Anti-IL-20 Antibody Protects against Ischemia/Reperfusion-Impaired Myocardial Function through Modulation of Oxidative Injuries, Inflammation and Cardiac Remodeling

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Reagents
2.2. Hypoxia/Reoxygenation (H/R)
2.3. Antibody Preparation
2.4. Animal Model for Ischemia-Reperfusion (I/R) Injury
2.5. Extraction of Proteins from Tissues, Western Blotting Assay, and Plasma Preparation
2.6. Antibodies
2.7. Isolation of mRNA and Quantitative Real-Time Polymerase Chain Reaction (PCR)
2.8. Enzyme-Linked Immunosorbent Assay (ELISA) and Antioxidant Enzyme Activity Assay
2.9. Determination of Cardiac Functional Parameters
2.10. Apoptotic Assay
2.11. Masson’s Trichrome Staining
2.12. Measurement of ROS Production
2.13. Statistical Analysis
3. Results
3.1. IL-20 Antibody Reduces I/R-Induced Oxidative Damages by Modulating Expression of NADPH Oxidase Subunits
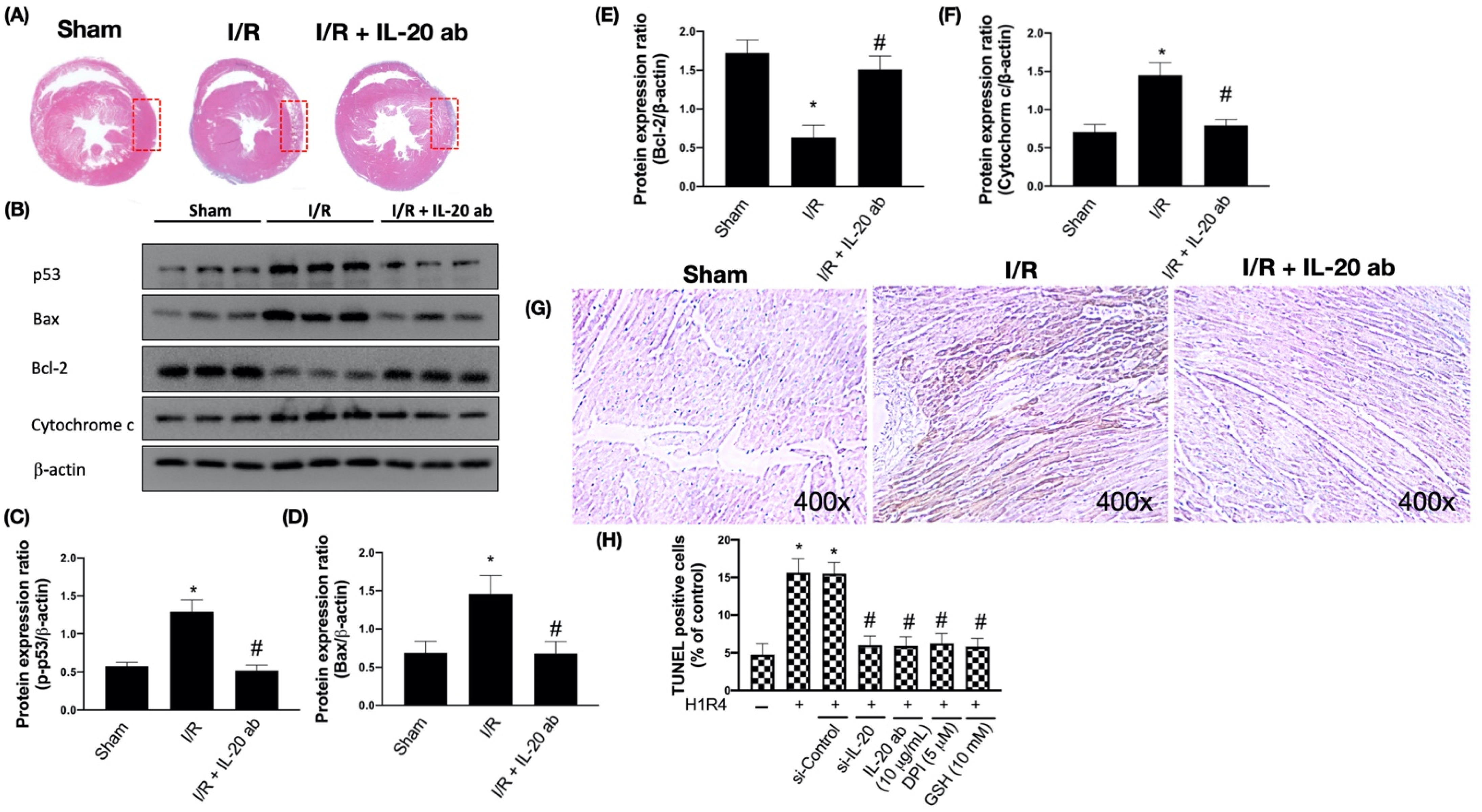
3.2. IL-20 Antibody Mitigates I/R-Caused Cardiac Apoptosis via Modulation of the Mitochondria-Dependent Pathway
3.3. IL-20 Antibody Reduces I/R-Mediated Activation of Proinflammatory Responses
3.4. IL-20 Antibody Attenuates Activation of I/R-Associated Fibrotic Factors
3.5. IL-20 Antibody Diminishes the I/R-Caused Myocardial Remodeling-Associated Molecule Expression and Cardiac Fibrosis
3.6. Treatment with Anti-IL-20 Antibody Reduces Myocardial Infarction-Caused Heart Function Impairment
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lauridsen, M.D.; Rorth, R.; Lindholm, M.G.; Kjaergaard, J.; Schmidt, M.; Moller, J.E.; Hassager, C.; Torp-Pedersen, C.; Gislason, G.; Kober, L.; et al. Trends in first-time hospitalization, management, and short-term mortality in acute myocardial infarction-related cardiogenic shock from 2005 to 2017: A nationwide cohort study. Am. Heart J. 2020, 229, 127–137. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, S.; Liu, J.; Wang, Y.; Cai, H.; Wang, D.; Fang, S.; Yu, B. D-dimer and the incidence of heart failure and mortality after acute myocardial infarction. Heart 2021, 107, 237–244. [Google Scholar] [CrossRef]
- Mahowald, M.K.; Alqahtani, F.; Alkhouli, M. Comparison of Outcomes of Coronary Revascularization for Acute Myocardial Infarction in Men Versus Women. Am. J. Cardiol. 2020, 132, 1–7. [Google Scholar] [CrossRef]
- Smolders, V.F.; Zodda, E.; Quax, P.H.A.; Carini, M.; Barbera, J.A.; Thomson, T.M.; Tura-Ceide, O.; Cascante, M. Metabolic Alterations in Cardiopulmonary Vascular Dysfunction. Front. Mol. Biosci. 2018, 5, 120. [Google Scholar] [CrossRef]
