Abstract
Cardiac surgeries have been improved by accompanying developing cardioplegia solutions. However, the cardioplegia application presents an ongoing challenge with a view of a sufficiently restored cardiac function. In this review, we focus on the cardioplegia-induced mechanism and summarize the findings of studies undertaken to improve cardioprotective strategies. Currently, and somewhat surprisingly, relatively little is known about cardiac electrolyte regulation through channel physiology. We hope that an improved understanding of the electrolyte transport through ion channels/transporters and modulations of water channel aquaporins will provide an insight into cardiac channel physiology and a channel-based cardiac pathology of a cardiochannelopathy.
1. Role of Cardioplegia and Potential Injuries
During cardiac surgery, hyperkalemic cardioplegic solutions are the essential gold standard to prevent cardiac depolarization and temporarily sustain a cardiac arrest through the reduction of cardiac loading. In detail, elevated extracellular K+ concentrations (10–40 mM) of hyperkalemic cardioplegic solutions increase the resting myocyte membrane potentials (Em) from −85 mV to between −65 and −40 mV, which inactivates fast Na+ channels [1]. These higher potentials block the conduction of myocardial action potentials, induce a depolarized arrest, and minimize tissue damage during cardiac surgery. Various cardioplegic solutions based on the component, temperature, or period of delivery have been developed as protective strategies.
Although cardioplegic solutions to protect cardiac cell death from ischemia, detrimental injury may occur during intraoperative ischemia due to multi-dose infusions of a cardioplegic solution or the misdistribution of a solution distal to total coronary occlusions [2]. In addition, there is the potential for a reperfusion injury during each infusion and after the aortic cross-clamp removal [2]. In the case of del Nido cardioplegia, the intracellular pH is maintained and the influx of Ca2+ during ischemic arrest is reduced; this has been used to improve myocardial protection [3,4]. However, although del Nido cardioplegia does not lead to clinical side effects, there is an adverse effect. When del Nido cardioplegia was used in an adult aortic root surgery, the ischemic time of the patient was increased compared with blood-based cardioplegia, thereby increasing the myocardial injury Although cardioplegic solutions possess advantages such as successful cardiac surgery to protect an IR injury, diabetic hearts have been shown to have worse clinical outcomes compared with non-diabetic hearts [5,6,7]. Thus, we considered that disease states including diabetes potentially affect a restored cardiac function. In this review, we highlight various potential threat factors of cardiac injuries, describe the related molecular mechanisms in cardioplegia-exposed cardiac tissues for a sufficiently restored cardiac function, and involve several approaches to improve the pathological mechanisms.
2. Cardioplegia Solution Characteristics, Risk Factors, and Related Mechanisms
2.1. Myocardial Ischemia/Reperfusion Injuries
Myocardial ischemia/reperfusion (IR) injuries occur during cardiac surgery and cardioplegic solutions have a protective effect although their effects are limited [8]. In the ischemic state, a lack of oxygen in cells results in ATP and lactic acid production by anaerobic glycolysis and, thus, a reduction in the intracellular pH [9]. Furthermore, the lactic acid generated inhibits the functions of mitochondrial permeability transition pores (mPTPs, a non-selective channel in the mitochondria) and myofibril contracture [10,11]. During a myocardial reperfusion, Ca2+ levels increase in the cardiomyocytes due to ROS-induced sarcoplasmic reticulum (SR) dysfunction [11,12], resulting in a reperfusion injury. The pH recovery mediates the opening of mPTPs, which interferes with mitochondrial membrane potentials [10]. Reoxygenation by reperfusion also increases the cytosolic Ca2+ levels in cardiomyocytes, causing their hypercontraction and subsequent cell death [12,13,14]. A schematic illustration of the mechanism of an IR injury is represented in Figure 1.
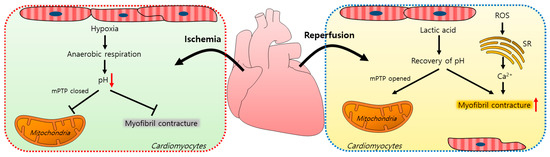
Figure 1.
Schematic illustration of the mechanism of an ischemia/reperfusion injury in cardiomyocytes. Ischemia-induced hypoxia results in anaerobic respiration and reduces the intracellular pH, which closes mPTPs and reduces myofibril contracture. During reperfusion, lactic acid accumulates, the intracellular pH increases, and mPTPs open. ROS increase the intracellular Ca2+ release from the SR and induce myofibril contracture. mPTP: mitochondrial permeability transition pore; ROS: reactive oxygen species; SR: sarcoplasmic reticulum.
2.2. Cardioplegia-Mediated Apoptosis
In this section, we discuss the mechanism of cardioplegia application and associated injuries after a cardioplegic arrest and summarize the numerous studies undertaken to improve the cardioprotective methods against an IR injury over the past several decades. Cold crystalloid or blood cardioplegic solutions have been widely used for cardiac surgery for myocardial protection. However, it has been addressed that cold crystalloid cardioplegia induces early apoptosis signaling events in the myocardial endothelium and cardiomyocytes [15,16]. Two types of cardioplegia methods have been evaluated after a cardioplegic arrest on myocardial injuries in dogs. Both cold blood and cold crystalloid cardioplegia resulted in higher cardiomyocyte apoptosis percentages and an increased caspase-3 expression [17]. To reduce the threat points posed by a cardioplegic arrest, the NHE-1 inhibitor eniporide was applied in a cold cardioplegic or a Krebs–Ringer solution to guinea pigs and was found to improve the relationship of the diastolic left ventricular pressure intracellular Ca2+ levels [18]. The cardioplegia group treated with eniporide showed reduced Ca2+ loading and infarct sizes [18]. Furthermore, the application of cyclosporine A, a mitochondrial permeability transition pore inhibitor, in the presence of cold crystalloid cardioplegia prevented mitochondrial permeability, the mitochondrial translocation of Bax, and protected the mitochondrial structures in a newborn piglet myocardium [16]. Bax (a member of the Bcl-2 family) triggers the permeabilization of the mitochondria to form pores on the mitochondrial outer membrane; this was increased by cold crystalloid cardioplegia. This translocation of Bax was inhibited by a pretreatment with cyclosporine A. The role of AMP-activated protein kinase (AMPK) on endoplasmic reticulum (ER) stress has been evaluated in a cardioplegic-induced hypoxia/reoxygenation (H/R) injury [19]. AMPK activation dramatically attenuated the H/R-induced apoptosis of cardiomyocytes by increasing the anti-apoptotic (e.g., Grp 78 protein) levels and suppressing the pro-apoptotic signals (e.g., caspase-12 and GADD153 (growth arrest and DNA damage-inducible gene 153)) [19]. The administration of bradykinin (BK) during cold crystalloid cardioplegia decreased oxygen consumption and reduced the myocardial metabolic demands of mild-to-moderate hypothermia [20,21] as well as IR-induced inflammation and apoptosis whereas the inhibition of the BK receptor or nitric oxide synthase diminished the inflammatory and apoptotic responses of rabbit cardiomyocytes [22]. Furthermore, nitric oxide (NO) modulation during a cardioplegic arrest might improve the anti-apoptotic signal profile through the NF-κB/Akt/Bcl-2/Bax axis [19]. More recently, the potential protective effects of the flavonoids astragalin and dihydromyricetin in ischemia during a cold crystalloid cardioplegic arrest were evaluated [23]. A cold crystalloid cardioplegic solution in the presence of astragalin or dihydromyricetin enhanced the anti-inflammatory pathways and attenuated the oxidative signals [23]. In addition to mitochondrial stress, the expression of urocortin (a cardioprotective protein) was investigated under a warm blood cardioplegic arrest [24,25]. More recently, in the heart of a diabetic patient, the downregulation of urocortin was associated with cardiomyocyte apoptosis and the alteration of the nuclear location of PKC-δ [26,27]. Cardioplegia techniques were also compared with the aim of reducing the risk of an IR injury. Retrograde cardioplegia involves the cannulation of the solution into the coronary sinus whereas antegrade cardioplegia involves cannulation into the aortic root [28]. Retrograde cardioplegia causes greater cardiomyocyte apoptosis and caspase-3 activation than antegrade cardioplegia after IR [29]. Although changes in the strategies of cardioplegic solutions have been addressed, there are no clear advantages of one cardioplegic strategy over another. Accordingly, we propose an understanding of the cardioplegia-occurring molecular mechanism with a view of a cellular pathological response.
