
Redox Regulation of PTEN by Peroxiredoxins
by
 , , ,
, , ,
Thang Nguyen Huu
1,2 ,
,
Jiyoung Park
3,
Ying Zhang
4,
Iha Park
1,2,
Hyun Joong Yoon
1,
Hyun Ae Woo
3,* and
Seung-Rock Lee
1,2,* 1
Department of Biochemistry, Research Center for Aging and Geriatrics, Research Institute of Medical Sciences, Chonnam National University Medical School, Gwangju 501-190, Korea
2
Department of Biomedical Sciences, Research Center for Aging and Geriatrics, Research Institute of Medical Sciences, Chonnam National University Medical School, Gwangju 501-190, Korea
3
College of Pharmacy, Graduate School of Pharmaceutical Sciences, Ewha Womans University, Seoul 120-750, Korea
4
Department of Cell Biology, School of Medicine, Jiangsu University, Zhenjiang 212013, China
*
Authors to whom correspondence should be addressed.
Antioxidants 2021, 10(2), 302; https://doi.org/10.3390/antiox10020302
Submission received: 31 December 2020
/
Revised: 8 February 2021
/
Accepted: 10 February 2021
/
Published: 16 February 2021
(This article belongs to the Special Issue Peroxiredoxin)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Phosphatase and tensin homolog deleted on chromosome 10 (PTEN) is known as a tumor suppressor gene that is frequently mutated in numerous human cancers and inherited syndromes. PTEN functions as a negative regulator of PI3K/Akt signaling pathway by dephosphorylating phosphatidylinositol (3, 4, 5)-trisphosphate (PIP3) to phosphatidylinositol (4, 5)-bisphosphate (PIP2), which leads to the inhibition of cell growth, proliferation, cell survival, and protein synthesis. PTEN contains a cysteine residue in the active site that can be oxidized by peroxides, forming an intramolecular disulfide bond between Cys124 and Cys71. Redox regulation of PTEN by reactive oxygen species (ROS) plays a crucial role in cellular signaling. Peroxiredoxins (Prxs) are a superfamily of peroxidase that catalyzes reduction of peroxides and maintains redox homeostasis. Mammalian Prxs have 6 isoforms (I-VI) and can scavenge cellular peroxides. It has been demonstrated that Prx I can preserve and promote the tumor-suppressive function of PTEN by preventing oxidation of PTEN under benign oxidative stress via direct interaction. Also, Prx II-deficient cells increased PTEN oxidation and insulin sensitivity. Furthermore, Prx III has been shown to protect PTEN from oxidation induced by 15s-HpETE and 12s-HpETE, these are potent inflammatory and pro-oxidant mediators. Understanding the tight connection between PTEN and Prxs is important for providing novel therapies. Herein, we summarized recent studies focusing on the relationship of Prxs and the redox regulation of PTEN.
1. Introduction
The phosphoinositide-3-kinase/protein kinase B (PI3K/Akt) pathway is the critical intracellular signaling in controlling a variety of cellular processes [1]. PI3Ks are intracellular lipid kinases that are conserved from yeast to mammals. It catalyzes the phosphorylation reaction of phosphatidylinositol (PI) at hydroxyl groups to form phosphoinositides [2]. There are three classes of PI3Ks: class I, II, and III, depending on the structure and substrate specificity. Among three classes, class I PI3K has the primary function of inducing the accumulation of PIP3 by PIP2 phosphorylation [2,3,4]. Class I PI3K is also classified into 2 subgroups, called class IA and class IB, depending on the signaling receptors that activate them. While the activation of class IA is induced by growth factor receptor tyrosine kinases (RTKs), the activation of class IB is induced by G protein-coupled receptors (GPCRs) [2,3,4]. All the members of class I have the heterodimeric structure that contains a regulatory subunit and a catalytic subunit: class IA with a p85 regulatory subunit and a p110 catalytic subunit, class IB with two regulatory subunits (p101, p84/p87PIKAP) and a p110γ catalytic subunit [2,3,4]. Triggering of the serine/threonine-specific protein kinase Akt after PIP3 generation leads to the activation of an abundance of the downstream targets [5], which is depicted in Figure 1. Akt PH domain binds to PIP3 after recruitment to the membrane [6] and Akt activation is fully completed when it is phosphorylated by both phosphoinositide-dependent kinase 1 (PDK1) at Threonine 308 in the activation loop [7,8] and the rapamycin complex-2 (mTORC2) at Serine 473 [9]. Other kinases, such as mitogen-activated protein kinase-activated kinase 2 (MAPKAPK-2), integrin-linked kinase, and PKB itself, are also known to induce Akt activation [10]. On the contrary, Akt activity can be inhibited by dephosphorylation via protein phosphatase 2A (PP2A) and PH domain leucine-rich repeat protein phosphatases (PHLPP) [11,12].
ROS are predominantly generated as by-products during various physiological processes and have both advantageous and disadvantageous effects in mammals [13]. At low concentrations, ROS have positive effects by means of increasing cellular antioxidative defense systems which enables cells to survive against higher levels of oxidative stress. ROS are also required for the regulation of intracellular signaling, which affects various cellular processes, including proliferation, cell survival, and other important events [14,15,16]. The signaling messenger function is exerted by triggering the reversible oxidation of the regulatory proteins’ cysteines. As the cells lack enzymes to remove hydroxyl radicals, further reactions can happen that leads to irreversibe oxidation and degradation of protein functions [17,18], which contributes to various diseases, such as diabetes, obesity, and cancer [19,20].
PTEN has an antagonizing function in the PI3K/Akt pathway by inhibiting the downstream signaling molecules via dephosphorylation of PIP3 to PIP2. Because PTEN possesses a cysteine in the phosphatase domain, it becomes a target of ROS, especially hydrogen peroxide (H2O2), which can oxidize and inactivate PTEN phosphatase activity, as mentioned by numerous studies [21,22]. In addition to thioredoxin (Trx), a system that can use its active cysteine residues for reducing oxidized proteins (contains disulfide bonds), Prxs are members of the thiol-dependent antioxidant family that acts as a scavenger of cytosolic or mitochondrial peroxides such as H2O2. Prxs undergo modifications in their active site, especially the cysteine residues, which enables the protective function through protein-protein interactions, or subcellular protein targeting. The spotlight of this review is to show the connection between Prxs and redox regulation of PTEN.
2. Characterization of PTEN
PTEN, located at chromosome 10q23, was firstly identified in 1997. PTEN mutations were found in an assortment of human diseases, for example, brain, breast, and prostate tumors. The report has shown that mutated PTEN accounted for 31% of glioblastoma cell lines and xenografts, 100% of prostate cancer cell lines, 6% of breast cancer cell lines and xenografts, and 17% of primary glioblastomas [24].
PTEN encodes a protein of 403 amino acids that consists of five essential domains: an N-terminal PIP2-binding domain (PBD), a phosphatase domain, a C2 domain, a carboxy-terminal tail domain, and a PDZ-binding motif [25,26]. The PBD domain localizes at the N-terminal of PTEN and is significant for PTEN catalytic function and membrane localization [27,28]. The phosphatase domain (the catalytic domain) comprises a conserved motif, called HCXXGXXR, which is found as a homology of the catalytic sequence in protein tyrosine phosphatases (PTPs) [29]. When compared to PTPs, the PTEN phosphatase sequence is similar to dual-specificity protein phosphatases (DUSPs) [24,30]. Furthermore, the N-terminal first 190 amino acids are also homologous to the actin-binding protein tensin 1 (TNS1) and auxilin, which is not linked with the PTEN catalytic function [24,30]. The C2 domain (amino acids 186–351) can bind phospholipid membrane independent of calcium because it lacks the canonical Ca2+ chelating residues in vitro, which makes PTEN inhibit cell migration [31]. The C-terminal domain (amino acid 353–403) of PTEN consists of 2 PEST sequences, including phosphorylated serine-threonine spots and a PDZ (PSD-95, DLG, ZO-1) binding motif [32]. This domain was also found to be mutated in tumors. The phosphorylated serine-threonine spots contain Ser362, Thr366, Ser370, Ser380, Thr382, Thr383, and Ser385 residues. There are some kinases inducing C-terminal domain phosphorylation: casein kinase 2 (CK2), GSK3β, LKB1, and MAST [33]. When the C-terminal domain is phosphorylated, PTEN stability is increased while PTEN phosphatase activity is decreased [34]. The PTEN PDZ-binding motif is also deleted in some tumors. This motif participates in the inhibition of cell migration and protein synthesis; it also stabilizes PTEN at the plasma membrane [2,35,36,37].
