
Epigallocatechin-3-Gallate Suppresses BMP-6-Mediated SMAD1/5/8 Transactivation of Hepcidin Gene by Inducing SMILE in Hepatocytes



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Plasmid DNA and Recombinant Adenovirus Construction
2.3. Cell Culture and Transient Transfection
2.4. Cell Viability Assay
2.5. Quantitative PCR (Q-PCR) Analysis
2.6. Western Blot (WB) Analysis
2.7. Immunocytochemical Analysis
2.8. Co-Immunoprecipitation (co-IP) Analysis
2.9. Chromatin Immunoprecipitation (ChIP) Assay
2.10. Measurement of Hepcidin Levels
2.11. Statistics
3. Results
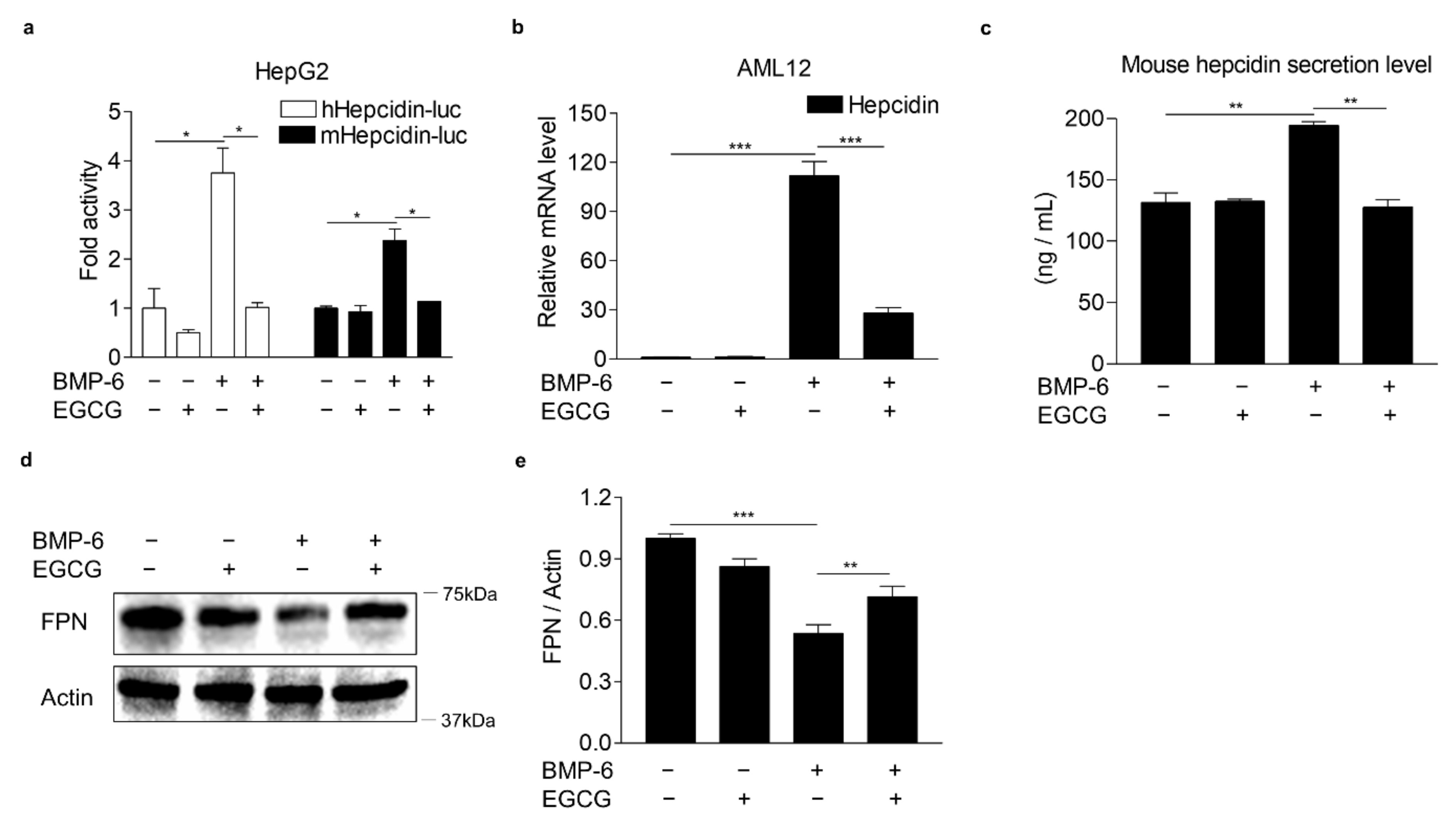
3.1. EGCG Suppresses BMP-6 Receptor Signaling in Hepatocytes
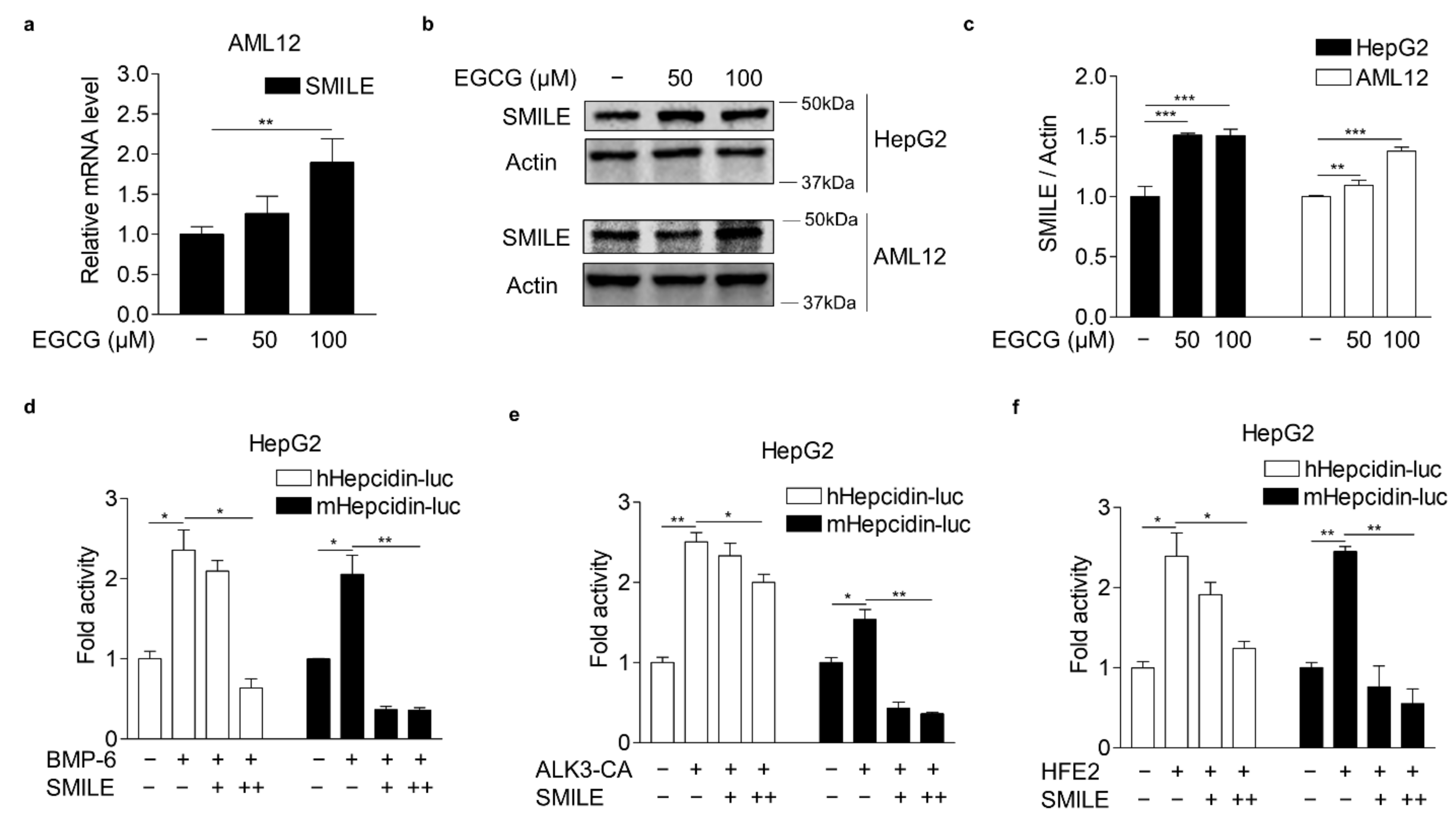
3.2. EGCG-Inducible SMILE Represses BMP-6 Receptor Signaling
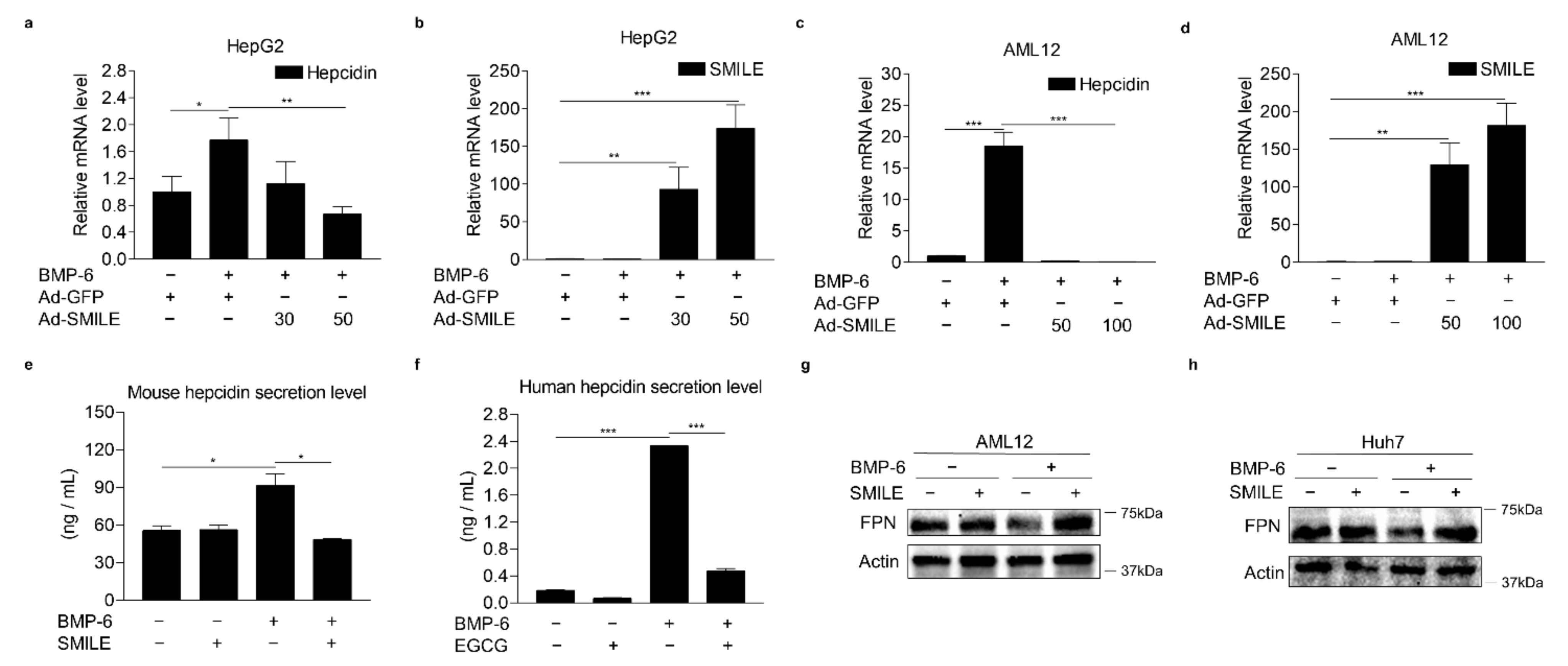
3.3. SMILE Inhibits BMP-6-Induced Hepcidin Expression
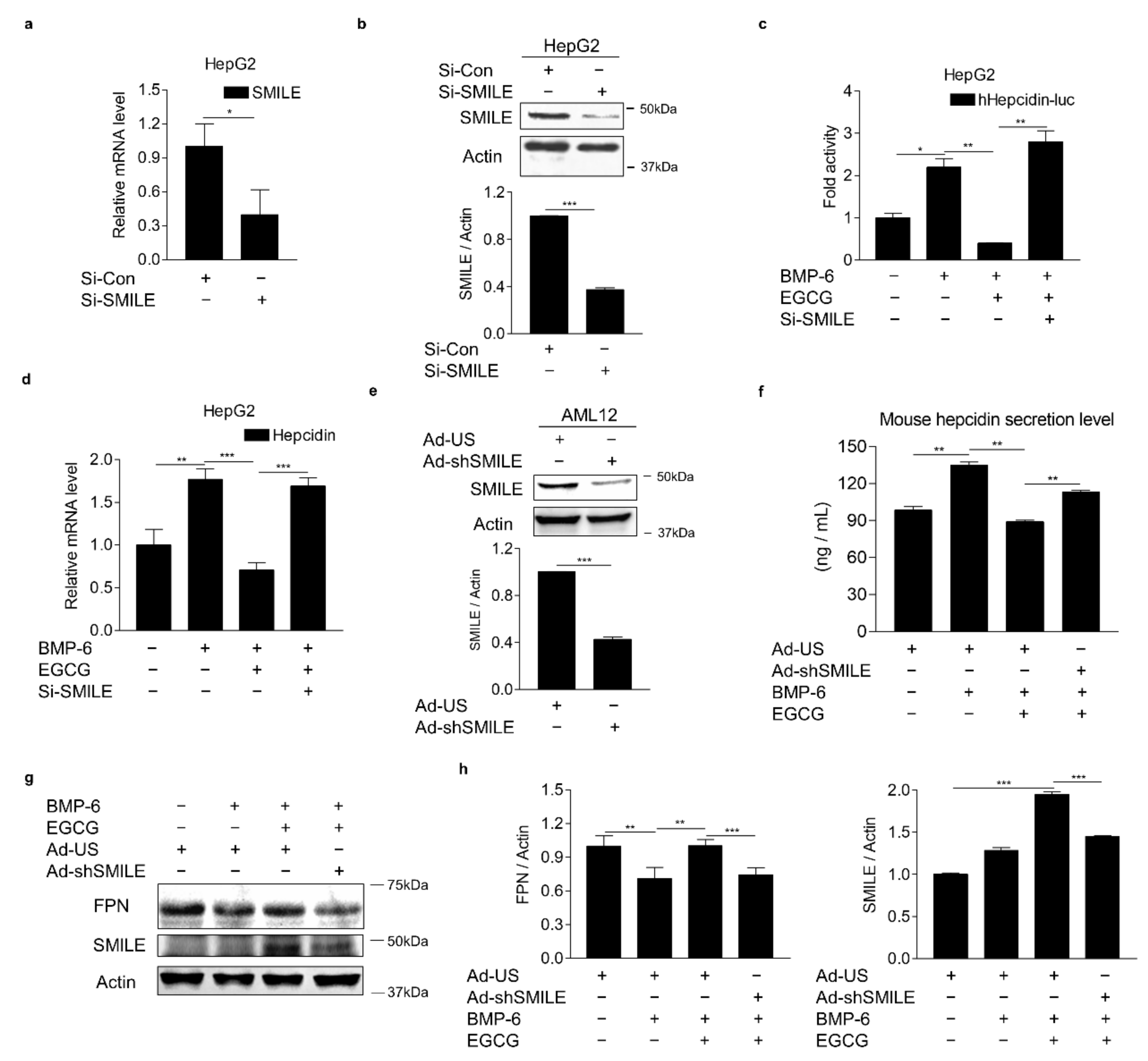
3.4. SMILE Is Required for the Inhibitory Effect of EGCG on BMP-6-Mediated Hepcidin Expression
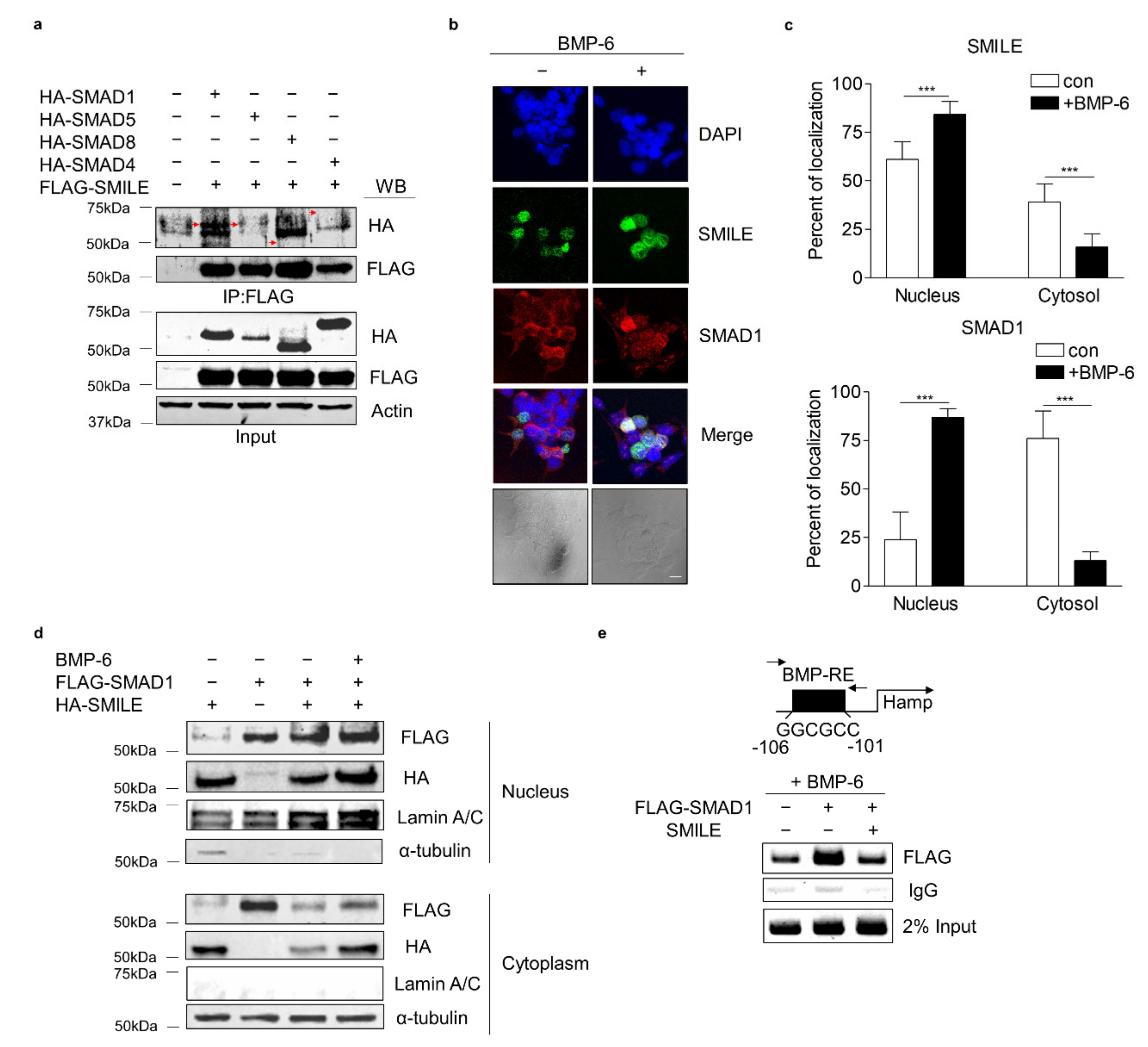
3.5. SMILE Interacts with SMAD1 and Inhibits Its DNA-Binding to Hepcidin Gene Promoter
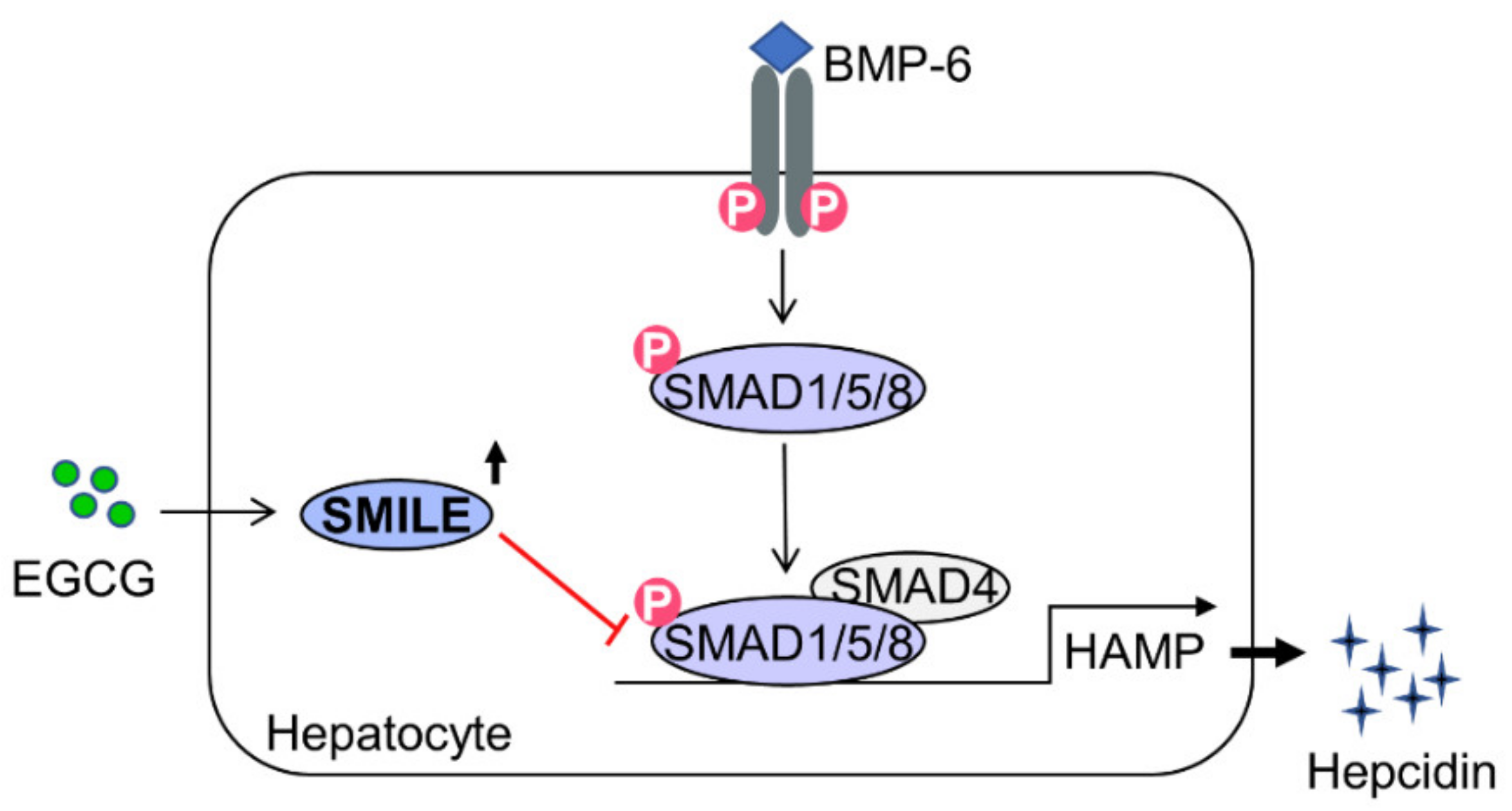
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| EGCG | Epigallocatechin-3-gallate |
| SMILE | Small heterodimer partner-interacting leucine zipper protein |
| BMP-6 | Bone morphogenetic protein 6 |
| ALK3 | Activin receptor-like kinase 3 |
| HJV | Hemojuvelin |
| SMAD | Small mothers against decapentaplegic homolog protein |
| FPN | Ferroportin |
| BMP-RE | BMP-response element |
| MOI | Multiplicity of infection |
| IRIDA | Iron-refractory iron deficiency anemia |
| IL-6 | Interleukin-6 |
| ERRγ | Estrogen-related receptor γ |
| ROS | Reactive oxygen species |
| FOXO1 | Forkhead box protein O1 |
| PCR | Polymerase chain reaction |
| ELISA | Enzyme-linked immunosorbent assay |
| Q-PCR | Quantitative PCR |
| WB | Western blot |
| HNF4α | Hepatocyte nuclear factor 4α |
| AMPK | AMP-dependent kinase |
| LKB1 | Liver kinase B1 |
References
- Ganz, T.; Nemeth, E. Hepcidin and iron homeostasis. Biochim. Biophys. Acta Mol. Cell Res. 2012, 1823, 1434–1443. [Google Scholar] [CrossRef] [Green Version]
