
Glyoxalase-I Is Upregulated in Acute Cerulein-Induced Pancreatitis: A New Mechanism in Pancreatic Inflammation?


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal Experiments
2.2. Cell Culture and Cerulein Treatment
2.3. Immunohistochemistry
2.4. qRT-PCR
2.5. Western Blot Analysis
2.6. Measurement of Specific Glo-I Activity
2.7. Measurement of Amylase Secretion, TNF-α and MGO by ELISA
2.8. Glo-I Plasmid Generation
2.9. Transfection
2.10. Statistical Analysis
3. Results
3.1. Glo-I Is Upregulated in CN-Induced AP in Mice
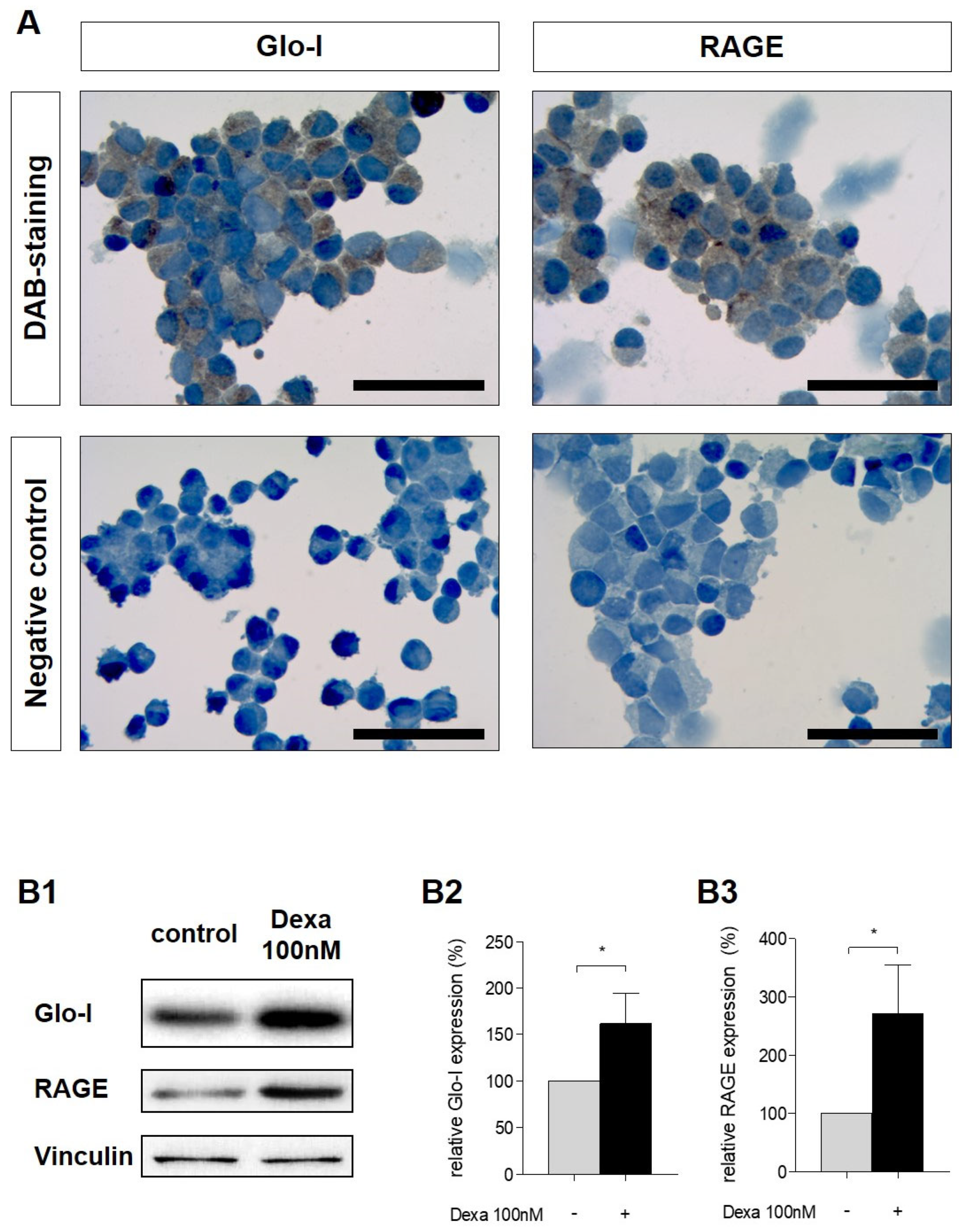
3.2. Glo-I and RAGE Expression in AR42J Cells
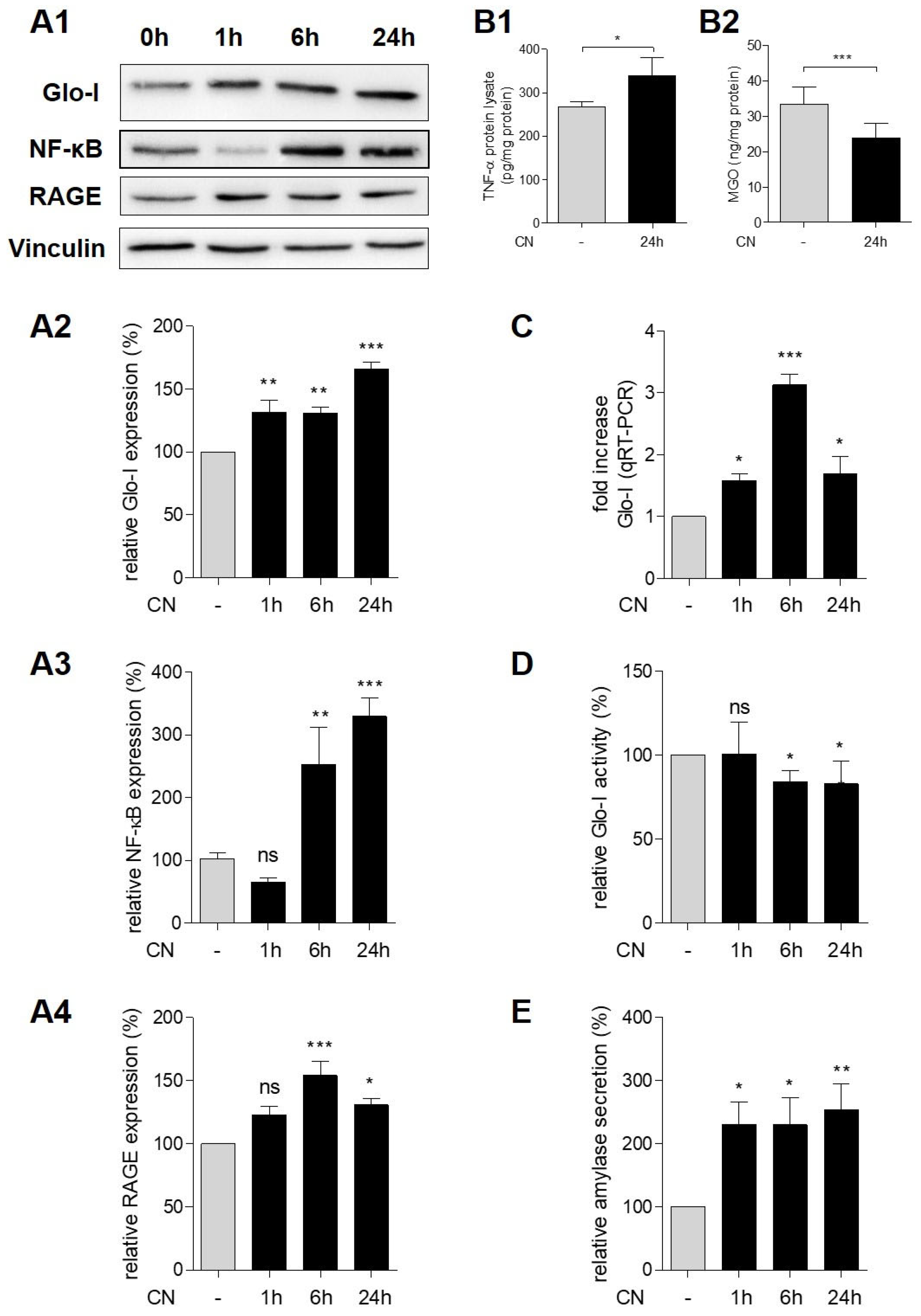
3.3. CN Induced Glo-I, RAGE, NF-κB and TNF-α
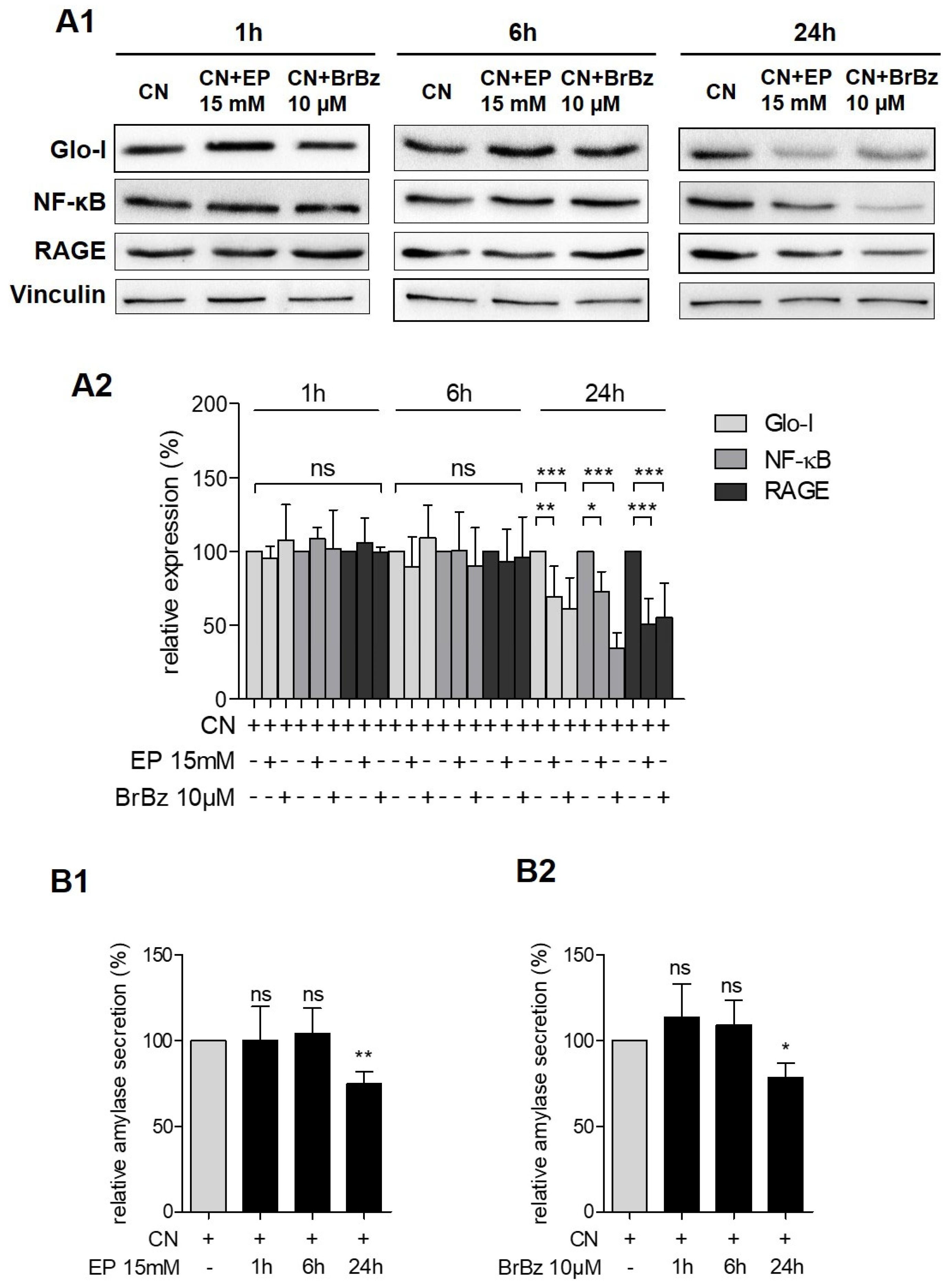
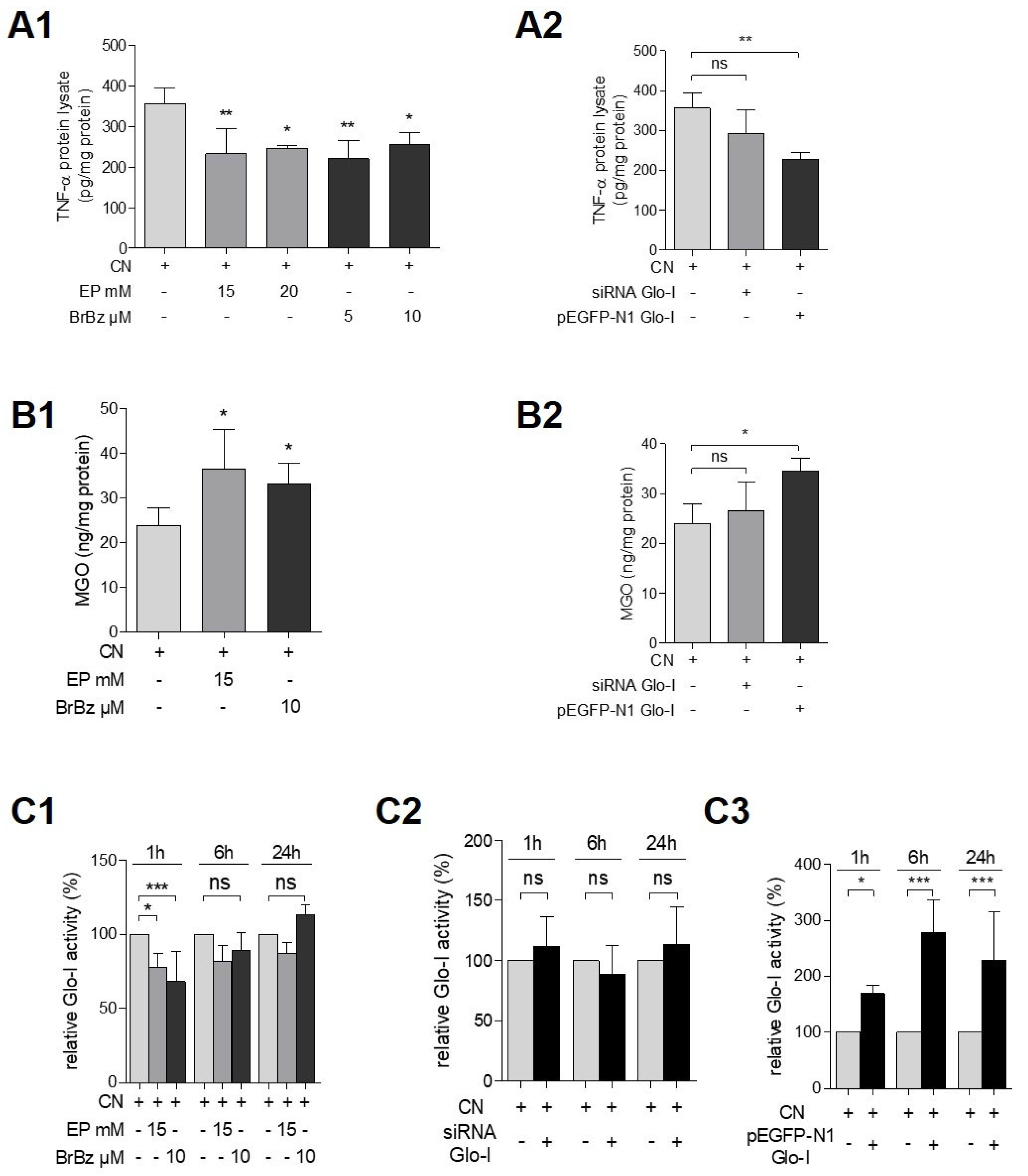
3.4. Pharmacological Modulation of Glo-I Attenuated CN-Induced Expression of Pro-Inflammatory Markers
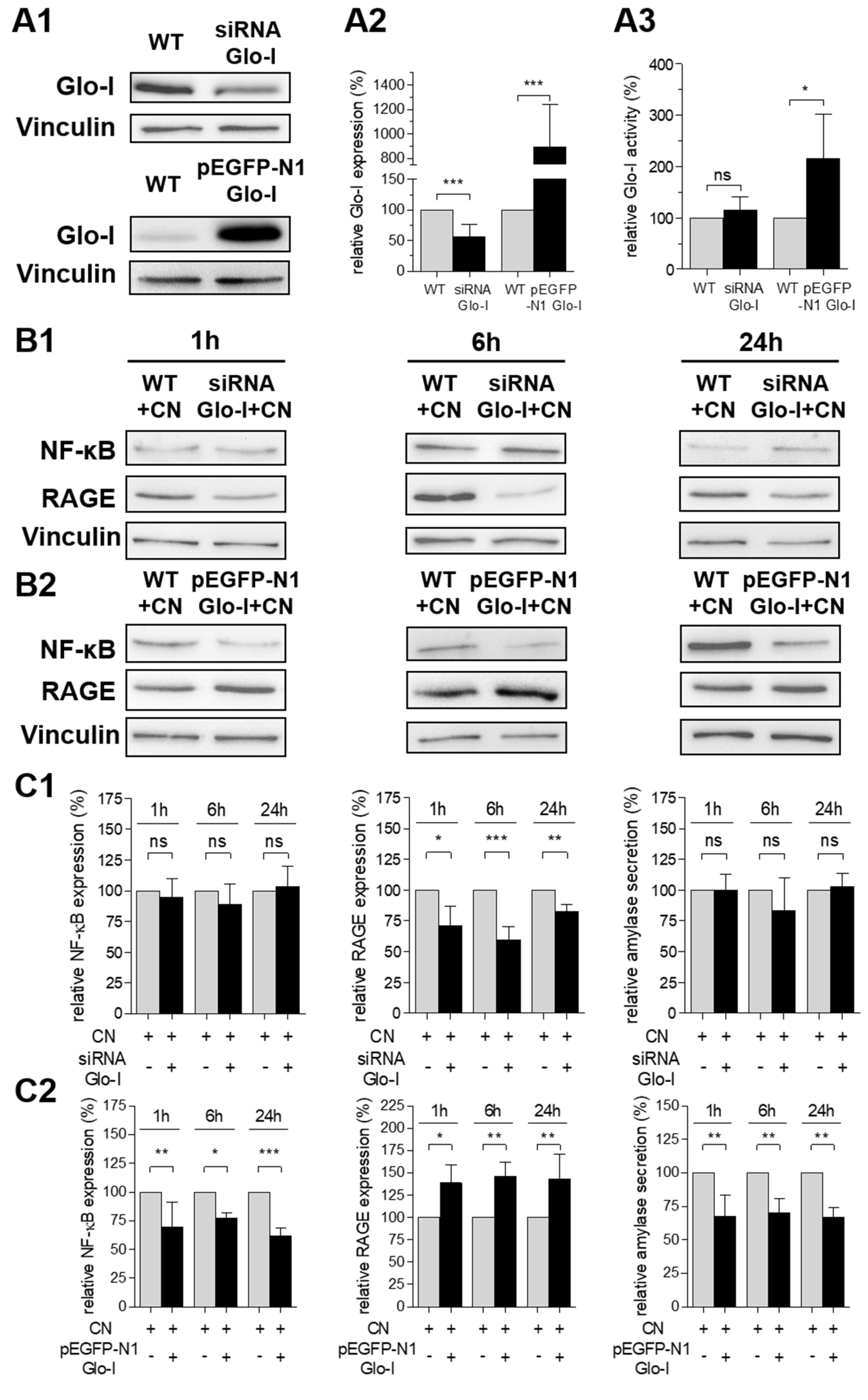
3.5. Glo-I Overexpression Reduced CN-Induced Increase of NF-κB, RAGE and Amylase
3.6. Effects of Pharmacological Modulation and Overexpression of Glo-I Are Mediated via Regulation of MGO
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AGE | advanced glycation end-products |
| AB | antibody |
| AP | acute pancreatitis |
| bw | body weight |
| BrBz | BrBzGSHCP2 (S-p-bromobenzylglutathione cyclopentyl diester) |
| CN | cerulein |
| DAB | Diaminobenzidin |
| DAMP | damage-associated molecular pattern |
| Dexa | dexamethasone |
| EP | ethyl pyruvate |
| FCS | fetal calf serum |
| Glo-I | Glyoxalase-I |
| Glo-II | Glyoxalase-II |
| GSH | L-gluthatione |
| MGO | methylglyoxal |
| NLRP3 | nucleotide-binding oligomerization domain-like receptor protein 3 |
| P/S | penicillin and streptomycin |
| PVDF | polyvinylidene fluoride |
| qRT-PCR | quantitative real-time polymerase chain reaction |
| RAGE | receptor for advanced glycation end-products |
| ROS | oxidative stress |
| SDS-PAGE | sodium dodecyl sulfate polyacrylamide gel electrophoresis |