2.3. Cardioplegia-Induced Inflammation and Dysfunction
Cytokine levels play critical roles in inflammatory responses. It is well-known that a cardiopulmonary bypass (CPB) contributes to postoperative complications [30]. The levels of tumor necrosis factor (TNF)-α, interleukin (IL)-6/8, and IL-10 in the blood of patients that had received cold crystalloid or warm blood cardioplegia were compared [30]. They reported that the serum levels of TNF-α, IL-6, and IL-8—excluding IL-10—were higher in the cold crystalloid than in the warm blood-exposed group. Based on their comparative study of cardioplegia trials, it would seem that warm blood cardioplegia may reduce the inflammatory response more effectively than cold crystalloid cardioplegia. Furthermore, an additional supplementation of an amino acid such as L-arginine in cardioplegia reduced the cytokine release; this was significant for IL-6 [31]. In addition to the inflammatory response, hyperkalemic cardioplegia induced cardiomyocyte swelling and reduced contractility in various models [32,33,34]. Treatment with the adenosine triphosphate-sensitive potassium channel (KATP) opener diazoxide also reduced cardiomyocyte swelling and inhibited cardioplegia-mediated reduced contractility [33]. It was revealed that the maintenance of the cardiomyocyte volume was associated with improved contractility and stunning.
2.4. Cardioplegia-Induced Oxidative Stress
Oxidative stress induced by CPB mediates myocardium inflammation and damage. To reduce the effect of oxidative stress, a pyruvate-enriched cardioplegia was investigated in pigs [35]. A combination of blood and cold crystalloid cardioplegia with pyruvate increased the tissue inhibitor of metalloproteinase-2 (TIMP-2) contents (a known stimulator of cell migration and apoptosis) and inhibited the metalloproteinase-9 (MMP-9) activity [35]. In addition, the application of pyruvate during cardioplegia reduced CPB-induced myocardial inflammation by increasing the myocardial GSH redox state (GSH/GSSG) and TIMP-2 levels [35]. GSH and GSSG are the mediators of oxidation/reduction reactions and the GSH/GSSG ratio is used as a marker of oxidative stress [36]. Cardioplegia with pyruvate decreased this ratio in the coronary sinus and finally reduced oxidative stress [35]. Moreover, a comparison of inflammatory cytokines after the administration of two types of cardioplegia solution, i.e., a “del Nido” or a “modified St. Thomas” solution, revealed no detectable difference, suggesting that the consideration of the administration intervals is more important than the cardioplegia solution type [37].
Various studies have been performed to improve the arrested status and to abolish the risk factors of cardioplegia such as inflammation, swelling, stunning, and oxidative stress (summarized in Table 1). However, a more effective strategy is still required to properly address these complications. The precise study of the mechanisms responsible for the effects of administered compounds on cardioplegia is required. In particular, an understanding of the changes in electrolytes through ion channels/transporters and the modulations of water channels would aid the understanding of heart damage. In the following section, we focus on the basic concept of electrolyte homeostasis as well as the changes of channel activity in cardiac tissue.

Table 1.
Characteristics of the modified cardioplegia solutions.
3. Altered Electrolyte Windows in the Cardiac Tissue
Na+-H+ exchangers (NHEs) are integral membrane glycoproteins that are expressed in most mammalian cells [39]. Members of the NHE family primarily regulate the intracellular pH but also regulate cell proliferation differentiation, apoptosis, migration, and volume control [40,41,42]. NHE contains 9 subfamilies (NHE-1 to NHE-9), which are composed of 600~900 amino acids [43]. In the heart, NHEs affect contractions by changing the intracellular pH. Furthermore, Na+ influx and H+ efflux through NHEs importantly restore the intracellular pH following ischemic acidification [2,44,45]. An Na+/H+ exchange transports Na+ in the forward direction under normal conditions, driven by an inwardly directed transmembrane Na+ gradient and, in exchange, expels a single proton; thus, it maintains the intracellular pH and regulates the intracellular Na+ levels [46]. However, during ischemia, an intracellular H+ accumulation provides a greater driving force. As a result, H+ leaks to the extracellular compartment and Na+ accumulates in the intracellular milieu. During ischemia, the transmembrane gradient was reduced by the accumulation of H+ in the extracellular compartment and this led to a decrease in the intracellular Na+ accumulation and NHE activity. During a reperfusion, the transmembrane gradient was restored by the removal of H+ in the extracellular compartment and H+ was excreted through the involvement of NHEs. This process arose from a sequential ion movement-like cascade reaction. Thus, the NHE transporters became reactivated during the reperfusion, which resulted in the intracellular Na+ accumulation [47] and stimulated the Ca2+ efflux through the Na+/Ca2+ exchanger (NCX). NCXs are plasma membrane-associated proteins that are driven by electrochemical gradients and allow Na+ to enter cells and Ca2+ to be released at a ratio of 3:1 [48]. Of the three NCX isoforms, NCX1 is expressed ubiquitously [49] whereas NCX2 and 3 are particularly expressed in the brain and skeletal muscles [50,51,52]. The three isoforms regulate Ca2+ homeostasis in a variety of cells and NCX1 is primarily involved with the heart [49,53]. Increased NCX1 levels in heart tissues are associated with human primary pulmonary hypertension and, when overexpressed, NCX1 decreases the SR Ca2+ load levels in heart failure [53]. Several studies have indicated that the NCX expression is associated with heart failure. NCX was overexpressed in heart failure animal models and a prolonged repolarization was revealed through monophasic action potentials (MAPs) in rodent chronic heart failure (CHF) [54,55]. Moreover, the overexpression of NCX prolonged the action potential duration [56] and this prolongation in heart failure sustained the time for the Ca2+ channel reopening, which may occur with a higher frequency of Ca2+ release from the SR [56,57]. In addition, the action potentials were shortened by inhibiting NCX with SEA0400 (a specific NCX1 inhibitor) [56]. In addition, inhibiting NCX shortened the MAP duration, reduced dispersion, and increased protection against the repolarization reserve and ventricular tachyarrhythmias (VTs) in rabbit heart failure [56]. In a forward mode, NCX releases Ca2+ whereas in a reverse mode, it enables Ca2+ to enter cells [58]. This reverse mode of NCX is induced by low intracellular Ca2+ levels and high intracellular Na+ levels in the failing heart [59,60]. Systolic and diastolic dysfunction and irreversible tissue injury induced by IR are associated with this intracellular Ca2+ accumulation [2,61,62]. Briefly, during ischemia, a reduced intracellular pH and ATP induced an excessive activity of NHE [63]. As a result, the intracellular Na+ was increased and cell swelling was induced whereas NCX was suppressed [61]. H+ was extruded during a reperfusion, resulting in a rapid increase in NHE activity and a reversed NCX was activated. The intracellular Ca2+ was increased again and an irreversible tissue injury occurred during the IR [63].
4. Bicarbonate Transporters and Their Associated Enzymes
In addition to cations such as H+ and Ca2+, the HCO3− anion also plays an important role in heart tissues that express HCO3− transporters such as the Cl−/HCO3− exchanger SLC26A6, which is involved in HCO3− influx and efflux [5,64]. SLC26A6 is a Cl−/HCO3− exchanger and is mainly expressed in the kidneys and exocrine glands [65,66,67]. SLC26A6 is encoded by the SLC26A6 gene and mediates the Cl− influx and HCO3− efflux. When SLC26A6 acts as an acid loader, it causes intracellular acidification by releasing HCO3− from cells [68]. Mouse heart tissues predominantly express anion exchanger Slc26a6 [68]. In a previous study, we found the expression and activity of Slc26a6 were more increased in the cardiac tissues of db/db mice (a type 2 diabetes mellitus mouse model) than in normal cardiac tissues and that Slc26a6 enhancement induced an exacerbated intracellular acidification after a cardioplegia-induced arrest [5]. In another study, it was reported that Slc26a6-depleted cardiomyocytes exhibited sarcomere shortening and that the cardiac contractility was reduced [69].
