
Downregulation of Glutathione-Mediated Detoxification Capacity by Binge Drinking Aggravates Acetaminophen-Induced Liver Injury through IRE1α ER Stress Signaling


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal and Treatments
2.2. Examination of Serum Biochemical Parameters
2.3. Examination of Histological Mouse Liver Tissue
2.4. Examination of Hepatic Sulfur-Containing Metabolites
2.5. Examination of Hepatic Lipid Peroxidation
2.6. Measurement of Hepatic ROS Generation
2.7. Immunoblotting Analysis
2.8. Statistical Analysis
3. Results
3.1. Pretreatment with Alcohol Binges Potentiates APAP-Induced Hepatotoxicity in Mice
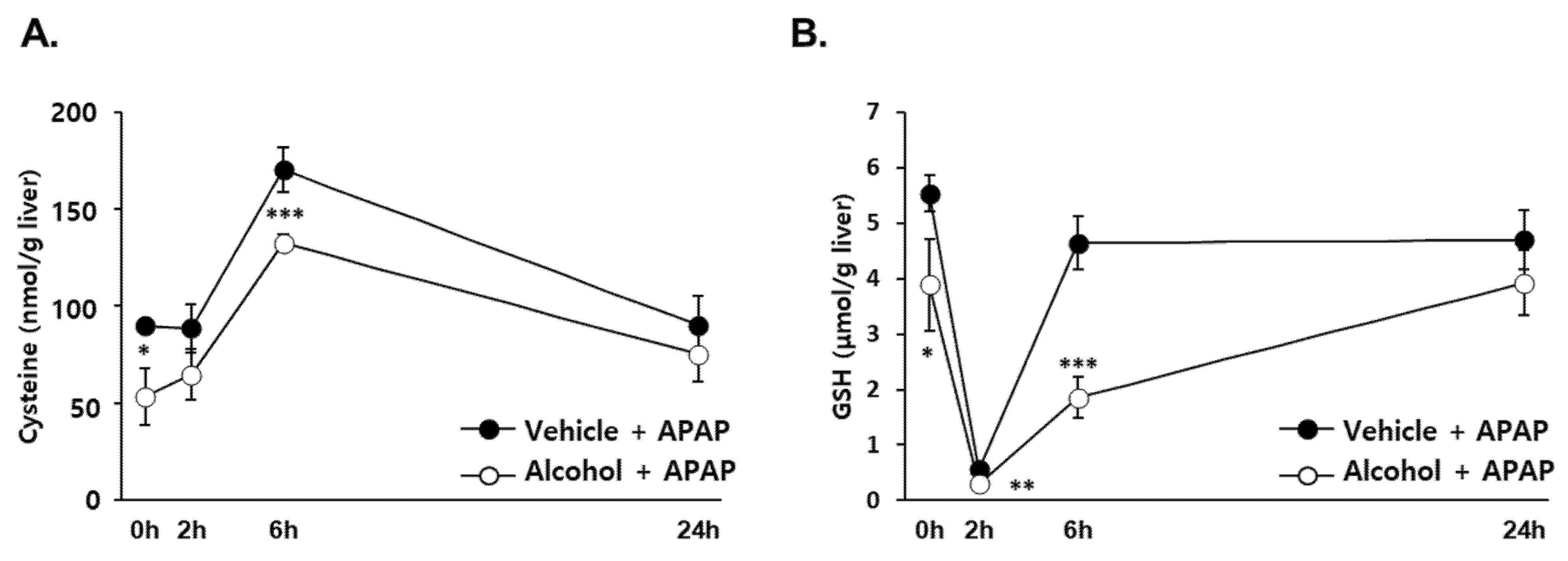
3.2. Effects of Alcohol Binges on Hepatic Cysteine and GSH Concentration in Mice
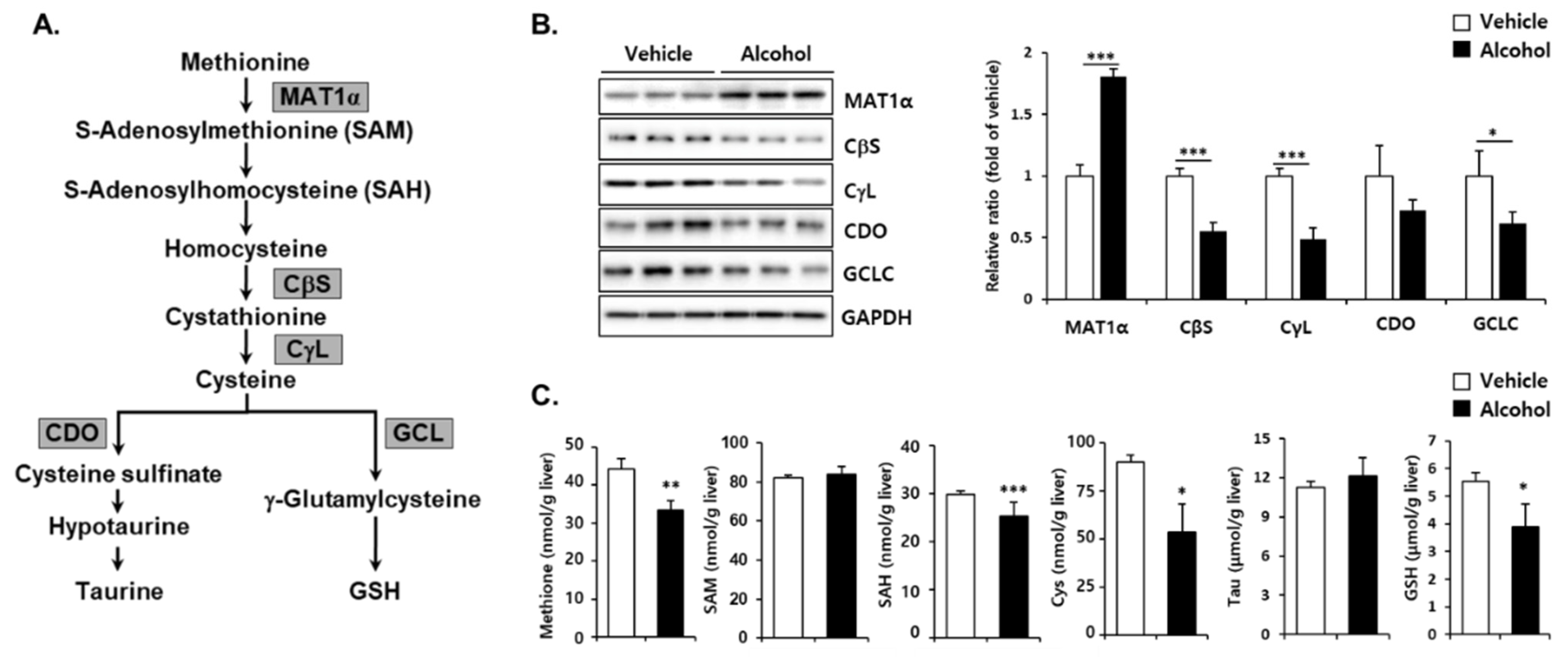
3.3. Effects of Alcohol Binges on Hepatic Sulfur Amino Acid Metabolism in Mice
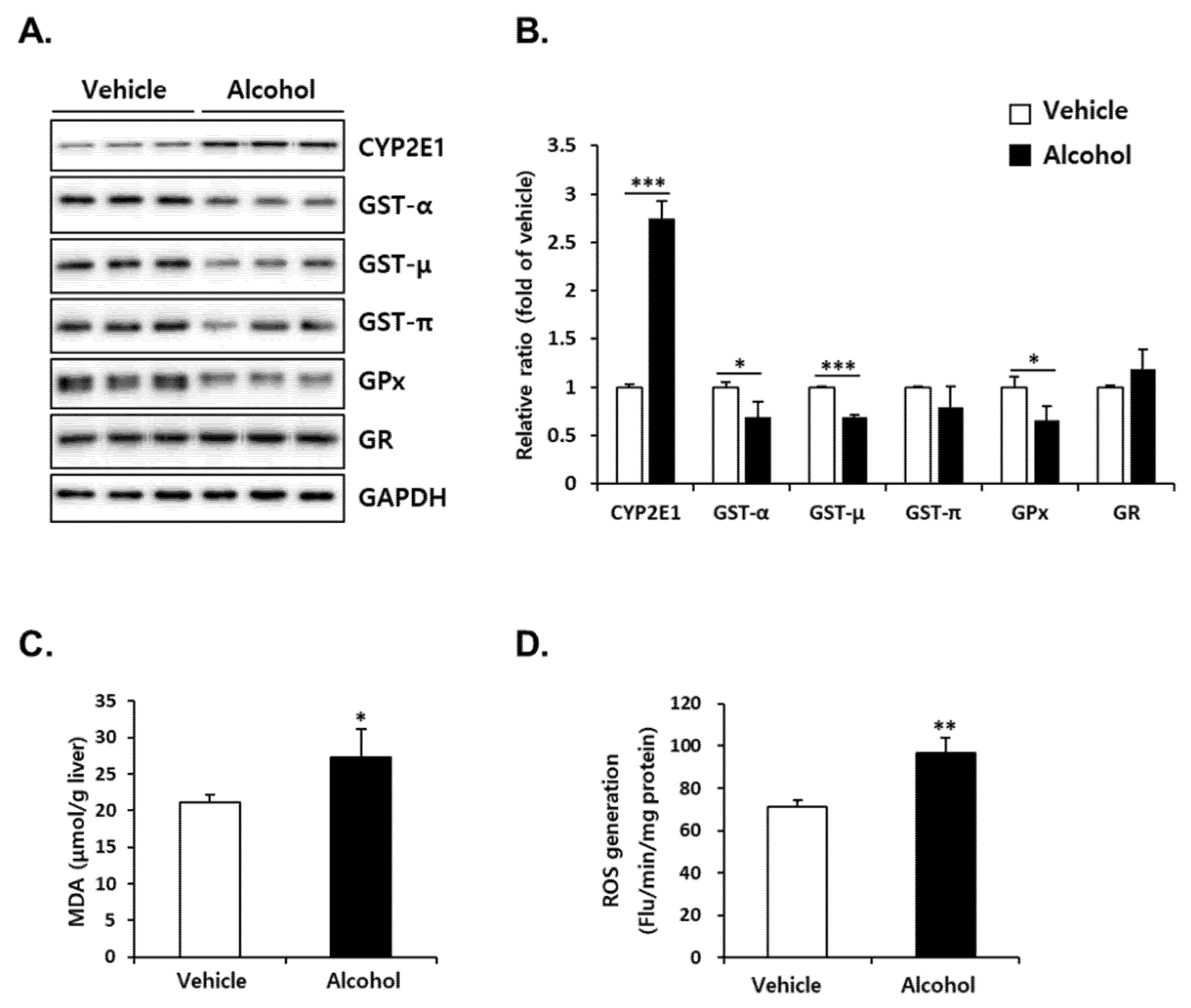
3.4. Alcohol Binges Induced Oxidative Stress in Mice Liver
3.5. Alcohol Binges Induced ER Stress in Mice Liver
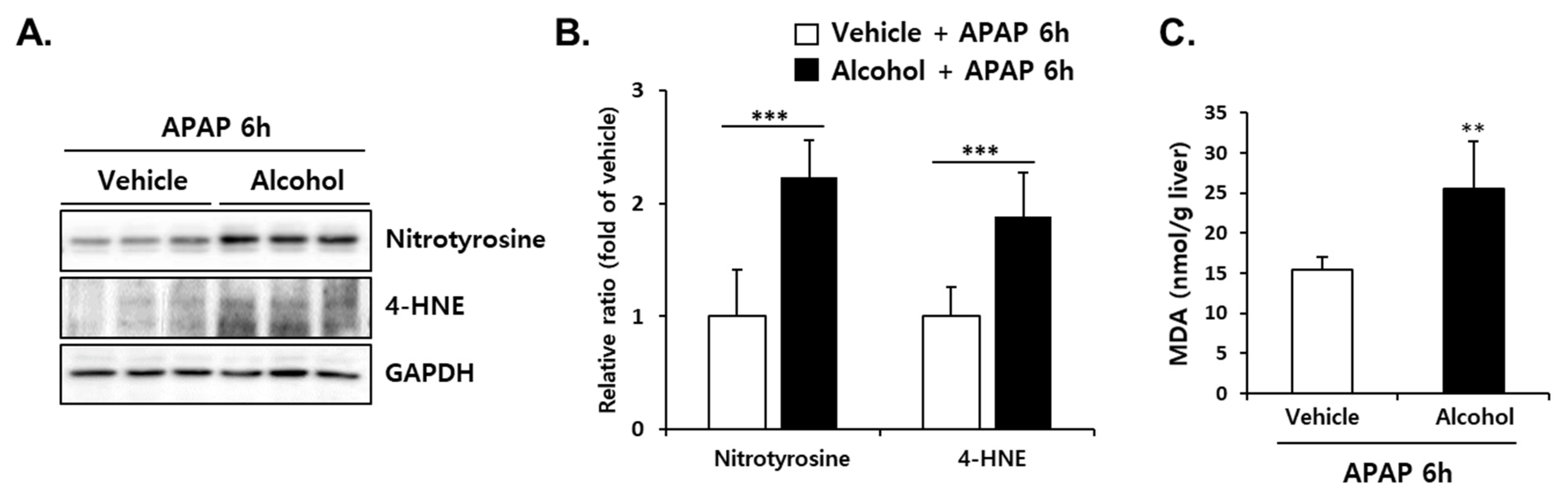
3.6. Pretreatment with Alcohol Binges Amplified APAP-Induced Oxidative Stress in Mice Liver
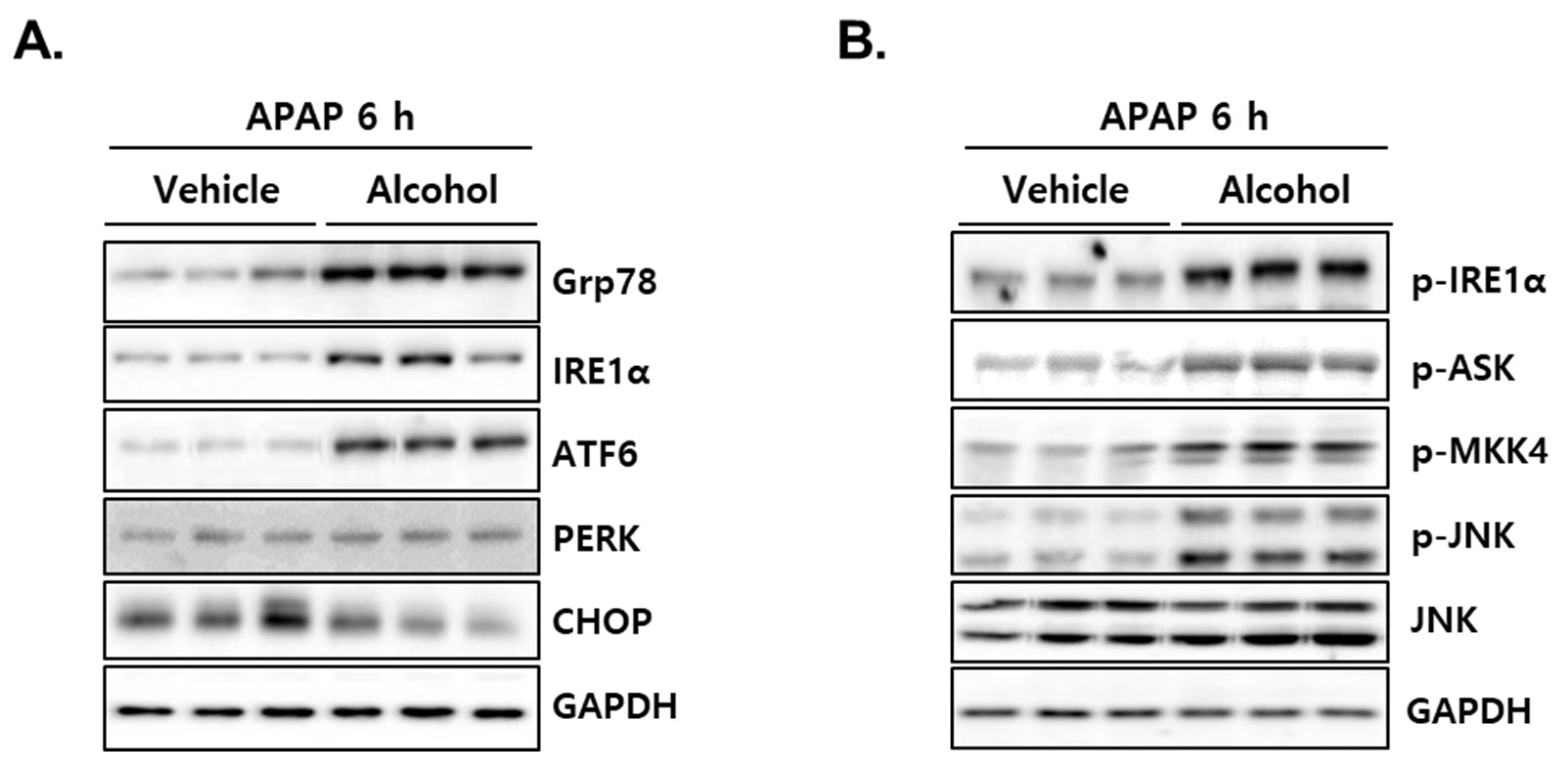
3.7. Administration of APAP in Mice with Pretreated Alcohol Binges Activates IRE1α/ASK1/MKK4/JNK Signal
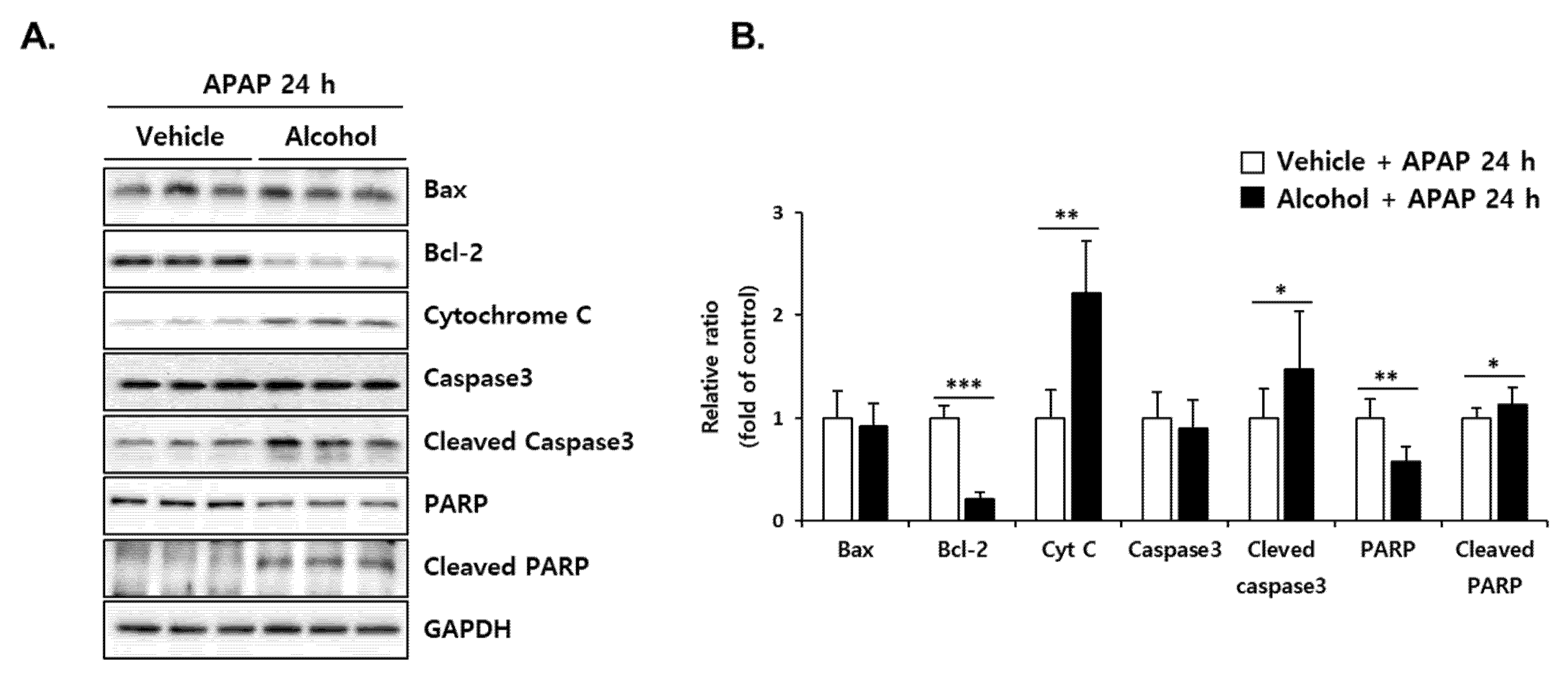
3.8. Pretreatment with Alcohol Binges Potentiates APAP-Induced Apoptosis in Mice Liver
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tredger, J.M.; Smith, H.M.; Read, R.B.; Portmann, B.; Williams, R. Effects of Ethanol Ingestion on the Hepatotoxicity and Metabolism of Paracetamol in Mice. Toxicology 1985, 36, 341–352. [Google Scholar] [CrossRef]