- Bentzon, J.F.; Otsuka, F.; Virmani, R.; Falk, E. Mechanisms of plaque formation and rupture. Circ. Res. 2014, 114, 1852–1866. [Google Scholar] [CrossRef] [PubMed]
- Neri, M.; Riezzo, I.; Pascale, N.; Pomara, C.; Turillazzi, E. Ischemia/Reperfusion Injury following Acute Myocardial Infarction: A Critical Issue for Clinicians and Forensic Pathologists. Mediat. Inflamm. 2017, 2017, 7018393. [Google Scholar] [CrossRef]
- Yellon, D.M.; Hausenloy, D.J. Myocardial reperfusion injury. N. Engl. J. Med. 2007, 357, 1121–1135. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Montero, J.; Brito, R.; Gajardo, A.I.; Rodrigo, R. Myocardial reperfusion injury and oxidative stress: Therapeutic opportunities. World J. Cardiol. 2018, 10, 74–86. [Google Scholar] [CrossRef] [PubMed]
- Maejima, Y.; Kuroda, J.; Matsushima, S.; Ago, T.; Sadoshima, J. Regulation of myocardial growth and death by NADPH oxidase. J. Mol. Cell. Cardiol. 2011, 50, 408–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmeyer, M.R.; Jones, S.P.; Ross, C.R.; Sharp, B.; Grisham, M.B.; Laroux, F.S.; Stalker, T.J.; Scalia, R.; Lefer, D.J. Myocardial ischemia/reperfusion injury in NADPH oxidase-deficient mice. Circ. Res. 2000, 87, 812–817. [Google Scholar] [CrossRef] [Green Version]
- Ago, T.; Kuroda, J.; Pain, J.; Fu, C.; Li, H.; Sadoshima, J. Upregulation of Nox4 by hypertrophic stimuli promotes apoptosis and mitochondrial dysfunction in cardiac myocytes. Circ. Res. 2010, 106, 1253–1264. [Google Scholar] [CrossRef] [PubMed]
- Orogo, A.M.; Gustafsson, A.B. Cell death in the myocardium: My heart won’t go on. IUBMB Life 2013, 65, 651–656. [Google Scholar] [CrossRef]
- Talman, V.; Ruskoaho, H. Cardiac fibrosis in myocardial infarction-from repair and remodeling to regeneration. Cell Tissue Res. 2016, 365, 563–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, X.; Deng, L.; Wang, D.; Li, N.; Chen, X.; Cheng, X.; Yuan, J.; Gao, X.; Liao, M.; Wang, M.; et al. Mechanism of TNF-alpha autocrine effects in hypoxic cardiomyocytes: Initiated by hypoxia inducible factor 1alpha, presented by exosomes. J. Mol. Cell. Cardiol. 2012, 53, 848–857. [Google Scholar] [CrossRef]
- Wollert, K.C.; Drexler, H. The role of interleukin-6 in the failing heart. Heart Fail. Rev. 2001, 6, 95–103. [Google Scholar] [CrossRef]
- Prabhu, S.D.; Frangogiannis, N.G. The Biological Basis for Cardiac Repair After Myocardial Infarction: From Inflammation to Fibrosis. Circ. Res. 2016, 119, 91–112. [Google Scholar] [CrossRef] [PubMed]