HCO3− is produced by the carbonic anhydrase (CA) catalyzed reaction between CO2 and H2O [70]. HCO3− is a product of CA and a source of HCO3− transporters. CAs are zinc metalloenzymes that hydrate carbon dioxide to bicarbonate and protons [71,72]. The CA family is composed of 5 types: α-, β-, γ-, δ-, and ζ-CAs. The β-ζ families of CA are non-mammal; therefore, we focused on α-CA. In mammals, it comprises CA I, CA II, CA III, CA VII, and CA VIII expressed in the cytosol; CA IV, CA IX, CA XII, and CA XIV expressed in the plasma membrane; CA VA and CA VB expressed in the mitochondria; and extracellularly secreted CA VI in the saliva and breast milk [73,74,75,76]. Although experimental evidence regarding cardiac CAs is sparse, CA II and CA IV were reportedly increased in hypertrophic ventricles and failing hearts in humans [77]. CA XIV interacted with the Cl−/HCO3− exchanger in hearts and the CA XIV protein levels were enhanced in the hypertrophic hearts of rats [78]. In spontaneously hypertensive rats (SHR), CA XIV levels were reduced by benzolamide (a CA inhibitor), which reduced the HCO3− flux to normal heart levels [78]. The roles of cardiac CAs require further study, especially regarding the modulation of the HCO3− flux.
5. Understanding of Cardioplegia-Induced Water Transport
Cardiac water contents modulate the cardiac output [79] and a cardiac tissue edema is a likely risk factor of a reduced contractile force [80]. Furthermore, IR injury and inflammatory responses caused by CPB mediate myocardial edemas [81]. Thus, an understanding of the CPB-mediated water movement is required to properly address a cardiac injury. In an early study, elevated extracellular Ca2+ concentrations increased ischemic swelling in rat hearts and an inhibition of the Ca2+ channel with verapamil or bepridil reduced myocardial edemas [82]. Thus, an understanding of the water channels is needed in the heart field and how to regulate the water channels should be elucidated.
Aquaporins (AQPs) are traditionally integral plasma membrane proteins and water channels that move water or electrolytes through the cell membrane [83]. AQP-1 is expressed in mouse cardiac muscles, which also contain the mRNAs of AQP-3, -4, -5, -7, -9, -10, and -11 [79,84]. Briefly, AQP-3 transports water and glycerol and is expressed in most epithelial cells including those of the kidney, eye, brain, pancreas, respiratory tract, urinary tract, and the basal keratinocyte layer [85,86,87,88]. AQP-4 mediates the movement of water in the adult brain and neurons of the paraventricular and supraoptic nuclei [89] and is expressed in the kidneys, skeletal muscle, and stomach [90,91]. AQP-5 is present in the intercalated ducts and acinar cells of salivary glands [92,93,94] and is involved in salivary fluid secretion [95]. AQP-5 is expressed in sweet, lacrimal, and airway submucosal glands and in the lung and corneal epithelium [94,96,97]. AQP-7 is a 269-amino acid aquaglyceroporin and facilitates glycerol transport in adipose tissues [98,99]. AQP-9 is present in the liver and testis and facilitates the passage of water, glycerol, and urea [94,100]. AQP-7 and AQP-9 are associated with glycerol metabolism in adipose tissues and the liver [101]. AQP-10 is a 310-amino acid aquaglyceroporin expressed in the small intestine and transports water and glycerol [102,103]. AQP-11 is present in the proximal kidney and brain tubules [104,105] and plays an important role in ER-resident peroxiporin activity and transports glycerol and water through the ER membranes [106]. AQPs regulate the volume of cells in an osmolarity-dependent manner. Under hypotonic conditions, the cell volumes expand and subsequently rapidly decrease [107,108]. K+ efflux from cells is induced by K+ channel activation whereas water is released from the cells through AQPs by osmosis, resulting in a “regulatory volume decrease” [109], which protects the cells from rupture under hypotonic conditions. Under hypertonic conditions, the cells undergo shrinkage and then return to their original state, resulting in a “regulatory volume increase” [110]. The mechanism of cell shrinkage involves the activation of ion channels such as the Na+-H+ exchanger (NHE) and Na+-K+-Cl− (NKCC), which allows Na+ to enter the cells [107] and the accompanying osmotic regulation induces water influx into the cells [111].
AQP-1, AQP-4, AQP-7, and AQP-11 are found in the cardiac tissues of humans, rats, and mice; AQP-8 transcription was confirmed at the mRNA level in mice hearts [112,113,114,115,116]. Furthermore, AQP-1 expression in the sarcolemma was reported to be sensitive to ischemia-induced osmotic stress [79]. After a cardioplegic arrest in humans, cardioplegia induces a mild ischemic injury and reduces the mRNA levels of AQP-1 and glycosylation in the cardiac tissues [117]. AQP-4 expression was shown to be no different after ischemia in mouse hearts [79]. During CPB using an intermittent antegrade cold blood cardioplegia, the level of urine AQP-2 of the kidney collecting ducts was increased in patients with cardiac surgery [118]. Urine AQP-2 was associated with the arginine vasopression (AVP) level, which led to the activation of AQP-2 to promote the reabsorption of free water in the collecting duct. In HL-1 mouse cardiomyocyte cells, the expression of AQP-4 was increased under ischemia conditions and TGN-20, an AQP-4 inhibitor, decreased the mRNA and protein expression of AQP-4. In addition, the inhibition of AQP-4 reduced cardiomyocyte pyroptosis [119]. The treatment of cis-dichlorodiammineplatinum (Cisplatin), an anti-cancer drug, induced a myocardial edema as a side effect [120,121]. The expression of AQP-3 and AQP-4 was increased by cisplatin administration in the rat left ventricle and the expression of p-53 and BAX (apoptosis markers) was increased [122]. During CPB in an adult goat heart, an edema in heart tissues was associated with increases in AQP-1 mRNA and protein levels [123]; HgCl2 was suppressed by HgCl2, which did not affect the expression of AQP-1 and improved the cardiac function. Thus, the myocardial water content was more increased in the goat heart CPB groups than in the control groups [123]. The decrease of the intracellular pH of the myocardial tissue and plasma in patients with a severe aortic stenosis after CPB was associated with ischemia and revealed that the expression of AQP-1 and AQP-4 was increased and a myocardial edema was induced [124]. More recently, the coordination between AQP-1 and the tight junction protein connexin 43 was associated with the pathological development of a myocardial edema [123]. The regulation of the cellular volume was achieved using the water channels and several related transporters [125]. AQP-7 transports water and small molecules such as glycerol. During hyperkalemic cardioplegia using St. Thomas’ Hospital solution No. 2 (STH2) in aquaporin-7 KO mice, the left ventricle advanced pressure was found to be lower than in WT mice and the Troponin T levels were significantly reduced in isolated hearts [126], which suggested that an AQP-7 deficiency protected the myocardium and reduced the risks of complications caused by cardiac surgery during hyperkalemic cardioplegia. A schematic of the actions of the AQP channels in the cardiac tissue is provided in Figure 2. Currently, it is unclear whether the cardiac water channels work alone or with other transporters during CPB. Conceivably, further studies are required on the coordination between the water channels and the ion transporters in the context of a cardiac injury.
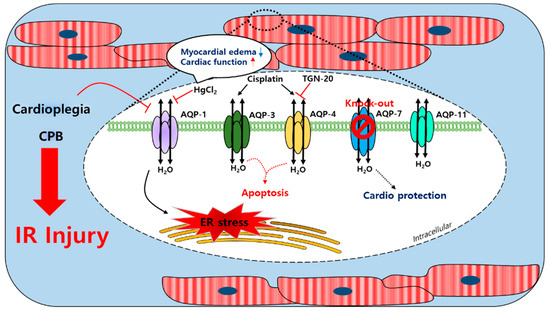
Figure 2.
Schematic illustration of the involved myocardial aquaporin channel induced by cardioplegia and CPB. An IR injury induced by cardioplegia and CPB regulated the expressions of myocardial AQPs. The AQP-1 expression was increased by CPB and affected the ER stress; the expression could be regulated by HgCl2. Cisplatin enhanced the AQP-3 and AQP-4 expression and apoptosis was induced. TGN-20 suppressed the ischemia-induced AQP-4 expression. Cardioplegia decreased the AQP-1 expression but did not affect the expressions of AQP-4 or AQP-11. An AQP-7 knockout study revealed its cardioprotective effect. CPB: cardiopulmonary bypass; AQP-1/3/4/7/11: aquaporin-1/3/4/7/11; IR injury: ischemia/reperfusion injury; ER stress: endoplasmic reticulum stress; cisplatin: cis-dichlorodiammineplatinum.
