
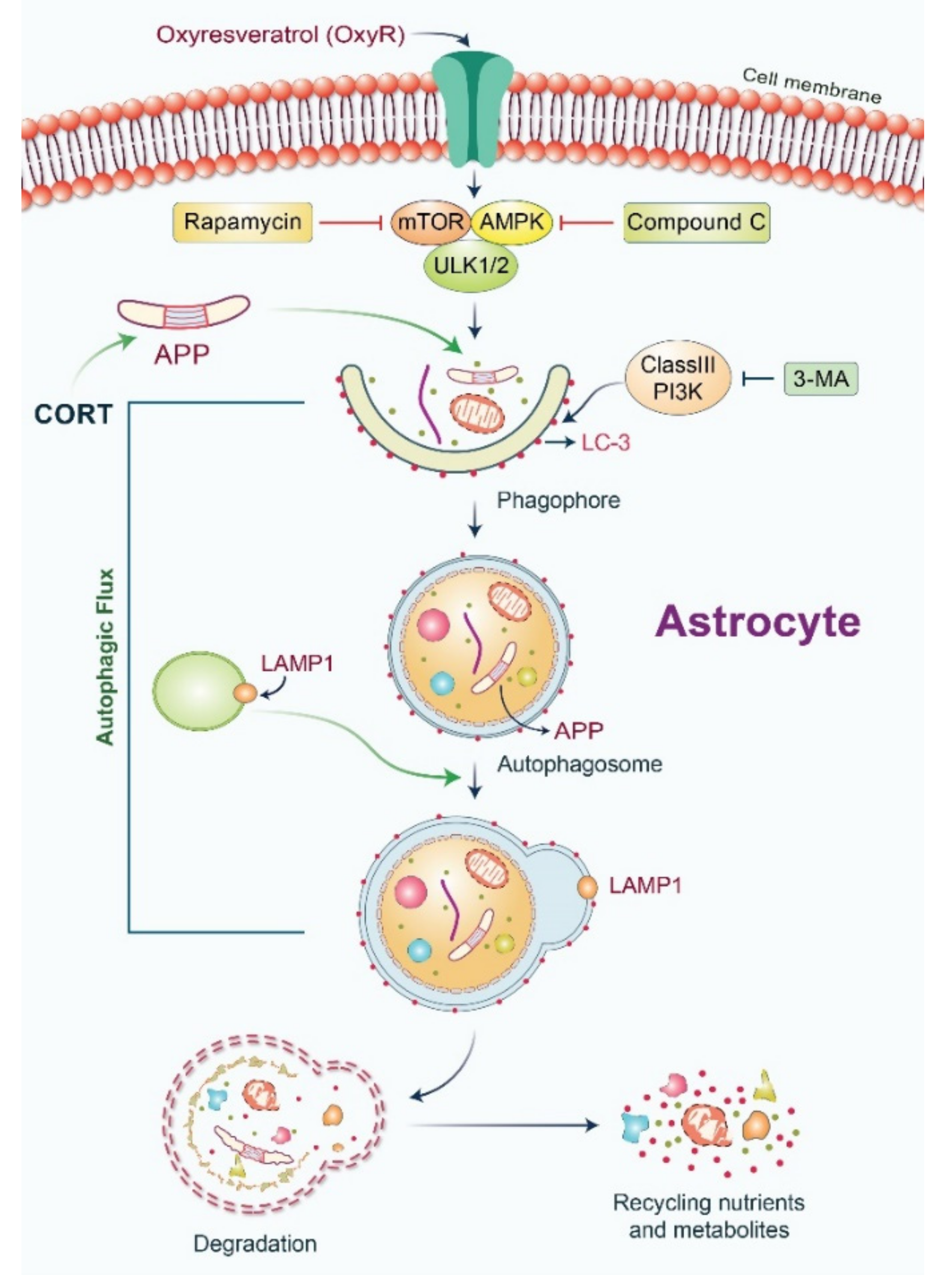
Antioxidant Compound, Oxyresveratrol, Inhibits APP Production through the AMPK/ULK1/mTOR-Mediated Autophagy Pathway in Mouse Cortical Astrocytes

Abstract
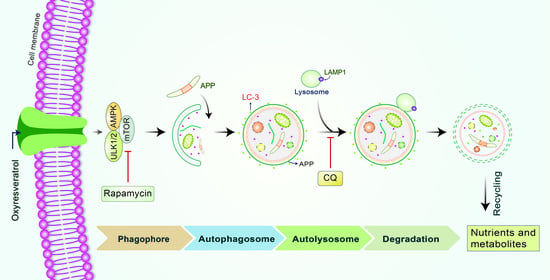
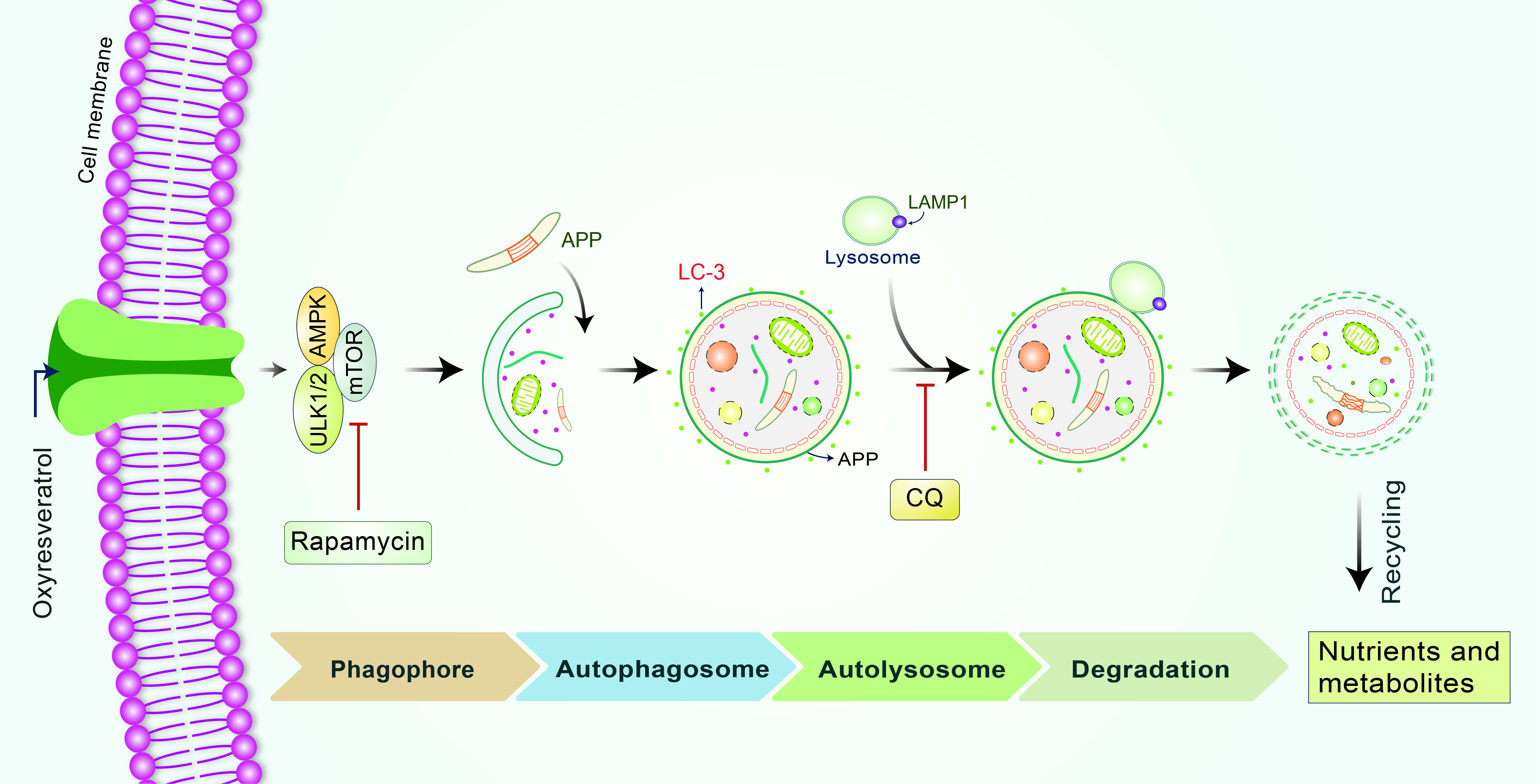
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Cortical Astrocyte Culture
2.3. Cortical Neuron Cultures
2.4. Immunocytochemical Analysis
2.5. ULK1 Small Interfering RNA (siRNA) Transfection
2.6. Immunoblotting
2.7. Autophagic Flux Evaluation
2.8. Statistical Analysis
3. Results
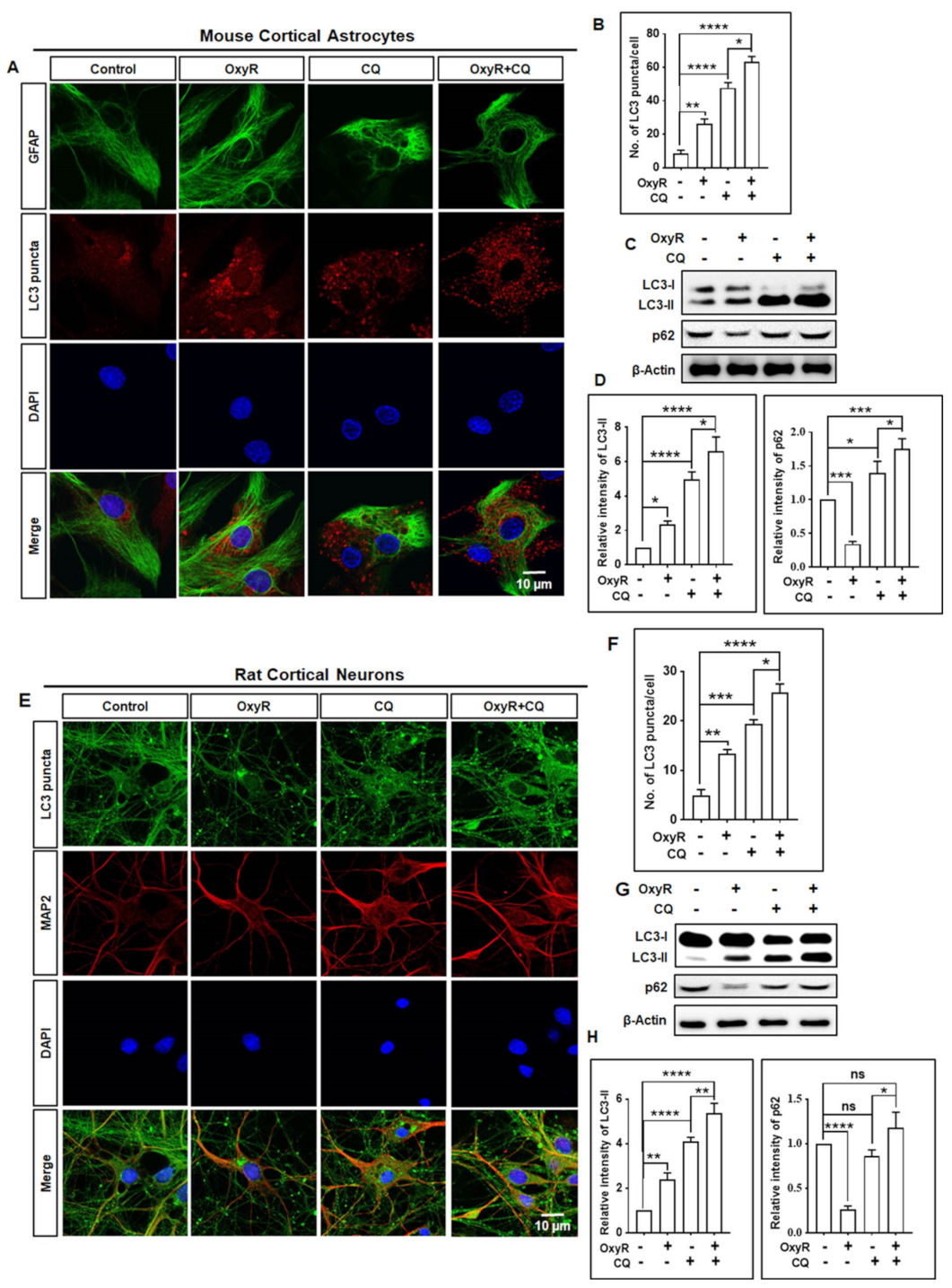
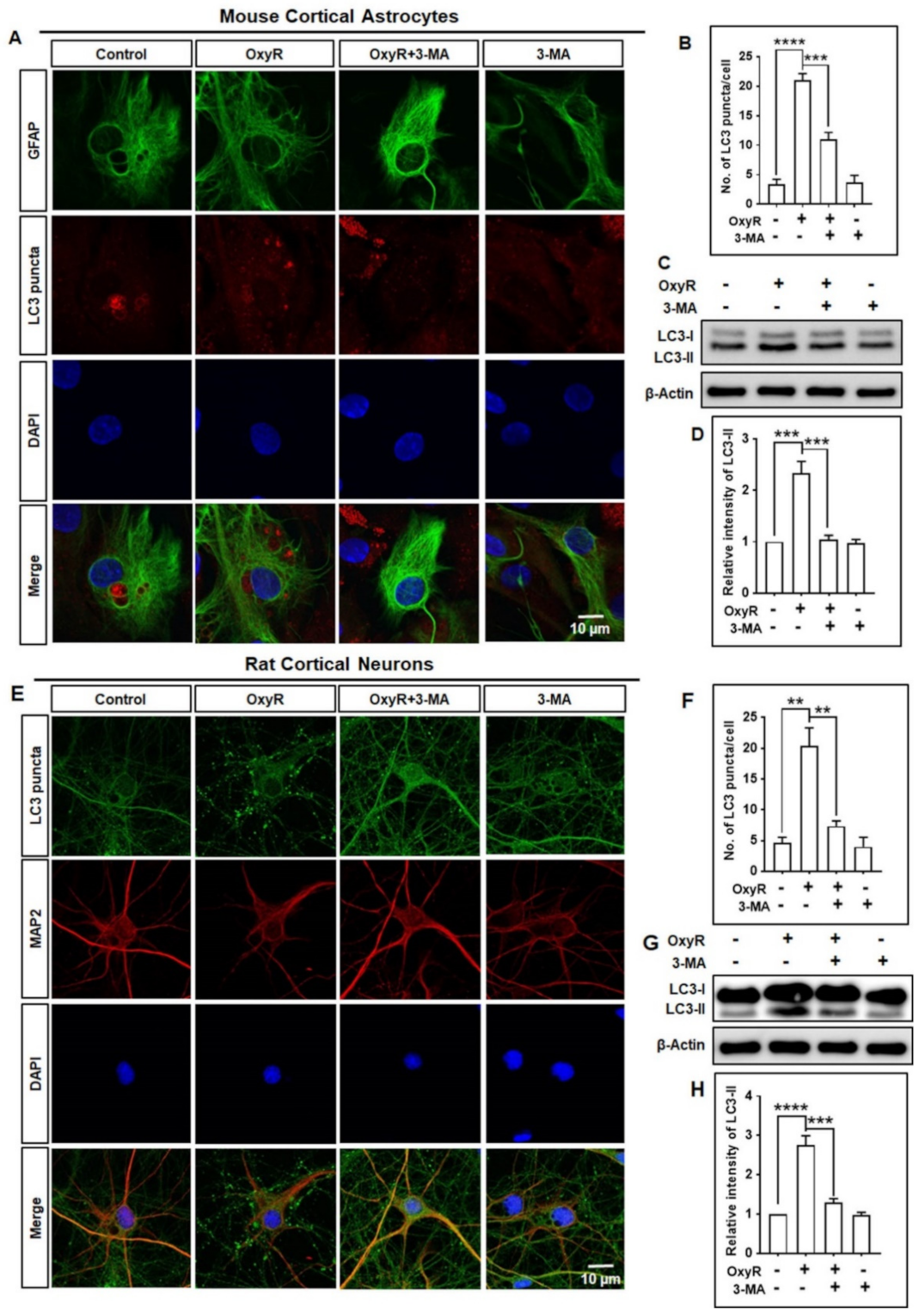
3.1. OxyR Treatment Activates Autophagic Flux in Mouse Cortical Astrocytes and Rat Cortical Neurons
3.2. OxyR Activates the Autophagic Pathway in Mouse Cortical Astrocytes and Rat Cortical Neurons
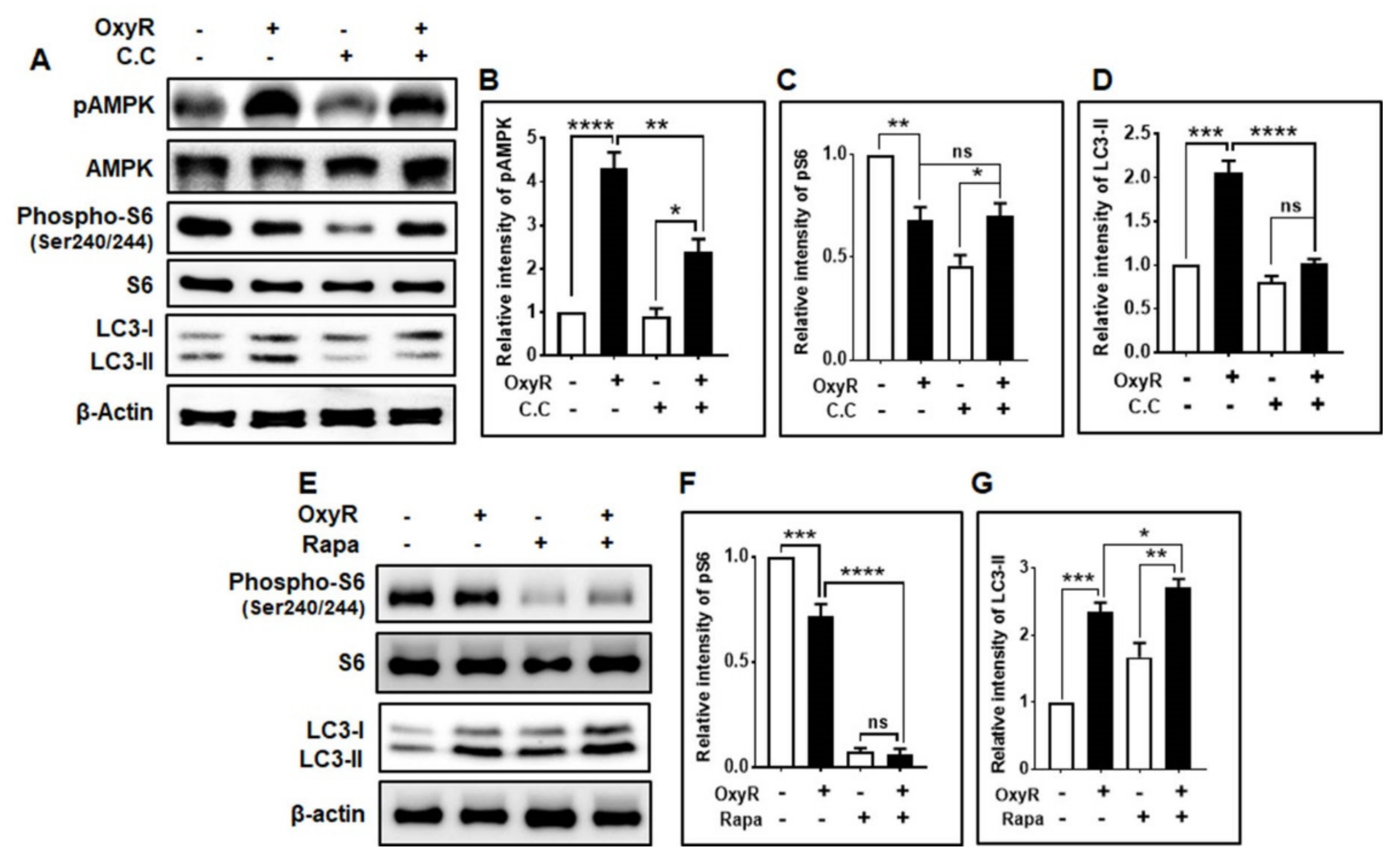
3.3. OxyR-Mediated Autophagy Is Dependent on the AMPK-mTOR Signaling Pathway in Mouse Cortical Astrocytes
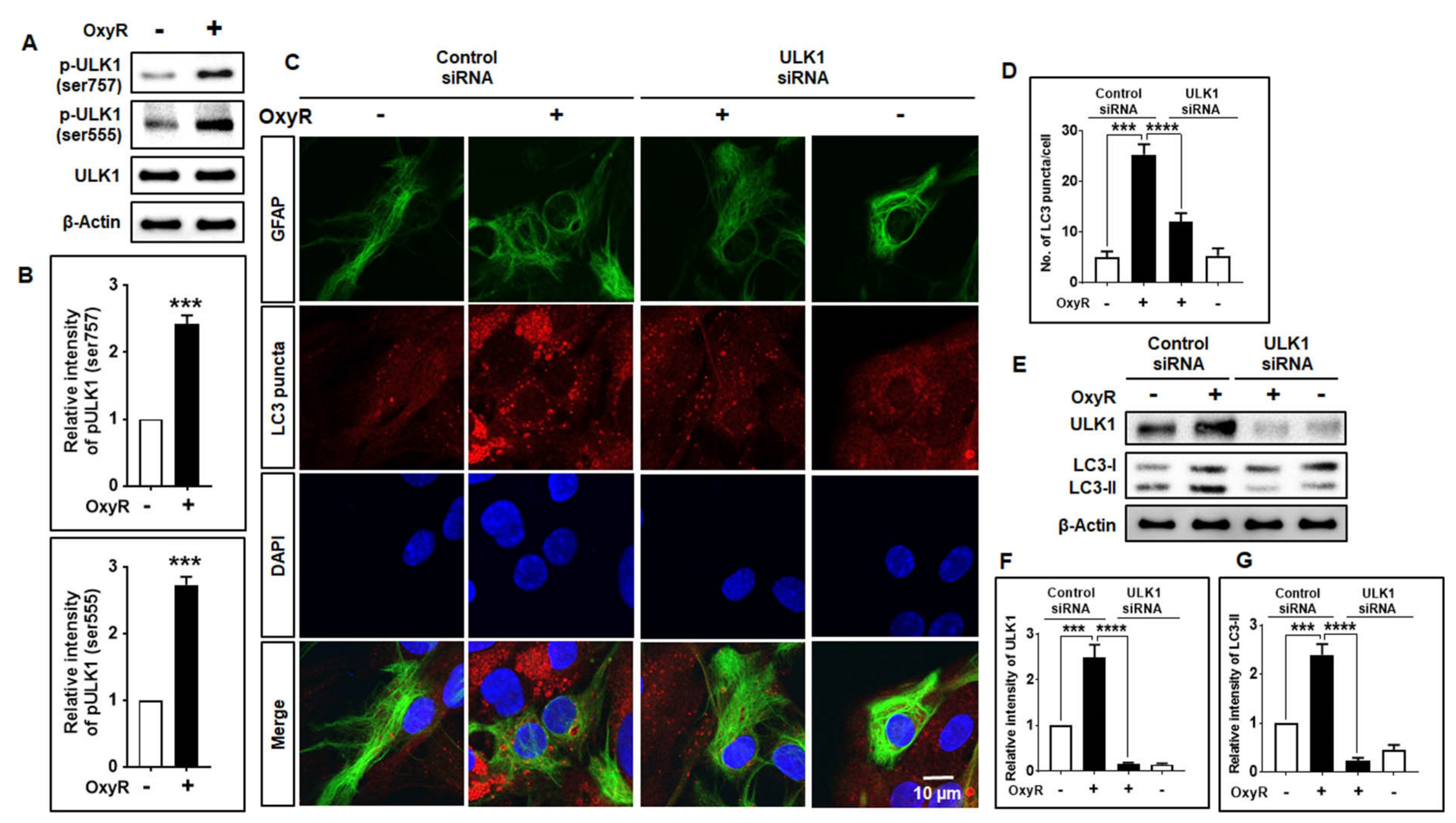
3.4. OxyR Activates the Autophagy Initiation Protein ULK1 in Mouse Cortical Astrocytes
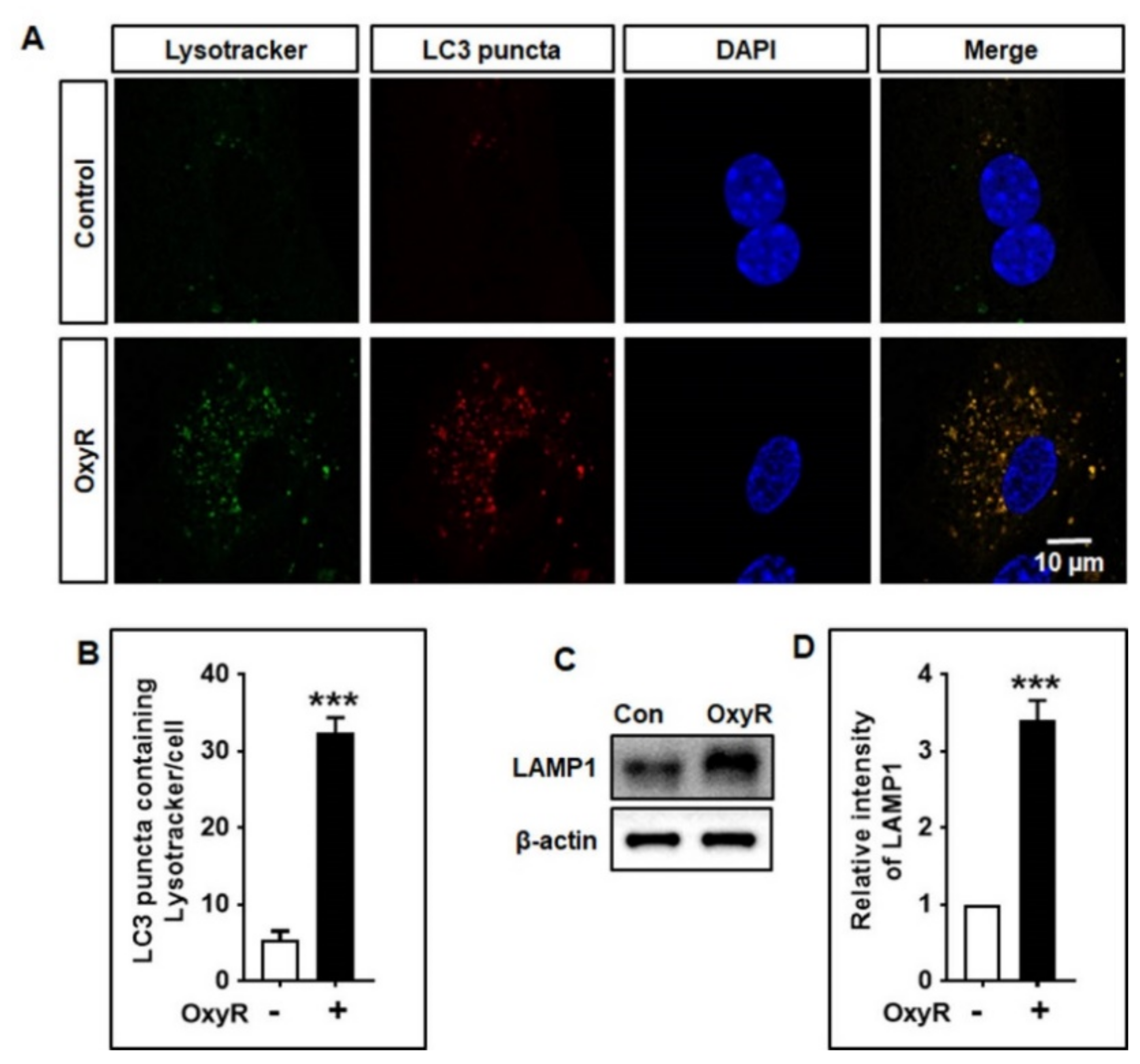
3.5. OxyR Activates Lysosomal Protein LAMP1 in Mouse Cortical Astrocytes
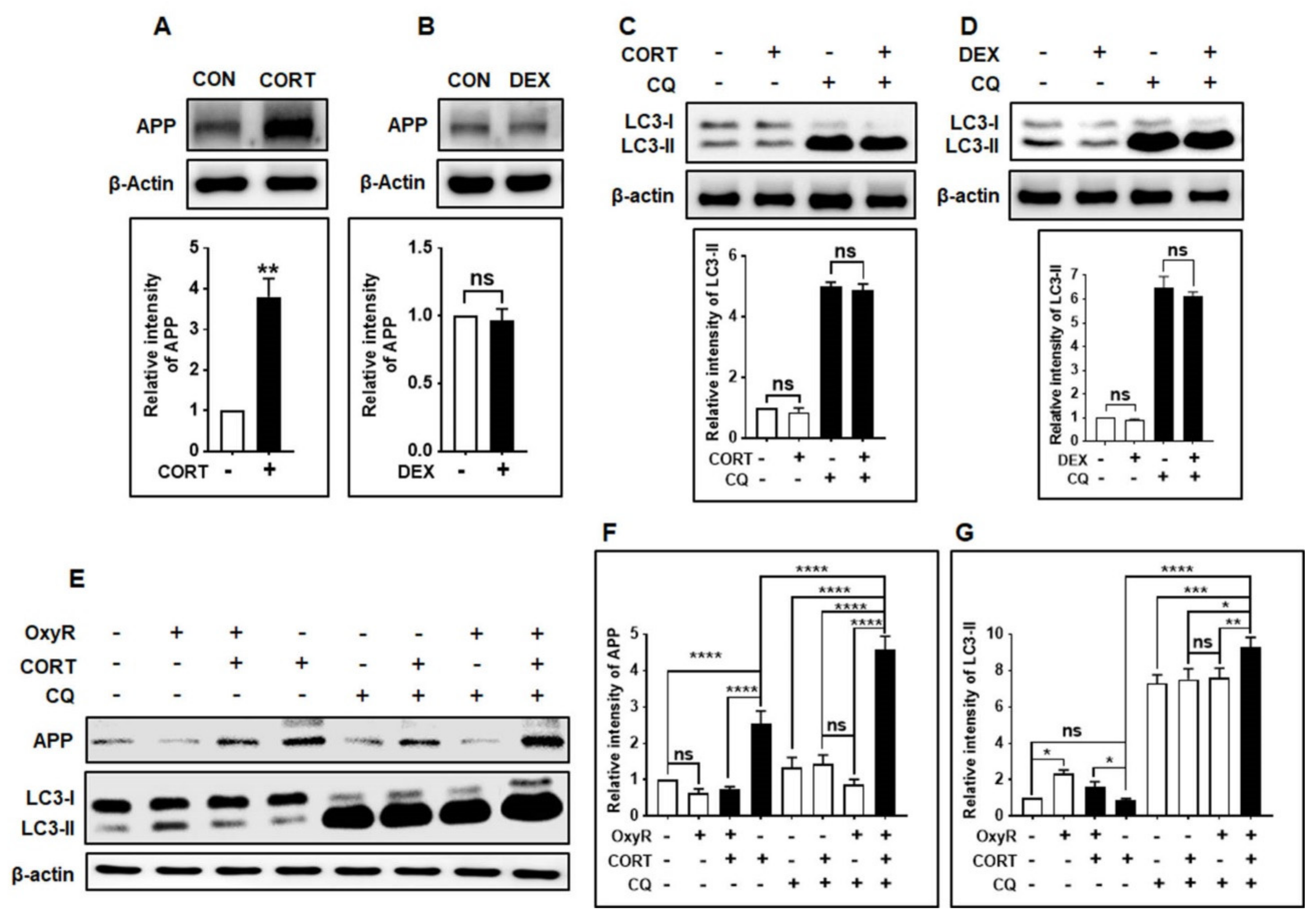
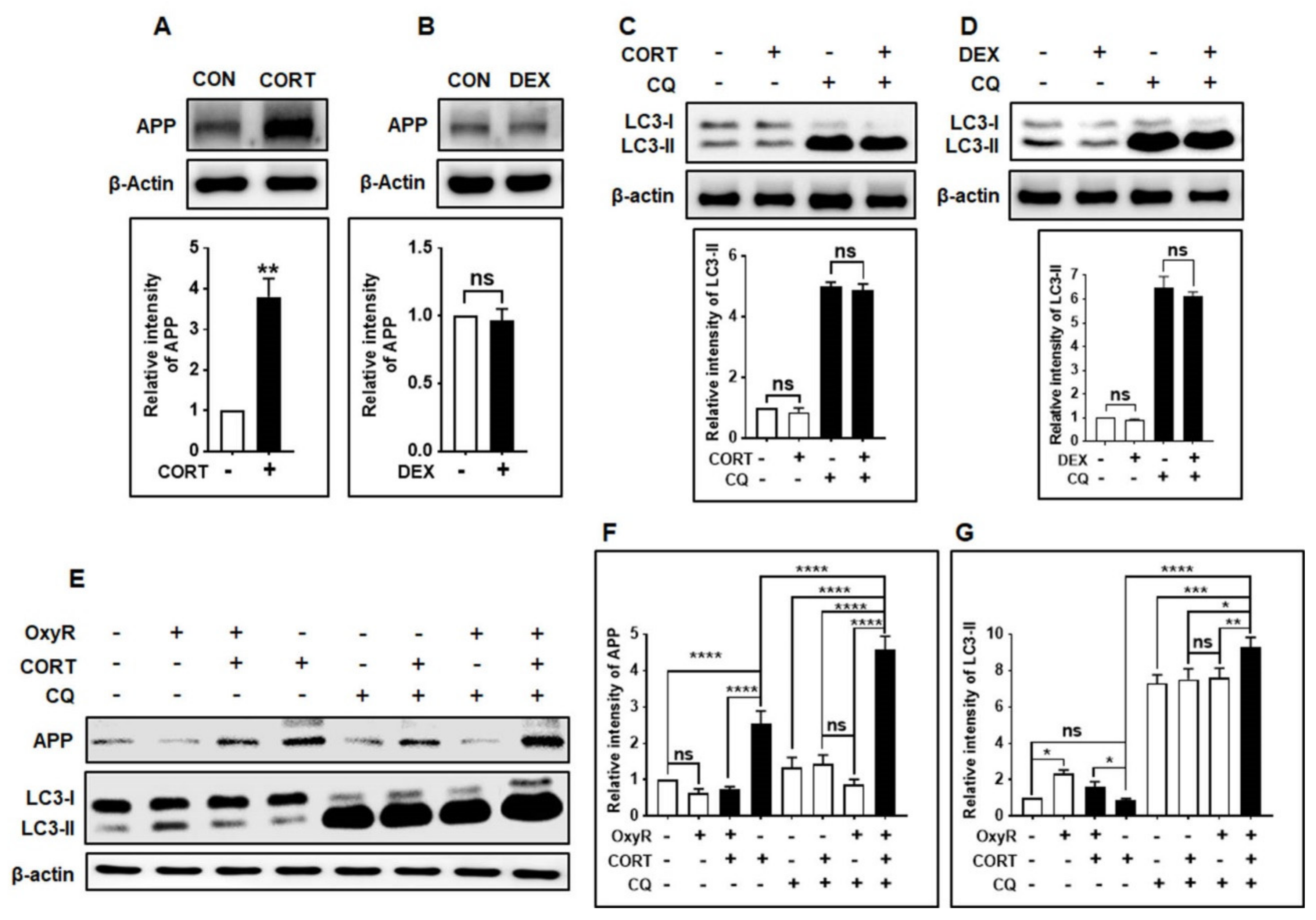
3.6. OxyR Decreases Glucocorticoid-Induced APP Expression via the Autophagy Pathway in Mouse Cortical Astrocytes
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Andrabi, A.S.; Spina, M.G.; Lorenz, P.; Ebmeyer, U.; Wolf, G.; Horn, T.F. Oxyresveratrol (trans-2,3′,4,5′-tetrahydroxystilbene) is neuroprotective and inhibits the apoptotic cell death in transient cerebral ischemia. Brain Res. 2004, 1017, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Qiu, F.; Komatsu, K.; Kawasaki, K.; Saito, K.; Yao, X.; Kano, Y. A Novel Stilbene Glucoside, Oxyresveratrol 3′-O-β-Glucopyranoside, from the Root Bark ofMorus alba. Planta Med. 1996, 62, 559–561. [Google Scholar] [CrossRef]