- Yoon, E.; Babar, A.; Choudhary, M.; Kutner, M.; Pyrsopoulos, N. Acetaminophen-Induced Hepatotoxicity: A Comprehensive Update. J. Clin. Transl. Hepatol. 2016, 4, 131–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazaleuskaya, L.L.; Sangkuhl, K.; Thorn, C.F.; FitzGerald, G.A.; Altman, R.B.; Klein, T.E. PharmGKB summary: Pathways of acetaminophen metabolism at the therapeutic versus toxic doses. Pharmacogenet. Genom. 2015, 25, 416–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davidson, D.G.; Eastham, W.N. Acute liver necrosis following overdose of paracetamol. Br. Med. J. 1966, 2, 497–499. [Google Scholar] [CrossRef] [Green Version]
- Addolorato, G.; Vassallo, G.A.; Antonelli, G.; Antonelli, M.; Tarli, C.; Mirijello, A.; Agyei-Nkansah, A.; Mentella, M.C.; Ferrarese, D.; Mora, V.; et al. Binge Drinking among adolescents is related to the development of Alcohol Use Disorders: Results from a Cross-Sectional Study. Sci. Rep. 2018, 8, 12624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stine, J.G.; Chalasani, N.P. Drug Hepatotoxicity: Environmental Factors. Clin. Liver Dis. 2017, 21, 103–113. [Google Scholar] [CrossRef]
- Caballería, J. Current concepts in alcohol metabolism. Ann. Hepatol. 2003, 2, 60–68. [Google Scholar] [CrossRef]
- Dupont, I.; Lucas, D.; Clot, P.; Ménez, C.; Albano, E. Cytochrome P4502E1 inducibility and hydroxyethyl radical formation among alcoholics. J. Hepatol. 1998, 28, 564–571. [Google Scholar] [CrossRef]
- Muller, A.; Sies, H. Role of alcohol dehydrogenase activity and the acetaldehyde in ethanol- induced ethane and pentane production by isolated perfused rat liver. Biochem. J. 1982, 206, 153–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Y.; Cederbaum, A.I. CYP2E1 and oxidative liver injury by alcohol. Free Radic. Biol. Med. 2008, 44, 723–738. [Google Scholar] [CrossRef] [Green Version]
- Lieber, C.S. Cytochrome P-4502E1: Its physiological and pathological role. Physiol. Rev. 1997, 77, 517–544. [Google Scholar] [CrossRef]
- Bolt, H.M.; Roos, P.H.; Thier, R. The cytochrome P-450 isoenzyme CYP2E1 in the biological processing of industrial chemicals: Consequences for occupational and environmental medicine. Int. Arch. Occup. Environ. Health 2003, 76, 174–185. [Google Scholar] [CrossRef]