Several recent studies have demonstrated that PTEN has two isoforms: PTEN-Long (PTEN-L) and PTEN-β [32]. The translation of PTEN-L starts from 519 base pairs upstream compared to the initiation site of canonical PTEN. Therefore, PTEN-L was found to have additional 173 amino acids than canonical PTEN. PTEN-L is considered as a PTEN variation. PTEN-L can be secreted from cells and quickly adopted by others [38]. Furthermore, PTEN-L is found in human blood, especially plasma and serum. Because of a poly-Arginine extension in the PTEN-L structure, the PI3K/Akt inhibition is promoted both in vitro as well as in vivo. Thus, in several studies, PTEN-L was utilized as a therapy to repress tumor progression. Other findings revealed the presence of PTEN-L in mitochondria where PTEN-L and canonical PTEN interacts, leading to the augmentation of PTEN-induced putative kinase 1 (PINK1) protein that is involved in the regulation of the mitochondrial function and energy production [39]. In contrast to PTEN-L, PTEN-β is mostly restricted to the nucleus and is found to interrelate with nucleolin. PTEN-β also affects nucleolin by dephosphorylation, which participates in the negative regulation of the transcription of DNA and biogenetic ribosome [40]. Since PTEN-L and PTEN β have a huge homologous sequence with canonical PTEN, they might be regulated by the same method.
It is well-known that the major function of PTEN is a lipid phosphatase, and its main substrate is intracellular PIP3. PTEN exerts its role as a negative regulator of the proto-oncogenic PI3K/Akt pathway, which leads to the inhibition of the downstream signaling [41,42]. It has been demonstrated that PTEN can autodephosphorylate and dephosphorylate some of its substrates, for example, focal adhesion kinase 1 (FAK), cAMP-responsive element-binding protein 1 (CREB1), proto-oncogene tyrosine-protein kinase SRC, and insulin receptor substrate 1 (IRS1) [43,44,45,46,47]. PTEN also exhibits scaffold functions in both the nucleus and cytoplasm, which suppresses tumors independent of PIP3 and the PI3K/Akt axis [25,48].
The PTEN phosphatase activity can be influenced by various factors, including PTEN post-translational modifications (PTMs) and PTEN-interacting proteins. It has been shown that several PTMs, including oxidation, S-nitrosylation, and acetylation, were found to regulate PTEN. PTEN is susceptible to oxidation since it harbors a cysteine residue at the catalytic site like other PTPs. This has been illustrated in various studies [22,49,50,51]. For instance, H2O2 can induce PTEN oxidation, which inactivates PTEN phosphatase function by establishing a Cys124-Cys71 disulfide bond [22]. However, oxidized PTEN form is gradually decreased by intracellular reducing systems, which is predominantly supported by Trxs [22]. Some organic peroxides and hydroperoxides such as cumen hydroperoxide (CuHP) and tert-butyl hydroperoxide (t-BHP) have been shown as tumor promoters. When cells were stimulated with CuHP or t-BHP at various concentrations, PTEN was quickly oxidized and the disulfide bond was formed [49,50]. In contrast to H2O2, CuHP and t-BHP induced irreversible PTEN oxidation, since the Trx was also targeted to oxidation and functional inhibition via dimerization [49,50]. Furthermore, the indirect inactivation of PTEN phosphatase following oxidation is regulated by PTEN interacting proteins. For example, the Parkinson disease protein 7 (PARK7) was found to repress the PTEN phosphatase function by binding to PTEN. When the affinity between PTEN and PARK7 increases, PTEN phosphatase activity is decreased [52].
S-nitrosylation is also an important mechanism that affects the redox status of PTEN. Numerous studies have illustrated that nitric oxide (NO) is an agent inducing S-nitrosylation of PTEN, leading to the repression of both PTEN phosphatase function and the Akt downregulation [53]. It has been shown that PTEN Cys83 is S-nitrosylated [54]. Recently, it has been found that the impairment of PARK2 can induce the suppression of PTEN by S-nitrosylation through increase the level of NO [55]. These data suggest that the S-nitrosylation is another PTM of PTEN and could be a potential target for therapeutic purposes.
Acetylation was also found to be one of the PTMs that control PTEN activity and function. Lys125 and Lys128 residues of PTEN are modified by acetyltransferase PCAF (or KAT2B) in response to growth factor stimulation [56], which leads to the PTEN phosphatase inhibition. In addition, p300-CREB-binding protein (CBP) has been shown to acetylate PTEN at Lys402 in the PDZ-binding motif. Acetylation of PTEN intervenes in the interaction between PTEN and its interacting proteins [57]. Moreover, NAD-dependent protein deacetylase sirtuin 1 (SIRT1) is reported to be involved in the deacetylation of PTEN reaction. Hyperacetylation of PTEN has been demonstrated in cells that lack SIRT1 [57,58]. PTEN is translocated from the nucleus to the cytosol upon SIRT1 depletion [58], suggesting that acetylation regulates its subcellular localization.
PTEN phosphatase activity can be regulated by protein-protein interactions, which affects its stability, subcellular localization, and affinity. Several studies have shown that PIP3-dependent Rac exchanger 2 protein (PREX2) [59], shank-interacting protein-like 1 (SIPL1; or SHARPIN) [60], and α-mannosidase 2C1 (MAN2C1) [61] can directly suppress PTEN lipid phosphatase activity by acting as negative regulators of PTEN. The phosphorylated Akt levels are also enhanced by these negative regulators, further indicating that the cellular activity of Akt depends on PTEN inhibition.
3. Characterization of Mammalian Peroxiredoxins
Peroxiredoxins (Prxs) are members of the small peroxidase family (22–27 kDa), which participate in the reduction of H2O2, organic hydroperoxides, and peroxynitrite since they possess thiol residues in their active sites [62,63,64,65,66]. Prxs are widely discovered in the mid-1990s [67], its scavenging role was long overshadowed by other protection systems, such as catalase and glutathione peroxidase (Gpx), but upgraded kinetics estimations presently demonstrate that Prxs are also crucial enzymes for reducing cellular peroxides [13,68]. At the NH2-terminal site of Prxs, it has a conserved peroxidatic cysteine residue (CP) that is easily oxidized by peroxides [69,70]. The other additional cysteine residue found in some Prxs, called resolving Cys (CR), is located at the COOH-terminal site of the molecules. Based on the presence or location of the CR residue, members of Prxs are divided into three groups: 2-Cys proteins, atypical 2-Cys proteins, and 1-Cys proteins [62]. There are a total of six Prxs isoforms in mammals: four 2-Cys Prx isoforms (Prx I to Prx IV), one atypical 2-Cys isoform (Prx V), and one 1-Cys Prx isoform (Prx VI) [65]. These isoforms differ in their subcellular localization and substrate preferences. In general, mammalian Prx I to Prx IV are more efficient in reducing H2O2, whereas Prx V is more effective against alkyl hydroperoxides and peroxynitrite compared to H2O2 and Prx VI is more active against alkyl hydroperoxides over other peroxides [71,72,73]. Despite each of these isoforms has a distinctive function in cellular redox protection, they all have the intracellular H2O2-reducing activity that can control the levels of intracellular H2O2 [74].
Prxs exist in a head-to-tail dimeric structure. During the reaction with H2O2, the CP-SH residue was transformed to cysteine sulfenic acid (CP-SOH). In the case of the 2-Cys Prxs, this unstable sulfenic acid forms an intermolecular disulfide bond with a CR-SH residue of the other molecule in the dimeric Prx structure. This disulfide bond is then reduced by the Trx system, which is firstly identified in yeast Prx (TSA) [75,76] when the crystal structure of rat Prx I was investigated and the intermolecular disulfide was found between Cys52 and Cys173 [77]. Mutant forms of Prx I to Prx IV that lack either CP or CR thus do not manifest Trx-dependent peroxidase activity [62]. Prx V and Prx VI have a similar initial step with 2-Cys Prxs in peroxidase cycles, which involves the oxidation of CP-SH (Cys47 for both human isoforms) into CP-SOH. In the case of Prx V, an intra-subunit disulfide bond forms between CP-SOH with Cys151 (CR-SH), and this disulfide bond is reduced by Trx [78]. Prx V dimeric structure with an intramolecular disulfide bond was determined in the Prx V crystal structure [72,79,80], which uncovered that Prx V dimers are not antiparallel and that Prx V interface-involved dimerization is different from that of Prx I to Prx IV and of Prx VI [72]. Prx VI is absent of CR, thus, Prx VI CP-SOH cannot be resolved within the dimer. A cysteine thiol (CR-SH) of the π isoform of glutathione S-transferase (πGST) was found to resolve the Prx VI–SOH [81,82,83]. The resulting heterodimeric disulfide (PrxVI–S–S–πGST) is reduced by two glutathione (GSH) molecules that are converted to oxidized glutathione (GSSH) [81,82,83]. Oxidized Prx VI is also reduced by dithiothreitol (DTT) but not by Trx. The Prx VI homodimer crystal structure is antiparallel to Cys47-SOH that was steadied by other surrounding residues [84]. Like Prx V, Prx VI functions as a monomer.