- Lorenz, P.; Roychowdhury, S.; Engelmann, M.; Wolf, G.; Horn, T.F. Oxyresveratrol and resveratrol are potent antioxidants and free radical scavengers: Effect on nitrosative and oxidative stress derived from microglial cells. Nitric Oxide 2003, 9, 64–76. [Google Scholar] [CrossRef] [PubMed]
- Rahman, A.; Bishayee, K.; Sadra, A.; Huh, S.-O. Oxyresveratrol activates parallel apoptotic and autophagic cell death pathways in neuroblastoma cells. Biochim. Biophys. Acta (BBA) Gen. Subj. 2017, 1861, 23–36. [Google Scholar] [CrossRef]
- Breuer, C.; Wolf, G.; Andrabi, S.A.; Lorenz, P.; Horn, T.F. Blood–brain barrier permeability to the neuroprotectant oxyresveratrol. Neurosci. Lett. 2006, 393, 113–118. [Google Scholar] [CrossRef]
- Ban, J.Y.; Jeon, S.-Y.; Nguyen, T.T.H.; Bae, K.; Song, K.-S.; Seonga, Y.H. Neuroprotective Effect of Oxyresveratrol from Smilacis Chinae Rhizome on Amyloid. BETA. Protein (25-35)-Induced Neurotoxicity in Cultured Rat Cortical Neurons. Biol. Pharm. Bull. 2006, 29, 2419–2424. [Google Scholar] [CrossRef] [Green Version]
- Jeon, S.-Y.; Kwon, S.-H.; Seong, Y.-H.; Bae, K.; Hur, J.-M.; Lee, Y.-Y.; Suh, D.-Y.; Song, K.-S. β-secretase (BACE1)-inhibiting stilbenoids from Smilax Rhizoma. Phytomedicine 2007, 14, 403–408. [Google Scholar] [CrossRef]
- Rahman, A.; Rahman, S.; Uddin, J.; Mamum-Or-Rashid, A.N.M.; Pang, M.-G.; Rhim, H. Emerging risk of environmental factors: Insight mechanisms of Alzheimer’s diseases. Environ. Sci. Pollut. Res. 2020, 27, 44659–44672. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, J.J.; Olabarria, M.; Chvatal, A.; Verkhratsky, A.; Rodr, J.J. Astroglia in dementia and Alzheimer’s disease. Cell Death Differ. 2008, 16, 378–385. [Google Scholar] [CrossRef] [Green Version]
