
Patient Endothelial Colony-Forming Cells to Model Coronary Artery Disease Susceptibility and Unravel the Role of Dysregulated Mitochondrial Redox Signalling

 , ,
, ,
Abstract
:1. Background
2. Methods
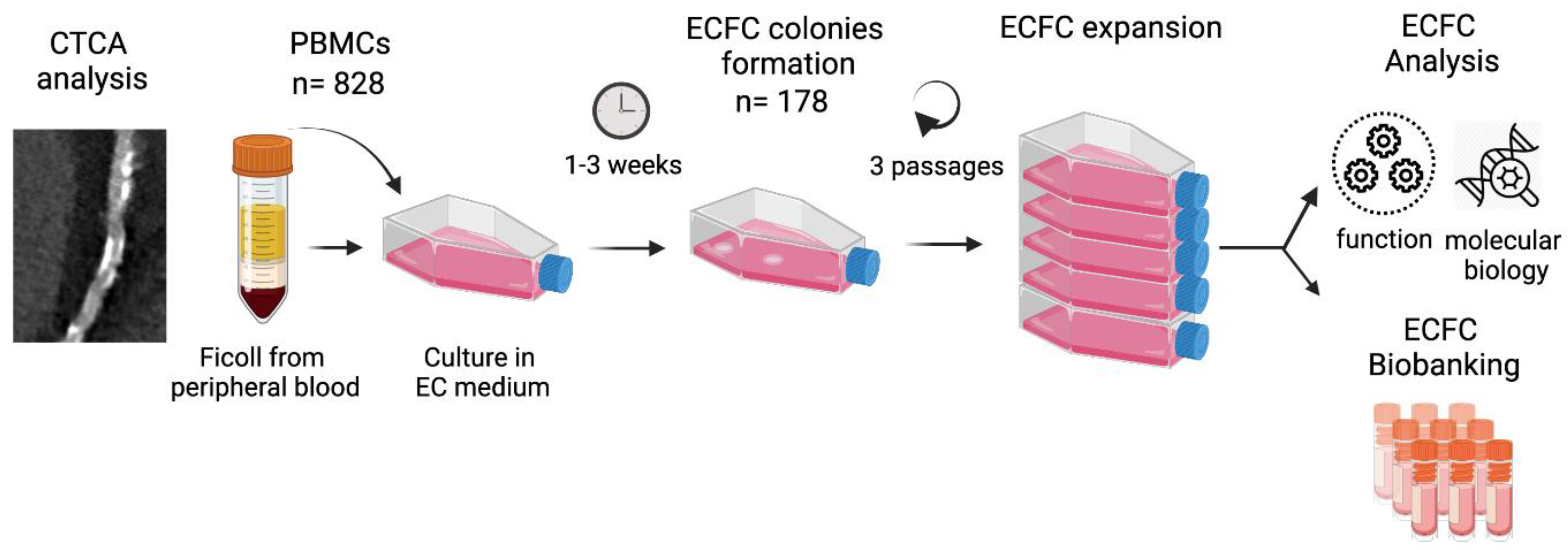
2.1. Study Population
2.2. Definition of Risk Factors
2.3. Imaging Analysis
2.4. Biological Samples and ECFC Growth
2.5. Assessment of ECFCs Phenotype
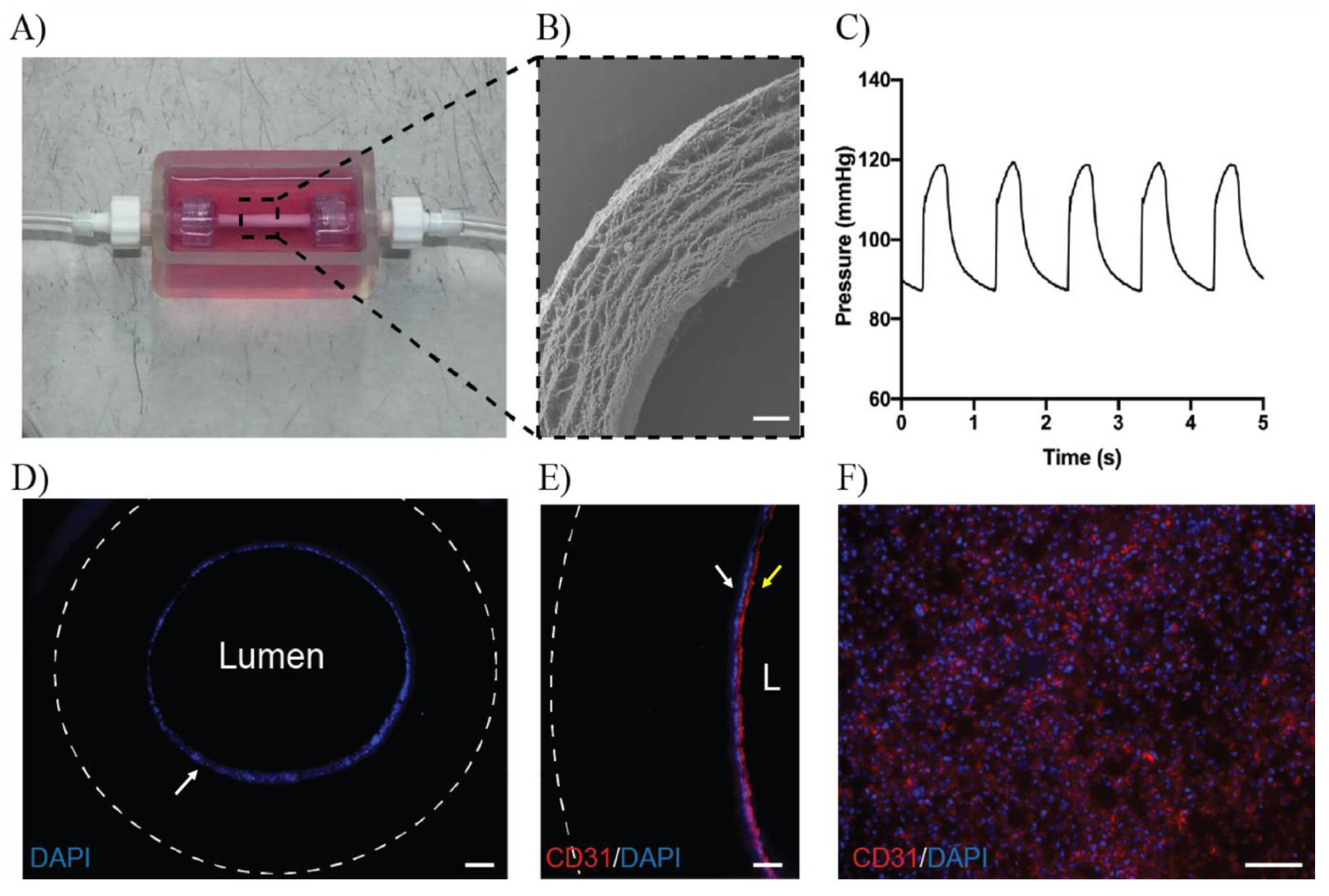
2.6. Culture of ECFCs in 3D Mini Vessels
2.7. Statistical Analysis
3. Results
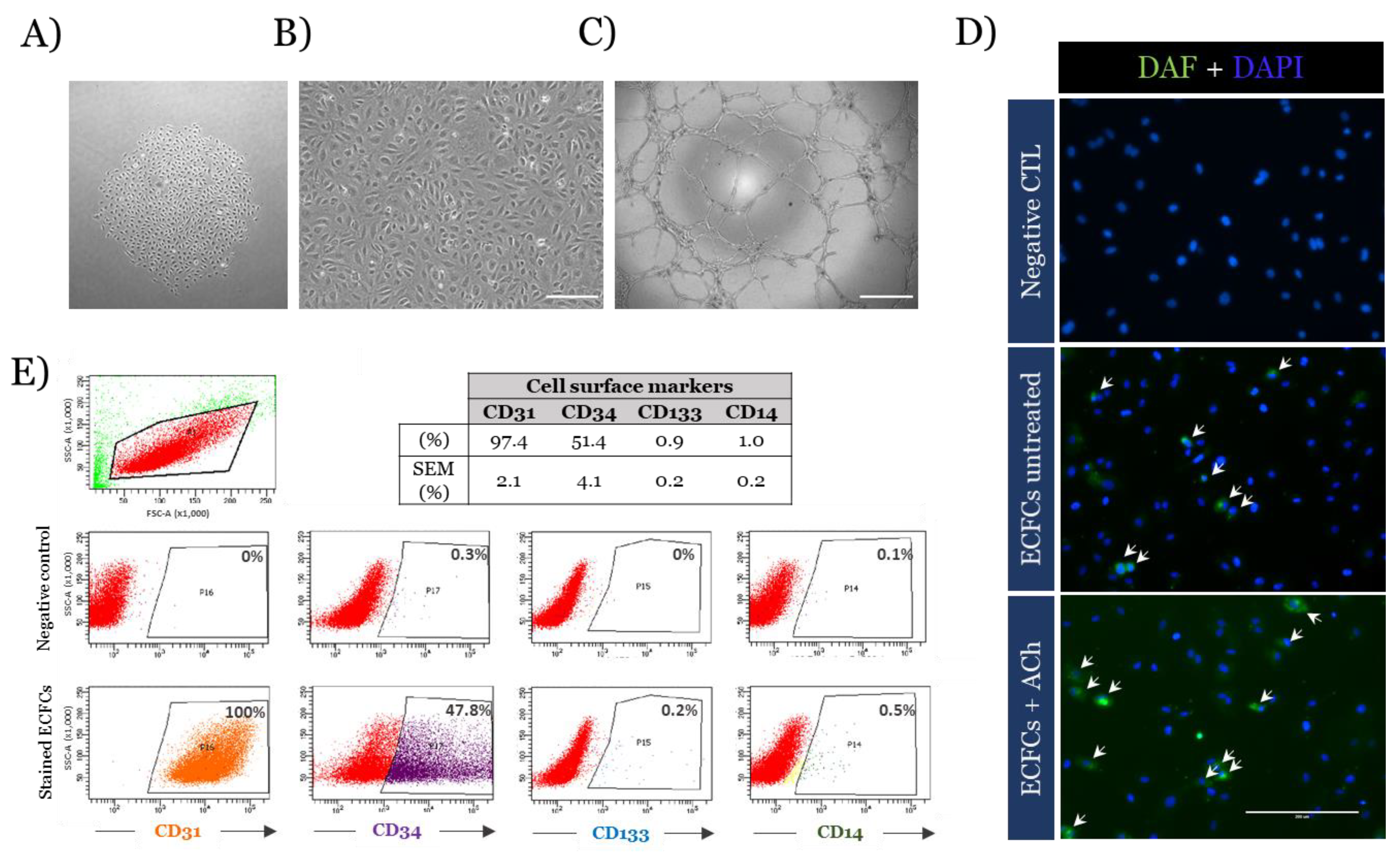
3.1. Baseline Characterisation of ECFCs
3.2. Spontaneous Growth and Effect of Clinical Characteristics
3.3. ECFC Phenotype Reflects the Coronary Artery Disease State of the Patient from Which They Were Derived
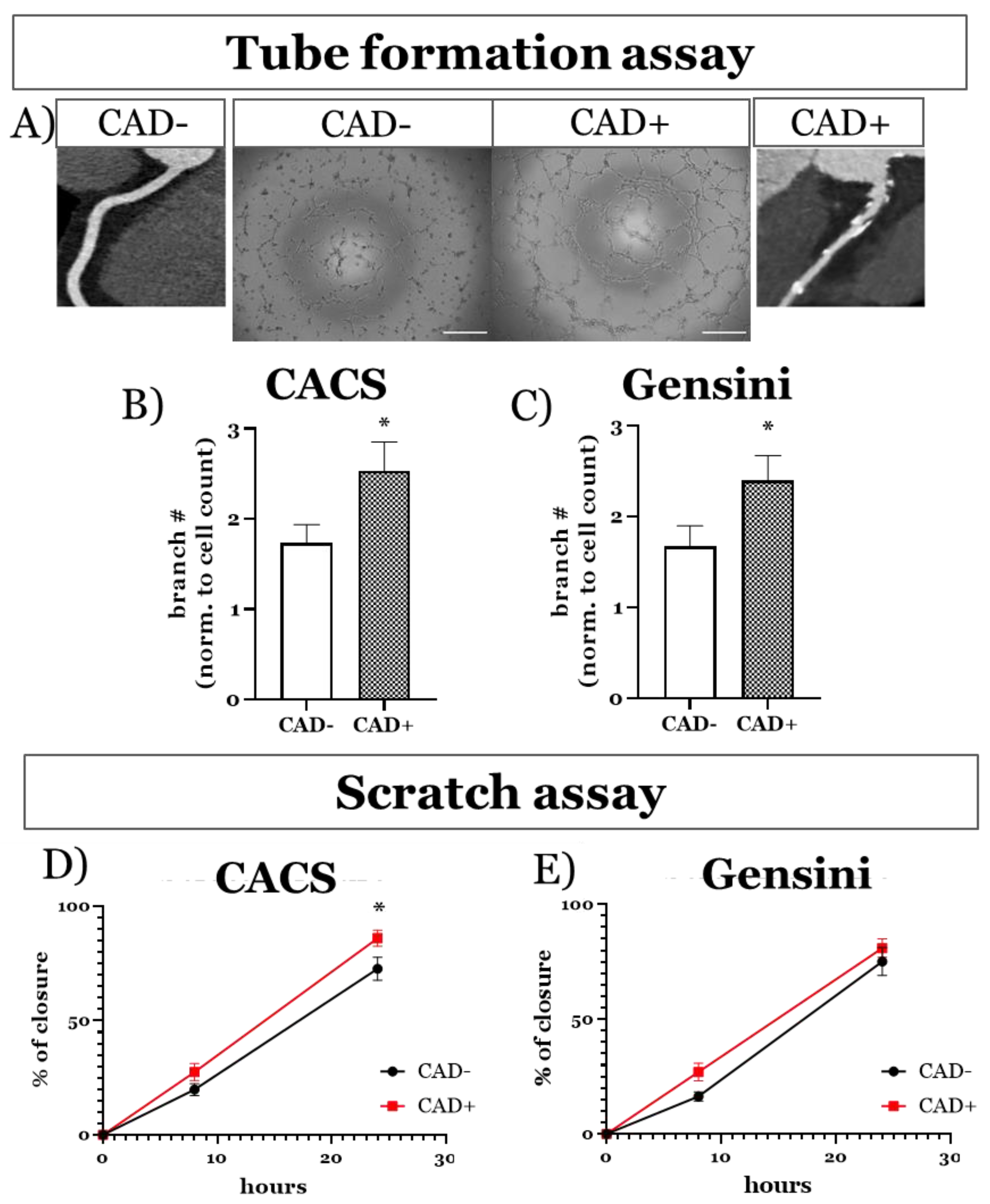
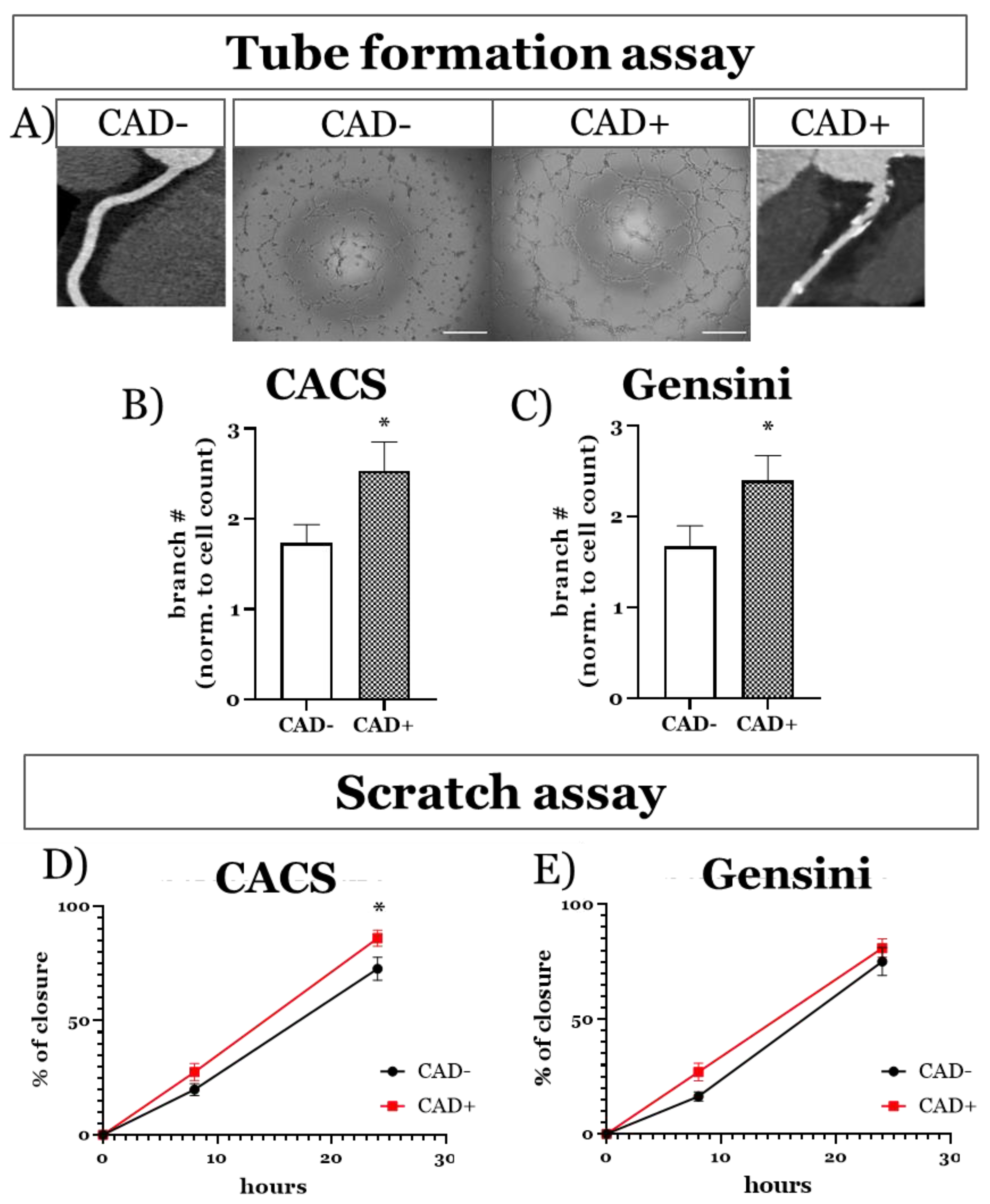
3.3.1. Functional Imprint of Coronary Artery Disease
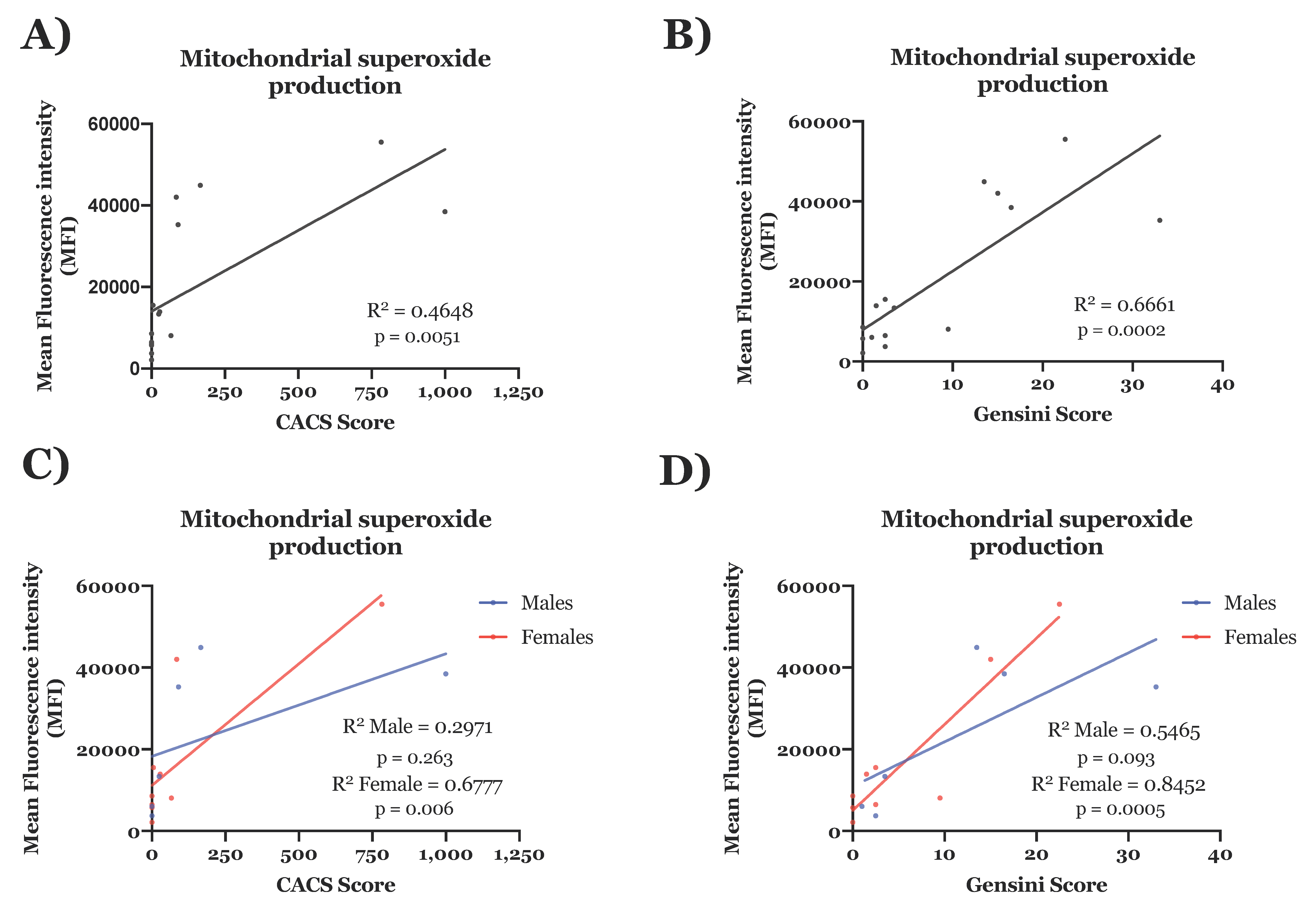
3.3.2. Molecular ECFC Phenotype Associated with Coronary Artery Disease Burden
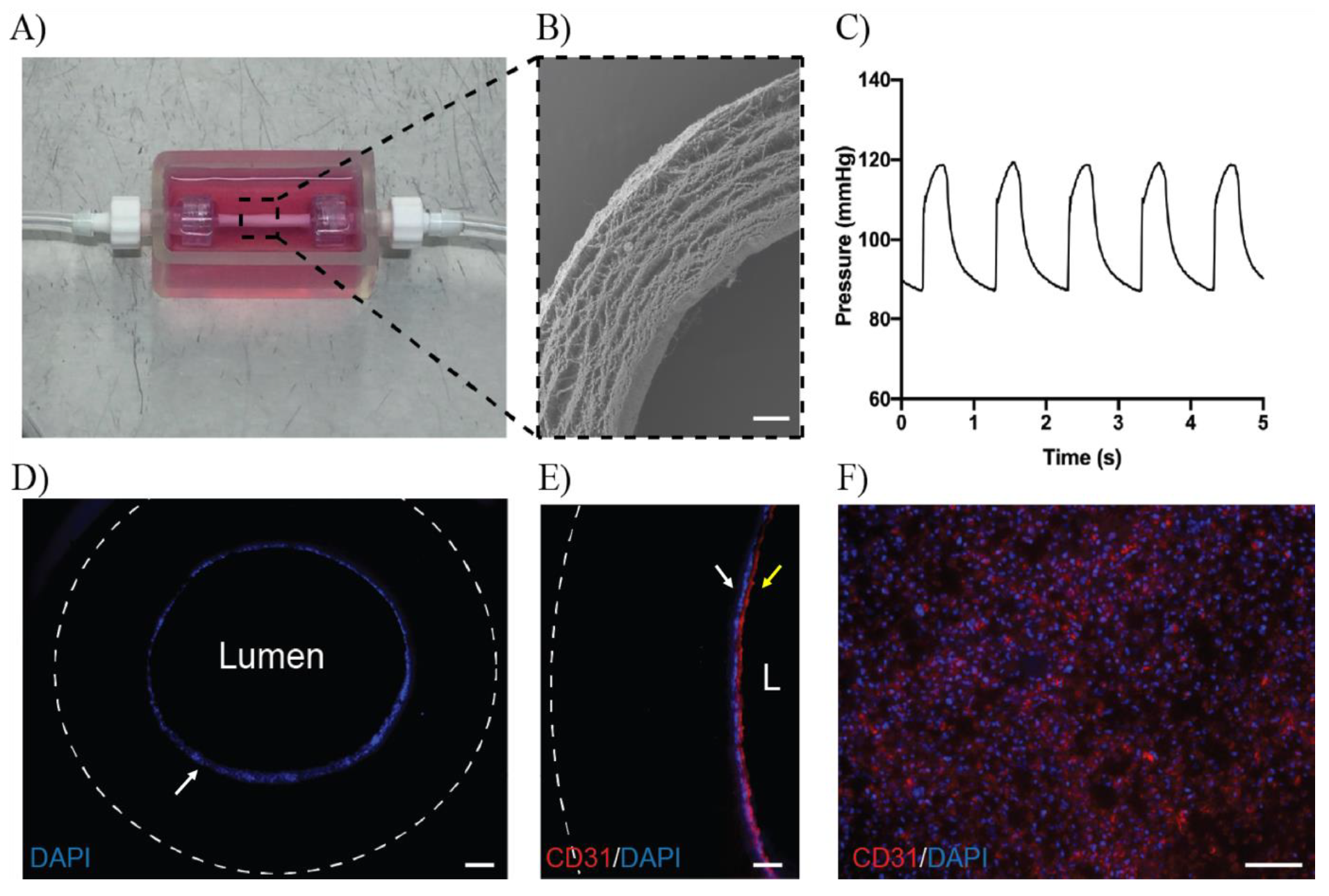
3.4. ECFCs Grown in 3D Mini Vessels
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vernon, S.T.; Coffey, S.; Bhindi, R.; Hoo, S.Y.S.; Nelson, G.I.; Ward, M.R.; Hansen, P.S.; Asrress, K.N.; Chow, C.K.; Celermajer, D.; et al. Increasing proportion of ST elevation myocardial infarction patients with coronary atherosclerosis poorly explained by standard modifiable risk factors. Eur. J. Prev. Cardiol. 2017, 24, 1824–1830. [Google Scholar] [CrossRef] [PubMed]
- Figtree, G.A.; Vernon, S.T.; Hadziosmanovic, N.; Sundström, J.; Alfredsson, J.; Arnott, C.; Delatour, V.; Leósdóttir, M.; Hagström, E. Mortality in STEMI patients without standard modifiable risk factors: A sex-disaggregated analysis of SWEDEHEART registry data. Lancet 2021, 397, 1085–1094. [Google Scholar] [CrossRef]
- Schiele, F.; Ecarnot, F.; Chopard, R. Coronary artery disease: Risk stratification and patient selection for more aggressive secondary prevention. Eur. J. Prev. Cardiol. 2017, 24, 88–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asahara, T.; Murohara, T.; Sullivan, A.; Silver, M.; van der Zee, R.; Li, T.; Witzenbichler, B.; Schatteman, G.; Isner, J.M. Isolation of putative progenitor endothelial cells for angiogenesis. Science 1997, 275, 964–966. [Google Scholar] [CrossRef]