Prx I belongs to the 2-Cys Prx group, therefore, when exposed to low levels of H2O2, the sulfenic acid (CP-SOH) was formed at the site of Cys51 after CP-SH oxidation. The intermolecular disulfide bond is formed after interacting with Cys172 of the other molecule in the Prx I dimeric structure. Eventually, this disulfide bond is reduced by Trx [62,76]. Upon exposure to higher levels of H2O2, a further oxidative reaction occurs that leads to oxidation of CP-SOH into CP-SO2H (sulfinic acid), subsequently inactivating its peroxidase activity. This sulfinic acid can undergo the reduction into thiol by Sulfiredoxin (Srx) system [85,86], whereas further oxidation of CP-SO2H to CPSO3H (sulfonic acid) is irreversible, which degrades Prx [85]. These reactions are shown in Figure 2. It has been shown that higher molecular weight complexes were preferred when Prx I is overoxidized because of oxidative stress or heat shock stress [87]. Accompanying the structural changes, the function of Prx I also alters from peroxidase to molecular chaperone activity [88,89].
The Prx peroxidase activity is regulated by numerous PTMs, such as phosphorylation, deacetylation, and S-nitrosylation [90,91,92]. It has been observed that membrane-associated Prx I is phosphorylated on Tyr194 in vitro upon stimulation of various receptors (Epidermal growth factor receptor—EGFR, platelet-derived growth factor—PDGF, B-cell receptor—BCR, and T-cell receptor—TCR) in several cell lines as well as in vivo following cutaneous injury in mice, thus contribute to remaining Nox-generated H2O2 levels for intracellular messenger function [90]. Furthermore, Prx I and Prx II have known as substrates of histone deacetylase 6 (HDAC6). Acetylated Prx I and Prx II increased in cell lines that lack HDAC6 and in HDAC6-knockdown conditions [91]. The acetylated form of Prx was also proved to be more effective in H2O2 reduction, which suggests potential therapeutic targets can be developed for the increase of acetylated Prx, including HDAC6 inhibitor [91]. Prx II was S-nitrosylated by nitric oxide (NO) both in vitro in human SH-SY5Y cells (cell-based models of Parkinson’s disease-PD) and in vivo in human PD brain tissue [92]. Prx II S-nitrosylation occurred at two cysteine residues (Cys51 and Cys172), which leads to the inactivation of Prx peroxidase activity [92]. Therefore, the relationship between NO and oxidative stress in neurodegenerative disorders was demonstrated partly through Prx II S-nitrosylation [92].
4. Redox regulation of PTEN by Peroxiredoxins
4.1. Regulation of PTEN by Prx I
It has been demonstrated that, under mild oxidative stress, PTEN tumor-suppressive activity was protected by Prx I via forming the intermolecular disulfide bond [93]. At the low level of H2O2 (25 µM), the PTEN lipid phosphatase activity was completely protected by Prx I in cells, as the interaction between Prx I and PTEN was found. On the other hand, at high levels of H2O2 (500 μM), the hyperoxidation forms of Prx I can be observed with the dissociation from PTEN. As a result, at the 1:1 ratio of Prx I and PTEN, the high concentrations of H2O2 can oxidize Prx I at Cys51 and promote unwinding of the PTEN-binding conformation of Prx I consequently inducing their dissociation [93]. In addition, the results also showed that the 1:1 ratio of Prx I and PTEN is the most effective ratio for protecting PTEN phosphatase activity, and additional amounts of Prx I could not increase the protection, suggesting the monomeric interaction between Prx I and PTEN [93]. Mutational analysis and computational analysis suggested that Prx I interacts within the C2 domain of PTEN (amino acid 186–274) and PTEN with the N-terminal of Prx I (amino acid 1–21) and the C- terminal of Prx I (amino acid 183–199) [93].
Exposure to H2O2 for various times resulted in the formation of hyperoxidized Prxs. In Hela cells untreated with H2O2, the Prx I dimeric forms were predominantly observed since Prxs are considered as dimers in the absence of NEM [94]. The Prx I hyperoxidation levels reached a peak after 5 min of exposure and then gradually declined. Besides, the increase of oxidized PTEN levels showed similar kinetics to H2O2-induced Prx I hyperoxidation, and the transient hyperoxidation-induced suppression of Prx weakened H2O2-scavenging activity [94].
4.2. Regulation of PTEN by Prx II
ROS has been demonstrated to participate in insulin signaling [95,96]. The downstream signaling of the PI3K/Akt pathway was activated when the cell was stimulated with insulin, which triggers the activation of specific receptors, including the insulin receptor (IR) and insulin receptor substrate (IRS). The GLUT4 was employed from the intracellular pool to the membrane surface after the insulin-induced activation of the downstream cascade, permitting the entry of glucose into the cell [97]. PTEN and PTPs all antagonize the insulin signaling as they directly interact with PI3K and IR [98], and both consist of a cysteine residue in the active site that is highly susceptible to H2O2-induced oxidation.
Prxs are well-known as peroxide-scavenging enzymes having a high affinity for H2O2, with half-maximal activity <20 µm [99]. Also, Prx II has shown involvement in various cellular processes, such as hemoglobin stability, hippocampal synaptic plasticity, osteoclasts, and maintaining the stemness of embryonic stem cells [100,101,102].
It has been demonstrated that in the Prx II-deficient MEFs treated with insulin, the downstream signaling of the PI3K/Akt pathway increased, which was accompanied by an increase in oxidized PTEN levels and ROS levels [103]. After insulin treatment (100 nM) of MEFs, the levels of phosphorylated insulin receptor β (IRβ) were almost 1.6-fold higher in Prx II−/− compared to in Wild type (WT) at 5 min [103]. In addition, the levels of PI3K and phosphorylated Akt in the Prx II-deficient MEFs were also higher than that of WT with 1.8-fold and 2.2-fold, respectively. Interestingly, PTP1B and PTEN oxidation levels in Prx II−/− increased over time compared to that in WT MEFs. ROS levels were also increased in Prx II−/− MEFs after exposure to insulin [103]. The participant of H2O2 in intracellular signaling by targeting PTEN and Prx II, and the regulation of H2O2 concentration by Prx II are depicted schematically in Figure 3.
4.3. Regulation of PTEN by Prx III
Lipoxygenases (LOX) are enzymes that catalyze the formation of hydroperoxyl-eicosatetraenoic acid (HpETE) from arachidonic acid (AA) and linoleic acid (LA), which could be important components in inflammatory and prooxidant mediators [104,105]. 15-Lipoxygenase (15-LOX) is a LOX family member that can promote the formation of a 15(s)-hydroperoxy-eicosatetraenoic acid (15s-HpETE) and a 15-hydroxyeicosatetraenoic acid (15s-HETE) from AA. There are two isoforms of human 15-LOX, called 15-LOX-1 and 15-LOX-2 [106,107]. 15s-HpETE is the main product after the catalysis of AA by 15-LOX-1. Small levels of 12s-HpETE are also synthesized in this catalysis. During the catalysis by 15-LOX-2, AA was metabolized mostly into 15s-HpETE but was not into 12s-HpETE [108]. The reduction and transformation of HpETEs then happened that causes the creation of eicosanoids, the vital lipid peroxides in the responsiveness of the immune system, and other physiological mechanisms. The increase of lipid peroxide levels is related to the pathological condition of differential human disorders and diseases, including neurodegeneration, atherosclerosis, type II diabetes, metabolic disorders, solid tumors, and hematologic malignancies, through the deterioration of biological oxidative processes [109,110,111,112].
In MEFs, it was shown that both 15s-HpETE and 12s-HpETE induce the oxidation of PTEN and that oxidized PTEN was gradually reversed to the reduced form by cellular antioxidants after 30 min of exposure [113]. Similar disulfide bond formation between Cys124 and Cys71 was observed in both H2O2- and 15s-HpETE-treated MEFs [113]. Moreover, Prx III-deficiency showed a higher level of PTEN oxidation compared to that of WT. Furthermore, Trx dimers were maintained for 60 min in Prx III−/− MEFs compared to 5 min in Prx III+/+ MEFs [113]. These data suggested that Prx III might play an important role in protecting PTEN and Trx system from oxidation by lipid peroxides [113], which is depicted in Figure 4.