References
- Peery, A.F.; Crockett, S.D.; Murphy, C.C.; Lund, J.L.; Dellon, E.S.; Williams, J.L.; Jensen, E.T.; Shaheen, N.J.; Barritt, A.S.; Lieber, S.R.; et al. Burden and Cost of Gastrointestinal, Liver, and Pancreatic Diseases in the United States: Update 2018. Gastroenterology 2019, 156, 254–272. [Google Scholar] [CrossRef] [Green Version]
- Mayerle, J.; Sendler, M.; Hegyi, E.; Beyer, G.; Lerch, M.M.; Sahin-Toth, M. Genetics, Cell Biology, and Pathophysiology of Pancreatitis. Gastroenterology 2019, 156, 1951–1968. [Google Scholar] [CrossRef] [Green Version]
- Hoque, R.; Sohail, M.; Malik, A.; Sarwar, S.; Luo, Y.; Shah, A.; Barrat, F.; Flavell, R.; Gorelick, F.; Husain, S.; et al. TLR9 and the NLRP3 inflammasome link acinar cell death with inflammation in acute pancreatitis. Gastroenterology 2011, 141, 358–369. [Google Scholar] [CrossRef] [Green Version]
- Sendler, M.; Weiss, F.U.; Golchert, J.; Homuth, G.; van den Brandt, C.; Mahajan, U.M.; Partecke, L.-I.; Döring, P.; Gukovsky, I.; Gukovskaya, A.S.; et al. Cathepsin B-Mediated Activation of Trypsinogen in Endocytosing Macrophages Increases Severity of Pancreatitis in Mice. Gastroenterology 2018, 154, 704–718. [Google Scholar] [CrossRef] [Green Version]
- Sendler, M.; Dummer, A.; Weiss, F.U.; Kruger, B.; Wartmann, T.; Scharffetter-Kochanek, K.; van Rooijen, N.; Malla, S.R.; Aghdassi, A.; Halangk, W.; et al. Tumour necrosis factor alpha secretion induces protease activation and acinar cell necrosis in acute experimental pancreatitis in mice. Gut 2013, 62, 430–439. [Google Scholar] [CrossRef]
- Dembinski, A.; Warzecha, Z.; Ceranowicz, P.; Stachura, J.; Tomaszewska, R.; Konturek, S.J.; Sendur, R.; Dembinski, M.; Pawlik, W.W. Pancreatic damage and regeneration in the course of ischemia-reperfusion induced pancreatitis in rats. J. Physiol. Pharmacol. 2001, 52, 221–235. [Google Scholar]