- Reichert, C.O.; da Cunha, J.; Levy, D.; Maselli, L.M.F.; Bydlowski, S.P.; Spada, C. Hepcidin: Homeostasis and Diseases Related to Iron Metabolism. Acta Haematol. 2017, 137, 220–236. [Google Scholar] [CrossRef]
- Ganz, T.; Nemeth, E. Iron homeostasis in host defence and inflammation. Nat. Rev. Immunol. 2015, 15, 500–510. [Google Scholar] [CrossRef] [Green Version]
- Díaz, V.; Gammella, E.; Recalcati, S.; Santambrogio, P.; Naldi, A.M.; Vogel, J.; Gassmann, M.; Cairo, G. Liver iron modulates hepcidin expression during chronically elevated erythropoiesis in mice. Hepatology 2013, 58, 2122–2132. [Google Scholar] [CrossRef]
- Mastrogiannaki, M.; Matak, P.; Mathieu, J.R.; Delga, S.; Mayeux, P.; Vaulont, S.; Peyssonnaux, C. Hepatic hypoxia-inducible factor-2 down-regulates hepcidin expression in mice through an erythropoietin-mediated increase in erythropoiesis. Haematologica 2012, 97, 827–834. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.H.; Li, C.; Xu, X.; Zheng, Y.; Xiao, C.; Zerfas, P.; Cooperman, S.; Eckhaus, M.; Rouault, T.; Mishra, L.; et al. A role of SMAD4 in iron metabolism through the positive regulation of hepcidin expression. Cell Metab. 2005, 2, 399–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babitt, J.L.; Huang, F.W.; Wrighting, D.M.; Xia, Y.; Sidis, Y.; Samad, T.A.; Campagna, J.A.; Chung, R.T.; Schneyer, A.L.; Woolf, C.J.; et al. Bone morphogenetic protein signaling by hemojuvelin regulates hepcidin expression. Nat. Genet. 2006, 38, 531–539. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Babitt, J.L.; Sidis, Y.; Chung, R.T.; Lin, H.Y. Hemojuvelin regulates hepcidin expression via a selective subset of BMP ligands and receptors independently of neogenin. Blood 2008, 111, 5195–5204. [Google Scholar] [CrossRef] [Green Version]
- Andriopoulos, B., Jr.; Corradini, E.; Xia, Y.; Faasse, S.A.; Chen, S.; Grgurevic, L.; Knutson, M.D.; Pietrangelo, A.; Vukicevic, S.; Lin, H.Y.; et al. BMP6 is a key endogenous regulator of hepcidin expression and iron metabolism. Nat. Genet. 2009, 41, 482–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meynard, D.; Kautz, L.; Darnaud, V.; Canonne-Hergaux, F.; Coppin, H.; Roth, M.P. Lack of the bone morphogenetic protein BMP6 induces massive iron overload. Nat. Genet. 2009, 41, 478–481. [Google Scholar] [CrossRef]