- Hsu, Y.H.; Chang, M.S. The therapeutic potential of anti-interleukin-20 monoclonal antibody. Cell Transplant. 2014, 23, 631–639. [Google Scholar] [CrossRef]
- Tsai, K.L.; Hsieh, P.L.; Chou, W.C.; Hung, C.H.; Yang, H.L.; Chang, Y.C.; Chu, P.M.; Chang, M.S.; Chan, S.H. IL-20 promotes hypoxia/reoxygenation-induced mitochondrial dysfunction and apoptosis in cardiomyocytes by upregulating oxidative stress by activating the PKC/NADPH oxidase pathway. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165684. [Google Scholar] [CrossRef]
- Wei, C.C.; Hsu, Y.H.; Li, H.H.; Wang, Y.C.; Hsieh, M.Y.; Chen, W.Y.; Hsing, C.H.; Chang, M.S. IL-20: Biological functions and clinical implications. J. Biomed. Sci. 2006, 13, 601–612. [Google Scholar] [CrossRef] [Green Version]
- Hsu, Y.H.; Hsing, C.H.; Li, C.F.; Chan, C.H.; Chang, M.C.; Yan, J.J.; Chang, M.S. Anti-IL-20 monoclonal antibody suppresses breast cancer progression and bone osteolysis in murine models. J. Immunol. 2012, 188, 1981–1991. [Google Scholar] [CrossRef] [Green Version]
- Hsu, Y.H.; Chen, W.Y.; Chan, C.H.; Wu, C.H.; Sun, Z.J.; Chang, M.S. Anti-IL-20 monoclonal antibody inhibits the differentiation of osteoclasts and protects against osteoporotic bone loss. J. Exp. Med. 2011, 208, 1849–1861. [Google Scholar] [CrossRef] [Green Version]
- Matsushima, S.; Tsutsui, H.; Sadoshima, J. Physiological and pathological functions of NADPH oxidases during myocardial ischemia-reperfusion. Trends Cardiovasc. Med. 2014, 24, 202–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, Y.; Chen, H.; Gao, J.; Liu, Y.; Li, J.; Wang, J. Molecular machinery and interplay of apoptosis and autophagy in coronary heart disease. J. Mol. Cell. Cardiol. 2019, 136, 27–41. [Google Scholar] [CrossRef] [Green Version]
- Levine, A.J. p53, the cellular gatekeeper for growth and division. Cell 1997, 88, 323–331. [Google Scholar] [CrossRef] [Green Version]
- Ong, S.B.; Samangouei, P.; Kalkhoran, S.B.; Hausenloy, D.J. The mitochondrial permeability transition pore and its role in myocardial ischemia reperfusion injury. J. Mol. Cell. Cardiol. 2015, 78, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Anversa, P.; Cheng, W.; Liu, Y.; Leri, A.; Redaelli, G.; Kajstura, J. Apoptosis and myocardial infarction. Basic Res. Cardiol. 1998, 93 (Suppl. 3), 8–12. [Google Scholar] [CrossRef]