- Söllvander, S.; Nikitidou, E.; Brolin, R.; Söderberg, L.; Sehlin, D.; Lannfelt, L.; Erlandsson, A. Accumulation of amyloid-β by astrocytes result in enlarged endosomes and microvesicle-induced apoptosis of neurons. Mol. Neurodegener. 2016, 11, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Rahman, A.; Hwang, H.; Cho, Y.; Rhim, H. Modulation of O-GlcNAcylation Regulates Autophagy in Cortical Astrocytes. Oxidative Med. Cell. Longev. 2019, 2019, 1–13. [Google Scholar] [CrossRef]
- Frost, G.R.; Li, Y.-M. The role of astrocytes in amyloid production and Alzheimer’s disease. Open Biol. 2017, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lööv, C.; Hillered, L.; Ebendal, T.; Erlandsson, A. Engulfing Astrocytes Protect Neurons from Contact-Induced Apoptosis following Injury. PLoS ONE 2012, 7, e33090. [Google Scholar] [CrossRef] [Green Version]
- Chung, W.-S.; Clarke, L.E.; Wang, G.X.; Stafford, B.K.; Sher, A.; Chakraborty, C.; Joung, J.; Foo, L.C.; Thompson, A.; Chen, C.; et al. Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. Nat. Cell Biol. 2013, 504, 394–400. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Connor, T.O.; Vassar, R. The contribution of activated astrocytes to Aβ production: Implications for Alzheimer’s disease pathogenesis. J. Neuroinflamm. 2011, 8, 150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, T.; Zhang, D.; Zeng, Y.; Huang, T.Y.; Xu, H.; Zhao, Y. Molecular and cellular mechanisms underlying the pathogenesis of Alzheimer’s disease. Mol. Neurodegener. 2020, 15, 1–37. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, M.; Tang, J.; Song, M.; Xu, X.; Xiong, J.; Li, J.; Bai, Y. Glucocorticoids Facilitate Astrocytic Amyloid-β Peptide Deposition by Increasing the Expression of APP and BACE1 and Decreasing the Expression of Amyloid-β-Degrading Proteases. Endocrinology 2011, 152, 2704–2715. [Google Scholar] [CrossRef] [Green Version]
- Rahman, M.; Hossain, M.; Biswas, P.; Islam, R.; Uddin, M.J.; Rahman, M.D.; Rhim, H. Molecular Insights into the Multifunctional Role of Natural Compounds: Autophagy Modulation and Cancer Prevention. Biomedicines 2020, 8, 517. [Google Scholar] [CrossRef] [PubMed]
- Tong, X.; Ganta, R.R.; Liu, Z. AMP-activated protein kinase (AMPK) regulates autophagy, inflammation and immunity and contributes to osteoclast differentiation and functionabs. Biol. Cell 2020, 112, 251–264. [Google Scholar] [CrossRef]
- Uddin, S.; Rahman, A.; Kabir, T.; Behl, T.; Mathew, B.; Perveen, A.; Barreto, G.E.; Bin-Jumah, M.N.; Abdel-Daim, M.M.; Ashraf, G.M. Multifarious roles of mTOR signaling in cognitive aging and cerebrovascular dysfunction of Alzheimer’s disease. IUBMB Life 2020, 72, 1843–1855. [Google Scholar] [CrossRef]
- Yan, Q.; Han, C.; Wang, G.; Waddington, J.L.; Zheng, L.; Zhen, X. Activation of AMPK/mTORC1-Mediated Autophagy by Metformin Reverses Clk1 Deficiency-Sensitized Dopaminergic Neuronal Death. Mol. Pharmacol. 2017, 92, 640–652. [Google Scholar] [CrossRef] [Green Version]
- Turco, E.; Fracchiolla, D.; Martens, S. Recruitment and Activation of the ULK1/Atg1 Kinase Complex in Selective Autophagy. J. Mol. Biol. 2020, 432, 123–134. [Google Scholar] [CrossRef]
- Singha, B.; Laski, J.; Valdés, Y.R.; Liu, E.; DiMattia, G.E.; Shepherd, T.G. Inhibiting ULK1 kinase decreases autophagy and cell viability in high-grade serous ovarian cancer spheroids. Am. J. Cancer Res. 2020, 10, 1384–1399. [Google Scholar]
