
Oxidative Stress and the Role of NADPH Oxidase in Glaucoma


Abstract
:1. Introduction
2. Pathogenesis of Glaucoma
2.1. Anatomical Structure of the Optic Nerve Head and Retina
2.2. Mechanical Theory of Glaucoma
2.3. Vascular Theory of Glaucoma
2.4. Immunological Theory of Glaucoma
3. Oxidative Stress in Glaucoma
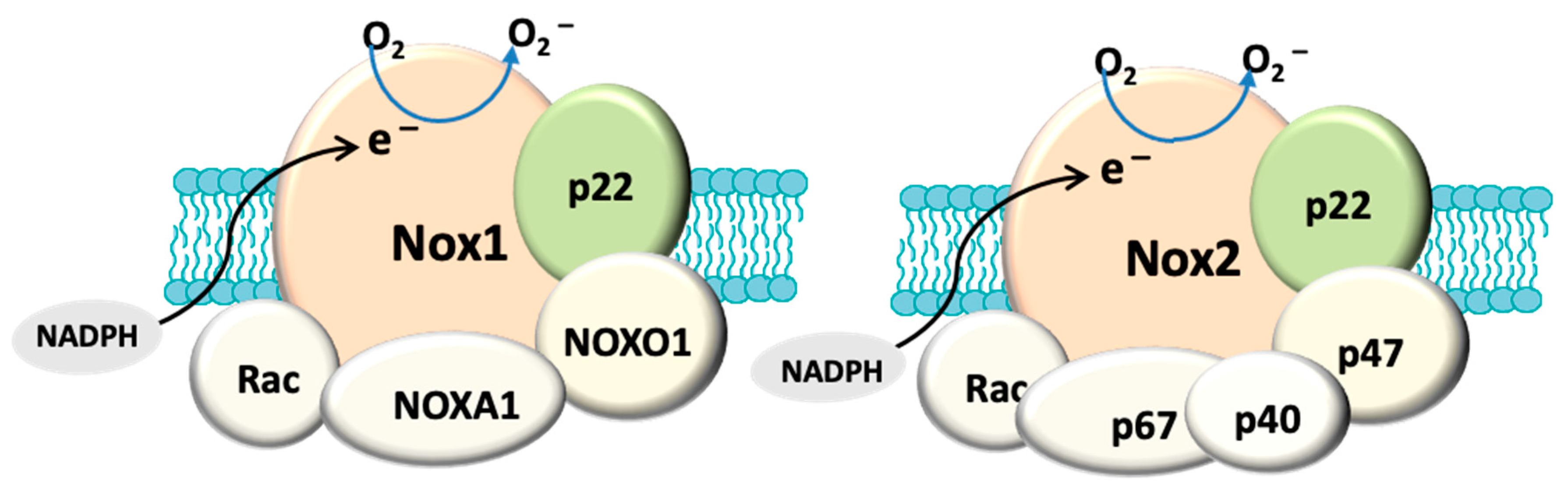
4. NADPH Oxidase and Oxidative Stress
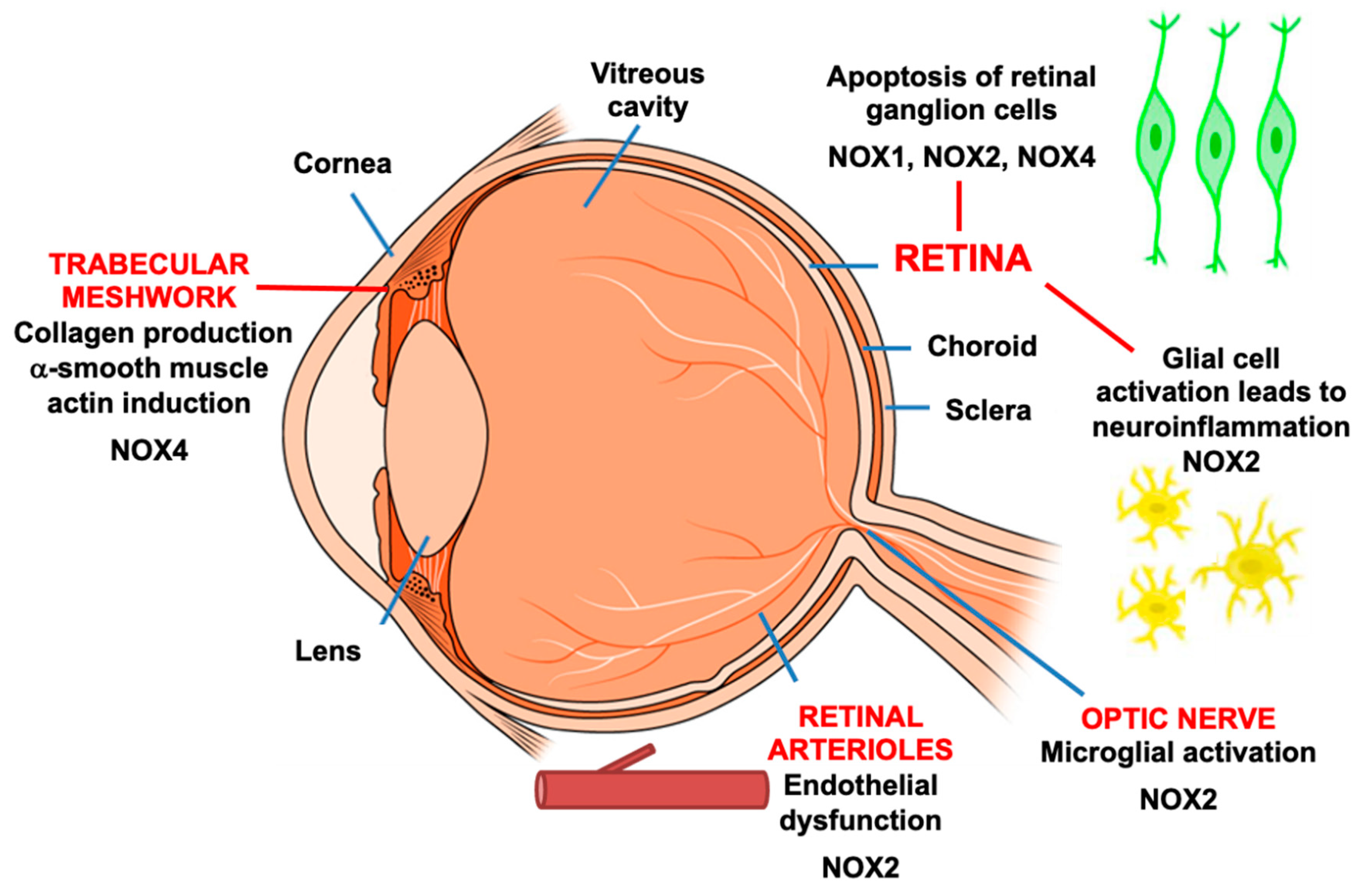
5. Expression of NADPH Oxidase and Glaucoma Pathology
5.1. Retinal Ischemia and Reperfusion
5.2. Ocular Hypertension
5.3. Optic Nerve Injury
6. Targeting Oxidative Stress for Glaucoma
6.1. Antioxidant Therapy
6.2. Pharmacological Inhibition of NADPH Oxidase
7. Future Horizons
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AGEs | Advanced glycation end products |
| CNS | central nervous system |
| EGCG | epigallocatechin-gallate |
| GLO | Glyoxalase |
| HSP | heat shock protein |
| NO | nitric oxide |
| NOX | NADPH oxidase |
| O2− | superoxide |
| ONH | optic nerve head |
| ONOO− | peroxynitrite |
| PERG | pattern electroretinogram |
| RGC | retinal ganglion cell |
| TGFβ | transforming growth factor β |
| TNFα | tumour necrosis factor α |
References
- Weinreb, R.N.; Khaw, P.T. Primary open-angle glaucoma. Lancet 2004, 363, 1711–1720. [Google Scholar] [CrossRef]
- Ju, W.K.; Liu, Q.; Kim, K.Y.; Crowston, J.G.; Lindsey, J.D.; Agarwal, N.; Ellisman, M.H.; Perkins, G.A.; Weinreb, R.N. Elevated hydrostatic pressure triggers mitochondrial fission and decreases cellular ATP in differentiated RGC-5 cells. Investig. Opthalmology Vis. Sci. 2007, 48, 2145–2151. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, M.R.; Agapova, O.A.; Yang, P.; Salvador-Silva, M.; Ricard, C.S.; Aoi, S. Differential gene expression in astrocytes from human normal and glaucomatous optic nerve head analyzed by cDNA microarray. Glia 2002, 38, 45–64. [Google Scholar] [CrossRef] [PubMed]
- Agapova, O.A.; Kaufman, P.L.; Hernandez, M.R. Androgen receptor and NFkB expression in human normal and glaucomatous optic nerve head astrocytes in vitro and in experimental glaucoma. Exp. Eye Res. 2006, 82, 1053–1059. [Google Scholar] [CrossRef]
- Katano, H.; Ishihara, M.; Shiraishi, Y.; Kawai, Y. Effects of aging on the electroretinogram during ischemia-reperfusion in rats. Jpn. J. Physiol. 2001, 51, 89–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leung, C.K.; Lindsey, J.D.; Crowston, J.G.; Lijia, C.; Chiang, S.; Weinreb, R.N. Longitudinal profile of retinal ganglion cell damage after optic nerve crush with blue-light confocal scanning laser ophthalmoscopy. Investig. Opthalmology Vis. Sci. 2008, 49, 4898–4902. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, S.; Yashar, B.M.; Hiriyanna, S.; Swaroop, A. Microarray analysis of gene expression in the aging human retina. Investig. Ophthalmol. Vis. Sci. 2002, 43, 2554–2560. [Google Scholar]
- Chrysostomou, V.; Rezania, F.; Trounce, I.A.; Crowston, J.G. Oxidative stress and mitochondrial dysfunction in glaucoma. Curr. Opin. Pharmacol. 2013, 13, 12–15. [Google Scholar] [CrossRef] [PubMed]
- Olver, J. Angioarchitecture of the Human Optic Nerve; Innovation-News-Communication: Rome, Italy, 1998. [Google Scholar]
- Hayreh, S.S. Blood supply of the optic nerve head. Ophthalmologica 1996, 210, 285–295. [Google Scholar] [CrossRef]
- Hayreh, S.S. Blood flow in the optic nerve head and factors that may influence it. Prog. Retin. Eye Res. 2001, 20, 595–624. [Google Scholar] [CrossRef]
- Anderson, D.R. Ultrastructure of human and monkey lamina cribrosa and optic nerve head. Arch. Ophthalmol. 1969, 82, 800–814. [Google Scholar] [CrossRef]
- Hernandez, M.R. The optic nerve head in glaucoma: Role of astrocytes in tissue remodeling. Prog. Retin. Eye Res. 2000, 19, 297–321. [Google Scholar] [CrossRef]
- Hofman, P.; Hoyng, P.; vanderWerf, F.; Vrensen, G.F.; Schlingemann, R.O. Lack of blood-brain barrier properties in microvessels of the prelaminar optic nerve head. Investig. Ophthalmol. Vis. Sci. 2001, 42, 895–901. [Google Scholar]
- Pournaras, C.J.; Riva, C.E. Studies of the hemodynamics of the optic head nerve using laser Doppler flowmetry. J. Français Ophtalmol. 2001, 24, 199–205. [Google Scholar]
- Flammer, J.; Orgul, S. Optic nerve blood-flow abnormalities in glaucoma. Prog. Retin. Eye Res. 1998, 17, 267–289. [Google Scholar] [CrossRef]
- Montezano, A.C.; Nguyen Dinh Cat, A.; Rios, F.J.; Touyz, R.M. Angiotensin II and vascular injury. Curr. Hypertens. Rep. 2014, 16, 431. [Google Scholar] [CrossRef]