- Paschalaki, K.E.; Randi, A.M. Recent advances in endothelial colony forming cells toward their use in clinical translation. Front. Med. 2018, 5, 295. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.; Weisdorf, D.J.; Solovey, A.; Hebbel, R.P. Origins of circulating endothelial cells and endothelial outgrowth from blood. J. Clin. Investig. 2000, 105, 71–77. [Google Scholar] [CrossRef] [Green Version]
- Fujisawa, T.; Tura-Ceide, O.; Hunter, A.; Mitchell, A.; Vesey, A.; Medine, C.; Gallogly, S.; Hadoke, P.W.; Keith, C.; Sproul, A.; et al. Endothelial progenitor cells do not originate from the bone marrow. Circulation 2019, 140, 1524–1526. [Google Scholar] [CrossRef]
- Medina, R.J.; Barber, C.L.; Sabatier, F.; Dignat-George, F.; Melero-Martin, J.M.; Khosrotehrani, K.; Ohneda, O.; Randi, A.M.; Chan, J.K.Y.; Yamaguchi, T.; et al. Endothelial progenitors: A consensus statement on nomenclature. Stem Cells Transl. Med. 2017, 6, 1316–1320. [Google Scholar] [CrossRef]
- Egorova, A.D.; DeRuiter, M.C.; De Boer, H.C.; Van De Pas, S.; Groot, A.C.G.-D.; Van Zonneveld, A.J.; Poelmann, R.E.; Hierck, B.P. Endothelial colony-forming cells show a mature transcriptional response to shear stress. In Vitro Cell. Dev. Biol.—Anim. 2011, 48, 21–29. [Google Scholar] [CrossRef]
- Prater, D.N.; Case, J.; Ingram, D.A.; Yoder, M. Working hypothesis to redefine endothelial progenitor cells. Leukemia 2007, 21, 1141–1149. [Google Scholar] [CrossRef] [Green Version]
- Ingram, D.A.; Mead, L.E.; Tanaka, H.; Meade, V.; Fenoglio, A.; Mortell, K.; Pollok, K.; Ferkowicz, M.J.; Gilley, D.; Yoder, M.C. Identification of a novel hierarchy of endothelial progenitor cells using human peripheral and umbilical cord blood. Blood 2004, 104, 2752–2760. [Google Scholar] [CrossRef] [PubMed]
- Gremmels, H.; Fledderus, J.O.; Van Balkom, B.W.; Verhaar, M.C. Transcriptome analysis in endothelial progenitor cell biology. Antioxid. Redox Signal. 2011, 15, 1029–1042. [Google Scholar] [CrossRef] [PubMed]
- Kutikhin, A.G.; Tupikin, A.E.; Matveeva, V.G.; Shishkova, D.K.; Antonova, L.V.; Kabilov, M.R.; Velikanova, E.A. Human peripheral blood-derived endothelial colony-forming cells are highly similar to mature vascular endothelial cells yet demonstrate a transitional transcriptomic signature. Cells 2020, 9, 876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leicht, S.F.; Schwarz, T.M.; Hermann, P.C.; Seissler, J.; Aicher, A.; Heeschen, C. Adiponectin pretreatment counteracts the detrimental effect of a diabetic environment on endothelial progenitors. Diabetes 2011, 60, 652–661. [Google Scholar] [CrossRef] [Green Version]