5. Conclusions
PTEN is well-known for the negative regulatory function of the PI3K/Akt pathway by dephosphorylating PIP3 to PIP2. However, ROS, which can be produced as a secondary messenger of intracellular signaling, has the ability to induce PTEN oxidation, forming an intramolecular disulfide bond and inactivating PTEN phosphatase function. In the cells, Prxs, the scavengers for peroxides, plays a crucial role in the redox regulation of PTEN and maintaining redox homeostasis. The redox regulation of PTEN by Prxs has been recently investigated in some contexts. The main focus of this review is on the redox regulation of PTEN concerning Prxs.
Prx I has been proved to preserve PTEN under mild oxidative stress by directly interacting with PTEN. However, under the high concentrations of H2O2, Prx I was hyperoxidized and dissociated from PTEN. Besides, Prx II-deficient MEFs induced PTEN oxidation and increased PI3K/Akt activation when exposed to insulin, which leads to an increase the insulin sensitivity. Additionally, the deficiency of Prx II in Hela cells increased the PIP3 accumulation and Akt activation following the stimulation of growth factors [21]. Growth factor stimulation also induced PTEN oxidation in Hela cells [21]. Therefore, the cytosolic Prx II may function to protect PTEN from oxidation. However, it has also been reported that direct interaction between PTEN and Prx II was not observed by immunoprecipitation [93,114]. Further studies are necessary to investigate the relation between redox regulation of PTEN and Prx II. Prx III-deficiency also induces the augmentation in both PTEN oxidation and Trx dimerization. This provides a new line of evidence regarding the role of Prxs in the redox regulation of PTEN. As Prx III localizes in the mitochondria, it can be speculated that Prx III directly reduces peroxides radicals, which decreases 12/15s-HpETE levels and subsequently declines PTEN oxidation levels. However, further investigation focusing on the mechanism of Prx III in regulating PTEN redox status is also needed.
Author Contributions
Conceptualization, H.A.W. and S.-R.L.; writing-original draft preparation, T.N.H.; writing-review and editing, T.N.H., J.P., Y.Z., I.P., H.J.Y.; supervision, S.-R.L.; funding acquisition, H.A.W., I.P., S.-R.L., J.P. All authors have read and agreed to the published version of the manuscript.
Funding
The work was supported by the National Research Foundation of Korea (2018R1D1A1B06051438, 2015R1D1A1A01059571, 2019R1I1A3A01062555, 2019R1A6C1010020 and 2020R1A6C103B074), Republic of Korea.
Acknowledgments
Figure was made in part using BioRender.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Vivanco, I.; Sawyers, C.L. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat. Rev. Cancer 2002, 2, 489–501. [Google Scholar] [CrossRef]
- Georgescu, M.M. PTEN Tumor Suppressor Network in PI3K-Akt Pathway Control. Genes Cancer 2010, 1, 1170–1177. [Google Scholar] [CrossRef]
- Engelman, J.A.; Luo, J.; Cantley, L.C. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat. Rev. Genet. 2006, 7, 606–619. [Google Scholar] [CrossRef]
- Chalhoub, N.; Baker, S.J. PTEN and the PI3-kinase pathway in cancer. Annu. Rev. Pathol. 2009, 4, 127–150. [Google Scholar] [CrossRef] [Green Version]
- Luo, J.; Manning, B.D.; Cantley, L.C. Targeting the PI3K-Akt pathway in human cancer: Rationale and promise. Cancer Cell 2003, 4, 257–262. [Google Scholar] [CrossRef] [Green Version]
- Franke, T.F. PI3K/Akt: Getting it right matters. Oncogene 2008, 27, 6473–6488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mora, A.; Komander, D.; van Aalten, D.M.; Alessi, D.R. PDK1, the master regulator of AGC kinase signal transduction. Semin. Cell Dev. Biol. 2004, 15, 161–170. [Google Scholar] [CrossRef] [Green Version]
- Alessi, D.R.; Kozlowski, M.T.; Weng, Q.P.; Morrice, N.; Avruch, J. 3-Phosphoinositide-dependent protein kinase 1 (PDK1) phosphorylates and activates the p70 S6 kinase in vivo and in vitro. Curr. Biol. 1998, 8, 69–81. [Google Scholar] [CrossRef] [Green Version]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, J.; Park, J.; Cron, P.; Hess, D.; Hemmings, B.A. Identification of a PKB/Akt hydrophobic motif Ser-473 kinase as DNA-dependent protein kinase. J. Biol. Chem. 2004, 279, 41189–41196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andjelković, M.; Jakubowicz, T.; Cron, P.; Ming, X.F.; Han, J.W.; Hemmings, B.A. Activation and phosphorylation of a pleckstrin homology domain containing protein kinase (RAC-PK/PKB) promoted by serum and protein phosphatase inhibitors. Proc. Natl. Acad. Sci. USA 1996, 93, 5699–5704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, T.; Furnari, F.; Newton, A.C. PHLPP: A phosphatase that directly dephosphorylates Akt, promotes apoptosis, and suppresses tumor growth. Mol. Cell. 2005, 18, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Winterbourn, C.C. Reconciling the chemistry and biology of reactive oxygen species. Nat. Chem. Biol. 2008, 4, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Burdon, R.H. Superoxide and hydrogen peroxide in relation to mammalian cell proliferation. Free Radic. Biol. Med. 1995, 18, 775–794. [Google Scholar] [CrossRef]
- Rhee, S.G.; Kang, S.W.; Jeong, W.; Chang, T.S.; Yang, K.S.; Woo, H.A. Intracellular messenger function of hydrogen peroxide and its regulation by peroxiredoxins. Curr. Opin. Cell Biol. 2005, 17, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.G. Redox signaling: Hydrogen peroxide as intracellular messenger. Exp. Mol. Med. 1999, 31, 53–59. [Google Scholar] [CrossRef] [Green Version]
- Dean, R.T.; Fu, S.; Stocker, R.; Davies, M.J. Biochemistry and pathology of radical-mediated protein oxidation. Biochem. J. 1997, 324, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, L.J.; Sohal, R.S. Mitochondrial adenine nucleotide translocase is modified oxidatively during aging. Proc. Natl. Acad. Sci. USA 1998, 95, 12896–12901. [Google Scholar] [CrossRef] [Green Version]
- Jones, D.P. Redefining oxidative stress. Antioxid. Redox Signal. 2006, 8, 1865–1879. [Google Scholar] [CrossRef]
- Roberts, C.K.; Sindhu, K.K. Oxidative stress and metabolic syndrome. Life Sci. 2009, 84, 705–712. [Google Scholar] [CrossRef]
- Kwon, J.; Lee, S.R.; Yang, K.S.; Ahn, Y.; Kim, Y.J.; Stadtman, E.R.; Rhee, S.G. Reversible oxidation and inactivation of the tumor suppressor PTEN in cells stimulated with peptide growth factors. Proc. Natl. Acad. Sci. USA 2004, 101, 16419–16424. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.R.; Yang, K.S.; Kwon, J.; Lee, C.; Jeong, W.; Rhee, S.G. Reversible inactivation of the tumor suppressor PTEN by H2O2. J. Biol. Chem. 2002, 277, 20336–20342. [Google Scholar] [CrossRef] [Green Version]
- Manning, B.D.; Cantley, L.C. AKT/PKB signaling: Navigating downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Yen, C.; Liaw, D.; Podsypanina, K.; Bose, S.; Wang, S.I.; Puc, J.; Miliaresis, C.; Rodgers, L.; McCombie, R.; et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 1997, 275, 1943–1947. [Google Scholar] [CrossRef] [PubMed]
- Song, M.S.; Salmena, L.; Pandolfi, P.P. The functions and regulation of the PTEN tumour suppressor. Nat. Rev. Mol. Cell Biol. 2012, 13, 283–296. [Google Scholar] [CrossRef]
- Lee, J.O.; Yang, H.; Georgescu, M.M.; Di Cristofano, A.; Maehama, T.; Shi, Y.; Dixon, J.E.; Pandolfi, P.; Pavletich, N.P. Crystal structure of the PTEN tumor suppressor: Implications for its phosphoinositide phosphatase activity and membrane association. Cell 1999, 99, 323–334. [Google Scholar] [CrossRef] [Green Version]