- Gottlieb, R.A.; Burleson, K.O.; Kloner, R.A.; Babior, B.M.; Engler, R.L. Reperfusion injury induces apoptosis in rabbit cardiomyocytes. J. Clin. Invest. 1994, 94, 1621–1628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saini, H.K.; Xu, Y.J.; Zhang, M.; Liu, P.P.; Kirshenbaum, L.A.; Dhalla, N.S. Role of tumour necrosis factor-alpha and other cytokines in ischemia-reperfusion-induced injury in the heart. Exp. Clin. Cardiol. 2005, 10, 213–222. [Google Scholar]
- Ueha, S.; Shand, F.H.; Matsushima, K. Cellular and molecular mechanisms of chronic inflammation-associated organ fibrosis. Front. Immunol. 2012, 3, 71. [Google Scholar] [CrossRef] [Green Version]
- Bujak, M.; Frangogiannis, N.G. The role of TGF-beta signaling in myocardial infarction and cardiac remodeling. Cardiovasc. Res. 2007, 74, 184–195. [Google Scholar] [CrossRef] [Green Version]
- Xie, L.; Law, B.K.; Chytil, A.M.; Brown, K.A.; Aakre, M.E.; Moses, H.L. Activation of the Erk pathway is required for TGF-beta1-induced EMT in vitro. Neoplasia 2004, 6, 603–610. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.L.; Hsieh, P.L.; Hung, C.H.; Cheng, H.C.; Chou, W.C.; Chu, P.M.; Chang, Y.C.; Tsai, K.L. Early Moderate Intensity Aerobic Exercise Intervention Prevents Doxorubicin-Caused Cardiac Dysfunction Through Inhibition of Cardiac Fibrosis and Inflammation. Cancers 2020, 12, 1102. [Google Scholar] [CrossRef] [PubMed]
- Kardami, E.; Jiang, Z.S.; Jimenez, S.K.; Hirst, C.J.; Sheikh, F.; Zahradka, P.; Cattini, P.A. Fibroblast growth factor 2 isoforms and cardiac hypertrophy. Cardiovasc. Res. 2004, 63, 458–466. [Google Scholar] [CrossRef]
- Heymans, S.; Luttun, A.; Nuyens, D.; Theilmeier, G.; Creemers, E.; Moons, L.; Dyspersin, G.D.; Cleutjens, J.P.; Shipley, M.; Angellilo, A.; et al. Inhibition of plasminogen activators or matrix metalloproteinases prevents cardiac rupture but impairs therapeutic angiogenesis and causes cardiac failure. Nat. Med. 1999, 5, 1135–1142. [Google Scholar] [CrossRef]
- Hackel, D.B.; Reimer, K.A.; Ideker, R.E.; Mikat, E.M.; Hartwell, T.D.; Parker, C.B.; Braunwald, E.B.; Buja, M.; Gold, H.K.; Jaffe, A.S.; et al. Comparison of enzymatic and anatomic estimates of myocardial infarct size in man. Circulation 1984, 70, 824–835. [Google Scholar] [CrossRef] [Green Version]
- Turer, A.T.; Hill, J.A. Pathogenesis of myocardial ischemia-reperfusion injury and rationale for therapy. Am. J. Cardiol. 2010, 106, 360–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Vries, D.K.; Kortekaas, K.A.; Tsikas, D.; Wijermars, L.G.; van Noorden, C.J.; Suchy, M.T.; Cobbaert, C.M.; Klautz, R.J.; Schaapherder, A.F.; Lindeman, J.H. Oxidative damage in clinical ischemia/reperfusion injury: A reappraisal. Antioxid. Redox Signal. 2013, 19, 535–545. [Google Scholar] [CrossRef] [Green Version]