- Paschalaki, K.; Starke, R.D.; Hu, Y.; Mercado, N.; Margariti, A.; Gorgoulis, V.; Randi, A.M.; Barnes, P.J. Dysfunction of endothelial progenitor cells from smokers and chronic obstructive pulmonary disease patients due to increased DNA damage and senescence. Stem Cells 2013, 31, 2813–2826. [Google Scholar] [CrossRef] [Green Version]
- Alvarado-Moreno, J.A.; Davila-Moreno, J.; Dominguez-Reyes, V.; Arreola-Diaz, R.; Isordia-Salas, I.; Chavez-Gonzalez, A.; Majluf-Cruz, A. Morphological and functional alterations in endothelial colony-forming cells from recovered COVID-19 patients. Thromb. Res. 2021, 206, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Kott, K.; Vernon, S.T.; Hansen, T.; Yu, C.; Bubb, K.J.; Coffey, S.; Sullivan, D.; Yang, J.; O’Sullivan, J.; Chow, C.; et al. Biobanking for discovery of novel cardiovascular biomarkers using imaging-quantified disease burden: Protocol for the longitudinal, prospective, BioHEART-CT cohort study. BMJ Open 2019, 9, e028649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vernon, S.T.; Hansen, T.; Kott, K.A.; Yang, J.; O’Sullivan, J.F.; Figtree, G.A. Utilizing state-of-the-art “omics” technology and bioinformatics to identify new biological mechanisms and biomarkers for coronary artery disease. Microcirculation 2018, 26, e12488. [Google Scholar] [CrossRef] [PubMed]
- Abbara, S.; Blanke, P.; Maroules, C.D.; Cheezum, M.; Choi, A.D.; Han, B.K.; Marwan, M.; Naoum, C.; Nørgaard, B.; Rubinshtein, R.; et al. SCCT guidelines for the performance and acquisition of coronary computed tomographic angiography: A report of the Society of Cardiovascular Computed Tomography Guidelines Committee. J. Cardiovasc. Comput. Tomogr. 2016, 10, 435–449. [Google Scholar] [CrossRef] [PubMed]
- Harden, S.; Bull, R.; Bury, R.; Castellano, E.; Clayton, B.; Hamilton, M.; Morgan-Hughes, G.; O’Regan, D.; Padley, S.; Roditi, G.; et al. The safe practice of CT coronary angiography in adult patients in UK imaging departments. Clin. Radiol. 2016, 71, 722–728. [Google Scholar] [CrossRef]
