Salens | EUK-134 | AKI | I/R injury | Rats | [134,135] |
| Paraquat | NRK-52E cells | [136] | ||||
| Lipopolysaccharide | Pigs | [138] | ||||
| EUK-8 | AKI | Lipopolysaccharide | Rats | [137] | ||
| EUK-118, EUK-134 | CKD | Uremic media | Endothelial cells | [139] | ||
| SOD mimics: Non-metal-based | Nitroxides | Tempol | AKI | Paraquat | NRK-52E cells | [136] |
| I/R injury | Rats | [142,143] | ||||
| Lipopolysaccharide | Rats | [144] | ||||
| DKD | Streptozotocin | Rats | [145,146,147] | |||
| KK/Ta-Akita mice | Mice | [42] | ||||
| Obesity | ZSF1 rats | Rats | [148] | |||
| HTN | Spontaneously hypertensive rats | Rats | [149,150] | |||
| Dahl salt-resistant rats | Rats | [152] | ||||
| Unilateral renal artery stenosis | Rats | [153] | ||||
| Angiotensin II | Rats | [154] | ||||
| Fructose | Rats | [151] | ||||
| CKD | 5/6 nephrectomy | Rats | [155] | |||
| 5/6 nephrectomy + IS | Rats | [161] | ||||
| Mito-TEMPO | AKI | Cecal ligation and puncture | Mice | [158] | ||
| CKD | 5/6 nephrectomy + IS | Rats | [161] | |||
| 5/6 nephrectomy | Mice | [160] | ||||
| Aldosterone | Mice | [159] | ||||
| GPx mimics | Organoselenium | Ebselen | AKI | Cisplatin | Rats, Mice, LLC-PK1 cells | [163,164,165,166] |
| Gentamycin | Rats | [167] | ||||
| I/R injury | Rats | [168] | ||||
| Radiocontrast | Rats | [169] | ||||
| Gentamycin | Rats | [167] | ||||
| CKD | Uremic media | Endothelial cells | [139] | |||
| DKD | Akita mice | Mice | [172] | |||
| ApoE/GPx1 dKO | Mice | [76,171] | ||||
| Zucker diabetic fat | Rats | [170] | ||||
| Diphenyl diselenide | AKI | Cisplatin | Rats | [178] | ||
| Mercuric chloride | Mice | [176] | ||||
| Glycerol | Rats | [175] | ||||
| DKD | Streptozotocin | Rats | [177] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hong, Y.A.; Park, C.W. Catalytic Antioxidants in the Kidney. Antioxidants 2021, 10, 130. https://doi.org/10.3390/antiox10010130
Hong YA, Park CW. Catalytic Antioxidants in the Kidney. Antioxidants. 2021; 10(1):130. https://doi.org/10.3390/antiox10010130
Chicago/Turabian StyleHong, Yu Ah, and Cheol Whee Park. 2021. "Catalytic Antioxidants in the Kidney" Antioxidants 10, no. 1: 130. https://doi.org/10.3390/antiox10010130
APA StyleHong, Y. A., & Park, C. W. (2021). Catalytic Antioxidants in the Kidney. Antioxidants, 10(1), 130. https://doi.org/10.3390/antiox10010130