
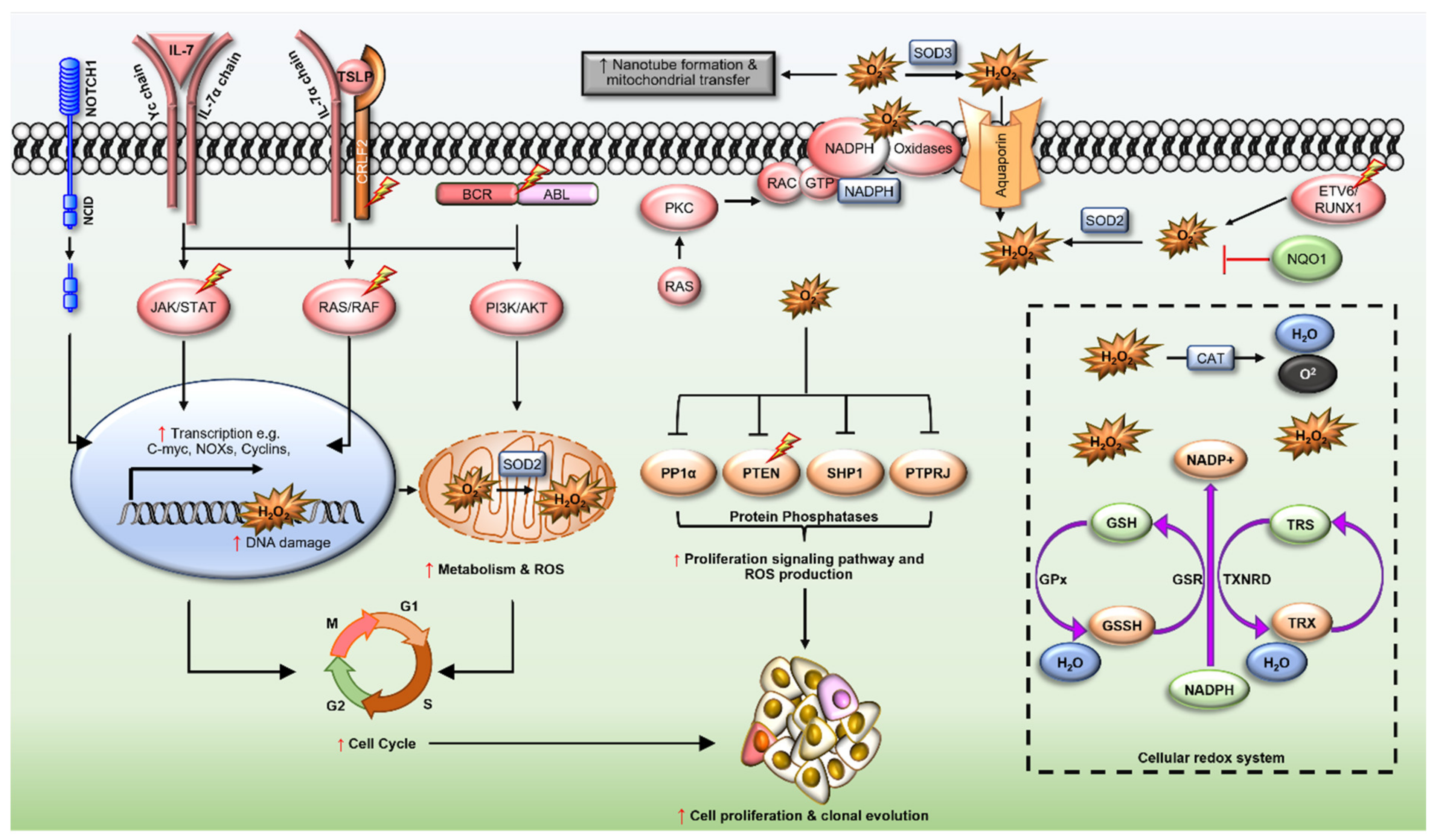
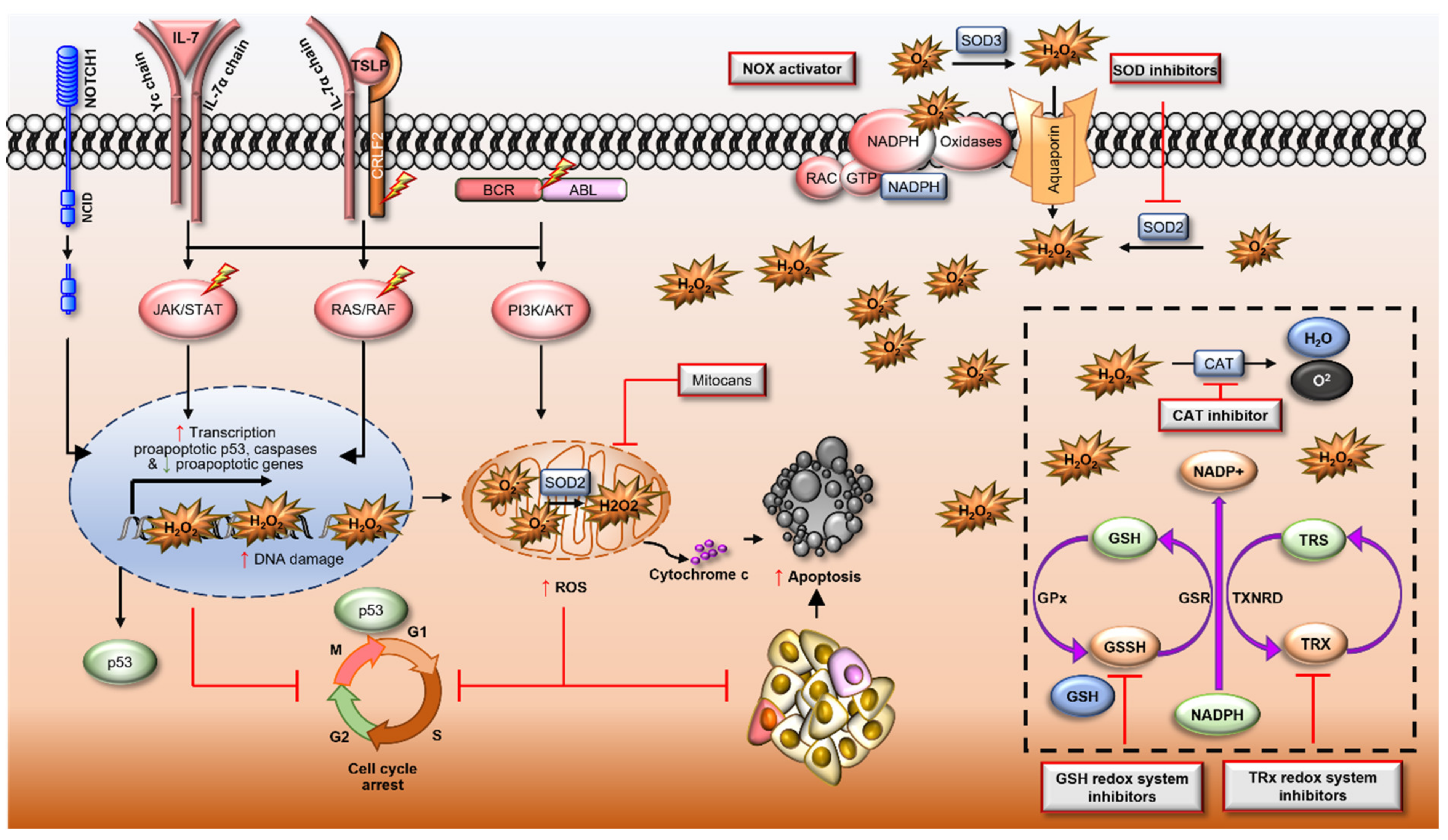
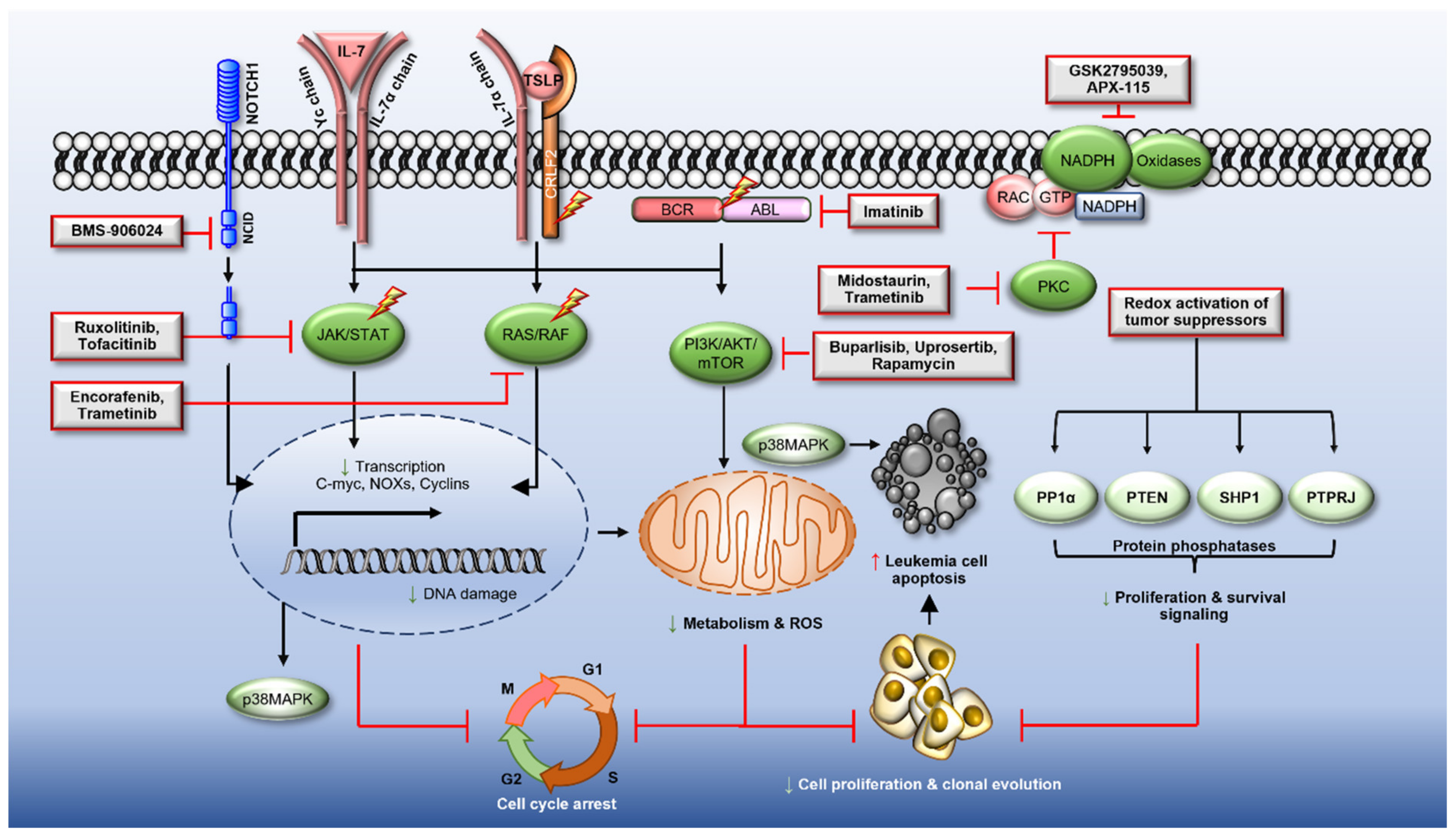
Reactive Oxygen Species in Acute Lymphoblastic Leukaemia: Reducing Radicals to Refine Responses

 , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
Abstract
Share and Cite
Mannan, A.; Germon, Z.P.; Chamberlain, J.; Sillar, J.R.; Nixon, B.; Dun, M.D. Reactive Oxygen Species in Acute Lymphoblastic Leukaemia: Reducing Radicals to Refine Responses. Antioxidants 2021, 10, 1616. https://doi.org/10.3390/antiox10101616
Mannan A, Germon ZP, Chamberlain J, Sillar JR, Nixon B, Dun MD. Reactive Oxygen Species in Acute Lymphoblastic Leukaemia: Reducing Radicals to Refine Responses. Antioxidants. 2021; 10(10):1616. https://doi.org/10.3390/antiox10101616
Chicago/Turabian StyleMannan, Abdul, Zacary P. Germon, Janis Chamberlain, Jonathan R. Sillar, Brett Nixon, and Matthew D. Dun. 2021. "Reactive Oxygen Species in Acute Lymphoblastic Leukaemia: Reducing Radicals to Refine Responses" Antioxidants 10, no. 10: 1616. https://doi.org/10.3390/antiox10101616
APA StyleMannan, A., Germon, Z. P., Chamberlain, J., Sillar, J. R., Nixon, B., & Dun, M. D. (2021). Reactive Oxygen Species in Acute Lymphoblastic Leukaemia: Reducing Radicals to Refine Responses. Antioxidants, 10(10), 1616. https://doi.org/10.3390/antiox10101616