- Travers, J.G.; Kamal, F.A.; Robbins, J.; Yutzey, K.E.; Blaxall, B.C. Cardiac Fibrosis: The Fibroblast Awakens. Circ Res 2016, 118, 1021–1040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- Fukui, T.; Yoshiyama, M.; Hanatani, A.; Omura, T.; Yoshikawa, J.; Abe, Y. Expression of p22-phox and gp91-phox, essential components of NADPH oxidase, increases after myocardial infarction. Biochem. Biophys. Res. Commun. 2001, 281, 1200–1206. [Google Scholar] [CrossRef]
- Krijnen, P.A.; Meischl, C.; Hack, C.E.; Meijer, C.J.; Visser, C.A.; Roos, D.; Niessen, H.W. Increased Nox2 expression in human cardiomyocytes after acute myocardial infarction. J. Clin. Pathol. 2003, 56, 194–199. [Google Scholar] [CrossRef] [Green Version]
- Looi, Y.H.; Grieve, D.J.; Siva, A.; Walker, S.J.; Anilkumar, N.; Cave, A.C.; Marber, M.; Monaghan, M.J.; Shah, A.M. Involvement of Nox2 NADPH oxidase in adverse cardiac remodeling after myocardial infarction. Hypertension 2008, 51, 319–325. [Google Scholar] [CrossRef] [Green Version]
- Shanmugasundaram, K.; Nayak, B.K.; Friedrichs, W.E.; Kaushik, D.; Rodriguez, R.; Block, K. NOX4 functions as a mitochondrial energetic sensor coupling cancer metabolic reprogramming to drug resistance. Nat. Commun. 2017, 8, 997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuroda, J.; Ago, T.; Matsushima, S.; Zhai, P.; Schneider, M.D.; Sadoshima, J. NADPH oxidase 4 (Nox4) is a major source of oxidative stress in the failing heart. Proc. Natl. Acad. Sci. USA 2010, 107, 15565–15570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, J.; Xue, F.S.; Yuan, Y.J.; Wang, Q.; Liao, X.; Wang, W.L. Cholinergic anti-inflammatory pathway: A possible approach to protect against myocardial ischemia reperfusion injury. Chin. Med. J. (Engl.) 2010, 123, 2720–2726. [Google Scholar] [PubMed]
- Dutot, M.; Liang, H.; Pauloin, T.; Brignole-Baudouin, F.; Baudouin, C.; Warnet, J.M.; Rat, P. Effects of toxic cellular stresses and divalent cations on the human P2X7 cell death receptor. Mol. Vis. 2008, 14, 889–897. [Google Scholar] [PubMed]
- Li, C.; Browder, W.; Kao, R.L. Early activation of transcription factor NF-kappaB during ischemia in perfused rat heart. Am. J. Physiol. 1999, 276, H543–H552. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. The role of transforming growth factor (TGF)-beta in the infarcted myocardium. J. Thorac. Dis. 2017, 9, S52–S63. [Google Scholar] [CrossRef] [Green Version]