- Shi, Y.; Massagué, J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell 2003, 113, 685–700. [Google Scholar] [CrossRef] [Green Version]
- Core, A.B.; Canali, S.; Babitt, J.L. Hemojuvelin and bone morphogenetic protein (BMP) signaling in iron homeostasis. Front. Pharmacol. 2014, 5, 104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silvestri, L.; Nai, A.; Dulja, A.; Pagani, A. Hepcidin and the BMP-SMAD pathway: An unexpected liaison. Vitam. Horm. 2019, 110, 71–99. [Google Scholar] [PubMed]
- Lu, R.; Misra, V. Zhangfei: A second cellular protein interacts with herpes simplex virus accessory factor HCF in a manner similar to Luman and VP16. Nucleic Acids Res. 2000, 28, 2446–2454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Y.B.; Nedumaran, B.; Choi, H.S. Molecular characterization of SMILE as a novel corepressor of nuclear receptors. Nucleic Acids Res. 2009, 37, 4100–4115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Y.B.; Park, J.H.; Kim, D.K.; Hwang, J.H.; Oh, S.; Park, S.B.; Shong, M.; Lee, I.K.; Choi, H.S. Transcriptional corepressor SMILE recruits SIRT1 to inhibit nuclear receptor estrogen receptor-related receptor gamma transactivation. J. Biol. Chem. 2009, 284, 28762–28774. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.M.; Han, H.S.; Jung, Y.S.; Harris, R.A.; Koo, S.H.; Choi, H.S. The SMILE transcriptional corepressor inhibits cAMP response element-binding protein (CREB)-mediated transactivation of gluconeogenic genes. J. Biol. Chem. 2018, 293, 13125–13133. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.M.; Gang, G.T.; Kim, D.K.; Kim, Y.D.; Koo, S.H.; Lee, C.H.; Choi, H.S. Ursodeoxycholic acid inhibits liver X receptor α-mediated hepatic lipogenesis via induction of the nuclear corepressor SMILE. J. Biol. Chem. 2014, 289, 1079–1091. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.M.; Seo, W.Y.; Han, H.S.; Oh, K.J.; Lee, Y.S.; Kim, D.K.; Choi, S.; Choi, B.H.; Harris, R.A.; Lee, C.H.; et al. Insulin-Inducible SMILE Inhibits Hepatic Gluconeogenesis. Diabetes 2016, 65, 62–73. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.-J.; Kim, K.-S.; Lim, D.; Yang, D.J.; Park, J.-I.; Kim, K.W.; Jeong, J.-H.; Choi, H.-S.; Kim, D.-K. Epigallocatechin-3-Gallate (EGCG)-Inducible SMILE Inhibits STAT3-Mediated Hepcidin Gene Expression. Antioxidants 2020, 9, 514. [Google Scholar] [CrossRef]