- Agatston, A.S.; Janowitz, F.W.R.; Hildner, F.J.; Zusmer, N.R.; Viamonte, M., Jr.; Detrano, R. Quantification of coronary artery calcium using ultrafast computed tomography. J. Am. Coll. Cardiol. 1990, 15, 827–832. [Google Scholar] [CrossRef] [Green Version]
- Leipsic, J.; Abbara, S.; Achenbach, S.; Cury, R.; Earls, J.P.; Mancini, G.J.; Nieman, K.; Pontone, G.; Raff, G.L. SCCT guidelines for the interpretation and reporting of coronary CT angiography: A report of the Society of Cardiovascular Computed Tomography Guidelines Committee. J. Cardiovasc. Comput. Tomogr. 2014, 8, 342–358. [Google Scholar] [CrossRef]
- Gensini, G.G. A more meaningful scoring system for determining the severity of coronary heart disease. Am. J. Cardiol. 1983, 51, 606. [Google Scholar] [CrossRef]
- Riedhammer, C.; Halbritter, D.; Weissert, R. Peripheral Blood Mononuclear Cells: Isolation, Freezing, Thawing, and Culture. Methods Mol. Biol. 2016, 1304, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Tan, R.P.; Chan, A.H.P.; Lennartsson, K.; Miravet, M.M.; Lee, S.-L.; Rnjak-Kovacina, J.; Clayton, Z.; Cooke, J.P.; Ng, M.K.C.; Patel, S.; et al. Integration of induced pluripotent stem cell-derived endothelial cells with polycaprolactone/gelatin-based electrospun scaffolds for enhanced therapeutic angiogenesis. Stem Cell Res. Ther. 2018, 9, 70. [Google Scholar] [CrossRef] [PubMed]
- Basile, D.P.; Yoder, M.C. Circulating and tissue resident endothelial progenitor cells. J. Cell. Physiol. 2014, 229, 10–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guzik, T.J.; Sadowski, J.; Guzik, B.; Jopek, A.; Kapelak, B.; Przybyłowski, P.; Wierzbicki, K.; Korbut, R.; Harrison, D.G.; Channon, K.M. Coronary Artery Superoxide Production and Nox Isoform Expression in Human Coronary Artery Disease. Arter. Thromb. Vasc. Biol. 2006, 26, 333–339. [Google Scholar] [CrossRef] [Green Version]
- Madamanchi, N.; Runge, M.S. Mitochondrial Dysfunction in Atherosclerosis. Circ. Res. 2007, 100, 460–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farcas, M.A.; Rouleau, L.; Fraser, R.; Leask, R.L. The development of 3-D, in vitro, endothelial culture models for the study of coronary artery disease. Biomed. Eng. Online 2009, 8, 30. [Google Scholar] [CrossRef] [Green Version]