- Wilkinson-Berka, J.L.; Tan, G.; Jaworski, K.; Harbig, J.; Miller, A.G. Identification of a retinal aldosterone system and the protective effects of mineralocorticoid receptor antagonism on retinal vascular pathology. Circ. Res. 2009, 104, 124–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno, M.C.; Campanelli, J.; Sande, P.; Sanez, D.A.; Keller Sarmiento, M.I.; Rosenstein, R.E. Retinal oxidative stress induced by high intraocular pressure. Free Radic. Biol. Med. 2004, 37, 803–812. [Google Scholar] [CrossRef] [PubMed]
- Quigley, H.A.; Addicks, E.M. Regional differences in the structure of the lamina cribrosa and their relation to glaucomatous optic nerve damage. Arch. Ophthalmol. 1981, 99, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Burgoyne, C.F.; Downs, J.C.; Bellezza, A.J.; Suh, J.K.; Hart, R.T. The optic nerve head as a biomechanical structure: A new paradigm for understanding the role of IOP-related stress and strain in the pathophysiology of glaucomatous optic nerve head damage. Prog. Retin. Eye Res. 2005, 24, 39–73. [Google Scholar] [CrossRef] [PubMed]
- Burgoyne, C.F.; Downs, J.C.; Bellezza, A.J.; Hart, R.T. Three-dimensional reconstruction of normal and early glaucoma monkey optic nerve head connective tissues. Investig. Ophthalmol. Vis. Sci. 2004, 45, 4388–4399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albon, J.; Purslow, P.P.; Karwatowski, W.S.; Easty, D.L. Age related compliance of the lamina cribrosa in human eyes. Br. J. Ophthalmol. 2000, 84, 318–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flammer, J. The vascular concept of glaucoma. Surv. Ophthalmol. 1994, 38, S3–S6. [Google Scholar] [CrossRef]
- Flammer, J.; Orgul, S.; Costa, V.P.; Orzalesi, N.; Krieglstein, G.K.; Serra, L.M.; Renard, J.P.; Stefansson, E. The impact of ocular blood flow in glaucoma. Prog. Retin. Eye Res. 2002, 21, 359–393. [Google Scholar] [CrossRef]
- Drance, S.; Anderson, D.R.; Schulzer, M.; Collaborative Normal-Tension Glaucoma Study Group. Risk factors for progression of visual field abnormalities in normal-tension glaucoma. Am. J. Ophthalmol. 2001, 131, 699–708. [Google Scholar] [CrossRef]
- Healey, P.R.; Mitchell, P.; Smith, W.; Wang, J.J. Optic disc hemorrhages in a population with and without signs of glaucoma. Ophthalmology 1998, 105, 216–223. [Google Scholar] [CrossRef]
- Gasser, P.; Flammer, J. Blood-cell velocity in the nailfold capillaries of patients with normal-tension and high-tension glaucoma. Am. J. Ophthalmol. 1991, 111, 585–588. [Google Scholar] [CrossRef]
- Nicolela, M.T.; Walman, B.E.; Buckley, A.R.; Drance, S.M. Ocular hypertension and primary open-angle glaucoma: A comparative study of their retrobulbar blood flow velocity. J. Glaucoma 1996, 5, 308–310. [Google Scholar] [CrossRef]
- Siegner, S.W.; Netland, P.A. Optic disc hemorrhages and progression of glaucoma. Ophthalmology 1996, 103, 1014–1024. [Google Scholar] [CrossRef]
- Klein, R.; Klein, B.E.; Moss, S.E.; Meuer, S.M. The epidemiology of retinal vein occlusion: The Beaver Dam Eye Study. Trans. Am. Ophthalmol. Soc. 2000, 98, 133–141. [Google Scholar]
- Sonnsjo, B.; Krakau, C.E. Arguments for a vascular glaucoma etiology. Acta Ophthalmol. 1993, 71, 433–444. [Google Scholar] [CrossRef] [PubMed]
- Rankin, S.J.; Drance, S.M. Peripapillary focal retinal arteriolar narrowing in open angle glaucoma. J. Glaucoma 1996, 5, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Cho, J.; Kim, M.H.; Guallar, E. The association of blood pressure and primary open-angle glaucoma: A meta-analysis. Am. J. Ophthalmol. 2014, 158, 615–627.e619. [Google Scholar] [CrossRef] [PubMed]
- Levine, R.M.; Yang, A.; Brahma, V.; Martone, J.F. Management of Blood Pressure in Patients with Glaucoma. Curr. Cardiol. Rep. 2017, 19, 109. [Google Scholar] [CrossRef] [PubMed]
- De Moraes, C.G.; Cioffi, G.A.; Weinreb, R.N.; Liebmann, J.M. New Recommendations for the Treatment of Systemic Hypertension and their Potential Implications for Glaucoma Management. J. Glaucoma 2018, 27, 567–571. [Google Scholar] [CrossRef] [PubMed]
- Schmidl, D.; Garhofer, G.; Schmetterer, L. The complex interaction between ocular perfusion pressure and ocular blood flow - relevance for glaucoma. Exp. Eye Res. 2011, 93, 141–155. [Google Scholar] [CrossRef] [PubMed]
- Skrzypecki, J.; Ufnal, M.; Szaflik, J.P.; Filipiak, K.J. Blood pressure and glaucoma: At the crossroads between cardiology and ophthalmology. Cardiol. J. 2019, 26, 8–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, R.W.; Drance, S.M. Chronic open-angle glaucoma and ocular hypertension. An epidemiological study. Br. J. Ophthalmol. 1975, 59, 211–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drance, S.M.; Sweeney, V.P.; Morgan, R.W.; Feldman, F. Studies of factors involved in the production of low tension glaucoma. Arch. Ophthalmol 1973, 89, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Song, B.J.; Aiello, L.P.; Pasquale, L.R. Presence and Risk Factors for Glaucoma in Patients with Diabetes. Curr. Diab. Rep. 2016, 16, 124. [Google Scholar] [CrossRef]