- Walker, S.M.; Leslie, N.R.; Perera, N.M.; Batty, I.H.; Downes, C.P. The tumour-suppressor function of PTEN requires an N-terminal lipid-binding motif. Biochem. J. 2004, 379, 301–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, R.B.; Liu, F.; Ross, A.H. Allosteric activation of PTEN phosphatase by phosphatidylinositol 4,5-bisphosphate. J. Biol. Chem. 2003, 278, 33617–33620. [Google Scholar] [CrossRef] [Green Version]
- Denu, J.M.; Stuckey, J.A.; Saper, M.A.; Dixon, J.E. Form and function in protein dephosphorylation. Cell 1996, 87, 361–364. [Google Scholar] [CrossRef] [Green Version]
- Steck, P.A.; Pershouse, M.A.; Jasser, S.A.; Yung, W.K.; Lin, H.; Ligon, A.H.; Langford, L.A.; Baumgard, M.L.; Hattier, T.; Davis, T.; et al. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat. Genet. 1997, 15, 356–362. [Google Scholar] [CrossRef]
- Hopkins, B.D.; Hodakoski, C.; Barrows, D.; Mense, S.M.; Parsons, R.E. PTEN function: The long and the short of it. Trends Biochem. Sci. 2014, 39, 183–190. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.R.; Chen, M.; Pandolfi, P.P. The functions and regulation of the PTEN tumour suppressor: New modes and prospects. Nat. Rev. Mol. Cell Biol. 2018, 19, 547–562. [Google Scholar] [CrossRef]
- Valiente, M.; Andrés-Pons, A.; Gomar, B.; Torres, J.; Gil, A.; Tapparel, C.; Antonarakis, S.E.; Pulido, R. Binding of PTEN to specific PDZ domains contributes to PTEN protein stability and phosphorylation by microtubule-associated serine/threonine kinases. J. Biol. Chem. 2005, 280, 28936–28943. [Google Scholar] [CrossRef] [PubMed]
- Vazquez, F.; Ramaswamy, S.; Nakamura, N.; Sellers, W.R. Phosphorylation of the PTEN tail regulates protein stability and function. Mol. Cell. Biol. 2000, 20, 5010–5018. [Google Scholar] [CrossRef] [Green Version]
- Leslie, N.R.; Yang, X.; Downes, C.P.; Weijer, C.J. PtdIns(3,4,5)P(3)-dependent and -independent roles for PTEN in the control of cell migration. Curr. Biol. 2007, 17, 115–125. [Google Scholar] [CrossRef] [Green Version]
- Mounir, Z.; Krishnamoorthy, J.L.; Robertson, G.P.; Scheuner, D.; Kaufman, R.J.; Georgescu, M.M.; Koromilas, A.E. Tumor suppression by PTEN requires the activation of the PKR-eIF2alpha phosphorylation pathway. Sci. Signal. 2009, 2, ra85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Hepner, K.; Castelino-Prabhu, S.; Do, D.; Kaye, M.B.; Yuan, X.J.; Wood, J.; Ross, C.; Sawyers, C.L.; Whang, Y.E. Evidence for regulation of the PTEN tumor suppressor by a membrane-localized multi-PDZ domain containing scaffold protein MAGI-2. Proc. Natl. Acad. Sci. USA 2000, 97, 4233–4238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hopkins, B.D.; Fine, B.; Steinbach, N.; Dendy, M.; Rapp, Z.; Shaw, J.; Pappas, K.; Yu, J.S.; Hodakoski, C.; Mense, S.; et al. A secreted PTEN phosphatase that enters cells to alter signaling and survival. Science 2013, 341, 399–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, H.; He, S.; Yang, J.; Jia, X.; Wang, P.; Chen, X.; Zhang, Z.; Zou, X.; McNutt, M.A.; Shen, W.H.; et al. PTENα, a PTEN isoform translated through alternative initiation, regulates mitochondrial function and energy metabolism. Cell Metab. 2014, 19, 836–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, H.; Chen, X.; Yin, Q.; Ruan, D.; Zhao, X.; Zhang, C.; McNutt, M.A.; Yin, Y. PTENβ is an alternatively translated isoform of PTEN that regulates rDNA transcription. Nat. Commun. 2017, 8, 14771. [Google Scholar] [CrossRef] [Green Version]
- Maehama, T.; Dixon, J.E. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J. Biol. Chem. 1998, 273, 13375–13378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stambolic, V.; Suzuki, A.; de la Pompa, J.L.; Brothers, G.M.; Mirtsos, C.; Sasaki, T.; Ruland, J.; Penninger, J.M.; Siderovski, D.P.; Mak, T.W. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell 1998, 95, 29–39. [Google Scholar] [CrossRef] [Green Version]
- Gu, T.; Zhang, Z.; Wang, J.; Guo, J.; Shen, W.H.; Yin, Y. CREB is a novel nuclear target of PTEN phosphatase. Cancer Res. 2011, 71, 2821–2825. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Wang, J.; Chandarlapaty, S.; Cross, J.; Thompson, C.; Rosen, N.; Jiang, X. PTEN is a protein tyrosine phosphatase for IRS1. Nat. Struct. Mol. Biol. 2014, 21, 522–527. [Google Scholar] [CrossRef]
- Tamura, M.; Gu, J.; Matsumoto, K.; Aota, S.; Parsons, R.; Yamada, K.M. Inhibition of cell migration, spreading, and focal adhesions by tumor suppressor PTEN. Science 1998, 280, 1614–1617. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Huang, W.C.; Li, P.; Guo, H.; Poh, S.B.; Brady, S.W.; Xiong, Y.; Tseng, L.M.; Li, S.H.; Ding, Z.; et al. Combating trastuzumab resistance by targeting SRC, a common node downstream of multiple resistance pathways. Nat. Med. 2011, 17, 461–469. [Google Scholar] [CrossRef]
- Zhang, X.C.; Piccini, A.; Myers, M.P.; Van Aelst, L.; Tonks, N.K. Functional analysis of the protein phosphatase activity of PTEN. Biochem. J. 2012, 444, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Salmena, L.; Carracedo, A.; Pandolfi, P.P. Tenets of PTEN tumor suppression. Cell 2008, 133, 403–414. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Han, S.J.; Park, I.; Kim, I.; Chay, K.O.; Kim, S.M.; Jang, D.I.; Lee, T.H.; Lee, S.R. Redox Regulation of the Tumor Suppressor PTEN by Hydrogen Peroxide and Tert-Butyl Hydroperoxide. Int. J. Mol. Sci. 2017, 18, 982. [Google Scholar] [CrossRef] [Green Version]
- Han, S.J.; Zhang, Y.; Kim, I.; Chay, K.O.; Yoon, H.J.; Jang, D.I.; Yang, S.Y.; Park, J.; Woo, H.A.; Park, I.; et al. Redox regulation of the tumor suppressor PTEN by the thioredoxin system and cumene hydroperoxide. Free Radic. Biol. Med. 2017, 112, 277–286. [Google Scholar] [CrossRef]
- Cho, S.H.; Lee, C.H.; Ahn, Y.; Kim, H.; Kim, H.; Ahn, C.Y.; Yang, K.S.; Lee, S.R. Redox regulation of PTEN and protein tyrosine phosphatases in H(2)O(2) mediated cell signaling. FEBS Lett. 2004, 560, 7–13. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.C.; Kitaura, H.; Taira, T.; Iguchi-Ariga, S.M.; Ariga, H. Oxidation of DJ-1-dependent cell transformation through direct binding of DJ-1 to PTEN. Int. J. Oncol. 2009, 35, 1331–1341. [Google Scholar] [PubMed]
- Kwak, Y.D.; Ma, T.; Diao, S.; Zhang, X.; Chen, Y.; Hsu, J.; Lipton, S.A.; Masliah, E.; Xu, H.; Liao, F.F. NO signaling and S-nitrosylation regulate PTEN inhibition in neurodegeneration. Mol. Neurodegener. 2010, 5, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Numajiri, N.; Takasawa, K.; Nishiya, T.; Tanaka, H.; Ohno, K.; Hayakawa, W.; Asada, M.; Matsuda, H.; Azumi, K.; Kamata, H.; et al. On-off system for PI3-kinase-Akt signaling through S-nitrosylation of phosphatase with sequence homology to tensin (PTEN). Proc. Natl. Acad. Sci. USA 2011, 108, 10349–10354. [Google Scholar] [CrossRef] [Green Version]
- Gupta, A.; Anjomani-Virmouni, S.; Koundouros, N.; Dimitriadi, M.; Choo-Wing, R.; Valle, A.; Zheng, Y.; Chiu, Y.H.; Agnihotri, S.; Zadeh, G.; et al. PARK2 Depletion Connects Energy and Oxidative Stress to PI3K/Akt Activation via PTEN S-Nitrosylation. Mol. Cell. 2017, 65, 999–1013.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okumura, K.; Mendoza, M.; Bachoo, R.M.; DePinho, R.A.; Cavenee, W.K.; Furnari, F.B. PCAF modulates PTEN activity. J. Biol. Chem. 2006, 281, 26562–26568. [Google Scholar] [CrossRef] [Green Version]
- Ikenoue, T.; Inoki, K.; Zhao, B.; Guan, K.L. PTEN acetylation modulates its interaction with PDZ domain. Cancer Res. 2008, 68, 6908–6912. [Google Scholar] [CrossRef] [Green Version]