- Vasa, M.; Fichtlscherer, S.; Aicher, A.; Adler, K.; Urbich, C.; Martin, H.; Zeiher, A.M.; Dimmeler, S. Number and migratory activity of circulating endothelial progenitor cells inversely correlate with risk factors for coronary artery disease. Circ. Res. 2001, 89, E1–E7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, S.; Xia, W.; Chen, C.; Robinson, E.A.; Tao, J. Endothelial progenitor cells and hypertension: Current concepts and future implications. Clin. Sci. 2016, 130, 2029–2042. [Google Scholar] [CrossRef] [PubMed]
- Coppolino, G.; Bolignano, D.; Campo, S.; Loddo, S.; Teti, D.; Buemi, M. Circulating progenitor cells after cold pressor test in hypertensive and uremic patients. Hypertens. Res. 2008, 31, 717–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.-W.; Lo, H.-H.; Chiu, Y.-L.; Chang, S.-J.; Huang, P.-H.; Liao, K.-H.; Tasi, C.-F.; Wu, C.-H.; Tsai, T.-N.; Cheng, C.-C.; et al. Dysregulated miR-361-5p/VEGF axis in the plasma and endothelial progenitor cells of patients with coronary artery disease. PLoS ONE 2014, 9, e98070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, W.-C.; Chiang, W.-H.; Wu, C.-H.; Li, Y.-C.; Campbell, M.; Huang, P.-H.; Lin, M.-W.; Lin, C.-H.; Cheng, S.-M.; Chang, P.-C.; et al. miR-548aq-3p is a novel target of Far infrared radiation which predicts coronary artery disease endothelial colony forming cell responsiveness. Sci. Rep. 2020, 10, 6805. [Google Scholar] [CrossRef] [Green Version]
- Stroncek, J.D.; Grant, B.S.; Brown, M.A.; Povsic, T.J.; Truskey, G.A.; Reichert, W.M. Comparison of endothelial cell phenotypic markers of late-outgrowth endothelial progenitor cells isolated from patients with coronary artery disease and healthy volunteers. Tissue Eng. Part A 2009, 15, 3473–3486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dikalov, S. Cross talk between mitochondria and NADPH oxidases. Free. Radic. Biol. Med. 2011, 51, 1289–1301. [Google Scholar] [CrossRef] [Green Version]
- Mercer, J.R.; Yu, E.; Figg, N.; Cheng, K.-K.; Prime, T.A.; Griffin, J.L.; Masoodi, M.; Vidal-Puig, A.; Murphy, M.; Bennett, M.R. The mitochondria-targeted antioxidant MitoQ decreases features of the metabolic syndrome in ATM+/−/ApoE−/− mice. Free Radic. Biol. Med. 2012, 52, 841–849. [Google Scholar] [CrossRef]
- Park, S.-Y.; Pekas, E.J.; Headid, R.J.; Son, W.-M.; Wooden, T.K.; Song, J.; Layec, G.; Yadav, S.K.; Mishra, P.K.; Pipinos, I.I. Acute mitochondrial antioxidant intake improves endothelial function, antioxidant enzyme activity, and exercise tolerance in patients with peripheral artery disease. Am. J. Physiol. Circ. Physiol. 2020, 319, H456–H467. [Google Scholar] [CrossRef]