- Lonardo, A.; Grisendi, A.; Bonilauri, S.; Rambaldi, M.; Selmi, I.; Tondelli, E. Ischaemic necrotizing pancreatitis after cardiac surgery. A case report and review of the literature. Ital. J. Gastroenterol. Hepatol. 1999, 31, 872–875. [Google Scholar]
- Sakorafas, G.H.; Tsiotos, G.G.; Bower, T.C.; Sarr, M.G. Ischemic necrotizing pancreatitis. Two case reports and review of the literature. Int. J. Pancreatol. 1998, 24, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Waldner, H. Vascular mechanisms to induce acute pancreatitis. Eur. Surg. Res. 1992, 24 (Suppl. 1), 62–67. [Google Scholar] [CrossRef] [PubMed]
- Warzecha, Z.; Dembinski, A.; Jaworek, J.; Ceranowicz, P.; Szlachcic, A.; Walocha, J.; Konturek, S.J. Role of sensory nerves in pancreatic secretion and caerulein-induced pancreatitis. J. Physiol. Pharmacol. 1997, 48, 43–58. [Google Scholar] [PubMed]
- Hollenbach, M. The Role of Glyoxalase-I (Glo-I), Advanced Glycation Endproducts (AGEs), and Their Receptor (RAGE) in Chronic Liver Disease and Hepatocellular Carcinoma (HCC). Int. J. Mol. Sci. 2017, 18, 2466. [Google Scholar] [CrossRef] [Green Version]
- Antognelli, C.; Talesa, V.N. Glyoxalases in Urological Malignancies. Int. J. Mol. Sci. 2018, 19, 415. [Google Scholar] [CrossRef] [Green Version]
- Ohmori, S.; Mori, M.; Shiraha, K.; Kawase, M. Biosynthesis and degradation of methylglyoxal in animals. Prog. Clin. Biol. Res. 1989, 290, 397–412. [Google Scholar] [PubMed]
- Ray, S.; Ray, M. Formation of methylglyoxal from aminoacetone by amine oxidase from goat plasma. J. Biol. Chem. 1983, 258, 3461–3462. [Google Scholar] [CrossRef]
- Casazza, J.P.; Felver, M.E.; Veech, R.L. The metabolism of acetone in rat. J. Biol. Chem. 1984, 259, 231–236. [Google Scholar] [CrossRef]
- Phillips, S.A.; Thornalley, P.J. The formation of methylglyoxal from triose phosphates. Investigation using a specific assay for methylglyoxal. Eur. J. Biochem. 1993, 212, 101–105. [Google Scholar] [CrossRef]
- Vaca, C.E.; Fang, J.L.; Conradi, M.; Hou, S.M. Development of a 32P-postlabelling method for the analysis of 2′-deoxyguanosine-3′-monophosphate and DNA adducts of methylglyoxal. Carcinogenesis 1994, 15, 1887–1894. [Google Scholar] [CrossRef]