- Rahman, A.; Saha, S.K.; Rahman, S.; Uddin, J.; Uddin, S.; Pang, M.-G.; Rhim, H.; Cho, S.-G. Molecular Insights Into Therapeutic Potential of Autophagy Modulation by Natural Products for Cancer Stem Cells. Front. Cell Dev. Biol. 2020, 8. [Google Scholar] [CrossRef]
- Rahman, A.; Hwang, H.; Nah, S.-Y.; Rhim, H. Gintonin stimulates autophagic flux in primary cortical astrocytes. J. Ginseng Res. 2020, 44, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Rahman, A.; Cho, Y.; Hwang, H.; Rhim, H. Pharmacological Inhibition of O-GlcNAc Transferase Promotes mTOR-Dependent Autophagy in Rat Cortical Neurons. Brain Sci. 2020, 10, 958. [Google Scholar] [CrossRef] [PubMed]
- Honda, S.; Arakawa, S.; Yamaguchi, H.; Torii, S.; Sakurai, H.T.; Tsujioka, M.; Murohashi, M.; Shimizu, S. Association Between Atg5-independent Alternative Autophagy and Neurodegenerative Diseases. J. Mol. Biol. 2020, 432, 2622–2632. [Google Scholar] [CrossRef]
- Fujikake, N.; Shin, M.; Shimizu, S. Association Between Autophagy and Neurodegenerative Diseases. Front. Neurosci. 2018, 12, 255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, Y.; Hwang, H.; Rahman, A.; Chung, C.; Rhim, H. Elevated O-GlcNAcylation induces an antidepressant-like phenotype and decreased inhibitory transmission in medial prefrontal cortex. Sci. Rep. 2020, 10, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Park, D.; Lee, Y. Biphasic Activity of Chloroquine in Human Colorectal Cancer Cells. Dev. Reprod. 2014, 18, 225–231. [Google Scholar] [CrossRef] [Green Version]
- Mauthe, M.; Orhon, I.; Rocchi, C.; Zhou, X.; Luhr, M.; Hijlkema, K.-J.; Coppes, R.P.; Engedal, N.; Mari, M.; Reggiori, F. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 2018, 14, 1435–1455. [Google Scholar] [CrossRef]