- Matthew, J.R.; Jessica, R.S.-P.; Chelsea, A.C.S.; Nina, Z.B.; Lauren, M.C.; Hannah, L.R.; Kayla, A.; Chonchol, M.W.; Rachel, A.G.-R.; Michael, P.M.; et al. Chronic Supplementation with a Mitochondrial Antioxidant (MitoQ) Improves Vascular Func-tion in Healthy Older Adults. Hypertension 2018, 71, 1056–1063. [Google Scholar] [CrossRef]
- Figtree, G.A.; Broadfoot, K.; Casadei, B.; Califf, R.; Crea, F.; Drummond, G.R.; Freedman, J.E.; Guzik, T.J.; Harrison, D.; Hausenloy, D.J.; et al. A call to action for new global approaches to cardiovascular disease drug solutions. Eur. Heart J. 2021, in press. [Google Scholar] [CrossRef]
- Figtree, G.A.; Broadfoot, K.; Casadei, B.; Califf, R.; Crea, F.; Drummond, G.R.; Freedman, J.E.; Guzik, T.J.; Harrison, D.; Hausenloy, D.J.; et al. A call to action for new global approaches to cardiovascular disease drug solutions. Circulation 2021, in press. [Google Scholar] [CrossRef]
- Zeng, L.; Talukdar, H.A.; Koplev, S.; Giannarelli, C.; Ivert, T.; Gan, L.-M.; Ruusalepp, A.; Schadt, E.E.; Kovacic, J.C.; Lusis, A.J.; et al. Contribution of Gene Regulatory Networks to Heritability of Coronary Artery Disease. J. Am. Coll. Cardiol. 2019, 73, 2946–2957. [Google Scholar] [CrossRef] [PubMed]
- Talukdar, H.A.; Asl, H.F.; Jain, R.K.; Ermel, R.; Ruusalepp, A.; Franzén, O.; Kidd, B.A.; Readhead, B.; Giannarelli, C.; Kovacic, J.C.; et al. Cross-Tissue Regulatory Gene Networks in Coronary Artery Disease. Cell Syst. 2016, 2, 196–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, K.; Shameer, K.; Glicksberg, B.; Readhead, B.; Sengupta, P.P.; Björkegren, J.L.; Kovacic, J.C.; Dudley, J.T. Enabling Precision Cardiology Through Multiscale Biology and Systems Medicine. JACC Basic Transl. Sci. 2017, 2, 311–327. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Cheng, H.; Hong, X.; Di Narzo, A.F.; Franzen, O.; Peng, S.; Ruusalepp, A.; Kovacic, J.C.; Bjorkegren, J.L.M.; Wang, X.; et al. EnsembleCNV: An ensemble machine learning algorithm to identify and genotype copy number variation using SNP array data. Nucleic Acids Res. 2019, 47, e39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franzén, O.; Ermel, R.; Sukhavasi, K.; Jain, R.; Jain, A.; Betsholtz, C.; Giannarelli, C.; Kovacic, J.C.; Ruusalepp, A.; Skogsberg, J.; et