- Koop, D.R. Oxidative and reductive metabolism by cytochrome P450 2E1. FASEB J. 1992, 6, 724–730. [Google Scholar] [CrossRef]
- Gonzalez, F.J. Role of cytochromes P450 in chemical toxicity and oxidative stress: Studies with CYP2E1. Mutat. Res. 2005, 569, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Lieber, C.S. Microsomal Ethanol-Oxidizing System (MEOS): The First 30 Years (1968-1998)-A Review. Alcohol. Clin. Exp. Res. 1999, 23, 991–1007. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.; Abril, E.R.; Bethea, N.W.; McCuskey, R.S. Ethanol binging enhances hepatic microvascular responses to acetaminophen in mice. Microcirculation 2004, 11, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Thummel, K.E.; Slattery, J.T.; Nelson, S.D. Mechanism by which ethanol diminishes the hepatotoxicity of acetaminophen. J. Pharmacol. Exp. Ther. 1988, 245, 129–136. [Google Scholar] [PubMed]
- Wong, L.T.; Whitehouse, L.W.; Solomonraj, G.; Paul, C.J. Effect of a concomitant single dose of ethanol on the hepatotoxicity and metabolism of acetaminophen in mice. Toxicology 1980, 17, 297–309. [Google Scholar] [CrossRef]
- Lee, S.M.; Cho, T.S.; Kim, D.J.; Cha, Y.N. Protective effect of ethanol against acetaminophen-induced hepatotoxicity in mice. Biochem. Pharmacol. 1999, 58, 1547–1555. [Google Scholar] [CrossRef]
- Seeff, L.B.; Cuccherini, B.A.; Zimmerman, H.J.; Adler, E.; Benjamin, S.B. Acetaminophen hepatotoxicity in alcoholics. A therapeutic misadventure. Ann. Intern. Med. 1986, 104, 399–404. [Google Scholar] [CrossRef]
- Maddrey, W.C. Hepatic effects of acetaminophen. Enhanced toxicity in alcoholics. J. Clin. Gastroenterol. 1987, 9, 180–185. [Google Scholar] [CrossRef]
- Zimmerman, H.J.; Maddrey, W.C. Acetaminophen (paracetamol) hepatotoxicity with regular intake of alcohol: Analysis of instances of therapeutic misadventure. Hepatology 1995, 22, 767–773. [Google Scholar] [CrossRef]