- Dewald, O.; Ren, G.; Duerr, G.D.; Zoerlein, M.; Klemm, C.; Gersch, C.; Tincey, S.; Michael, L.H.; Entman, M.L.; Frangogiannis, N.G. Of mice and dogs: Species-specific differences in the inflammatory response following myocardial infarction. Am. J. Pathol. 2004, 164, 665–677. [Google Scholar] [CrossRef] [Green Version]
- Dobaczewski, M.; Chen, W.; Frangogiannis, N.G. Transforming growth factor (TGF)-beta signaling in cardiac remodeling. J. Mol. Cell. Cardiol. 2011, 51, 600–606. [Google Scholar] [CrossRef] [Green Version]
- Geiser, A.G.; Busam, K.J.; Kim, S.J.; Lafyatis, R.; O’Reilly, M.A.; Webbink, R.; Roberts, A.B.; Sporn, M.B. Regulation of the transforming growth factor-beta 1 and -beta 3 promoters by transcription factor Sp1. Gene 1993, 129, 223–228. [Google Scholar] [CrossRef]
- Duncan, M.R.; Frazier, K.S.; Abramson, S.; Williams, S.; Klapper, H.; Huang, X.; Grotendorst, G.R. Connective tissue growth factor mediates transforming growth factor beta-induced collagen synthesis: Down-regulation by cAMP. FASEB J. 1999, 13, 1774–1786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abel, E.D.; Litwin, S.E.; Sweeney, G. Cardiac remodeling in obesity. Physiol. Rev. 2008, 88, 389–419. [Google Scholar] [CrossRef] [PubMed]
- Ducharme, A.; Frantz, S.; Aikawa, M.; Rabkin, E.; Lindsey, M.; Rohde, L.E.; Schoen, F.J.; Kelly, R.A.; Werb, Z.; Libby, P.; et al. Targeted deletion of matrix metalloproteinase-9 attenuates left ventricular enlargement and collagen accumulation after experimental myocardial infarction. J. Clin. Invest. 2000, 106, 55–62. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsai, K.-L.; Chou, W.-C.; Cheng, H.-C.; Huang, Y.-T.; Chang, M.-S.; Chan, S.-H. Anti-IL-20 Antibody Protects against Ischemia/Reperfusion-Impaired Myocardial Function through Modulation of Oxidative Injuries, Inflammation and Cardiac Remodeling. Antioxidants 2021, 10, 275. https://doi.org/10.3390/antiox10020275
Tsai K-L, Chou W-C, Cheng H-C, Huang Y-T, Chang M-S, Chan S-H. Anti-IL-20 Antibody Protects against Ischemia/Reperfusion-Impaired Myocardial Function through Modulation of Oxidative Injuries, Inflammation and Cardiac Remodeling. Antioxidants. 2021; 10(2):275. https://doi.org/10.3390/antiox10020275
Chicago/Turabian StyleTsai, Kun-Ling, Wan-Ching Chou, Hui-Ching Cheng, Yu-Ting Huang, Ming-Shi Chang, and Shih-Hung Chan. 2021. "Anti-IL-20 Antibody Protects against Ischemia/Reperfusion-Impaired Myocardial Function through Modulation of Oxidative Injuries, Inflammation and Cardiac Remodeling" Antioxidants 10, no. 2: 275. https://doi.org/10.3390/antiox10020275