- Gerber, A.L.; Harris, A.; Siesky, B.; Lee, E.; Schaab, T.J.; Huck, A.; Amireskandari, A. Vascular Dysfunction in Diabetes and Glaucoma: A Complex Relationship Reviewed. J. Glaucoma 2015, 24, 474–479. [Google Scholar] [CrossRef] [PubMed]
- Trope, G.E.; Salinas, R.G.; Glynn, M. Blood viscosity in primary open-angle glaucoma. Can. J. Ophthalmol. 1987, 22, 202–204. [Google Scholar]
- Gramer, G.; Weber, B.H.; Gramer, E. Migraine and Vasospasm in Glaucoma: Age-Related Evaluation of 2027 Patients With Glaucoma or Ocular Hypertension. Investig. Opthalmol. Vis. Sci. 2015, 56, 7999–8007. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.Y.; Su, C.C.; Wang, T.H.; Tsai, I.J. Migraine and increased risk of developing open angle glaucoma: A population-based cohort study. BMC Ophthalmol 2019, 19, 50. [Google Scholar] [CrossRef] [PubMed]
- Konieczka, K.; Choi, H.J.; Koch, S.; Fankhauser, F.; Schoetzau, A.; Kim, D.M. Relationship between normal tension glaucoma and Flammer syndrome. EPMA J. 2017, 8, 111–117. [Google Scholar] [CrossRef]
- Nicolela, M.T.; Ferrier, S.N.; Morrison, C.A.; Archibald, M.L.; LeVatte, T.L.; Wallace, K.; Chauhan, B.C.; LeBlanc, R.P. Effects of cold-induced vasospasm in glaucoma: The role of endothelin-1. Investig. Opthalmol. Vis. Sci. 2003, 44, 2565–2572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Murugesan, P.; Huang, K.; Cai, H. NADPH oxidases and oxidase crosstalk in cardiovascular diseases: Novel therapeutic targets. Nat. Rev. Cardiol. 2020, 17, 170–194. [Google Scholar] [CrossRef] [PubMed]
- Henry, E.; Newby, D.E.; Webb, D.J.; O’Brien, C. Peripheral endothelial dysfunction in normal pressure glaucoma. Investig. Ophthalmol. Vis. Sci. 1999, 40, 1710–1714. [Google Scholar]
- Sugiyama, T.; Moriya, S.; Oku, H.; Azuma, I. Association of endothelin-1 with normal tension glaucoma: Clinical and fundamental studies. Surv. Ophthalmol. 1995, 39 (Suppl. S1), S49–S56. [Google Scholar] [CrossRef]
- Kaiser, H.J.; Flammer, J.; Wenk, M.; Luscher, T. Endothelin-1 plasma levels in normal-tension glaucoma: Abnormal response to postural changes. Graefes Arch. Clin. Exp. Ophthalmol. 1995, 233, 484–488. [Google Scholar] [CrossRef] [PubMed]
- Noske, W.; Hensen, J.; Wiederholt, M. Endothelin-like immunoreactivity in aqueous humor of patients with primary open-angle glaucoma and cataract. Graefes Arch. Clin. Exp. Ophthalmol. 1997, 235, 551–552. [Google Scholar] [CrossRef]
- Haufschild, T.; Shaw, S.G.; Kesselring, J.; Flammer, J. Increased endothelin-1 plasma levels in patients with multiple sclerosis. J. Neuroophthalmol. 2001, 21, 37–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 7–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wax, M.B.; Tezel, G.; Saito, I.; Gupta, R.S.; Harley, J.B.; Li, Z.; Romano, C. Anti-Ro/SS-A positivity and heat shock protein antibodies in patients with normal-pressure glaucoma. Am. J. Ophthalmol. 1998, 125, 145–157. [Google Scholar] [CrossRef]
- Tezel, G.; Wax, M.B. Glaucoma. Chem. Immunol. Allergy 2007, 92, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Wax, M.B.; Tezel, G. Immunoregulation of retinal ganglion cell fate in glaucoma. Exp. Eye Res. 2009, 88, 825–830. [Google Scholar] [CrossRef] [PubMed]
- Tezel, G. Fourth, ARVO/Pfizer Ophthalmics Research Institute Conference Working Group, and the immune system in glaucoma. Investig. Ophthalmol. Vis. Sci. 2009, 50, 1001–1012. [Google Scholar] [CrossRef] [PubMed]
- Neufeld, A.H. Microglia in the optic nerve head and the region of parapapillary chorioretinal atrophy in glaucoma. Arch. Ophthalmol. 1999, 117, 1050–1056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ju, K.R.; Kim, H.S.; Kim, J.H.; Lee, N.Y.; Park, C.K. Retinal glial cell responses and Fas/FasL activation in rats with chronic ocular hypertension. Brain Res. 2006, 1122, 209–221. [Google Scholar] [CrossRef]
- Bosco, A.; Steele, M.R.; Vetter, M.L. Early microglia activation in a mouse model of chronic glaucoma. J. Comp. Neurol. 2011, 519, 599–620. [Google Scholar] [CrossRef] [Green Version]
- Howell, G.R.; Soto, I.; Zhu, X.; Ryan, M.; Macalinao, D.G.; Sousa, G.L.; Caddle, L.B.; MacNicoll, K.H.; Barbay, J.M.; Porciatti, V.; et al. Radiation treatment inhibits monocyte entry into the optic nerve head and prevents neuronal damage in a mouse model of glaucoma. J. Clin. Investig. 2012, 122, 1246–1261. [Google Scholar] [CrossRef] [Green Version]