- Draganov, P.; Durrence, H.; Cox, C.; Reuben, A. Alcohol-acetaminophen syndrome. Even moderate social drinkers are at risk. Postgrad. Med. 2000, 107, 189–195. [Google Scholar] [CrossRef]
- Schmidt, L.E.; Dalhoff, K.; Poulsen, H.E. Acute versus chronic alcohol consumption in acetaminophen-induced hepatotoxicity. Hepatology 2002, 35, 876–882. [Google Scholar] [CrossRef]
- Carson, E.J.; Pruett, S.B. Development and characterization of a binge drinking model in mice for evaluation of the immunological effects of ethanol. Alcohol. Clin. Exp. Res. 1996, 20, 132–138. [Google Scholar] [CrossRef]
- Kirpich, I.; Ghare, S.; Zhang, J.; Gobejishvili, L.; Kharebava, G.; Barve, S.J.; Barker, D.; Moghe, A.; McClain, C.J.; Barve, S. Binge alcohol-induced microvesicular liver steatosis and injury are associated with down-regulation of hepatic Hdac 1, 7, 9, 10, 11 and up-regulation of Hdac 3. Alcohol. Clin. Exp. Res. 2012, 36, 1578–1586. [Google Scholar] [CrossRef] [PubMed]
- Reitman, S.; Frankel, S. A colorimetric method for the determination of serum glutamic oxalacetic and glutamic pyruvic transaminases. Am. J. Clin. Pathol. 1957, 28, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Jeong, T.B.; Kwon, D.; Son, S.W.; Kim, S.H.; Lee, Y.H.; Seo, M.S.; Kim, K.S.; Jung, Y.S. Weaning Mice and Adult Mice Exhibit Differential Carbon Tetrachloride-Induced Acute Hepatotoxicity. Antioxidants 2020, 9, 201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, Y.S.; Kim, S.J.; Kwon, D.Y.; Kim, Y.C. Comparison of the effects of buthioninesulfoximine and phorone on the metabolism of sulfur-containing amino acids in rat liver. Biochem. Biophys. Res. Commun. 2008, 368, 913–918. [Google Scholar] [CrossRef]
- Kwon, Y.H.; Stipanuk, M.H. Cysteine regulates expression of cysteine dioxygenase and gamma-glutamylcysteine synthetase in cultured rat hepatocytes. Am. J. Physiol. Endocrinol. Metab. 2001, 280, E804–E815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stipanuk, M.H. Metabolism of sulfur-containing amino acids. Annu. Rev. Nutr. 1986, 6, 179–209. [Google Scholar] [CrossRef] [PubMed]