- Lo, T.W.; Westwood, M.E.; McLellan, A.C.; Selwood, T.; Thornalley, P.J. Binding and modification of proteins by methylglyoxal under physiological conditions. A kinetic and mechanistic study with N alpha-acetylarginine, N alpha-acetylcysteine, and N alpha-acetyllysine, and bovine serum albumin. J. Biol. Chem. 1994, 269, 32299–32305. [Google Scholar] [CrossRef]
- Singh, R.; Barden, A.; Mori, T.; Beilin, L. Advanced glycation end-products: A review. Diabetologia 2001, 44, 129–146. [Google Scholar] [CrossRef] [Green Version]
- Thornalley, P.J. The glyoxalase system in health and disease. Mol. Aspects Med. 1993, 14, 287–371. [Google Scholar] [CrossRef]
- Mannervik, B.; Glyoxalase, I. Enzymatic Basis of Detoxification; Academic Press: New York, NY, USA, 1980; Volume 2, pp. 263–293. [Google Scholar]
- Hollenbach, M.; Hintersdorf, A.; Huse, K.; Sack, U.; Bigl, M.; Groth, M.; Santel, T.; Buchold, M.; Lindner, I.; Otto, A.; et al. Ethyl pyruvate and ethyl lactate down-regulate the production of pro-inflammatory cytokines and modulate expression of immune receptors. Biochem. Pharmacol. 2008, 76, 631–644. [Google Scholar] [CrossRef]
- Hollenbach, M.; Thonig, A.; Pohl, S.; Ripoll, C.; Michel, M.; Zipprich, A. Expression of glyoxalase-I is reduced in cirrhotic livers: A possible mechanism in the development of cirrhosis. PLoS ONE 2017, 12, e0171260. [Google Scholar] [CrossRef] [Green Version]
- Michel, M.; Hollenbach, M.; Pohl, S.; Ripoll, C.; Zipprich, A. Inhibition of Glyoxalase-I Leads to Reduced Proliferation, Migration and Colony Formation, and Enhanced Susceptibility to Sorafenib in Hepatocellular Carcinoma. Front. Oncol. 2019, 9, 785. [Google Scholar] [CrossRef] [Green Version]
- Santel, T.; Pflug, G.; Hemdan, N.Y.; Schafer, A.; Hollenbach, M.; Buchold, M.; Hintersdorf, A.; Lindner, I.; Otto, A.; Bigl, M.; et al. Curcumin inhibits glyoxalase 1: A possible link to its anti-inflammatory and anti-tumor activity. PLoS ONE 2008, 3, e3508. [Google Scholar] [CrossRef] [Green Version]