6. Conclusions and Future Works
Cardioplegia is used to maintain a normal cardiac function during cardiac surgery; however, the procedure has various side effects. As shown in Table 1, despite the efforts made, myocardial protection during a cardioplegia-induced arrest remains a challenging issue. Previously, we reported that an ion imbalance occurred in cardioplegia-exposed cardiac tissue and that this was more severe in diabetic hearts [5]. Furthermore, it appears that modulations of the electrolyte movements through ion channels such as SLC26A6 and its related enzyme, carbonic anhydrase, as well as through ion transporters such as NCX, NHE, and AQPs, play an important role in cardiac physiology. In our previous study in other tissues, the joint edema showed in rheumatoid arthritis (RA) patients was caused by the osmotic change of the synovium, where it was addressed that not only AQP-1 but also a combined ion transporter such as Na+-K+-2Cl− co-transporter 1 (NKCC1) and osmotic regulatory protein oxidative stress-responsive kinase 1 were involved [127,128]. A cytotoxic edema was induced by a pro-inflammatory cytokine such as IL-6 in RA synovial fluids from RA patients and NKCC1 was recruited to the plasma membrane of RA fibroblast-like synoviocytes and a combined volume regulation through AQP-1 was mediated [128]. In this regard, we considered the sufficient possibility that a cardiac edema caused by an IR injury after a cardiac arrest is associated with the convergent involvement of dynamic ion transporters and AQP. Conceivably, the severity of the IR injury might be enhanced by the involvement of dysregulated convergent channels/transporters. Although the field of cardiac channelopathy is relatively new and ion regulation through channels and transporters is complicated, we hope that this review provides a few unexpected clues and a medicinal approach. We believe that the delicate tuning of an ion imbalance might resolve the myocardial dysfunctions induced by cardioplegia and membrane-associated cardiac channels and transporters will provide favorable advanced strategies to achieve a clinical use in the cardiac field.
Author Contributions
J.H.H. and M.J.J. contributed to conception, design, and writing of the manuscript; M.J.J. drafted the article, collected the referenced articles, and prepared all animations; J.H.H. approved the version of the manuscript and accepts responsibility for all aspects of the work. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by the National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIT; 2019R1F1A1046785).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Kleber, A.G. Resting membrane potential, extracellular potassium activity, and intracellular sodium activity during acute global ischemia in isolated perfused guinea pig hearts. Circ. Res. 1983, 52, 442–450. [Google Scholar] [CrossRef]
- Muraki, S.; Morris, C.D.; Budde, J.M.; Zhao, Z.Q.; Guyton, R.A.; Vinten-Johansen, J. Blood cardioplegia supplementation with the sodium-hydrogen ion exchange inhibitor cariporide to attenuate infarct size and coronary artery endothelial dysfunction after severe regional ischemia in a canine model. J. Thorac. Cardiovasc. Surg. 2003, 125, 155–164. [Google Scholar] [CrossRef][Green Version]
- Govindapillai, A.; Hua, R.; Rose, R.; Friesen, C.H.; O’Blenes, S.B. Protecting the aged heart during cardiac surgery: Use of del Nido cardioplegia provides superior functional recovery in isolated hearts. J. Thorac. Cardiovasc. Surg. 2013, 146, 940–948. [Google Scholar] [CrossRef]
- Ad, N.; Holmes, S.D.; Massimiano, P.S.; Rongione, A.J.; Fornaresio, L.M.; Fitzgerald, D. The use of del Nido cardioplegia in adult cardiac surgery: A prospective randomized trial. J. Thorac. Cardiovasc. Surg. 2018, 155, 1011–1018. [Google Scholar] [CrossRef]
- Ji, M.; In Lee, S.; Lee, S.A.; Son, K.H.; Hong, J.H. Enhanced Activity by NKCC1 and Slc26a6 Mediates Acidic pH and Cl(-) Movement after Cardioplegia-Induced Arrest of db/db Diabetic Heart. Mediat. Inflamm. 2019, 2019, 7583760. [Google Scholar] [CrossRef]
- Dobson, G.P.; Faggian, G.; Onorati, F.; Vinten-Johansen, J. Hyperkalemic cardioplegia for adult and pediatric surgery: End of an era? Front. Physiol. 2013, 4, 228. [Google Scholar] [CrossRef]
- Feng, J.; Sellke, F. Microvascular dysfunction in patients with diabetes after cardioplegic arrest and cardiopulmonary bypass. Curr. Opin. Cardiol. 2016, 31, 618–624. [Google Scholar] [CrossRef]
- Chambers, D.J.; Fallouh, H.B. Cardioplegia and cardiac surgery: Pharmacological arrest and cardioprotection during global ischemia and reperfusion. Pharmacol. Ther. 2010, 127, 41–52. [Google Scholar] [CrossRef]
- Bernardi, P.; Vassanelli, S.; Veronese, P.; Colonna, R.; Szabo, I.; Zoratti, M. Modulation of the mitochondrial permeability transition pore. Effect of protons and divalent cations. J. Biol. Chem. 1992, 267, 2934–2939. [Google Scholar] [CrossRef]
- Crompton, M. The mitochondrial permeability transition pore and its role in cell death. Biochem. J. 1999, 341 Pt 2, 233–249. [Google Scholar] [CrossRef]
- Perrelli, M.G.; Pagliaro, P.; Penna, C. Ischemia/reperfusion injury and cardioprotective mechanisms: Role of mitochondria and reactive oxygen species. World J. Cardiol. 2011, 3, 186–200. [Google Scholar] [CrossRef]
- Ruiz-Meana, M.; Abellan, A.; Miro-Casas, E.; Garcia-Dorado, D. Opening of mitochondrial permeability transition pore induces hypercontracture in Ca2+ overloaded cardiac myocytes. Basic Res. Cardiol. 2007, 102, 542–552. [Google Scholar] [CrossRef]
- Piper, H.M.; Abdallah, Y.; Schafer, C. The first minutes of reperfusion: A window of opportunity for cardioprotection. Cardiovasc. Res. 2004, 61, 365–371. [Google Scholar] [CrossRef]
- Brdiczka, D.G.; Zorov, D.B.; Sheu, S.S. Mitochondrial contact sites: Their role in energy metabolism and apoptosis. Biochim. Biophys. Acta 2006, 1762, 148–163. [Google Scholar] [CrossRef]
- Fischer, U.M.; Klass, O.; Stock, U.; Easo, J.; Geissler, H.J.; Fischer, J.H.; Bloch, W.; Mehlhorn, U. Cardioplegic arrest induces apoptosis signal-pathway in myocardial endothelial cells and cardiac myocytes. Eur. J. Cardiothorac. Surg. 2003, 23, 984–990. [Google Scholar] [CrossRef][Green Version]
- Oka, N.; Wang, L.; Mi, W.; Zhu, W.; Honjo, O.; Caldarone, C.A. Cyclosporine A prevents apoptosis-related mitochondrial dysfunction after neonatal cardioplegic arrest. J. Thorac. Cardiovasc. Surg. 2008, 135, 123–130.e2. [Google Scholar] [CrossRef]
- Yeh, C.H.; Wang, Y.C.; Wu, Y.C.; Chu, J.J.; Lin, P.J. Continuous tepid blood cardioplegia can preserve coronary endothelium and ameliorate the occurrence of cardiomyocyte apoptosis. Chest 2003, 123, 1647–1654. [Google Scholar] [CrossRef]