- Butterfield, D.A.; Howard, B.J.; Yatin, S.; Allen, K.L.; Carney, J.M. Free radical oxidation of brain proteins in accelerated senescence and its modulation by N-tert-butyl-alpha-phenylnitrone. Proc. Natl. Acad. Sci. USA 1997, 94, 674–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cakatay, U.; Telci, A.; Kayali, R.; Tekeli, F.; Akcay, T.; Sivas, A. Relation of oxidative protein damage and nitrotyrosine levels in the aging rat brain. Exp. Gerontol. 2001, 36, 221–229. [Google Scholar] [CrossRef]
- Papaioannou, N.; Tooten, P.C.; van Ederen, A.M.; Bohl, J.R.; Rofina, J.; Tsangaris, T.; Gruys, E. Immunohistochemical investigation of the brain of aged dogs. I. Detection of neurofibrillary tangles and of 4-hydroxynonenal protein, an oxidative damage product, in senile plaques. Amyloid 2001, 8, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Sohal, R.S.; Agarwal, S.; Candas, M.; Forster, M.J.; Lal, H. Effect of age and caloric restriction on DNA oxidative damage in different tissues of C57BL/6 mice. Mech. Ageing Dev. 1994, 76, 215–224. [Google Scholar] [CrossRef]
- Jarrett, S.G.; Lin, H.; Godley, B.F.; Boulton, M.E. Mitochondrial DNA damage and its potential role in retinal degeneration. Prog. Retin. Eye Res. 2008, 27, 596–607. [Google Scholar] [CrossRef] [PubMed]
- Tezel, G. Oxidative stress in glaucomatous neurodegeneration: Mechanisms and consequences. Prog. Retin. Eye Res. 2006, 25, 490–513. [Google Scholar] [CrossRef] [Green Version]
- Griendling, K.K.; Sorescu, D.; Ushio-Fukai, M. NAD(P)H oxidase: Role in cardiovascular biology and disease. Circ. Res. 2000, 86, 494–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spiekermann, S.; Landmesser, U.; Dikalov, S.; Bredt, M.; Gamez, G.; Tatge, H.; Reepschlager, N.; Hornig, B.; Drexler, H.; Harrison, D.G. Electron spin resonance characterization of vascular xanthine and NAD(P)H oxidase activity in patients with coronary artery disease: Relation to endothelium-dependent vasodilation. Circulation 2003, 107, 1383–1389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landmesser, U.; Dikalov, S.; Price, S.R.; McCann, L.; Fukai, T.; Holland, S.M.; Mitch, W.E.; Harrison, D.G. Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J. Clin. Investig. 2003, 111, 1201–1209. [Google Scholar] [CrossRef]
- Fridovich, I. Mitochondria: Are they the seat of senescence? Aging Cell 2004, 3, 13–16. [Google Scholar] [CrossRef] [PubMed]
- Chidlow, G.; Wood, J.P.M.; Casson, R.J. Investigations into Hypoxia and Oxidative Stress at the Optic Nerve Head in a Rat Model of Glaucoma. Front. Neurosci. 2017, 11, 478. [Google Scholar] [CrossRef]
- Casson, R.J.; Chidlow, G.; Wood, J.P.M. Estimate of the adenosine triphosphate requirement of human retinal ganglion cells. Clin. Exp. Ophthalmol. 2019, 47, 683–684. [Google Scholar] [CrossRef]
- Lee, S.; Van Bergen, N.J.; Kong, G.Y.; Chrysostomou, V.; Waugh, H.S.; O’Neill, E.C.; Crowston, J.G.; Trounce, I.A. Mitochondrial dysfunction in glaucoma and emerging bioenergetic therapies. Exp. Eye Res. 2011, 93, 204–212. [Google Scholar] [CrossRef]
- Harris, J.J.; Attwell, D. The energetics of CNS white matter. J. Neurosci. 2012, 32, 356–371. [Google Scholar] [CrossRef]
- Minckler, D.S.; McLean, I.W.; Tso, M.O. Distribution of axonal and glial elements in the rhesus optic nerve head studied by electron microscopy. Am. J. Ophthalmol. 1976, 82, 179–187. [Google Scholar] [CrossRef]
- Li, Z.; Okamoto, K.; Hayashi, Y.; Sheng, M. The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell 2004, 119, 873–887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruan, Y.; Jiang, S.; Musayeva, A.; Gericke, A. Oxidative Stress and Vascular Dysfunction in the Retina: Therapeutic Strategies. Antioxidants 2020, 9, 761. [Google Scholar] [CrossRef] [PubMed]
- Dikalov, S.I.; Li, W.; Doughan, A.K.; Blanco, R.R.; Zafari, A.M. Mitochondrial reactive oxygen species and calcium uptake regulate activation of phagocytic NADPH oxidase. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2012, 302, R1134–R1142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, E.C.; Liu, G.S.; Dusting, G.J. Redox mechanisms in pathological angiogenesis in the retina: Roles for NADPH oxidase. Curr. Pharm. Des. 2015, 21, 5988–5998. [Google Scholar] [CrossRef]
- Chan, E.C.; Jiang, F.; Peshavariya, H.M.; Dusting, G.J. Regulation of cell proliferation by NADPH oxidase-mediated signaling: Potential roles in tissue repair, regenerative medicine and tissue engineering. Pharmacol. Ther. 2009, 122, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Berthoud, V.M.; Beyer, E.C. Oxidative stress, lens gap junctions, and cataracts. Antioxid. Redox Signal. 2009, 11, 339–353. [Google Scholar] [CrossRef] [Green Version]