- Antognelli, C.; Moretti, S.; Frosini, R.; Puxeddu, E.; Sidoni, A.; Talesa, V.N. Methylglyoxal Acts as a Tumor-Promoting Factor in Anaplastic Thyroid Cancer. Cells 2019, 8, 547. [Google Scholar] [CrossRef] [Green Version]
- Lerch, M.M.; Gorelick, F.S. Models of acute and chronic pancreatitis. Gastroenterology 2013, 144, 1180–1193. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Neuhofer, P.; Song, L.; Rabe, B.; Lesina, M.; Kurkowski, M.U.; Treiber, M.; Wartmann, T.; Regnér, S.; Thorlacius, H.; et al. IL-6 trans-signaling promotes pancreatitis-associated lung injury and lethality. J. Clin. Invest. 2013, 123, 1019–1031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Algul, H.; Treiber, M.; Lesina, M.; Nakhai, H.; Saur, D.; Geisler, F.; Pfeifer, A.; Paxian, S.; Schmid, R.M. Pancreas-specific RelA/p65 truncation increases susceptibility of acini to inflammation-associated cell death following cerulein pancreatitis. J. Clin. Invest. 2007, 117, 1490–1501. [Google Scholar] [CrossRef] [Green Version]
- Weis, S.; Heindl, M.; Carvalho, T.; Jentho, E.; Lorenz, J.; Sommerer, I.; Mössner, J.; Hoffmeister, A. Azithromycin does not improve disease severity in acute experimental pancreatitis. PLoS ONE 2019, 14, e0216614. [Google Scholar] [CrossRef] [PubMed]
- Weis, S.; Schlaich, T.C.; Dehghani, F.; Carvalho, T.; Sommerer, I.; Fricke, S.; Kahlenberg, F.; Mössner, J.; Hoffmeister, A. p8 Deficiency causes siderosis in spleens and lymphocyte apoptosis in acute pancreatitis. Pancreas 2014, 43, 1277–1285. [Google Scholar] [CrossRef] [PubMed]
- Heindl, M.; Tuennemann, J.; Sommerer, I.; Mossner, J.; Hoffmeister, A. Loss of Bace1 in mice does not alter the severity of caerulein induced pancreatitis. PLoS ONE 2015, 10, e0125556. [Google Scholar] [CrossRef] [Green Version]