- Dara, L.; Ji, C.; Kaplowitz, N. The contribution of endoplasmic reticulum stress to liver diseases. Hepatology 2011, 53, 1752–1763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, C.; Kaplowitz, N.; Lau, M.Y.; Kao, E.; Petrovic, L.M.; Lee, A.S. Liver-specific loss of glucose-regulated protein 78 perturbs the unfolded protein response and exacerbates a spectrum of liver diseases in mice. Hepatology 2011, 54, 229–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, C. Mechanisms of alcohol-induced endoplasmic reticulum stress and organ injuries. Biochem. Res. Int. 2012, 2012, 216450. [Google Scholar] [CrossRef] [Green Version]
- Kaplowitz, N.; Ji, C. Unfolding new mechanisms of alcoholic liver disease in the endoplasmic reticulum. J. Gastroenterol. Hepatol. 2006, 21 (Suppl. 3), S7–S9. [Google Scholar] [CrossRef]
- Ji, C.; Kaplowitz, N. Betaine decreases hyperhomocysteinemia, endoplasmic reticulum stress, and liver injury in alcohol-fed mice. Gastroenterology 2003, 124, 1488–1499. [Google Scholar] [CrossRef]
- Ji, C.; Kaplowitz, N. Hyperhomocysteinemia, endoplasmic reticulum stress, and alcoholic liver injury. World J. Gastroenterol. 2004, 10, 1699–1708. [Google Scholar] [CrossRef] [Green Version]
- Nagy, G.; Kardon, T.; Wunderlich, L.; Szarka, A.; Kiss, A.; Schaff, Z.; Banhegyi, G.; Mandl, J. Acetaminophen induces ER dependent signaling in mouse liver. Arch. Biochem. Biophys. 2007, 459, 273–279. [Google Scholar] [CrossRef]
- Kusama, H.; Kon, K.; Ikejima, K.; Arai, K.; Aoyama, T.; Uchiyama, A.; Yamashina, S.; Watanabe, S. Sodium 4-phenylbutyric acid prevents murine acetaminophen hepatotoxicity by minimizing endoplasmic reticulum stress. J. Gastroenterol. 2017, 52, 611–622. [Google Scholar] [CrossRef]