- Magnani, F.; Mattevi, A. Structure and mechanisms of ROS generation by NADPH oxidases. Curr. Opin. Struct. Biol. 2019, 59, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Nisimoto, Y.; Diebold, B.A.; Cosentino-Gomes, D.; Lambeth, J.D. Nox4: A hydrogen peroxide-generating oxygen sensor. Biochemistry 2014, 53, 5111–5120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takac, I.; Schroder, K.; Zhang, L.; Lardy, B.; Anilkumar, N.; Lambeth, J.D.; Shah, A.M.; Morel, F.; Brandes, R.P. The E-loop is involved in hydrogen peroxide formation by the NADPH oxidase Nox4. J. Biol. Chem. 2011, 286, 13304–13313. [Google Scholar] [CrossRef] [Green Version]
- Dvoriantchikova, G.; Grant, J.; Santos, A.R.; Hernandez, E.; Ivanov, D. Neuronal NAD(P)H oxidases contribute to ROS production and mediate RGC death after ischemia. Investig. Ophthalmol. Vis. Sci. 2012, 53, 2823–2830. [Google Scholar] [CrossRef] [Green Version]
- Geng, L.; Fan, L.M.; Liu, F.; Smith, C.; Li, J. Nox2 dependent redox-regulation of microglial response to amyloid-beta stimulation and microgliosis in aging. Sci. Rep. 2020, 10, 1582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soto, I.; Howell, G.R. The complex role of neuroinflammation in glaucoma. Cold Spring Harb. Perspect. Med. 2014, 4. [Google Scholar] [CrossRef]
- Sakai, M.; Sakai, H.; Nakamura, Y.; Fukuchi, T.; Sawaguchi, S. Immunolocalization of heat shock proteins in the retina of normal monkey eyes and monkey eyes with laser-induced glaucoma. Jpn. J. Ophthalmol. 2003, 47, 42–52. [Google Scholar] [CrossRef]
- Chen, F.; Pandey, D.; Chadli, A.; Catravas, J.D.; Chen, T.; Fulton, D.J. Hsp90 regulates NADPH oxidase activity and is necessary for superoxide but not hydrogen peroxide production. Antioxid. Redox Signal. 2011, 14, 2107–2119. [Google Scholar] [CrossRef] [Green Version]
- Aragones, G.; Rowan, S.; S, G.F.; Yang, W.; Weinberg, J.; Taylor, A.; Bejarano, E. Glyoxalase System as a Therapeutic Target against Diabetic Retinopathy. Antioxidants 2020, 9, 62. [Google Scholar] [CrossRef] [PubMed]
- Tezel, G.; Luo, C.; Yang, X. Accelerated aging in glaucoma: Immunohistochemical assessment of advanced glycation end products in the human retina and optic nerve head. Investig. Ophthalmol. Vis. Sci. 2007, 48, 1201–1211. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.G.; Tan, G.; Binger, K.J.; Pickering, R.J.; Thomas, M.C.; Nagaraj, R.H.; Cooper, M.E.; Wilkinson-Berka, J.L. Candesartan attenuates diabetic retinal vascular pathology by restoring glyoxalase-I function. Diabetes 2010, 59, 3208–3215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cepas, V.; Collino, M.; Mayo, J.C.; Sainz, R.M. Redox Signaling and Advanced Glycation Endproducts (AGEs) in Diet-Related Diseases. Antioxidants 2020, 9, 142. [Google Scholar] [CrossRef] [Green Version]
- Evangelho, K.; Mastronardi, C.A.; de-la-Torre, A. Experimental Models of Glaucoma: A Powerful Translational Tool for the Future Development of New Therapies for Glaucoma in Humans-A Review of the Literature. Medicina 2019, 55, 280. [Google Scholar] [CrossRef] [Green Version]
- Ramirez, A.I.; de Hoz, R.; Salobrar-Garcia, E.; Salazar, J.J.; Rojas, B.; Ajoy, D.; Lopez-Cuenca, I.; Rojas, P.; Trivino, A.; Ramirez, J.M. The Role of Microglia in Retinal Neurodegeneration: Alzheimer’s Disease, Parkinson, and Glaucoma. Front. Aging Neurosci. 2017, 9, 214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yokota, H.; Narayanan, S.P.; Zhang, W.; Liu, H.; Rojas, M.; Xu, Z.; Lemtalsi, T.; Nagaoka, T.; Yoshida, A.; Brooks, S.E.; et al. Neuroprotection from retinal ischemia/reperfusion injury by NOX2 NADPH oxidase deletion. Investig. Ophthalmol. Vis. Sci. 2011, 52, 8123–8131. [Google Scholar] [CrossRef] [PubMed]
- Sapienza, A.; Raveu, A.L.; Reboussin, E.; Roubeix, C.; Boucher, C.; Degardin, J.; Godefroy, D.; Rostene, W.; Reaux-Le Goazigo, A.; Baudouin, C.; et al. Bilateral neuroinflammatory processes in visual pathways induced by unilateral ocular hypertension in the rat. J. Neuroinflamm. 2016, 13, 44. [Google Scholar] [CrossRef]
- Kumar, A.; Barrett, J.P.; Alvarez-Croda, D.M.; Stoica, B.A.; Faden, A.I.; Loane, D.J. NOX2 drives M1-like microglial/macrophage activation and neurodegeneration following experimental traumatic brain injury. Brain Behav. Immun. 2016, 58, 291–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cherry, J.D.; Olschowka, J.A.; O’Banion, M.K. Neuroinflammation and M2 microglia: The good, the bad, and the inflamed. J. Neuroinflamm. 2014, 11, 98. [Google Scholar] [CrossRef] [Green Version]