- Kim, H.S.; Quon, M.J.; Kim, J.A. New insights into the mechanisms of polyphenols beyond antioxidant properties; lessons from the green tea polyphenol, epigallocatechin 3-gallate. Redox Biol. 2014, 2, 187–195. [Google Scholar] [CrossRef] [Green Version]
- Chakrawarti, L.; Agrawal, R.; Dang, S.; Gupta, S.; Gabrani, R. Therapeutic effects of EGCG: A patent review. Expert Opin. Ther. Pat. 2016, 26, 907–916. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wu, S. Epigallocatechin gallate suppresses hepatic cholesterol synthesis by targeting SREBP-2 through SIRT1/FOXO1 signaling pathway. Mol. Cell. Biochem. 2018, 448, 175–185. [Google Scholar] [CrossRef]
- Zhang, Q.; Yuan, H.; Zhang, C.; Guan, Y.; Wu, Y.; Ling, F.; Niu, Y.; Li, Y. Epigallocatechin gallate improves insulin resistance in HepG2 cells through alleviating inflammation and lipotoxicity. Diabetes Res. Clin. Pract. 2018, 142, 363–373. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.Y.; Li, Y.Q.; Guo, Z.W.; Zhou, X.H.; Lu, M.D.; Xue, T.C.; Gao, B. ERK1/2-HNF4α axis is involved in epigallocatechin-3-gallate inhibition of HBV replication. Acta Pharmacol. Sin. 2020, 41, 278–285. [Google Scholar] [CrossRef]
- Mleczko-Sanecka, K.; Casanovas, G.; Ragab, A.; Breitkopf, K.; Müller, A.; Boutros, M.; Dooley, S.; Hentze, M.W.; Muckenthaler, M.U. SMAD7 controls iron metabolism as a potent inhibitor of hepcidin expression. Blood 2010, 115, 2657–2665. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.K.; Jeong, J.H.; Lee, J.M.; Kim, K.S.; Park, S.H.; Kim, Y.D.; Koh, M.; Shin, M.; Jung, Y.S.; Kim, H.S.; et al. Inverse agonist of estrogen-related receptor γ controls Salmonella typhimurium infection by modulating host iron homeostasis. Nat. Med. 2014, 20, 419–424. [Google Scholar] [CrossRef]
- Xie, Y.B.; Lee, O.H.; Nedumaran, B.; Seong, H.A.; Lee, K.M.; Ha, H.; Lee, I.K.; Yun, Y.; Choi, H.S. SMILE, a new orphan nuclear receptor SHP-interacting protein, regulates SHP-repressed estrogen receptor transactivation. Biochem. J. 2008, 416, 463–473. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.K.; Kim, Y.H.; Jung, Y.S.; Kim, K.S.; Jeong, J.H.; Lee, Y.S.; Yuk, J.M.; Oh, B.C.; Choy, H.E.; Dooley, S.; et al. Orphan nuclear receptor SHP regulates iron metabolism through inhibition of BMP6-mediated hepcidin expression. Sci. Rep. 2016, 6, 34630. [Google Scholar] [CrossRef] [Green Version]