- Uzi, D.; Barda, L.; Scaiewicz, V.; Mills, M.; Mueller, T.; Gonzalez-Rodriguez, A.; Valverde, A.M.; Iwawaki, T.; Nahmias, Y.; Xavier, R.; et al. CHOP is a critical regulator of acetaminophen-induced hepatotoxicity. J. Hepatol. 2013, 59, 495–503. [Google Scholar] [CrossRef]
- Kera, Y.; Komura, S.; Ohbora, Y.; Kiriyama, T.; Inoue, K. Ethanol induced changes in lipid peroxidation and nonprotein sulfhydryl content. Different sensitivities in rat liver and kidney. Res. Commun. Chem. Pathol. Pharmacol. 1985, 47, 203–209. [Google Scholar]
- Videla, L.A.; Fernandez, V.; Ugarte, G.; Valenzuela, A. Effect of acute ethanol intoxication on the content of reduced glutathione of the liver in relation to its lipoperoxidative capacity in the rat. FEBS Lett. 1980, 111, 6–10. [Google Scholar] [CrossRef] [Green Version]
- Vina, J.; Estrela, J.M.; Guerri, C.; Romero, F.J. Effect of ethanol on glutathione concentration in isolated hepatocytes. Biochem. J. 1980, 188, 549–552. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Checa, J.C.; Ookhtens, M.; Kaplowitz, N. Effect of chronic ethanol feeding on rat hepatocytic glutathione. Compartmentation, efflux, and response to incubation with ethanol. J. Clin. Invest. 1987, 80, 57–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Speisky, H.; MacDonald, A.; Giles, G.; Orrego, H.; Israel, Y. Increased loss and decreased synthesis of hepatic glutathione after acute ethanol administration. Turnover studies. Biochem. J. 1985, 225, 565–572. [Google Scholar] [CrossRef] [Green Version]
- Choi, D.W.; Kim, S.Y.; Kim, S.K.; Kim, Y.C. Factors involved in hepatic glutathione depletion induced by acute ethanol administration. J. Toxicol. Environ. Health Part A 2000, 60, 459–469. [Google Scholar] [CrossRef]