- Camara, A.K.; An, J.; Chen, Q.; Novalija, E.; Varadarajan, S.G.; Schelling, P.; Stowe, D.F. Na+/H+ exchange inhibition with cardioplegia reduces cytosolic [Ca2+] and myocardial damage after cold ischemia. J. Cardiovasc. Pharmacol. 2003, 41, 686–698. [Google Scholar] [CrossRef]
- Yeh, C.H.; Chen, T.P.; Wang, Y.C.; Lin, Y.M.; Fang, S.W. AMP-activated protein kinase activation during cardioplegia-induced hypoxia/reoxygenation injury attenuates cardiomyocytic apoptosis via reduction of endoplasmic reticulum stress. Mediat. Inflamm. 2010, 2010, 130636. [Google Scholar] [CrossRef]
- Nardi, P.; Vacirca, S.R.; Russo, M.; Colella, D.F.; Bassano, C.; Scafuri, A.; Pellegrino, A.; Melino, G.; Ruvolo, G. Cold crystalloid versus warm blood cardioplegia in patients undergoing aortic valve replacement. J. Thorac. Dis. 2018, 10, 1490–1499. [Google Scholar] [CrossRef]
- Geissler, H.J.; Mehlhorn, U. Cold crystalloid cardioplegia. Multimed. Man Cardiothorac. Surg. 2006, 2006, mmcts2004001040. [Google Scholar] [CrossRef]
- Yeh, C.H.; Chen, T.P.; Wang, Y.C.; Lin, Y.M.; Fang, S.W. Cardiomyocytic apoptosis limited by bradykinin via restoration of nitric oxide after cardioplegic arrest. J. Surg. Res. 2010, 163, e1–e9. [Google Scholar] [CrossRef]
- Wang, D.; Zhang, X.; Qu, D.; Han, J.; Meng, F.; Xu, M.; Zheng, Q. Astragalin and dihydromyricetin as adjuncts to histidinetryptophanketoglutarate cardioplegia enhances protection during cardioplegic arrest. Mol. Med. Rep. 2018, 18, 2929–2936. [Google Scholar] [CrossRef]
- Scarabelli, T.M.; Pasini, E.; Stephanou, A.; Comini, L.; Curello, S.; Raddino, R.; Ferrari, R.; Knight, R.; Latchman, D.S. Urocortin promotes hemodynamic and bioenergetic recovery and improves cell survival in the isolated rat heart exposed to ischemia/reperfusion. J. Am. Coll. Cardiol. 2002, 40, 155–161. [Google Scholar] [CrossRef][Green Version]
- Scarabelli, T.M.; Pasini, E.; Ferrari, G.; Ferrari, M.; Stephanou, A.; Lawrence, K.; Townsend, P.; Chen-Scarabelli, C.; Gitti, G.; Saravolatz, L.; et al. Warm blood cardioplegic arrest induces mitochondrial-mediated cardiomyocyte apoptosis associated with increased urocortin expression in viable cells. J. Thorac. Cardiovasc. Surg. 2004, 128, 364–371. [Google Scholar] [CrossRef]
- Chen-Scarabelli, C.; Faggian, G.; Yuan, Z.; Tessari, M.; Rungatscher, A.; Di Rezze, J.; Scarabelli, G.M.; Abounit, K.; McCauley, R.; Saravolatz, L.; et al. Warm-blood cardioplegic arrest induces selective mitochondrial translocation of protein kinase Cepsilon followed by interaction with 6.1 inwardly rectifying potassium channel subunit in viable myocytes overexpressing urocortin. J. Thorac. Cardiovasc. Surg. 2009, 138, 1213–1221. [Google Scholar] [CrossRef]
- Chen-Scarabelli, C.; Knight, R.; Stephanou, A.; Scarabelli, G.; Onorati, F.; Tessari, M.; Rungatscher, A.; Narula, J.; Saravolatz, L.; Mazzucco, A.; et al. Diabetic hearts have lower basal urocortin levels that fail to increase after cardioplegic arrest: Association with increased apoptosis and postsurgical cardiac dysfunction. J. Thorac. Cardiovasc. Surg. 2014, 148, 2296–2308. [Google Scholar] [CrossRef]
- Buckberg, G.D. Antegrade/retrograde blood cardioplegia to ensure cardioplegic distribution: Operative techniques and objectives. J. Card. Surg. 1989, 4, 216–238. [Google Scholar] [CrossRef]
- Vahasilta, T.; Saraste, A.; Kyto, V.; Malmberg, M.; Kiss, J.; Kentala, E.; Kallajoki, M.; Savunen, T. Cardiomyocyte apoptosis after antegrade and retrograde cardioplegia. Ann. Thorac. Surg. 2005, 80, 2229–2234. [Google Scholar] [CrossRef]
- Wan, S.; Yim, A.P.; Arifi, A.A.; Lee, T.W.; Huynh, C.H.; DeSmet, J.M.; LeClerc, J.L.; Vincent, J.L. Can cardioplegia management influence cytokine responses during clinical cardiopulmonary bypass? Ann. Thorac. Cardiovasc. Surg. 1999, 5, 81–85. [Google Scholar]
- Colagrande, L.; Formica, F.; Porta, F.; Martino, A.; Sangalli, F.; Avalli, L.; Paolini, G. Reduced cytokines release and myocardial damage in coronary artery bypass patients due to L-arginine cardioplegia supplementation. Ann. Thorac. Surg. 2006, 81, 1256–1261. [Google Scholar] [CrossRef]
- Drewnowska, K.; Clemo, H.F.; Baumgarten, C.M. Prevention of myocardial intracellular edema induced by St. Thomas’ Hospital cardioplegic solution. J. Mol. Cell. Cardiol. 1991, 23, 1215–1221. [Google Scholar] [CrossRef]
- Mizutani, S.; Al-Dadah, A.S.; Bloch, J.B.; Prasad, S.M.; Diodato, M.D.; Schuessler, R.B.; Damiano, R.J., Jr.; Lawton, J.S. Hyperkalemic cardioplegia-induced myocyte swelling and contractile dysfunction: Prevention by diazoxide. Ann. Thorac. Surg. 2006, 81, 154–159. [Google Scholar] [CrossRef]
- Starr, J.P.; Jia, C.X.; Amirhamzeh, M.M.; Rabkin, D.G.; Hart, J.P.; Hsu, D.T.; Fisher, P.E.; Szabolcs, M.; Spotnitz, H.M. Coronary perfusate composition influences diastolic properties, myocardial water content, and histologic characteristics of the rat left ventricle. Ann. Thorac. Surg. 1999, 68, 925–930. [Google Scholar] [CrossRef]
- Ryou, M.G.; Flaherty, D.C.; Hoxha, B.; Gurji, H.; Sun, J.; Hodge, L.M.; Olivencia-Yurvati, A.H.; Mallet, R.T. Pyruvate-enriched cardioplegia suppresses cardiopulmonary bypass-induced myocardial inflammation. Ann. Thorac. Surg. 2010, 90, 1529–1535. [Google Scholar] [CrossRef]
- Jones, D.P. Redox potential of GSH/GSSG couple: Assay and biological significance. Methods Enzymol. 2002, 348, 93–112. [Google Scholar] [PubMed]
- Gorjipour, F.; Dehaki, M.G.; Totonchi, Z.; Hajimiresmaiel, S.J.; Azarfarin, R.; Pazoki-Toroudi, H.; Mahdavi, M.; Korbi, M.; Dehaki, M.G.; Soltani, B.; et al. Inflammatory cytokine response and cardiac troponin I changes in cardiopulmonary bypass using two cardioplegia solutions; del Nido and modified St. Thomas’: A randomized controlled trial. Perfusion 2017, 32, 394–402. [Google Scholar] [CrossRef]
- Han, C.K.; Kuo, W.W.; Shen, C.Y.; Chen, T.S.; Pai, P.; Tsai, C.H.; Lo, F.Y.; Ju, D.T.; Huang, C.Y. Dilong prevents the high-KCl cardioplegic solution administration-induced apoptosis in H9c2 cardiomyoblast cells mediated by MEK. Am. J. Chin. Med. 2014, 42, 1507–1519. [Google Scholar] [CrossRef] [PubMed]
- Cingolani, H.E.; Ennis, I.L. Sodium-hydrogen exchanger, cardiac overload, and myocardial hypertrophy. Circulation 2007, 115, 1090–1100. [Google Scholar] [CrossRef]
- Cardone, R.A.; Casavola, V.; Reshkin, S.J. The role of disturbed pH dynamics and the Na+/H+ exchanger in metastasis. Nat. Rev. Cancer 2005, 5, 786–795. [Google Scholar] [CrossRef]
- Baumgartner, M.; Patel, H.; Barber, D.L. Na(+)/H(+) exchanger NHE1 as plasma membrane scaffold in the assembly of signaling complexes. Am. J. Physiol. Cell Physiol. 2004, 287, C844–C850. [Google Scholar] [CrossRef]