- Hollenbach, M.; Sonnenberg, S.; Sommerer, I.; Lorenz, J.; Hoffmeister, A. Pitfalls in AR42J-model of cerulein-induced acute pancreatitis. PLoS ONE 2021, 16, e0242706. [Google Scholar] [CrossRef] [PubMed]
- Charafe-Jauffret, E.; Tarpin, C.; Bardou, V.J.; Bertucci, F.; Ginestier, C.; Braud, A.C.; Puig, B.; Geneix, J.; Hassoun, J.; Birnbaum, D.; et al. Immunophenotypic analysis of inflammatory breast cancers: Identification of an ‘inflammatory signature’. J. Pathol. 2004, 202, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Ceranowicz, P.; Warzecha, Z.; Dembinski, A.; Cieszkowski, J.; Dembinski, M.; Sendur, R.; Kusnierz-Cabala, B.; Tomaszewska, R.; Kuwahara, A.; Kato, I. Pretreatment with obestatin inhibits the development of cerulein-induced pancreatitis. J. Physiol. Pharmacol. 2009, 60, 95–101. [Google Scholar] [PubMed]
- Arai, M.; Nihonmatsu-Kikuchi, N.; Itokawa, M.; Rabbani, N.; Thornalley, P.J. Measurement of glyoxalase activities. Biochem. Soc. Trans. 2014, 42, 491–494. [Google Scholar] [CrossRef]
- Bergmans, H.E.; van Die, I.M.; Hoekstra, W.P. Transformation in Escherichia coli: Stages in the process. J. Bacteriol. 1981, 146, 564–570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Logsdon, C.D.; Moessner, J.; Williams, J.A.; Goldfine, I.D. Glucocorticoids increase amylase mRNA levels, secretory organelles, and secretion in pancreatic acinar AR42J cells. J. Cell Biol. 1985, 100, 1200–1208. [Google Scholar] [CrossRef] [Green Version]
- Rabbani, N.; Thornalley, P.J. Methylglyoxal, glyoxalase 1 and the dicarbonyl proteome. Amino Acids. 2012, 42, 1133–1142. [Google Scholar] [CrossRef]
- Rabbani, N.; Xue, M.; Thornalley, P.J. Dicarbonyls and glyoxalase in disease mechanisms and clinical therapeutics. Glycoconj. J. 2016, 33, 513–525. [Google Scholar] [CrossRef] [Green Version]
- Thornalley, P.J. The glyoxalase system: New developments towards functional characterization of a metabolic pathway fundamental to biological life. Biochem. J. 1990, 269, 1–11. [Google Scholar] [CrossRef]