- Misra, J.; Chanda, D.; Kim, D.K.; Li, T.; Koo, S.H.; Back, S.H.; Chiang, J.Y.L.; Choi, H.S. Curcumin differentially regulates endoplasmic reticulum stress through transcriptional corepressor SMILE (small heterodimer partner-interacting leucine zipper protein)-mediated inhibition of CREBH (cAMP responsive element-binding protein H). J. Biol. Chem. 2011, 286, 41972–41984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.Y.; Kim, H.J.; Kim, K.T.; Park, Y.Y.; Seong, H.A.; Park, K.C.; Lee, I.K.; Ha, H.; Shong, M.; Park, S.C.; et al. Orphan nuclear receptor small heterodimer partner represses hepatocyte nuclear factor 3/Foxa transactivation via inhibition of its DNA binding. Mol. Endocrinol. 2004, 18, 2880–2894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.K.; Kim, Y.H.; Hynx, D.; Wang, Y.; Yang, K.J.; Ryu, D.; Kim, K.S.; Yoo, E.K.; Kim, J.S.; Koo, S.H.; et al. PKB/Akt phosphorylation of ERRγ contributes to insulin-mediated inhibition of hepatic gluconeogenesis. Diabetologia 2014, 57, 2576–2585. [Google Scholar] [CrossRef] [Green Version]
- Nemeth, E.; Tuttle, M.S.; Powelson, J.; Vaughn, M.B.; Donovan, A.; Ward, D.M.; Ganz, T.; Kaplan, J. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 2004, 306, 2090–2093. [Google Scholar] [CrossRef] [Green Version]
- Mayeur, C.; Lohmeyer, L.K.; Leyton, P.; Kao, S.M.; Pappas, A.E.; Kolodziej, S.A.; Spagnolli, E.; Yu, B.; Galdos, R.L.; Yu, P.B.; et al. The type I BMP receptor Alk3 is required for the induction of hepatic hepcidin gene expression by interleukin-6. Blood 2014, 123, 2261–2268. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.G.; Wang, Y.; Wu, Q.; Cheng, W.H.; Liu, W.; Zhao, Y.; Mayeur, C.; Schmidt, P.J.; Yu, P.B.; Wang, F.; et al. HFE interacts with the BMP type I receptor ALK3 to regulate hepcidin expression. Blood 2014, 124, 1335–1343. [Google Scholar] [CrossRef] [Green Version]
- Girelli, D.; Ugolini, S.; Busti, F.; Marchi, G.; Castagna, A. Modern iron replacement therapy: Clinical and pathophysiological insights. Int. J. Hematol. 2018, 107, 16–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinbicker, A.U.; Muckenthaler, M.U. Out of balance--systemic iron homeostasis in iron-related disorders. Nutrients 2013, 5, 3034–3061. [Google Scholar] [CrossRef]