- Fliegel, L. The Na+/H+ exchanger isoform 1. Int. J. Biochem. Cell Biol. 2005, 37, 33–37. [Google Scholar] [CrossRef]
- De Vito, P. The sodium/hydrogen exchanger: A possible mediator of immunity. Cell Immunol. 2006, 240, 69–85. [Google Scholar] [CrossRef]
- Karmazyn, M.; Moffat, M.P. Role of Na+/H+ exchange in cardiac physiology and pathophysiology: Mediation of myocardial reperfusion injury by the pH paradox. Cardiovasc. Res. 1993, 27, 915–924. [Google Scholar] [CrossRef] [PubMed]
- Karmazyn, M. The myocardial sodium-hydrogen exchanger (NHE) and its role in mediating ischemic and reperfusion injury. Keio J. Med. 1998, 47, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Frolich, O.; Karmazyn, M. The Na-H exchanger revisited: An update on Na-H exchange regulation and the role of the exchanger in hypertension and cardiac function in health and disease. Cardiovasc. Res. 1997, 36, 138–148. [Google Scholar] [CrossRef]
- Avkiran, M. Rational basis for use of sodium-hydrogen exchange inhibitors in myocardial ischemia. Am. J. Cardiol. 1999, 83, 10G–17G, discussion 17G–18G. [Google Scholar] [CrossRef]
- Reeves, J.P.; Hale, C.C. The stoichiometry of the cardiac sodium-calcium exchange system. J. Biol. Chem. 1984, 259, 7733–7739. [Google Scholar] [CrossRef]
- Blaustein, M.P.; Lederer, W.J. Sodium/calcium exchange: Its physiological implications. Physiol. Rev. 1999, 79, 763–854. [Google Scholar] [CrossRef]
- Lee, S.L.; Yu, A.S.; Lytton, J. Tissue-specific expression of Na(+)-Ca2+ exchanger isoforms. J. Biol. Chem. 1994, 269, 14849–14852. [Google Scholar] [CrossRef]
- Nicoll, D.A.; Quednau, B.D.; Qui, Z.; Xia, Y.R.; Lusis, A.J.; Philipson, K.D. Cloning of a third mammalian Na+-Ca2+ exchanger, NCX3. J. Biol. Chem. 1996, 271, 24914–24921. [Google Scholar] [CrossRef] [PubMed]
- Hasenfuss, G.; Schillinger, W.; Lehnart, S.E.; Preuss, M.; Pieske, B.; Maier, L.S.; Prestle, J.; Minami, K.; Just, H. Relationship between Na+-Ca2+-exchanger protein levels and diastolic function of failing human myocardium. Circulation 1999, 99, 641–648. [Google Scholar] [CrossRef] [PubMed]
- Khananshvili, D. Sodium-calcium exchangers (NCX): Molecular hallmarks underlying the tissue-specific and systemic functions. Pflug. Arch. 2014, 466, 43–60. [Google Scholar] [CrossRef] [PubMed]
- Pogwizd, S.M.; Bers, D.M. Cellular basis of triggered arrhythmias in heart failure. Trends Cardiovasc. Med. 2004, 14, 61–66. [Google Scholar] [CrossRef]
- Mork, H.K.; Sjaastad, I.; Sande, J.B.; Periasamy, M.; Sejersted, O.M.; Louch, W.E. Increased cardiomyocyte function and Ca2+ transients in mice during early congestive heart failure. J. Mol. Cell. Cardiol. 2007, 43, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Milberg, P.; Pott, C.; Frommeyer, G.; Fink, M.; Ruhe, M.; Matsuda, T.; Baba, A.; Klocke, R.; Quang, T.H.; Nikol, S.; et al. Acute inhibition of the Na(+)/Ca(2+) exchanger reduces proarrhythmia in an experimental model of chronic heart failure. Heart Rhythm 2012, 9, 570–578. [Google Scholar] [CrossRef]
- Sipido, K.R.; Volders, P.G.; de Groot, S.H.; Verdonck, F.; Van de Werf, F.; Wellens, H.J.; Vos, M.A. Enhanced Ca(2+) release and Na/Ca exchange activity in hypertrophied canine ventricular myocytes: Potential link between contractile adaptation and arrhythmogenesis. Circulation 2000, 102, 2137–2144. [Google Scholar] [CrossRef]
- Tykocki, N.R.; Jackson, W.F.; Watts, S.W. Reverse-mode Na+/Ca2+ exchange is an important mediator of venous contraction. Pharmacol. Res. 2012, 66, 544–554. [Google Scholar] [CrossRef]
- Pieske, B.; Maier, L.S.; Piacentino, V., 3rd; Weisser, J.; Hasenfuss, G.; Houser, S. Rate dependence of [Na+]i and contractility in nonfailing and failing human myocardium. Circulation 2002, 106, 447–453. [Google Scholar] [CrossRef]
- Bers, D.M. Cardiac excitation-contraction coupling. Nature 2002, 415, 198–205. [Google Scholar] [CrossRef]
- Pike, M.M.; Luo, C.S.; Clark, M.D.; Kirk, K.A.; Kitakaze, M.; Madden, M.C.; Cragoe, E.J., Jr.; Pohost, G.M. NMR measurements of Na+ and cellular energy in ischemic rat heart: Role of Na(+)-H+ exchange. Am. J. Physiol. 1993, 265, H2017–H2026. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.H.; Allen, D.G. Activity of the Na(+)/H(+) exchanger is critical to reperfusion damage and preconditioning in the isolated rat heart. Cardiovasc. Res. 2000, 48, 244–253. [Google Scholar] [CrossRef]
- Thind, G.S.; Agrawal, P.R.; Hirsh, B.; Saravolatz, L.; Chen-Scarabelli, C.; Narula, J.; Scarabelli, T.M. Mechanisms of myocardial ischemia-reperfusion injury and the cytoprotective role of minocycline: Scope and limitations. Future Cardiol. 2015, 11, 61–76. [Google Scholar] [CrossRef] [PubMed]
- Prasad, V.; Bodi, I.; Meyer, J.W.; Wang, Y.; Ashraf, M.; Engle, S.J.; Doetschman, T.; Sisco, K.; Nieman, M.L.; Miller, M.L.; et al. Impaired cardiac contractility in mice lacking both the AE3 Cl-/HCO3-exchanger and the NKCC1 Na+-K+-2Cl-cotransporter: Effects on Ca2+ handling and protein phosphatases. J. Biol. Chem. 2008, 283, 31303–31314. [Google Scholar] [CrossRef] [PubMed]
- Lohi, H.; Kujala, M.; Kerkela, E.; Saarialho-Kere, U.; Kestila, M.; Kere, J. Mapping of five new putative anion transporter genes in human and characterization of SLC26A6, a candidate gene for pancreatic anion exchanger. Genomics 2000, 70, 102–112. [Google Scholar] [CrossRef] [PubMed]
- Mukaibo, T.; Munemasa, T.; George, A.T.; Tran, D.T.; Gao, X.; Herche, J.L.; Masaki, C.; Shull, G.E.; Soleimani, M.; Melvin, J.E. The apical anion exchanger Slc26a6 promotes oxalate secretion by murine submandibular gland acinar cells. J. Biol. Chem. 2018, 293, 6259–6268. [Google Scholar] [CrossRef]
- Munemasa, T.; Mukaibo, T.; Melvin, J.E. Slc26a6 is an apical membrane anion exchanger that drives HCO3(-)-dependent fluid secretion in murine pancreatic acinar cells. Am. J. Physiol. Cell Physiol. 2019, 317, C1153–C1160. [Google Scholar] [CrossRef]
- Alvarez, B.V.; Kieller, D.M.; Quon, A.L.; Markovich, D.; Casey, J.R. Slc26a6: A cardiac chloride-hydroxyl exchanger and predominant chloride-bicarbonate exchanger of the mouse heart. J. Physiol. 2004, 561, 721–734. [Google Scholar] [CrossRef]
- Sirish, P.; Ledford, H.A.; Timofeyev, V.; Thai, P.N.; Ren, L.; Kim, H.J.; Park, S.; Lee, J.H.; Dai, G.; Moshref, M.; et al. Action Potential Shortening and Impairment of Cardiac Function by Ablation of Slc26a6. Circ. Arrhythm. Electrophysiol. 2017, 10, e005267. [Google Scholar] [CrossRef]
- Wang, H.S.; Chen, Y.; Vairamani, K.; Shull, G.E. Critical role of bicarbonate and bicarbonate transporters in cardiac function. World J. Biol. Chem. 2014, 5, 334–345. [Google Scholar] [CrossRef]