- Heckmann, B.L.; Yang, X.; Zhang, X.; Liu, J. The autophagic inhibitor 3-methyladenine potently stimulates PKA-dependent lipolysis in adipocytes. Br. J. Pharmacol. 2012, 168, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G. AMP-activated/SNF1 protein kinases: Conserved guardians of cellular energy. Nat. Rev. Mol. Cell Biol. 2007, 8, 774–785. [Google Scholar] [CrossRef] [PubMed]
- Meley, D.; Bauvy, C.; Houben-Weerts, J.H.; Dubbelhuis, P.F.; Helmond, M.T.; Codogno, P.; Meijer, A.J. AMP-activated Protein Kinase and the Regulation of Autophagic Proteolysis. J. Biol. Chem. 2006, 281, 34870–34879. [Google Scholar] [CrossRef] [Green Version]
- Alers, S.; Löffler, A.S.; Wesselborg, S.; Stork, B. Role of AMPK-mTOR-Ulk1/2 in the Regulation of Autophagy: Cross Talk, Shortcuts, and Feedbacks. Mol. Cell. Biol. 2011, 32, 2–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Chhipa, R.R.; Nakano, I.; Dasgupta, B. The AMPK Inhibitor Compound C Is a Potent AMPK-Independent Antiglioma Agent. Mol. Cancer Ther. 2014, 13, 596–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yim, W.W.-Y.; Mizushima, N. Lysosome biology in autophagy. Cell Discov. 2020, 6, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, X.; Godar, R.J.; Liu, H.; Diwan, A. Enhancing lysosome biogenesis attenuates BNIP3-induced cardiomyocyte death. Autophagy 2012, 8, 297–309. [Google Scholar] [CrossRef] [Green Version]
- Blasko, I.; Veerhuis, R.; Stampfer-Kountchev, M.; Saurwein-Teissl, M.; Eikelenboom, P.; Grubeck-Loebenstein, B. Costimulatory Effects of Interferon-γ and Interleukin-1β or Tumor Necrosis Factor α on the Synthesis of Aβ1-40 and Aβ1-42 by Human Astrocytes. Neurobiol. Dis. 2000, 7, 682–689. [Google Scholar] [CrossRef] [Green Version]