- Katsarou, A.; Pantopoulos, K. Hepcidin Therapeutics. Pharmaceuticals 2018, 11, 127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finberg, K.E.; Heeney, M.M.; Campagna, D.R.; Aydinok, Y.; Pearson, H.A.; Hartman, K.R.; Mayo, M.M.; Samuel, S.M.; Strouse, J.J.; Markianos, K.; et al. Mutations in TMPRSS6 cause iron-refractory iron deficiency anemia (IRIDA). Nat. Genet. 2008, 40, 569–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mu, M.; An, P.; Wu, Q.; Shen, X.; Shao, D.; Wang, H.; Zhang, Y.; Zhang, S.; Yao, H.; Min, J.; et al. The dietary flavonoid myricetin regulates iron homeostasis by suppressing hepcidin expression. J. Nutr. Biochem. 2016, 30, 53–61. [Google Scholar] [CrossRef]
- Koonyosying, P.; Kongkarnka, S.; Uthaipibull, C.; Svasti, S.; Fucharoen, S.; Srichairatanakool, S. Green tea extract modulates oxidative tissue injury in beta-thalassemic mice by chelation of redox iron and inhibition of lipid peroxidation. Biomed. Pharm. 2018, 108, 1694–1702. [Google Scholar] [CrossRef] [PubMed]
- Henning, S.M.; Niu, Y.; Lee, N.H.; Thames, G.D.; Minutti, R.R.; Wang, H.; Go, V.L.; Heber, D. Bioavailability and antioxidant activity of tea flavanols after consumption of green tea, black tea, or a green tea extract supplement. Am. J. Clin. Nutr. 2004, 80, 1558–1564. [Google Scholar] [CrossRef] [PubMed]
- Chow, H.H.; Cai, Y.; Hakim, I.A.; Crowell, J.A.; Shahi, F.; Brooks, C.A.; Dorr, R.T.; Hara, Y.; Alberts, D.S. Pharmacokinetics and safety of green tea polyphenols after multiple-dose administration of epigallocatechin gallate and polyphenon E in healthy individuals. Clin. Cancer Res. 2003, 9, 3312–3319. [Google Scholar] [PubMed]
- Ullmann, U.; Haller, J.; Decourt, J.P.; Girault, N.; Girault, J.; Richard-Caudron, A.S.; Pineau, B.; Weber, P. A single ascending dose study of epigallocatechin gallate in healthy volunteers. J. Int. Med. Res. 2003, 31, 88–101. [Google Scholar] [CrossRef]
- Chen, I.J.; Liu, C.Y.; Chiu, J.P.; Hsu, C.H. Therapeutic effect of high-dose green tea extract on weight reduction: A randomized, double-blind, placebo-controlled clinical trial. Clin. Nutr. 2016, 35, 592–599. [Google Scholar] [CrossRef]