al. Global analysis of A-to-I RNA editing reveals association with common disease variants. PeerJ 2018, 6, e4466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olin, J.W.; Di Narzo, A.F.; D’Escamard, V.; Kadian-Dodov, D.; Cheng, H.; Georges, A.; King, A.; Thomas, A.; Barwari, T.; Michelis, K.C.; et al. A plasma proteogenomic signature for fibromuscular dysplasia. Cardiovasc. Res. 2020, 116, 63–77. [Google Scholar] [CrossRef]


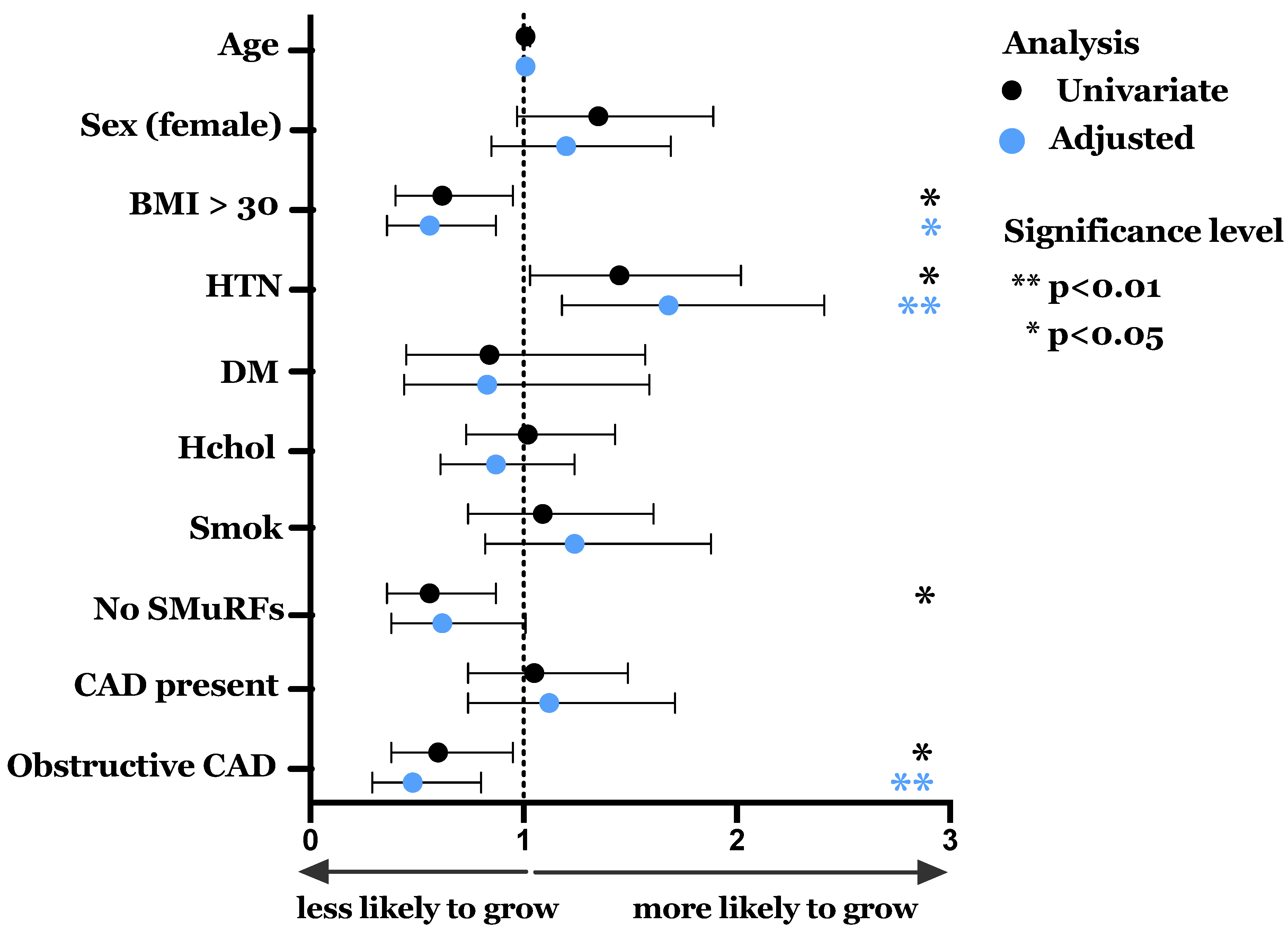


{kind=link}
{kind=link}
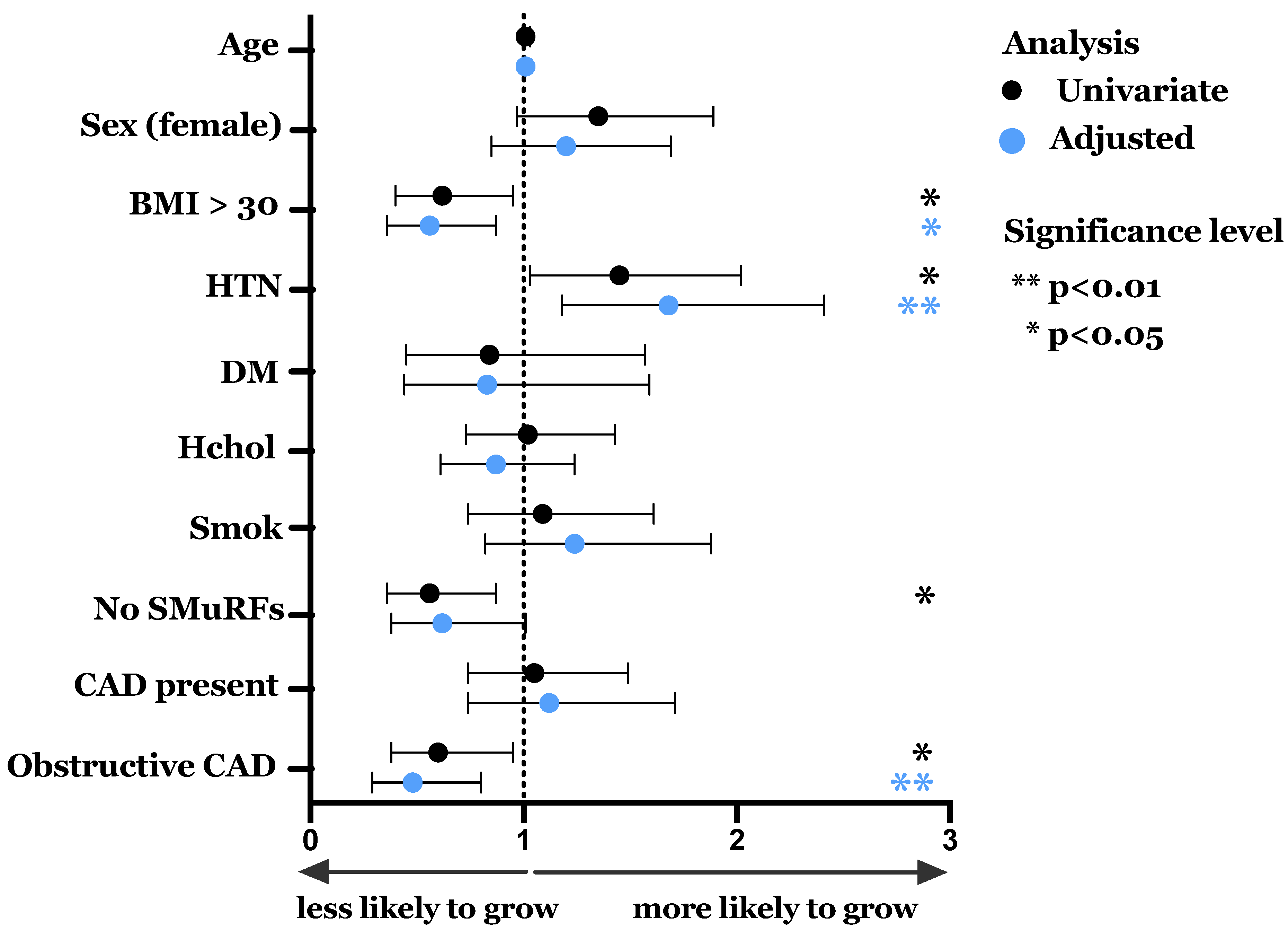{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | Whole Cohort (n = 828) | ECFC Growth (n = 178) | No ECFC Growth (n = 650) | p Value |
|---|---|---|---|---|
| Age, mean (SD) | 60.9 (11.9) | 62.1 (11.9) | 60.6 (11.9) | 0.14 |
| Female, n (%) | 379 (45.8) | 92 (51.7) | 287 (44.2) | 0.07 |
| Hypertension, n (%) | 319 (38.5) | 81 (45.5) | 238 (36.7) | 0.03 |
| Diabetes mellitus, n (%) | 69 (8.3) | 13 (7.3) | 56 (9.4) | 0.58 |
| Hypercholesterolaemia, n (%) | 476 (57.5) | 103 (57.9) | 373 (57.4) | 0.91 |
| Significant smoking history, n (%) | 190 (22.9) | 43 (24.2) | 147 (22.6) | 0.67 |
| Current smoker, n (%) | 59 (7.1) | 10 (5.6) | 49 (7.5) | 0.38 |
| BMI, mean, (SD) | 27.1 (5.0) | 27.3 (5.1) | 26.3 (4.8) | 0.02 |
| BMI >30 kg/m2, n (%) | 191 (23.1) | 30 (16.9) | 161 (24.8) | 0.03 |
| Significant family history CAD, n (%) | 163 (19.7) | 29 (16.3) | 134 (20.6) | 0.20 |
| SMuRFs—mean, (SD) | 1.27 (0.96) | 1.35 (0.87) | 1.25 (0.98) | 0.24 |
| 0 SMuRFs, n (%) | 185 (22.3) | 27 (15.2) | 158 (24.3) | <0.01 |
| Coronary artery calcium score—median, (IQR) | 10 (0–177) | 7.97 (0–140) | 11 (0-193) | 0.67 |
| Calcified plaque present (CACS > 0) | 494 (59.7) | 109 (61.2) | 385 (59.2) | 0.63 |
| Gensini score—median, IQR | 4 (0–13) | 3 (0–10) | 4 (0–14) | 0.27 |
| CAD present (Gensini > 0) | 547 (66.1) | 119 (66.9) | 428 (65.9) | 0.80 |
| Obstructive disease > 50% stenosis—n, (%) | 165 (19.9) | 25 (14.0) | 140 (21.5) | 0.03 |
| Medication use: | ||||
| Anti-coagulant—n, (%) | 76 (9.2) | 18 (10.1) | 58 (8.9) | 0.63 |
| Anti-platelet agent—n, (%) | 145 (17.5) | 32 (18.0) | 113 (17.4) | 0.85 |
| Statin—n, (%) | 282 (34.1) | 66 (37.1) | 216 (33.2) | 0.34 |
| Beta-blocker—n, (%) | 122 (14.7) | 21 (11.8) | 101 (15.6) | 0.21 |