- Supuran, C.T. Carbonic anhydrases: Novel therapeutic applications for inhibitors and activators. Nat. Rev. Drug Discov. 2008, 7, 168–181. [Google Scholar] [CrossRef]
- Supuran, C.T.; Scozzafava, A.; Casini, A. Carbonic anhydrase inhibitors. Med. Res. Rev. 2003, 23, 146–189. [Google Scholar] [CrossRef]
- De Simone, G.; Di Fiore, A.; Supuran, C.T. Are carbonic anhydrase inhibitors suitable for obtaining antiobesity drugs? Curr. Pharm. Des. 2008, 14, 655–660. [Google Scholar] [CrossRef]
- Krungkrai, J.; Supuran, C.T. The alpha-carbonic anhydrase from the malaria parasite and its inhibition. Curr. Pharm. Des. 2008, 14, 631–640. [Google Scholar] [CrossRef]
- Scozzafava, A.; Menabuoni, L.; Mincione, F.; Briganti, F.; Mincione, G.; Supuran, C.T. Carbonic anhydrase inhibitors. Synthesis of water-soluble, topically effective, intraocular pressure-lowering aromatic/heterocyclic sulfonamides containing cationic or anionic moieties: Is the tail more important than the ring? J. Med. Chem. 1999, 42, 2641–2650. [Google Scholar] [CrossRef]
- Supuran, C.T. Carbonic anhydrase inhibitors. Bioorg. Med. Chem. Lett. 2010, 20, 3467–3474. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, B.V.; Quon, A.L.; Mullen, J.; Casey, J.R. Quantification of carbonic anhydrase gene expression in ventricle of hypertrophic and failing human heart. BMC Cardiovasc. Disord. 2013, 13, 2. [Google Scholar] [CrossRef] [PubMed]
- Vargas, L.A.; Alvarez, B.V. Carbonic anhydrase XIV in the normal and hypertrophic myocardium. J. Mol. Cell. Cardiol. 2012, 52, 741–752. [Google Scholar] [CrossRef] [PubMed]
- Butler, T.L.; Au, C.G.; Yang, B.; Egan, J.R.; Tan, Y.M.; Hardeman, E.C.; North, K.N.; Verkman, A.S.; Winlaw, D.S. Cardiac aquaporin expression in humans, rats, and mice. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H705–H713. [Google Scholar] [CrossRef] [PubMed]
- Jia, C.X.; Rabkin, D.G.; Hart, J.P.; Dean, D.A.; Cabreriza, S.A.; Weinberg, A.D.; Spotnitz, H.M. Regional variation in myocardial water content in the edematous pig heart. J. Surg. Res. 2002, 106, 70–75. [Google Scholar] [CrossRef]
- Palmer, B.S.; Hadziahmetovic, M.; Veci, T.; Angelos, M.G. Global ischemic duration and reperfusion function in the isolated perfused rat heart. Resuscitation 2004, 62, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Askenasy, N. Is cytotoxic cellular edema real? The effect of calcium ion on water homeostasis in the rat heart. Cardiovasc. Toxicol. 2001, 1, 21–34. [Google Scholar] [CrossRef]
- Denker, B.M.; Smith, B.L.; Kuhajda, F.P.; Agre, P. Identification, purification, and partial characterization of a novel Mr 28,000 integral membrane protein from erythrocytes and renal tubules. J. Biol. Chem. 1988, 263, 15634–15642. [Google Scholar] [CrossRef]
- Au, C.G.; Cooper, S.T.; Lo, H.P.; Compton, A.G.; Yang, N.; Wintour, E.M.; North, K.N.; Winlaw, D.S. Expression of aquaporin 1 in human cardiac and skeletal muscle. J. Mol. Cell. Cardiol. 2004, 36, 655–662. [Google Scholar] [CrossRef] [PubMed]
- Hara-Chikuma, M.; Verkman, A.S. Roles of aquaporin-3 in the epidermis. J. Investig. Dermatol. 2008, 128, 2145–2151. [Google Scholar] [CrossRef] [PubMed]
- Spector, D.A.; Wade, J.B.; Dillow, R.; Steplock, D.A.; Weinman, E.J. Expression, localization, and regulation of aquaporin-1 to -3 in rat urothelia. Am. J. Physiol. Ren. Physiol. 2002, 282, F1034–F1042. [Google Scholar] [CrossRef]
- King, L.S.; Nielsen, S.; Agre, P. Aquaporins in complex tissues. I. Developmental patterns in respiratory and glandular tissues of rat. Am. J. Physiol. 1997, 273, C1541–C1548. [Google Scholar] [CrossRef]
- Liu, Y.L.; Matsuzaki, T.; Nakazawa, T.; Murata, S.; Nakamura, N.; Kondo, T.; Iwashina, M.; Mochizuki, K.; Yamane, T.; Takata, K.; et al. Expression of aquaporin 3 (AQP3) in normal and neoplastic lung tissues. Hum. Pathol. 2007, 38, 171–178. [Google Scholar] [CrossRef]
- Jung, J.S.; Bhat, R.V.; Preston, G.M.; Guggino, W.B.; Baraban, J.M.; Agre, P. Molecular characterization of an aquaporin cDNA from brain: Candidate osmoreceptor and regulator of water balance. Proc. Natl. Acad. Sci. USA 1994, 91, 13052–13056. [Google Scholar] [CrossRef]
- Verkman, A.S. Aquaporins in clinical medicine. Annu. Rev. Med. 2012, 63, 303–316. [Google Scholar] [CrossRef]
- Papadopoulos, M.C.; Verkman, A.S. Aquaporin 4 and neuromyelitis optica. Lancet Neurol. 2012, 11, 535–544. [Google Scholar] [CrossRef]
- Agre, P.; Brown, D.; Nielsen, S. Aquaporin water channels: Unanswered questions and unresolved controversies. Curr. Opin. Cell Biol. 1995, 7, 472–483. [Google Scholar] [CrossRef]
- He, X.; Tse, C.M.; Donowitz, M.; Alper, S.L.; Gabriel, S.E.; Baum, B.J. Polarized distribution of key membrane transport proteins in the rat submandibular gland. Pflug. Arch. 1997, 433, 260–268. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, T.; Suzuki, T.; Koyama, H.; Tanaka, S.; Takata, K. Aquaporin-5 (AQP5), a water channel protein, in the rat salivary and lacrimal glands: Immunolocalization and effect of secretory stimulation. Cell Tissue Res. 1999, 295, 513–521. [Google Scholar] [CrossRef]
- Song, Y.; Verkman, A.S. Aquaporin-5 dependent fluid secretion in airway submucosal glands. J. Biol. Chem. 2001, 276, 41288–41292. [Google Scholar] [CrossRef]
- Borok, Z.; Li, X.; Fernandes, V.F.; Zhou, B.; Ann, D.K.; Crandall, E.D. Differential regulation of rat aquaporin-5 promoter/enhancer activities in lung and salivary epithelial cells. J. Biol. Chem. 2000, 275, 26507–26514. [Google Scholar] [CrossRef]
- Nielsen, S.; King, L.S.; Christensen, B.M.; Agre, P. Aquaporins in complex tissues. II. Subcellular distribution in respiratory and glandular tissues of rat. Am. J. Physiol. 1997, 273, C1549–C1561. [Google Scholar] [CrossRef]
- Suzuki-Toyota, F.; Ishibashi, K.; Yuasa, S. Immunohistochemical localization of a water channel, aquaporin 7 (AQP7), in the rat testis. Cell Tissue Res. 1999, 295, 279–285. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, I.V.; Soveral, G. Aquaporins in Obesity. Adv. Exp. Med. Biol. 2017, 969, 227–238. [Google Scholar] [CrossRef]
- Elkjaer, M.; Vajda, Z.; Nejsum, L.N.; Kwon, T.; Jensen, U.B.; Amiry-Moghaddam, M.; Frokiaer, J.; Nielsen, S. Immunolocalization of AQP9 in liver, epididymis, testis, spleen, and brain. Biochem. Biophys. Res. Commun. 2000, 276, 1118–1128. [Google Scholar] [CrossRef]
- Maeda, N.; Hibuse, T.; Funahashi, T. Role of aquaporin-7 and aquaporin-9 in glycerol metabolism; involvement in obesity. Handb. Exp. Pharmacol. 2009, 233–249. [Google Scholar] [CrossRef]
- Ishibashi, K.; Morinaga, T.; Kuwahara, M.; Sasaki, S.; Imai, M. Cloning and identification of a new member of water channel (AQP10) as an aquaglyceroporin. Biochim. Biophys. Acta 2002, 1576, 335–340. [Google Scholar] [CrossRef]