- van Buul, J.D.; Hordijk, P.L. Signaling in leukocyte transendothelial migration. Arter. Thromb. Vasc. Biol. 2004, 24, 824–833. [Google Scholar] [CrossRef]
- Rashid, K.; Akhtar-Schaefer, I.; Langmann, T. Microglia in Retinal Degeneration. Front. Immunol. 2019, 10, 1975. [Google Scholar] [CrossRef] [Green Version]
- Gericke, A.; Mann, C.; Zadeh, J.K.; Musayeva, A.; Wolff, I.; Wang, M.; Pfeiffer, N.; Daiber, A.; Li, H.; Xia, N.; et al. Elevated Intraocular Pressure Causes Abnormal Reactivity of Mouse Retinal Arterioles. Oxid. Med. Cell. Longev. 2019, 2019, 9736047. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Pagano, P.J. Microvascular NADPH oxidase in health and disease. Free Radic. Biol. Med. 2017, 109, 33–47. [Google Scholar] [CrossRef] [PubMed]
- Vital, S.A.; Terao, S.; Nagai, M.; Granger, D.N. Mechanisms underlying the cerebral microvascular responses to angiotensin II-induced hypertension. Microcirculation 2010, 17, 641–649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Z.; Zhang, S.; Lee, C.; Kumar, A.; Arjunan, P.; Li, Y.; Zhang, F.; Li, X. An optic nerve crush injury murine model to study retinal ganglion cell survival. J. Vis. Exp. 2011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, K.; Maruyama, K.; Himori, N.; Omodaka, K.; Yokoyama, Y.; Shiga, Y.; Morin, R.; Nakazawa, T. The novel Rho kinase (ROCK) inhibitor K-115: A new candidate drug for neuroprotective treatment in glaucoma. Investig. Ophthalmol. Vis. Sci. 2014, 55, 7126–7136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehrabian, Z.; Guo, Y.; Weinreich, D.; Bernstein, S.L. Oligodendrocyte death, neuroinflammation, and the effects of minocycline in a rodent model of nonarteritic anterior ischemic optic neuropathy (rNAION). Mol. Vis. 2017, 23, 963–976. [Google Scholar]
- Gu, X.J.; Liu, X.; Chen, Y.Y.; Zhao, Y.; Xu, M.; Han, X.J.; Liu, Q.P.; Yi, J.L.; Li, J.M. Involvement of NADPH oxidases in alkali burn-induced corneal injury. Int. J. Mol. Med. 2016, 38, 75–82. [Google Scholar] [CrossRef] [Green Version]
- Singh, L.; Saini, N.; Pushker, N.; Sen, S.; Sharma, A.; Kashyap, S. Prognostic significance of NADPH oxidase-4 as an indicator of reactive oxygen species stress in human retinoblastoma. Int. J. Clin. Oncol. 2016, 21, 651–657. [Google Scholar] [CrossRef]
- Falsini, B.; Marangoni, D.; Salgarello, T.; Stifano, G.; Montrone, L.; Di Landro, S.; Guccione, L.; Balestrazzi, E.; Colotto, A. Effect of epigallocatechin-gallate on inner retinal function in ocular hypertension and glaucoma: A short-term study by pattern electroretinogram. Graefes Arch. Clin. Exp. Ophthalmol. 2009, 247, 1223–1233. [Google Scholar] [CrossRef]
- Harris, A.; Gross, J.; Moore, N.; Do, T.; Huang, A.; Gama, W.; Siesky, B. The effects of antioxidants on ocular blood flow in patients with glaucoma. Acta Ophthalmol. 2018, 96, e237–e241. [Google Scholar] [CrossRef]
- Garcia-Medina, J.J.; Garcia-Medina, M.; Garrido-Fernandez, P.; Galvan-Espinosa, J.; Garcia-Maturana, C.; Zanon-Moreno, V.; Pinazo-Duran, M.D. A two-year follow-up of oral antioxidant supplementation in primary open-angle glaucoma: An open-label, randomized, controlled trial. Acta Ophthalmol. 2015, 93, 546–554. [Google Scholar] [CrossRef] [PubMed]
- Quaranta, L.; Riva, I.; Biagioli, E.; Rulli, E.; Rulli, E.; Poli, D.; Legramandi, L.; CoQun Study, G. Evaluating the Effects of an Ophthalmic Solution of Coenzyme Q10 and Vitamin E in Open-Angle Glaucoma Patients: A Study Protocol. Adv. Ther. 2019, 36, 2506–2514. [Google Scholar] [CrossRef]
- Drummond, G.R.; Selemidis, S.; Griendling, K.K.; Sobey, C.G. Combating oxidative stress in vascular disease: NADPH oxidases as therapeutic targets. Nat. Rev. Drug Discov. 2011, 10, 453–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, I.; de Serna, D.G.; Gutierrez, A.; Schade, D.S. Vitamin E in humans: An explanation of clinical trial failure. Endocr. Pract. 2006, 12, 576–582. [Google Scholar] [CrossRef]
- Urner, S.; Ho, F.; Jha, J.C.; Ziegler, D.; Jandeleit-Dahm, K. NADPH Oxidase Inhibition: Preclinical and Clinical Studies in Diabetic Complications. Antioxid. Redox Signal. 2020, 33, 415–434. [Google Scholar] [CrossRef] [PubMed]
- Deliyanti, D.; Wilkinson-Berka, J.L. Inhibition of NOX1/4 with GKT137831: A potential novel treatment to attenuate neuroglial cell inflammation in the retina. J. Neuroinflamm. 2015, 12, 136. [Google Scholar] [CrossRef] [Green Version]