| ACE/ARB agent—n, (%) | 256 (30.9) | 63 (35.4) | 193 (29.7) | 0.15 |
| Target (n CAD− vs. n CAD+) | CAD− (mean ± SEM) | CAD+ (mean ± SEM) | p Value |
|---|---|---|---|
| NOX2: - CACS (14 vs. 17) - Gensini (6 vs. 19) | 1.00 ± 0.21 1.00 ± 0.26 | 0.93 ± 0.23 1.19 ± 0.23 | 0.92 0.68 |
| NOX4: - CACS (11 vs. 14) - Gensini (6 vs. 19) | 1.00 ± 0.27 1.00 ± 0.47 | 1.48 ± 0.36 1.26 ± 0.26 | 0.30 0.63 |
| eNOS:- CACS (14 vs. 16) - Gensini (9 vs. 21) | 1.00 ± 0.30 1.00 ± 0.34 | 1.34 ± 0.45 1.77 ± 0.49 | 0.54 0.34 |
| AKT: - CACS (13 vs. 18) - Gensini (8 vs. 23) | 1.00 ± 0.47 1.00 ± 0.60 | 0.93 ± 0.27 0.70 ± 0.18 | 0.90 0.53 |
| pAKT: - CACS (13 vs. 18) - Gensini (8 vs. 23) | 1.00 ± 0.17 1.00 ± 0.21 | 1.43 ± 0.37 1.09 ± 0.26 | 0.31 0.84 |
| pAKT/AKT: - CACS (13 vs. 18) - Gensini (8 vs. 23) | 1.00 ± 0.24 1.00 ± 0.30 | 0.66 ± 0.13 0.84 ± 0.16 | 0.16 0.62 |
| ERK:- CACS (12 vs. 18) - Gensini (7 vs. 23) | 1.00 ± 0.13 1.00 ± 0.18 | 1.36 ± 0.25 1.20 ± 0.19 | 0.28 0.60 |
| pERK: - CACS (12 vs. 18) - Gensini (7 vs. 23) | 1.00 ± 0.41 1.00 ± 0.63 | 1.82 ± 0.67 1.69 ± 0.55 | 0.31 0.52 |
| pERK/ERK: - CACS (12 vs. 18) - Gensini (7 vs. 23) | 1.00 ± 0.35 1.00 ± 0.50 | 1.26 ± 0.46 1.26 ± 0.39 | 0.69 0.73 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Besnier, M.; Finemore, M.; Yu, C.; Kott, K.A.; Vernon, S.T.; Seebacher, N.A.; Genetzakis, E.; Furman, A.; Tang, O.; Davis, R.L.; et al. Patient Endothelial Colony-Forming Cells to Model Coronary Artery Disease Susceptibility and Unravel the Role of Dysregulated Mitochondrial Redox Signalling. Antioxidants 2021, 10, 1547. https://doi.org/10.3390/antiox10101547
Besnier M, Finemore M, Yu C, Kott KA, Vernon ST, Seebacher NA, Genetzakis E, Furman A, Tang O, Davis RL, et al. Patient Endothelial Colony-Forming Cells to Model Coronary Artery Disease Susceptibility and Unravel the Role of Dysregulated Mitochondrial Redox Signalling. Antioxidants. 2021; 10(10):1547. https://doi.org/10.3390/antiox10101547
Chicago/Turabian StyleBesnier, Marie, Meghan Finemore, Christine Yu, Katharine A. Kott, Stephen T. Vernon, Nicole A. Seebacher, Elijah Genetzakis, Anamarija Furman, Owen Tang, Ryan L. Davis, and et al. 2021. "Patient Endothelial Colony-Forming Cells to Model Coronary Artery Disease Susceptibility and Unravel the Role of Dysregulated Mitochondrial Redox Signalling" Antioxidants 10, no. 10: 1547. https://doi.org/10.3390/antiox10101547