- Hatakeyama, S.; Yoshida, Y.; Tani, T.; Koyama, Y.; Nihei, K.; Ohshiro, K.; Kamiie, J.I.; Yaoita, E.; Suda, T.; Hatakeyama, K.; et al. Cloning of a new aquaporin (AQP10) abundantly expressed in duodenum and jejunum. Biochem. Biophys. Res. Commun. 2001, 287, 814–819. [Google Scholar] [CrossRef]
- Atochina-Vasserman, E.N.; Biktasova, A.; Abramova, E.; Cheng, D.S.; Polosukhin, V.V.; Tanjore, H.; Takahashi, S.; Sonoda, H.; Foye, L.; Venkov, C.; et al. Aquaporin 11 insufficiency modulates kidney susceptibility to oxidative stress. Am. J. Physiol. Ren. Physiol. 2013, 304, F1295–F1307. [Google Scholar] [CrossRef] [PubMed]
- Morishita, Y.; Matsuzaki, T.; Hara-chikuma, M.; Andoo, A.; Shimono, M.; Matsuki, A.; Kobayashi, K.; Ikeda, M.; Yamamoto, T.; Verkman, A.; et al. Disruption of aquaporin-11 produces polycystic kidneys following vacuolization of the proximal tubule. Mol. Cell. Biol. 2005, 25, 7770–7779. [Google Scholar] [CrossRef] [PubMed]
- Fruhbeck, G.; Balaguer, I.; Mendez-Gimenez, L.; Valenti, V.; Becerril, S.; Catalan, V.; Gomez-Ambrosi, J.; Silva, C.; Salvador, J.; Calamita, G.; et al. Aquaporin-11 Contributes to TGF-beta1-Induced Endoplasmic Reticulum Stress in Human Visceral Adipocytes: Role in Obesity-Associated Inflammation. Cells 2020, 9, 1403. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, E.K.; Lambert, I.H.; Pedersen, S.F. Physiology of cell volume regulation in vertebrates. Physiol. Rev. 2009, 89, 193–277. [Google Scholar] [CrossRef] [PubMed]
- Kitchen, P.; Oberg, F.; Sjohamn, J.; Hedfalk, K.; Bill, R.M.; Conner, A.C.; Conner, M.T.; Tornroth-Horsefield, S. Plasma Membrane Abundance of Human Aquaporin 5 Is Dynamically Regulated by Multiple Pathways. PLoS ONE 2015, 10, e0143027. [Google Scholar] [CrossRef]
- Yang, B.; Verkman, A.S. Water and glycerol permeabilities of aquaporins 1-5 and MIP determined quantitatively by expression of epitope-tagged constructs in Xenopus oocytes. J. Biol. Chem. 1997, 272, 16140–16146. [Google Scholar] [CrossRef]
- Miyazaki, H.; Shiozaki, A.; Niisato, N.; Marunaka, Y. Physiological significance of hypotonicity-induced regulatory volume decrease: Reduction in intracellular Cl- concentration acting as an intracellular signaling. Am. J. Physiol. Ren. Physiol. 2007, 292, F1411–F1417. [Google Scholar] [CrossRef] [PubMed]
- Kapus, A.; Grinstein, S.; Wasan, S.; Kandasamy, R.; Orlowski, J. Functional characterization of three isoforms of the Na+/H+ exchanger stably expressed in Chinese hamster ovary cells. ATP dependence, osmotic sensitivity, and role in cell proliferation. J. Biol. Chem. 1994, 269, 23544–23552. [Google Scholar] [CrossRef]
- Hasegawa, H.; Lian, S.C.; Finkbeiner, W.E.; Verkman, A.S. Extrarenal tissue distribution of CHIP28 water channels by in situ hybridization and antibody staining. Am. J. Physiol. 1994, 266, C893–C903. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Ma, T.; Verkman, A.S. cDNA cloning, gene organization, and chromosomal localization of a human mercurial insensitive water channel. Evidence for distinct transcriptional units. J. Biol. Chem. 1995, 270, 22907–22913. [Google Scholar] [CrossRef]
- Ishibashi, K.; Yamauchi, K.; Kageyama, Y.; Saito-Ohara, F.; Ikeuchi, T.; Marumo, F.; Sasaki, S. Molecular characterization of human Aquaporin-7 gene and its chromosomal mapping. Biochim. Biophys. Acta 1998, 1399, 62–66. [Google Scholar] [CrossRef]
- Ma, T.; Yang, B.; Verkman, A.S. Gene structure, cDNA cloning, and expression of a mouse mercurial-insensitive water channel. Genomics 1996, 33, 382–388. [Google Scholar] [CrossRef]
- Ma, T.; Yang, B.; Verkman, A.S. Cloning of a novel water and urea-permeable aquaporin from mouse expressed strongly in colon, placenta, liver, and heart. Biochem. Biophys. Res. Commun. 1997, 240, 324–328. [Google Scholar] [CrossRef]
- Rutkovskiy, A.; Bliksoen, M.; Hillestad, V.; Amin, M.; Czibik, G.; Valen, G.; Vaage, J.; Amiry-Moghaddam, M.; Stenslokken, K.O. Aquaporin-1 in cardiac endothelial cells is downregulated in ischemia, hypoxia and cardioplegia. J. Mol. Cell. Cardiol. 2013, 56, 22–33. [Google Scholar] [CrossRef]
- Yokoyama, S.; Imamura, T.; Yamashita, S.; Doi, T.; Fukahara, K.; Yoshimura, N.; Kinugawa, K. Peak Lag Between Plasma Vasopressin and Urine Aquaporin-2 Following Cardiac Surgery. Int. Heart J. 2021, 62, 1057–1061. [Google Scholar] [CrossRef]
- Jiang, Q.; Dong, X.; Hu, D.; Chen, L.; Luo, Y. Aquaporin 4 inhibition alleviates myocardial ischemia-reperfusion injury by restraining cardiomyocyte pyroptosis. Bioengineered 2021, 12, 9021–9030. [Google Scholar] [CrossRef]
- Varga, Z.V.; Ferdinandy, P.; Liaudet, L.; Pacher, P. Drug-induced mitochondrial dysfunction and cardiotoxicity. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H1453–H1467. [Google Scholar] [CrossRef]
- Sanchez-Gonzalez, P.D.; Lopez-Hernandez, F.J.; Lopez-Novoa, J.M.; Morales, A.I. An integrative view of the pathophysiological events leading to cisplatin nephrotoxicity. Crit. Rev. Toxicol. 2011, 41, 803–821. [Google Scholar] [CrossRef]
- Koral, L.; Ovali, M.A.; Tufekcioglu, N.K.; Karakilic, E.; Adali, Y.; Uzun, M. The role of AQP3 and AQP4 channels in cisplatin-induced cardiovascular edema and the protective effect of melatonin. Mol. Biol. Rep. 2021, 48, 7457–7465. [Google Scholar] [CrossRef] [PubMed]
- Ding, F.B.; Yan, Y.M.; Huang, J.B.; Mei, J.; Zhu, J.Q.; Liu, H. The involvement of AQP1 in heart oedema induced by global myocardial ischemia. Cell Biochem. Funct. 2013, 31, 60–64. [Google Scholar] [CrossRef] [PubMed]
- Politi, M.T.; Ochoa, F.; Netti, V.; Ferreyra, R.; Bortman, G.; Sanjuan, N.; Morales, C.; Piazza, A.; Capurro, C. Changes in cardiac Aquaporin expression during aortic valve replacement surgery with cardiopulmonary bypass. Eur. J. Cardiothorac. Surg. 2020, 57, 556–564. [Google Scholar] [CrossRef]
- Hwang, S.; Kang, J.Y.; Kim, M.J.; Shin, D.M.; Hong, J.H. Carbonic anhydrase 12 mutation modulates membrane stability and volume regulation of aquaporin 5. J. Enzyme Inhib. Med. Chem. 2019, 34, 179–188. [Google Scholar] [CrossRef]
- Fujii, M.; Ota, K.; Bessho, R. Cardioprotective effect of hyperkalemic cardioplegia in an aquaporin 7-deficient murine heart. Gen. Thorac. Cardiovasc. Surg. 2020, 68, 578–584. [Google Scholar] [CrossRef] [PubMed]
- Alessi, D.R.; Zhang, J.; Khanna, A.; Hochdorfer, T.; Shang, Y.; Kahle, K.T. The WNK-SPAK/OSR1 pathway: Master regulator of cation-chloride cotransporters. Sci. Signal. 2014, 7, re3. [Google Scholar] [CrossRef]
- Ji, M.J.; Ryu, H.J.; Hong, J.H. Synovial Fluid of Patient with Rheumatoid Arthritis Enhanced Osmotic Sensitivity through the Cytotoxic Edema Module in Synoviocytes. Front. Cell Dev. Biol. 2021, 9, 700879. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).