- Misra, K.; Banerjee, A.B.; Ray, S.; Ray, M. Glyoxalase III from Escherichia coli: A single novel enzyme for the conversion of methylglyoxal into D-lactate without reduced glutathione. Biochem. J. 1995, 305 Pt 3, 999–1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgenstern, J.; Fleming, T.; Schumacher, D.; Eckstein, V.; Freichel, M.; Herzig, S.; Nawroth, P. Loss of Glyoxalase 1 Induces Compensatory Mechanism to Achieve Dicarbonyl Detoxification in Mammalian Schwann Cells. J. Biol. Chem. 2017, 292, 3224–3238. [Google Scholar] [CrossRef] [Green Version]
- Hollenbach, M.; Thonig, A.; Pohl, S.; Ripoll, C.; Zipprich, A. Expression of glyoxalase-I is reduced in cirrhotic livers: A mechanism that could explain higher concentration of reactive oxygen species in the development of cirrhosis. J. Hepatol. 2015, 62, S340–S341. [Google Scholar] [CrossRef]
- Zuin, A.; Vivancos, A.P.; Sanso, M.; Takatsume, Y.; Ayte, J.; Inoue, Y.; Hidalgo, E. The glycolytic metabolite methylglyoxal activates Pap1 and Sty1 stress responses in Schizosaccharomyces pombe. J. Biol. Chem. 2005, 280, 36708–36713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakamoto, H.; Mashima, T.; Yamamoto, K.; Tsuruo, T. Modulation of heat-shock protein 27 (Hsp27) anti-apoptotic activity by methylglyoxal modification. J. Biol. Chem. 2002, 277, 45770–45775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thornalley, P.J.; Edwards, L.G.; Kang, Y.; Wyatt, C.; Davies, N.; Ladan, M.J.; Double, J. Antitumour activity of S-p-bromobenzylglutathione cyclopentyl diester in vitro and in vivo. Inhibition of glyoxalase I and induction of apoptosis. Biochem. Pharmacol. 1996, 51, 1365–1372. [Google Scholar] [CrossRef]
- Laga, M.; Cottyn, A.; van Herreweghe, F.; Vanden, B.W.; Haegeman, G.; van Oostveldt, P.; Vandekerckhove, J.; Vancompernolle, K. Methylglyoxal suppresses TNF-alpha-induced NF-kappaB activation by inhibiting NF-kappaB DNA-binding. Biochem. Pharmacol. 2007, 74, 579–589. [Google Scholar] [CrossRef]