- Kim, S.K.; Seo, J.M.; Jung, Y.S.; Kwak, H.E.; Kim, Y.C. Alterations in hepatic metabolism of sulfur-containing amino acids induced by ethanol in rats. Amino Acids 2003, 24, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Coles, B.; Wilson, I.; Wardman, P.; Hinson, J.A.; Nelson, S.D.; Ketterer, B. The spontaneous and enzymatic reaction of N-acetyl-p-benzoquinonimine with glutathione: A stopped-flow kinetic study. Arch. Biochem. Biophys. 1988, 264, 253–260. [Google Scholar] [CrossRef]
- Jaw, S.; Jeffery, E.H. Interaction of caffeine with acetaminophen. 1. Correlation of the effect of caffeine on acetaminophen hepatotoxicity and acetaminophen bioactivation following treatment of mice with various cytochrome P450 inducing agents. Biochem. Pharmacol. 1993, 46, 493–501. [Google Scholar] [CrossRef]
- Rutkowski, D.T.; Kaufman, R.J. A trip to the ER: Coping with stress. Trends Cell Biol. 2004, 14, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Hwang, C.; Sinskey, A.J.; Lodish, H.F. Oxidized redox state of glutathione in the endoplasmic reticulum. Science 1992, 257, 1496–1502. [Google Scholar] [CrossRef]
- Cribb, A.E.; Peyrou, M.; Muruganandan, S.; Schneider, L. The endoplasmic reticulum in xenobiotic toxicity. Drug Metab. Rev. 2005, 37, 405–442. [Google Scholar] [CrossRef]
- Loeper, J.; Descatoire, V.; Maurice, M.; Beaune, P.; Feldmann, G.; Larrey, D.; Pessayre, D. Presence of functional cytochrome P-450 on isolated rat hepatocyte plasma membrane. Hepatology 1990, 11, 850–858. [Google Scholar] [CrossRef]
- Hur, K.Y.; So, J.S.; Ruda, V.; Frank-Kamenetsky, M.; Fitzgerald, K.; Koteliansky, V.; Iwawaki, T.; Glimcher, L.H.; Lee, A.H. IRE1alpha activation protects mice against acetaminophen-induced hepatotoxicity. J. Exp. Med. 2012, 209, 307–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hotamisligil, G.S. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell 2010, 140, 900–917. [Google Scholar] [CrossRef] [Green Version]
- Malhi, H.; Kaufman, R.J. Endoplasmic reticulum stress in liver disease. J. Hepatol. 2011, 54, 795–809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hollien, J.; Weissman, J.S. Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science 2006, 313, 104–107. [Google Scholar] [CrossRef] [Green Version]