- Chae, H.D.; Broxmeyer, H.E. SIRT1 deficiency downregulates PTEN/JNK/FOXO1 pathway to block reactive oxygen species-induced apoptosis in mouse embryonic stem cells. Stem Cells Dev. 2011, 20, 1277–1285. [Google Scholar] [CrossRef]
- Fine, B.; Hodakoski, C.; Koujak, S.; Su, T.; Saal, L.H.; Maurer, M.; Hopkins, B.; Keniry, M.; Sulis, M.L.; Mense, S.; et al. Activation of the PI3K pathway in cancer through inhibition of PTEN by exchange factor P-REX2a. Science 2009, 325, 1261–1265. [Google Scholar] [CrossRef] [Green Version]
- He, L.; Ingram, A.; Rybak, A.P.; Tang, D. Shank-interacting protein-like 1 promotes tumorigenesis via PTEN inhibition in human tumor cells. J. Clin. Investig. 2010, 120, 2094–2108. [Google Scholar] [CrossRef]
- He, L.; Fan, C.; Kapoor, A.; Ingram, A.J.; Rybak, A.P.; Austin, R.C.; Dickhout, J.; Cutz, J.C.; Scholey, J.; Tang, D. α-Mannosidase 2C1 attenuates PTEN function in prostate cancer cells. Nat. Commun. 2011, 2, 307. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.G.; Kang, S.W.; Chang, T.S.; Jeong, W.; Kim, K. Peroxiredoxin, a novel family of peroxidases. IUBMB Life 2001, 52, 35–41. [Google Scholar] [CrossRef]
- Rhee, S.G.; Chae, H.Z.; Kim, K. Peroxiredoxins: A historical overview and speculative preview of novel mechanisms and emerging concepts in cell signaling. Free Radic. Biol. Med. 2005, 38, 1543–1552. [Google Scholar] [CrossRef]
- Poole, L.B.; Hall, A.; Nelson, K.J. Overview of peroxiredoxins in oxidant defense and redox regulation. Curr. Protoc. Toxicol. 2011, 49, 7.9.1–7.9.15. [Google Scholar] [CrossRef] [Green Version]
- Perkins, A.; Nelson, K.J.; Parsonage, D.; Poole, L.B.; Karplus, P.A. Peroxiredoxins: Guardians against oxidative stress and modulators of peroxide signaling. Trends Biochem. Sci. 2015, 40, 435–445. [Google Scholar] [CrossRef] [Green Version]
- Rhee, S.G.; Woo, H.A.; Kil, I.S.; Bae, S.H. Peroxiredoxin functions as a peroxidase and a regulator and sensor of local peroxides. J. Biol. Chem. 2012, 287, 4403–4410. [Google Scholar] [CrossRef] [Green Version]
- Chae, H.Z.; Robison, K.; Poole, L.B.; Church, G.; Storz, G.; Rhee, S.G. Cloning and sequencing of thiol-specific antioxidant from mammalian brain: Alkyl hydroperoxide reductase and thiol-specific antioxidant define a large family of antioxidant enzymes. Proc. Natl. Acad. Sci. USA 1994, 91, 7017–7021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adimora, N.J.; Jones, D.P.; Kemp, M.L. A model of redox kinetics implicates the thiol proteome in cellular hydrogen peroxide responses. Antioxid. Redox Signal. 2010, 13, 731–743. [Google Scholar] [CrossRef] [Green Version]
- Wood, Z.A.; Schröder, E.; Robin Harris, J.; Poole, L.B. Structure, mechanism and regulation of peroxiredoxins. Trends Biochem. Sci. 2003, 28, 32–40. [Google Scholar] [CrossRef]
- Wood, Z.A.; Poole, L.B.; Karplus, P.A. Peroxiredoxin evolution and the regulation of hydrogen peroxide signaling. Science 2003, 300, 650–653. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.G.; Woo, H.A. Multiple functions of peroxiredoxins: Peroxidases, sensors and regulators of the intracellular messenger H2O2, and protein chaperones. Antioxid. Redox Signal. 2011, 15, 781–794. [Google Scholar] [CrossRef] [PubMed]
- Knoops, B.; Goemaere, J.; Van der Eecken, V.; Declercq, J.P. Peroxiredoxin 5: Structure, mechanism, and function of the mammalian atypical 2-Cys peroxiredoxin. Antioxid. Redox Signal. 2011, 15, 817–829. [Google Scholar] [CrossRef]
- Fisher, A.B. Peroxiredoxin 6: A bifunctional enzyme with glutathione peroxidase and phospholipase A2 activities. Antioxid. Redox Signal. 2011, 15, 831–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhee, S.G. Cell signaling. H2O2, a necessary evil for cell signaling. Science 2006, 312, 1882–1883. [Google Scholar] [CrossRef]
- Chae, H.Z.; Chung, S.J.; Rhee, S.G. Thioredoxin-dependent peroxide reductase from yeast. J. Biol. Chem. 1994, 269, 27670–27678. [Google Scholar] [CrossRef]
- Chae, H.Z.; Uhm, T.B.; Rhee, S.G. Dimerization of thiol-specific antioxidant and the essential role of cysteine 47. Proc. Natl. Acad. Sci. USA 1994, 91, 7022–7026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirotsu, S.; Abe, Y.; Okada, K.; Nagahara, N.; Hori, H.; Nishino, T.; Hakoshima, T. Crystal structure of a multifunctional 2-Cys peroxiredoxin heme-binding protein 23 kDa/proliferation-associated gene product. Proc. Natl. Acad. Sci. USA 1999, 96, 12333–12338. [Google Scholar] [CrossRef] [Green Version]
- Seo, M.S.; Kang, S.W.; Kim, K.; Baines, I.C.; Lee, T.H.; Rhee, S.G. Identification of a new type of mammalian peroxiredoxin that forms an intramolecular disulfide as a reaction intermediate. J. Biol. Chem. 2000, 275, 20346–20354. [Google Scholar] [CrossRef] [Green Version]
- Declercq, J.P.; Evrard, C.; Clippe, A.; Stricht, D.V.; Bernard, A.; Knoops, B. Crystal structure of human peroxiredoxin 5, a novel type of mammalian peroxiredoxin at 1.5 A resolution. J. Mol. Biol. 2001, 311, 751–759. [Google Scholar] [CrossRef] [Green Version]
- Smeets, A.; Marchand, C.; Linard, D.; Knoops, B.; Declercq, J.P. The crystal structures of oxidized forms of human peroxiredoxin 5 with an intramolecular disulfide bond confirm the proposed enzymatic mechanism for atypical 2-Cys peroxiredoxins. Arch. Biochem. Biophys. 2008, 477, 98–104. [Google Scholar] [CrossRef]
- Manevich, Y.; Feinstein, S.I.; Fisher, A.B. Activation of the antioxidant enzyme 1-CYS peroxiredoxin requires glutathionylation mediated by heterodimerization with pi GST. Proc. Natl. Acad. Sci. USA 2004, 101, 3780–3785. [Google Scholar] [CrossRef] [Green Version]
- Ralat, L.A.; Manevich, Y.; Fisher, A.B.; Colman, R.F. Direct evidence for the formation of a complex between 1-cysteine peroxiredoxin and glutathione S-transferase pi with activity changes in both enzymes. Biochemistry 2006, 45, 360–372. [Google Scholar] [CrossRef] [PubMed]
- Ralat, L.A.; Misquitta, S.A.; Manevich, Y.; Fisher, A.B.; Colman, R.F. Characterization of the complex of glutathione S-transferase pi and 1-cysteine peroxiredoxin. Arch. Biochem. Biophys. 2008, 474, 109–118. [Google Scholar] [CrossRef] [Green Version]
- Choi, H.J.; Kang, S.W.; Yang, C.H.; Rhee, S.G.; Ryu, S.E. Crystal structure of a novel human peroxidase enzyme at 2.0 A resolution. Nat. Struct. Biol. 1998, 5, 400–406. [Google Scholar] [CrossRef]
- Yang, K.S.; Kang, S.W.; Woo, H.A.; Hwang, S.C.; Chae, H.Z.; Kim, K.; Rhee, S.G. Inactivation of human peroxiredoxin I during catalysis as the result of the oxidation of the catalytic site cysteine to cysteine-sulfinic acid. J. Biol. Chem. 2002, 277, 38029–38036. [Google Scholar] [CrossRef] [Green Version]
- Chang, T.S.; Jeong, W.; Woo, H.A.; Lee, S.M.; Park, S.; Rhee, S.G. Characterization of mammalian sulfiredoxin and its reactivation of hyperoxidized peroxiredoxin through reduction of cysteine sulfinic acid in the active site to cysteine. J. Biol. Chem. 2004, 279, 50994–51001. [Google Scholar] [CrossRef] [Green Version]
- Jang, H.H.; Lee, K.O.; Chi, Y.H.; Jung, B.G.; Park, S.K.; Park, J.H.; Lee, J.R.; Lee, S.S.; Moon, J.C.; Yun, J.W.; et al. Two enzymes in one; two yeast peroxiredoxins display oxidative stress-dependent switching from a peroxidase to a molecular chaperone function. Cell 2004, 117, 625–635. [Google Scholar] [CrossRef]