- Wilkinson-Berka, J.L.; Deliyanti, D.; Rana, I.; Miller, A.G.; Agrotis, A.; Armani, R.; Szyndralewiez, C.; Wingler, K.; Touyz, R.M.; Cooper, M.E.; et al. NADPH oxidase, NOX1, mediates vascular injury in ischemic retinopathy. Antioxid. Redox Signal. 2014, 20, 2726–2740. [Google Scholar] [CrossRef] [PubMed]
- Augsburger, F.; Filippova, A.; Rasti, D.; Seredenina, T.; Lam, M.; Maghzal, G.; Mahiout, Z.; Jansen-Durr, P.; Knaus, U.G.; Doroshow, J.; et al. Pharmacological characterization of the seven human NOX isoforms and their inhibitors. Redox. Biol. 2019, 26, 101272. [Google Scholar] [CrossRef] [PubMed]
- Elbatreek, M.H.; Mucke, H.; Schmidt, H. NOX Inhibitors: From Bench to Naxibs to Bedside. Handb. Exp. Pharmacol. 2020. [Google Scholar] [CrossRef]
- Hirano, K.; Chen, W.S.; Chueng, A.L.; Dunne, A.A.; Seredenina, T.; Filippova, A.; Ramachandran, S.; Bridges, A.; Chaudry, L.; Pettman, G.; et al. Discovery of GSK2795039, a Novel Small Molecule NADPH Oxidase 2 Inhibitor. Antioxid. Redox Signal. 2015, 23, 358–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teixeira, G.; Szyndralewiez, C.; Molango, S.; Carnesecchi, S.; Heitz, F.; Wiesel, P.; Wood, J.M. Therapeutic potential of NADPH oxidase 1/4 inhibitors. Br. J. Pharmacol. 2017, 174, 1647–1669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buck, A.; Sanchez Klose, F.P.; Venkatakrishnan, V.; Khamzeh, A.; Dahlgren, C.; Christenson, K.; Bylund, J. DPI Selectively Inhibits Intracellular NADPH Oxidase Activity in Human Neutrophils. Immunohorizons 2019, 3, 488–497. [Google Scholar] [CrossRef]
- Chai, Y.; Cao, Z.; Yu, R.; Liu, Y.; Yuan, D.; Lei, L. Dexmedetomidine Attenuates LPS-Induced Monocyte-Endothelial Adherence via Inhibiting Cx43/PKC-alpha/NOX2/ROS Signaling Pathway in Monocytes. Oxid. Med. Cell Longev. 2020, 2020, 2930463. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Luo, L. An Effective NADPH Oxidase 2 Inhibitor Provides Neuroprotection and Improves Functional Outcomes in Animal Model of Traumatic Brain Injury. Neurochem. Res. 2020, 45, 1097–1106. [Google Scholar] [CrossRef] [PubMed]
- Anvari, E.; Wikstrom, P.; Walum, E.; Welsh, N. The novel NADPH oxidase 4 inhibitor GLX351322 counteracts glucose intolerance in high-fat diet-treated C57BL/6 mice. Free Radic. Res. 2015, 49, 1308–1318. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Elksnis, A.; Wikstrom, P.; Walum, E.; Welsh, N.; Carlsson, P.O. The novel NADPH oxidase 4 selective inhibitor GLX7013114 counteracts human islet cell death in vitro. PLoS ONE 2018, 13, e0204271. [Google Scholar] [CrossRef] [PubMed]
- Dionysopoulou, S.; Wikstrom, P.; Walum, E.; Thermos, K. Effect of NADPH oxidase inhibitors in an experimental retinal model of excitotoxicity. Exp. Eye Res. 2020, 200, 108232. [Google Scholar] [CrossRef]
- Prendes, M.A.; Harris, A.; Wirostko, B.M.; Gerber, A.L.; Siesky, B. The role of transforming growth factor beta in glaucoma and the therapeutic implications. Br. J. Ophthalmol. 2013, 97, 680–686. [Google Scholar] [CrossRef]
- Fleenor, D.L.; Shepard, A.R.; Hellberg, P.E.; Jacobson, N.; Pang, I.H.; Clark, A.F. TGFbeta2-induced changes in human trabecular meshwork: Implications for intraocular pressure. Investig. Ophthalmol. Vis. Sci. 2006, 47, 226–234. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Luna, C.; Liton, P.B.; Navarro, I.; Epstein, D.L.; Gonzalez, P. Sustained stress response after oxidative stress in trabecular meshwork cells. Mol. Vis. 2007, 13, 2282–2288. [Google Scholar]
- Goetz, R.K.; Irnaten, M.; O’Brien, C.J. TGF-β induces NOX4 and fibrotic genes in trabecular meshwork cells: Role in glaucoma. Investig. Ophthalmol. Vis. Sci. 2019, 60, 3800. [Google Scholar]
- Rao, V.R.; Stubbs, E.B. TGF-β2 selectively increases NADPH oxidase type 4 (NOX4) expression in human trabecular meshwork (TM) cells. In Proceedings of the the Association for Research in Vision and Ophthalmology (ARVO), Virtual Meeting, Baltimore, MD, USA, 3–7 May 2020. [Google Scholar]
- Brown, K.D.; Shah, M.H.; Liu, G.S.; Chan, E.C.; Crowston, J.G.; Peshavariya, H.M. Transforming Growth Factor beta1-Induced NADPH Oxidase-4 Expression and Fibrotic Response in Conjunctival Fibroblasts. Investig. Ophthalmol. Vis. Sci. 2017, 58, 3011–3017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, M.H.; Chan, E.C.; Van Bergen, N.J.; Pandav, S.S.; Ng, S.; Crowston, J.G.; Peshavariya, H.M. Nox4 Facilitates TGFbeta1-Induced Fibrotic Response in Human Tenon’s Fibroblasts and Promotes Wound Collagen Accumulation in Murine Model of Glaucoma Filtration Surgery. Antioxidants 2020, 9, 1126. [Google Scholar] [CrossRef]