- Matsuzawa, A.; Nishitoh, H.; Tobiume, K.; Takeda, K.; Ichijo, H. Physiological roles of ASK1-mediated signal transduction in oxidative stress- and endoplasmic reticulum stress-induced apoptosis: Advanced findings from ASK1 knockout mice. Antioxid. Redox Signal. 2002, 4, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, H.; Maeda, S.; Hikiba, Y.; Ohmae, T.; Shibata, W.; Yanai, A.; Sakamoto, K.; Ogura, K.; Noguchi, T.; Karin, M.; et al. Deletion of apoptosis signal-regulating kinase 1 attenuates acetaminophen-induced liver injury by inhibiting c-Jun N-terminal kinase activation. Gastroenterology 2008, 135, 1311–1321. [Google Scholar] [CrossRef]
- Kato, H.; Nakajima, S.; Saito, Y.; Takahashi, S.; Katoh, R.; Kitamura, M. mTORC1 serves ER stress-triggered apoptosis via selective activation of the IRE1-JNK pathway. Cell Death Differ. 2012, 19, 310–320. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, S.H.; Choi, H.J.; Seo, H.; Kwon, D.; Yun, J.; Jung, Y.-S. Downregulation of Glutathione-Mediated Detoxification Capacity by Binge Drinking Aggravates Acetaminophen-Induced Liver Injury through IRE1α ER Stress Signaling. Antioxidants 2021, 10, 1949. https://doi.org/10.3390/antiox10121949
Kim SH, Choi HJ, Seo H, Kwon D, Yun J, Jung Y-S. Downregulation of Glutathione-Mediated Detoxification Capacity by Binge Drinking Aggravates Acetaminophen-Induced Liver Injury through IRE1α ER Stress Signaling. Antioxidants. 2021; 10(12):1949. https://doi.org/10.3390/antiox10121949
Chicago/Turabian StyleKim, Sou Hyun, Hun Ji Choi, Hyeji Seo, Doyoung Kwon, Jaesuk Yun, and Young-Suk Jung. 2021. "Downregulation of Glutathione-Mediated Detoxification Capacity by Binge Drinking Aggravates Acetaminophen-Induced Liver Injury through IRE1α ER Stress Signaling" Antioxidants 10, no. 12: 1949. https://doi.org/10.3390/antiox10121949