- Lesné, S.; Docagne, F.; Gabriel, C.; Liot, G.; Lahiri, D.K.; Buée, L.; Plawinski, L.; Delacourte, A.; MacKenzie, E.T.; Buisson, A.; et al. Transforming Growth Factor-β1 Potentiates Amyloid-β Generation in Astrocytes and in Transgenic Mice. J. Biol. Chem. 2003, 278, 18408–18418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, K.N.; Billings, L.M.; Roozendaal, B.; McGaugh, J.L.; LaFerla, F.M. Glucocorticoids Increase Amyloid-beta and Tau Pathology in a Mouse Model of Alzheimer’s Disease. J. Neurosci. 2006, 26, 9047–9056. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N.; Yoshimori, T.; Ohsumi, Y. The Role of Atg Proteins in Autophagosome Formation. Annu. Rev. Cell Dev. Biol. 2011, 27, 107–132. [Google Scholar] [CrossRef]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.-L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, K.; Klionsky, D.J. AMPK Activates Autophagy by Phosphorylating ULK1. Circ. Res. 2011, 108, 787–788. [Google Scholar] [CrossRef] [Green Version]
- Oddo, S. The role of mTOR signaling in Alzheimer disease. Front. Biosci. 2012, S4, 941–952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Wang, H.; Zhang, D.; Luo, W.; Liu, R.; Xu, D.; Diao, L.; Liao, L.; Liu, Z. Phosphorylation of ULK1 affects autophagosome fusion and links chaperone-mediated autophagy to macroautophagy. Nat. Commun. 2018, 9, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Zachari, M.; Ganley, I.G. The mammalian ULK1 complex and autophagy initiation. Essays Biochem. 2017, 61, 585–596. [Google Scholar] [CrossRef] [Green Version]
- Egan, D.F.; Kim, J.; Shaw, R.J.; Guan, K.-L. The autophagy initiating kinase ULK1 is regulated via opposing phosphorylation by AMPK and mTOR. Autophagy 2011, 7, 643–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, A.; Bishayee, K.; Habib, K.; Sadra, A.; Huh, S.-O. 18α-Glycyrrhetinic acid lethality for neuroblastoma cells via de-regulating the Beclin-1/Bcl-2 complex and inducing apoptosis. Biochem. Pharmacol. 2016, 117, 97–112. [Google Scholar] [CrossRef]