- Lavi, S.; Yang, E.H.; Prasad, A.; Mathew, V.; Barsness, G.W.; Rihal, C.S.; Lerman, L.O.; Lerman, A. The interaction between coronary endothelial dysfunction, local oxidative stress, and endogenous nitric oxide in humans. Hypertension 2008, 51, 127–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiffrin, E.L. Oxidative stress, nitric oxide synthase, and superoxide dismutase: A matter of imbalance underlies endothelial dysfunction in the human coronary circulation. Hypertension 2008, 51, 31–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunter, J.J.; Merigan, W.H.; Schallek, J.B. Imaging Retinal Activity in the Living Eye. Annu. Rev. Vis. Sci. 2019, 5, 15–45. [Google Scholar] [CrossRef] [PubMed]
- Costa, C.; Gimenez-Capitan, A.; Karachaliou, N.; Rosell, R. Comprehensive molecular screening: From the RT-PCR to the RNA-seq. Transl. Lung Cancer Res. 2013, 2, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Lukowski, S.W.; Lo, C.Y.; Sharov, A.; Nguyen, Q.H.; Fang, L.; Hung, S.S.C.; Zhu, L.; Zhang, T.; Nguyen, T.; Senabouth, A.; et al. Generation of human neural retina transcriptome atlas by single cell RNA sequencing. bioRxiv 2019, 425223. [Google Scholar] [CrossRef]
- Scimone, C.; Alibrandi, S.; Scalinci, S.Z.; Trovato Battagliola, E.; D’Angelo, R.; Sidoti, A.; Donato, L. Expression of Pro-Angiogenic Markers Is Enhanced by Blue Light in Human RPE Cells. Antioxidants 2020, 9, 1154. [Google Scholar] [CrossRef] [PubMed]
- Donato, L.; Scimone, C.; Alibrandi, S.; Nicocia, G.; Rinaldi, C.; Sidoti, A.; D’Angelo, R. Discovery of GLO1 New Related Genes and Pathways by RNA-Seq on A2E-Stressed Retinal Epithelial Cells Could Improve Knowledge on Retinitis Pigmentosa. Antioxidants 2020, 9, 416. [Google Scholar] [CrossRef] [PubMed]
- Owen, N.; Moosajee, M. RNA-sequencing in ophthalmology research: Considerations for experimental design and analysis. Ther. Adv. Ophthalmol. 2019, 11, 2515841419835460. [Google Scholar] [CrossRef]
- Donato, L.; Scimone, C.; Alibrandi, S.; Pitruzzella, A.; Scalia, F.; D’Angelo, R.; Sidoti, A. Possible A2E Mutagenic Effects on RPE Mitochondrial DNA from Innovative RNA-Seq Bioinformatics Pipeline. Antioxidants 2020, 9, 1158. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Animal Model | Tissue Basal Expression of NOX | Tissue NOX Induction upon Injury | Localisation of NOX | Site of ROS Production | NOX Association to Retinal Injury |
|---|---|---|---|---|---|
| Retinal ischemia and reperfusion (6 h) [98] | mRNA: NOX1, NOX2, NOX3, NOX4, p22PHOX and p47PHOX in retinas | mRNA: NOX2 and p22PHOX in retinas | Not performed | Inner retina | Apoptosis of retinal ganglion cells Glial cell activation |
| Retinal ischemia and reperfusion (3 h) [87] | Protein: NOX1, NOX2, NOX4, NOXO1 and p47PHOX in retinas | Protein: NOX1 and NOXO1 in retinas | Retinal ganglion cells | Inner retina | No evaluation of cell injury in the eye section |
| Optic nerve crush [108] | mRNA: NOX1, NOX2, NOX4 in retinas | mRNA: NOX1, NOX2, NOX4 in retinas | Not performed | Retinal ganglion layer | Reduction in the survival of retinal ganglion cells |
| Photocoagulation of trabecular meshwork [73] | NOX2 mRNA and protein in optic nerve head | NOX2 mRNA and protein in optic nerve head | Microglia in optic nerve head | Optic nerve head | Microglial activation in optic nerve head |
| Cauterization of episcleral vein [99] | NOX2 mRNA in retinas | NOX2 mRNA in retinas | Not performed | Not measured | Microglial activation in retinas |
| Cauterization of episcleral vein [104] | NOX1 mRNA NOX2 mRNA and protein in retinas | NOX2 mRNA and protein in retinas | Retinal ganglion layer Retinal arterioles | Retinal ganglion layer Retinal arterioles | Impaired endothelial function |
| Laser-induced injury on optic nerve [109] | NOX2 protein in optic nerve | NOX2 protein in optic nerve | Not performed | Not measured | Microglial activation in the optic nerve |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fan Gaskin, J.C.; Shah, M.H.; Chan, E.C. Oxidative Stress and the Role of NADPH Oxidase in Glaucoma. Antioxidants 2021, 10, 238. https://doi.org/10.3390/antiox10020238
Fan Gaskin JC, Shah MH, Chan EC. Oxidative Stress and the Role of NADPH Oxidase in Glaucoma. Antioxidants. 2021; 10(2):238. https://doi.org/10.3390/antiox10020238
Chicago/Turabian StyleFan Gaskin, Jennifer C., Manisha H. Shah, and Elsa C. Chan. 2021. "Oxidative Stress and the Role of NADPH Oxidase in Glaucoma" Antioxidants 10, no. 2: 238. https://doi.org/10.3390/antiox10020238
APA StyleFan Gaskin, J. C., Shah, M. H., & Chan, E. C. (2021). Oxidative Stress and the Role of NADPH Oxidase in Glaucoma. Antioxidants, 10(2), 238. https://doi.org/10.3390/antiox10020238