- Suganuma, M.; Okabe, S.; Kai, Y.; Sueoka, N.; Sueoka, E.; Fujiki, H. Synergistic effects of (−)-epigallocatechin gallate with (−)-epicatechin, sulindac, or tamoxifen on cancer-preventive activity in the human lung cancer cell line PC-9. Cancer Res. 1999, 59, 44–47. [Google Scholar]
- Casanovas, G.; Mleczko-Sanecka, K.; Altamura, S.; Hentze, M.W.; Muckenthaler, M.U. Bone morphogenetic protein (BMP)-responsive elements located in the proximal and distal hepcidin promoter are critical for its response to HJV/BMP/SMAD. J. Mol. Med. 2009, 87, 471–480. [Google Scholar] [CrossRef]
- Steinbicker, A.U.; Sachidanandan, C.; Vonner, A.J.; Yusuf, R.Z.; Deng, D.Y.; Lai, C.S.; Rauwerdink, K.M.; Winn, J.C.; Saez, B.; Cook, C.M.; et al. Inhibition of bone morphogenetic protein signaling attenuates anemia associated with inflammation. Blood 2011, 117, 4915–4923. [Google Scholar] [CrossRef] [Green Version]
- Radhakrishnan, K.; Kim, Y.-H.; Jung, Y.S.; Kim, J.; Kim, D.-K.; Cho, S.J.; Lee, I.-K.; Dooley, S.; Lee, C.-H.; Choi, H.-S. Orphan Nuclear Receptor ERRγ Is a Novel Transcriptional Regulator of IL-6 Mediated Hepatic BMP6 Gene Expression in Mice. Int. J. Mol. Sci. 2020, 21, 7148. [Google Scholar] [CrossRef]
- Galaris, D.; Barbouti, A.; Pantopoulos, K. Iron homeostasis and oxidative stress: An intimate relationship. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 118535. [Google Scholar] [CrossRef]
- Bayele, H.K.; Balesaria, S.; Srai, S.K. Phytoestrogens modulate hepcidin expression by Nrf2: Implications for dietary control of iron absorption. Free Radic. Biol. Med. 2015, 89, 1192–1202. [Google Scholar] [CrossRef] [Green Version]
- Lim, P.J.; Duarte, T.L.; Arezes, J.; Garcia-Santos, D.; Hamdi, A.; Pasricha, S.R.; Armitage, A.E.; Mehta, H.; Wideman, S.; Santos, A.G.; et al. Nrf2 controls iron homeostasis in haemochromatosis and thalassaemia via Bmp6 and hepcidin. Nat. Metab. 2019, 1, 519–531. [Google Scholar] [CrossRef]
- Charlebois, E.; Pantopoulos, K. Iron overload inhibits BMP/SMAD and IL-6/STAT3 signaling to hepcidin in cultured hepatocytes. PLoS ONE 2021, 16, e0253475. [Google Scholar] [CrossRef] [PubMed]
- Al-Basher, G.I. Green tea activity and iron overload induced molecular fibrogenesis of rat liver. Saudi J. Biol. Sci. 2019, 26, 531–540. [Google Scholar] [CrossRef] [PubMed]