- Lim, J.C.; Choi, H.I.; Park, Y.S.; Nam, H.W.; Woo, H.A.; Kwon, K.S.; Kim, Y.S.; Rhee, S.G.; Kim, K.; Chae, H.Z. Irreversible oxidation of the active-site cysteine of peroxiredoxin to cysteine sulfonic acid for enhanced molecular chaperone activity. J. Biol. Chem. 2008, 283, 28873–28880. [Google Scholar] [CrossRef] [Green Version]
- Rhee, S.G.; Jeong, W.; Chang, T.S.; Woo, H.A. Sulfiredoxin, the cysteine sulfinic acid reductase specific to 2-Cys peroxiredoxin: Its discovery, mechanism of action, and biological significance. Kidney Int. Suppl. 2007, 72, S3–S8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woo, H.A.; Yim, S.H.; Shin, D.H.; Kang, D.; Yu, D.Y.; Rhee, S.G. Inactivation of peroxiredoxin I by phosphorylation allows localized H(2)O(2) accumulation for cell signaling. Cell 2010, 140, 517–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parmigiani, R.B.; Xu, W.S.; Venta-Perez, G.; Erdjument-Bromage, H.; Yaneva, M.; Tempst, P.; Marks, P.A. HDAC6 is a specific deacetylase of peroxiredoxins and is involved in redox regulation. Proc. Natl. Acad. Sci. USA 2008, 105, 9633–9638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, J.; Nakamura, T.; Cho, D.H.; Gu, Z.; Lipton, S.A. S-nitrosylation of peroxiredoxin 2 promotes oxidative stress-induced neuronal cell death in Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2007, 104, 18742–18747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, J.; Schulte, J.; Knight, A.; Leslie, N.R.; Zagozdzon, A.; Bronson, R.; Manevich, Y.; Beeson, C.; Neumann, C.A. Prdx1 inhibits tumorigenesis via regulating PTEN/AKT activity. EMBO J. 2009, 28, 1505–1517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Park, J.; Han, S.J.; Yang, S.Y.; Yoon, H.J.; Park, I.; Woo, H.A.; Lee, S.R. Redox regulation of tumor suppressor PTEN in cell signaling. Redox Biol. 2020, 34, 101553. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Tseng, Y.; White, M.F. Insulin signaling meets mitochondria in metabolism. Trends Endocrinol. Metab. 2010, 21, 589–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loh, K.; Deng, H.; Fukushima, A.; Cai, X.; Boivin, B.; Galic, S.; Bruce, C.; Shields, B.J.; Skiba, B.; Ooms, L.M.; et al. Reactive oxygen species enhance insulin sensitivity. Cell Metab. 2009, 10, 260–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cline, G.W.; Petersen, K.F.; Krssak, M.; Shen, J.; Hundal, R.S.; Trajanoski, Z.; Inzucchi, S.; Dresner, A.; Rothman, D.L.; Shulman, G.I. Impaired glucose transport as a cause of decreased insulin-stimulated muscle glycogen synthesis in type 2 diabetes. N. Engl. J. Med. 1999, 341, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Carracedo, A.; Pandolfi, P.P. The PTEN-PI3K pathway: Of feedbacks and cross-talks. Oncogene 2008, 27, 5527–5541. [Google Scholar] [CrossRef] [Green Version]
- Zhao, W.; Fan, G.C.; Zhang, Z.G.; Bandyopadhyay, A.; Zhou, X.; Kranias, E.G. Protection of peroxiredoxin II on oxidative stress-induced cardiomyocyte death and apoptosis. Basic Res. Cardiol. 2009, 104, 377–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, Y.H.; Kim, S.U.; Kwon, T.H.; Lee, D.S.; Ha, H.L.; Park, D.S.; Woo, E.J.; Lee, S.H.; Kim, J.M.; Chae, H.B.; et al. Peroxiredoxin II is essential for preventing hemolytic anemia from oxidative stress through maintaining hemoglobin stability. Biochem. Biophys. Res. Commun. 2012, 426, 427–432. [Google Scholar] [CrossRef]
- Kim, S.U.; Jin, M.H.; Kim, Y.S.; Lee, S.H.; Cho, Y.S.; Cho, K.J.; Lee, K.S.; Kim, Y.I.; Kim, G.W.; Kim, J.M.; et al. Peroxiredoxin II preserves cognitive function against age-linked hippocampal oxidative damage. Neurobiol. Aging 2011, 32, 1054–1068. [Google Scholar] [CrossRef]
- Kim, S.U.; Park, Y.H.; Kim, J.M.; Sun, H.N.; Song, I.S.; Huang, S.M.; Lee, S.H.; Chae, J.I.; Hong, S.; Sik Choi, S.; et al. Dominant role of peroxiredoxin/JNK axis in stemness regulation during neurogenesis from embryonic stem cells. Stem Cells 2014, 32, 998–1011. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Park, S.J.; Chae, U.; Seong, J.; Lee, H.S.; Lee, S.R.; Lee, S.; Lee, D.S. Peroxiredoxin 2 mediates insulin sensitivity of skeletal muscles through regulation of protein tyrosine phosphatase oxidation. Int. J. Biochem. Cell Biol. 2018, 99, 80–90. [Google Scholar] [CrossRef]
- Brash, A.R. Lipoxygenases: Occurrence, functions, catalysis, and acquisition of substrate. J. Biol. Chem. 1999, 274, 23679–23682. [Google Scholar] [CrossRef] [Green Version]
- Wisastra, R.; Dekker, F.J. Inflammation, Cancer and Oxidative Lipoxygenase Activity are Intimately Linked. Cancers 2014, 6, 1500–1521. [Google Scholar] [CrossRef] [Green Version]
- Sigal, E.; Craik, C.S.; Highland, E.; Grunberger, D.; Costello, L.L.; Dixon, R.A.; Nadel, J.A. Molecular cloning and primary structure of human 15-lipoxygenase. Biochem. Biophys. Res. Commun. 1988, 157, 457–464. [Google Scholar] [CrossRef]
- Brash, A.R.; Boeglin, W.E.; Chang, M.S. Discovery of a second 15S-lipoxygenase in humans. Proc. Natl. Acad. Sci. USA 1997, 94, 6148–6152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shappell, S.B.; Boeglin, W.E.; Olson, S.J.; Kasper, S.; Brash, A.R. 15-lipoxygenase-2 (15-LOX-2) is expressed in benign prostatic epithelium and reduced in prostate adenocarcinoma. Am. J. Pathol. 1999, 155, 235–245. [Google Scholar] [CrossRef] [Green Version]
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting cancer cells by ROS-mediated mechanisms: A radical therapeutic approach? Nat. Rev. Drug Discov. 2009, 8, 579–591. [Google Scholar] [CrossRef]
- Klil-Drori, A.J.; Ariel, A. 15-Lipoxygenases in cancer: A double-edged sword? Prostaglandins Other Lipid Mediat. 2013, 106, 16–22. [Google Scholar] [CrossRef]
- Middleton, M.K.; Zukas, A.M.; Rubinstein, T.; Jacob, M.; Zhu, P.; Zhao, L.; Blair, I.; Puré, E. Identification of 12/15-lipoxygenase as a suppressor of myeloproliferative disease. J. Exp. Med. 2006, 203, 2529–2540. [Google Scholar] [CrossRef] [PubMed]
- Mahipal, S.V.; Subhashini, J.; Reddy, M.C.; Reddy, M.M.; Anilkumar, K.; Roy, K.R.; Reddy, G.V.; Reddanna, P. Effect of 15-lipoxygenase metabolites, 15-(S)-HPETE and 15-(S)-HETE on chronic myelogenous leukemia cell line K-562: Reactive oxygen species (ROS) mediate caspase-dependent apoptosis. Biochem. Pharmacol. 2007, 74, 202–214. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Park, J.; Han, S.J.; Lim, Y.; Park, I.; Kim, J.S.; Woo, H.A.; Lee, S.R. Peroxiredoxin III Protects Tumor Suppressor PTEN from Oxidation by 15-Hydroperoxy-eicosatetraenoic Acid. Oxid. Med. Cell Longev. 2019, 2019, 2828493. [Google Scholar] [CrossRef] [PubMed]
- Castaldo, S.A.; Ajime, T.; Serrão, G.; Anastácio, F.; Rosa, J.T.; Giacomantonio, C.A.; Howarth, A.; Hill, R.; Madureira, P.A. Annexin A2 Regulates AKT Upon H2O2-Dependent Signaling Activation in Cancer Cells. Cancers 2019, 11, 492. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
PTEN and PI3K/Akt signaling pathway. Triggering of PI3K by ligands-activated receptors (RTK, GPCR) leads to PIP3 generation. Akt is then activated through phosphorylation by mTORC2 and PDK1, which triggers the downstream signaling through a series of phosphorylation reactions. Activated Akt promotes translation of glucose transporter 4 (GLUT4) through direct inhibition of AS160 (Akt substrate of 160 kDa), which leads to the increase of glucose uptake. Furthermore, through phosphorylation, Akt inactivates forkhead box protein O (FOXO), tuberous sclerosis complex 1/2 (TSC1/2), and glycogen synthase kinase 3 (GSK3), which increases cell survival, cell growth, and proliferation [23]. PTEN dephosphorylates PIP3 to PIP2 and reduces PIP3 accumulation. Black arrows (activating), red arrows (blocking).