- Xue, M.; Rabbani, N.; Momiji, H.; Imbasi, P.; Anwar, M.M.; Kitteringham, N.; Park, B.K.; Souma, T.; Moriguchi, T.; Yamamoto, M.; et al. Transcriptional control of glyoxalase 1 by Nrf2 provides a stress-responsive defence against dicarbonyl glycation. Biochem. J. 2012, 443, 213–222. [Google Scholar] [CrossRef] [Green Version]
- Palsamy, P.; Bidasee, K.R.; Ayaki, M.; Augusteyn, R.C.; Chan, J.Y.; Shinohara, T. Methylglyoxal induces endoplasmic reticulum stress and DNA demethylation in the Keap1 promoter of human lens epithelial cells and age-related cataracts. Free Radic. Biol. Med. 2014, 72, 134–148. [Google Scholar] [CrossRef] [Green Version]
- Reiniger, N.; Lau, K.; McCalla, D.; Eby, B.; Cheng, B.; Lu, Y.; Qu, W.; Quadri, N.; Ananthakrishnan, R.; Furmansky, M.; et al. Deletion of the receptor for advanced glycation end products reduces glomerulosclerosis and preserves renal function in the diabetic OVE26 mouse. Diabetes 2010, 59, 2043–2054. [Google Scholar] [CrossRef] [Green Version]
- Jang, S.; Kwon, D.M.; Kwon, K.; Park, C. Generation and characterization of mouse knockout for glyoxalase 1. Biochem. Biophys. Res. Commun. 2017, 490, 460–465. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hollenbach, M.; Sonnenberg, S.; Sommerer, I.; Lorenz, J.; Hoffmeister, A. Glyoxalase-I Is Upregulated in Acute Cerulein-Induced Pancreatitis: A New Mechanism in Pancreatic Inflammation? Antioxidants 2021, 10, 1574. https://doi.org/10.3390/antiox10101574
Hollenbach M, Sonnenberg S, Sommerer I, Lorenz J, Hoffmeister A. Glyoxalase-I Is Upregulated in Acute Cerulein-Induced Pancreatitis: A New Mechanism in Pancreatic Inflammation? Antioxidants. 2021; 10(10):1574. https://doi.org/10.3390/antiox10101574
Chicago/Turabian StyleHollenbach, Marcus, Sebastian Sonnenberg, Ines Sommerer, Jana Lorenz, and Albrecht Hoffmeister. 2021. "Glyoxalase-I Is Upregulated in Acute Cerulein-Induced Pancreatitis: A New Mechanism in Pancreatic Inflammation?" Antioxidants 10, no. 10: 1574. https://doi.org/10.3390/antiox10101574