- Murase, T.; Misawa, K.; Haramizu, S.; Hase, T. Catechin-induced activation of the LKB1/AMP-activated protein kinase pathway. Biochem. Pharmacol. 2009, 78, 78–84. [Google Scholar] [CrossRef]
- Lee, J.; Yang, G.; Kim, Y.J.; Tran, Q.H.; Choe, W.; Kang, I.; Kim, S.S.; Ha, J. Hydrogen-rich medium protects mouse embryonic fibroblasts from oxidative stress by activating LKB1-AMPK-FoxO1 signal pathway. Biochem. Biophys. Res. Commun. 2017, 491, 733–739. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Li, W.; Liu, Y.; Sun, Y.; Li, Y.; Yao, Q.; Li, J.; Zhang, Q.; Gao, Y.; Gao, L.; et al. Alpha-lipoic acid improves high-fat diet-induced hepatic steatosis by modulating the transcription factors SREBP-1, FoxO1 and Nrf2 via the SIRT1/LKB1/AMPK pathway. J. Nutr. Biochem. 2014, 25, 1207–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yun, H.; Park, S.; Kim, M.J.; Yang, W.K.; Im, D.U.; Yang, K.R.; Hong, J.; Choe, W.; Kang, I.; Kim, S.S.; et al. AMP-activated protein kinase mediates the antioxidant effects of resveratrol through regulation of the transcription factor FoxO1. FEBS J. 2014, 281, 4421–4438. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, Y.-J.; Park, W.-R.; Choi, B.; Choi, H.-S.; Kim, D.-K. Epigallocatechin-3-Gallate Suppresses BMP-6-Mediated SMAD1/5/8 Transactivation of Hepcidin Gene by Inducing SMILE in Hepatocytes. Antioxidants 2021, 10, 1590. https://doi.org/10.3390/antiox10101590
Kim Y-J, Park W-R, Choi B, Choi H-S, Kim D-K. Epigallocatechin-3-Gallate Suppresses BMP-6-Mediated SMAD1/5/8 Transactivation of Hepcidin Gene by Inducing SMILE in Hepatocytes. Antioxidants. 2021; 10(10):1590. https://doi.org/10.3390/antiox10101590
Chicago/Turabian StyleKim, Yu-Ji, Woo-Ram Park, Byungyoon Choi, Hueng-Sik Choi, and Don-Kyu Kim. 2021. "Epigallocatechin-3-Gallate Suppresses BMP-6-Mediated SMAD1/5/8 Transactivation of Hepcidin Gene by Inducing SMILE in Hepatocytes" Antioxidants 10, no. 10: 1590. https://doi.org/10.3390/antiox10101590
APA StyleKim, Y.-J., Park, W.-R., Choi, B., Choi, H.-S., & Kim, D.-K. (2021). Epigallocatechin-3-Gallate Suppresses BMP-6-Mediated SMAD1/5/8 Transactivation of Hepcidin Gene by Inducing SMILE in Hepatocytes. Antioxidants, 10(10), 1590. https://doi.org/10.3390/antiox10101590