Figure 1.
PTEN and PI3K/Akt signaling pathway. Triggering of PI3K by ligands-activated receptors (RTK, GPCR) leads to PIP3 generation. Akt is then activated through phosphorylation by mTORC2 and PDK1, which triggers the downstream signaling through a series of phosphorylation reactions. Activated Akt promotes translation of glucose transporter 4 (GLUT4) through direct inhibition of AS160 (Akt substrate of 160 kDa), which leads to the increase of glucose uptake. Furthermore, through phosphorylation, Akt inactivates forkhead box protein O (FOXO), tuberous sclerosis complex 1/2 (TSC1/2), and glycogen synthase kinase 3 (GSK3), which increases cell survival, cell growth, and proliferation [23]. PTEN dephosphorylates PIP3 to PIP2 and reduces PIP3 accumulation. Black arrows (activating), red arrows (blocking).
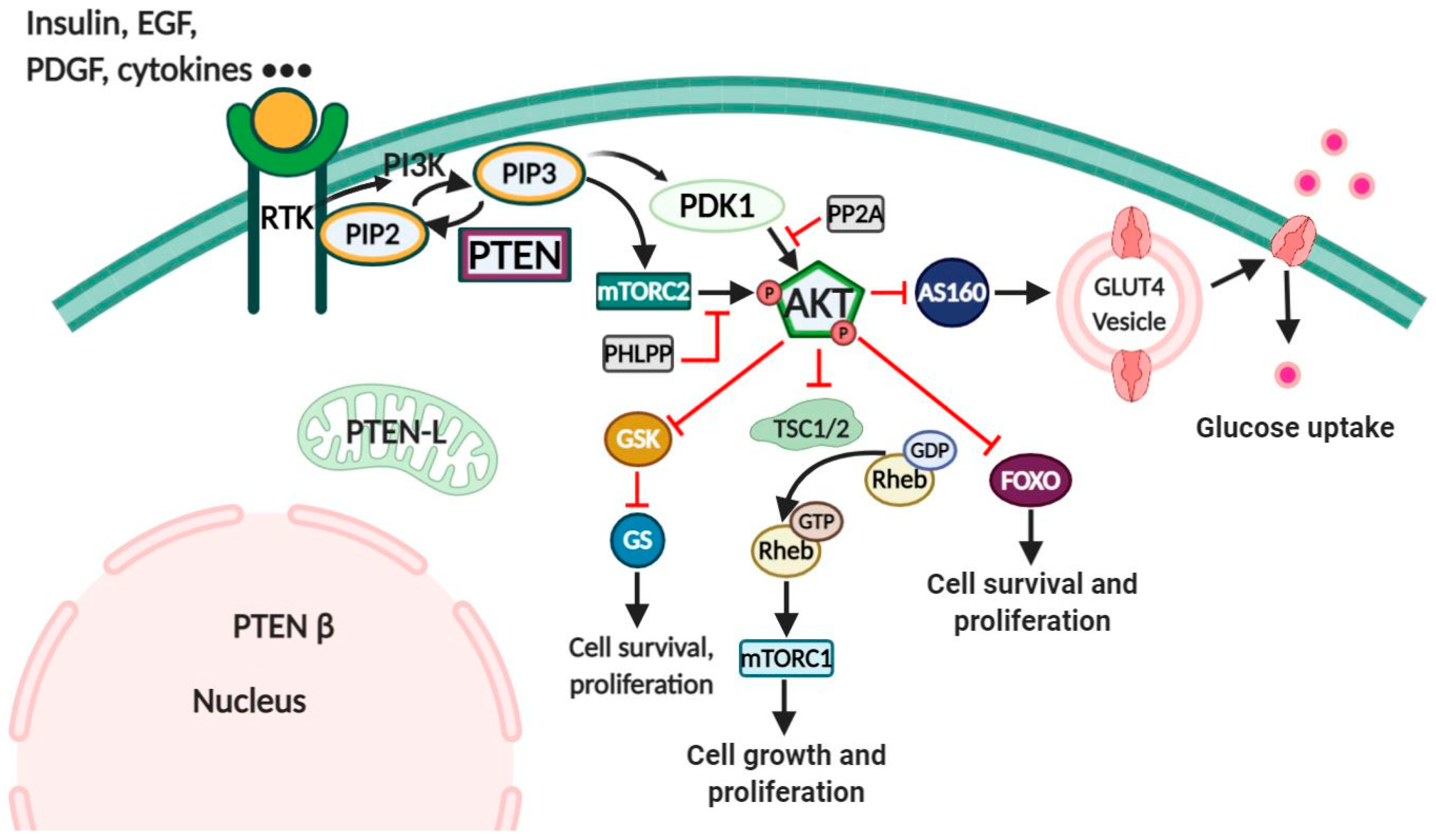
Figure 2.
Reactions between Prx I, Trx, Srx and H2O2.

Figure 3.
Role of Prx II in the redox regulation of PTEN. Insulin induced the activation of PI3K, resulting in the conversion of PIP2 to PIP3. PIP3 induced the production of H2O2 by activating the NOX complex. NOX-induced H2O2 oxidized PTEN and started the forming of the intramolecular disulfide bond, which inactivates PTEN lipid phosphatase. The presence of Prx II reduced PTEN oxidation levels. Black arrows (activating), red arrows (blocking).
Figure 3.
Role of Prx II in the redox regulation of PTEN. Insulin induced the activation of PI3K, resulting in the conversion of PIP2 to PIP3. PIP3 induced the production of H2O2 by activating the NOX complex. NOX-induced H2O2 oxidized PTEN and started the forming of the intramolecular disulfide bond, which inactivates PTEN lipid phosphatase. The presence of Prx II reduced PTEN oxidation levels. Black arrows (activating), red arrows (blocking).

Figure 4.
A schematic model for the effect of 12/15s-HpETE and Prx III on the redox regulation of PTEN. Prx III plays an important role in the control of endogenous lipid peroxide-induced redox regulation of PTEN. 12/15s-HpETE inhibits the Trx redox system by inducing dimerization of Trx, resulting in the delayed reduction of oxidized PTEN and oxidized Prx. Prx III prevents 12/15s-HpETE-mediated PTEN oxidation by catalyzing the reduction of lipid peroxide. Black arrows (activating), red arrows (blocking).
Figure 4.
A schematic model for the effect of 12/15s-HpETE and Prx III on the redox regulation of PTEN. Prx III plays an important role in the control of endogenous lipid peroxide-induced redox regulation of PTEN. 12/15s-HpETE inhibits the Trx redox system by inducing dimerization of Trx, resulting in the delayed reduction of oxidized PTEN and oxidized Prx. Prx III prevents 12/15s-HpETE-mediated PTEN oxidation by catalyzing the reduction of lipid peroxide. Black arrows (activating), red arrows (blocking).

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Nguyen Huu, T.; Park, J.; Zhang, Y.; Park, I.; Yoon, H.J.; Woo, H.A.; Lee, S.-R. Redox Regulation of PTEN by Peroxiredoxins. Antioxidants 2021, 10, 302. https://doi.org/10.3390/antiox10020302
AMA Style
Nguyen Huu T, Park J, Zhang Y, Park I, Yoon HJ, Woo HA, Lee S-R. Redox Regulation of PTEN by Peroxiredoxins. Antioxidants. 2021; 10(2):302. https://doi.org/10.3390/antiox10020302
Chicago/Turabian StyleNguyen Huu, Thang, Jiyoung Park, Ying Zhang, Iha Park, Hyun Joong Yoon, Hyun Ae Woo, and Seung-Rock Lee. 2021. "Redox Regulation of PTEN by Peroxiredoxins" Antioxidants 10, no. 2: 302. https://doi.org/10.3390/antiox10020302
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.