- Mauvezin, C.; Neisch, A.L.; Ayala, C.I.; Kim, J.; Beltrame, A.; Braden, C.R.; Gardner, M.K.; Hays, T.S.; Neufeld, T.P. Coordination of autophagosome–lysosome fusion and transport by a Klp98A–Rab14 complex inDrosophila. J. Cell Sci. 2016, 129, 971–982. [Google Scholar] [CrossRef] [Green Version]
- Du, H.; Ma, L.; Chen, G.; Li, S. The effects of oxyresveratrol abrogates inflammation and oxidative stress in rat model of spinal cord injury. Mol. Med. Rep. 2017, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rahman, M.A.; Cho, Y.; Nam, G.; Rhim, H. Antioxidant Compound, Oxyresveratrol, Inhibits APP Production through the AMPK/ULK1/mTOR-Mediated Autophagy Pathway in Mouse Cortical Astrocytes. Antioxidants 2021, 10, 408. https://doi.org/10.3390/antiox10030408
Rahman MA, Cho Y, Nam G, Rhim H. Antioxidant Compound, Oxyresveratrol, Inhibits APP Production through the AMPK/ULK1/mTOR-Mediated Autophagy Pathway in Mouse Cortical Astrocytes. Antioxidants. 2021; 10(3):408. https://doi.org/10.3390/antiox10030408
Chicago/Turabian StyleRahman, Md. Ataur, Yoonjeong Cho, Ghilsoo Nam, and Hyewhon Rhim. 2021. "Antioxidant Compound, Oxyresveratrol, Inhibits APP Production through the AMPK/ULK1/mTOR-Mediated Autophagy Pathway in Mouse Cortical Astrocytes" Antioxidants 10, no. 3: 408. https://doi.org/10.3390/antiox10030408
APA StyleRahman, M. A., Cho, Y., Nam, G., & Rhim, H. (2021). Antioxidant Compound, Oxyresveratrol, Inhibits APP Production through the AMPK/ULK1/mTOR-Mediated Autophagy Pathway in Mouse Cortical Astrocytes. Antioxidants, 10(3), 408. https://doi.org/10.3390/antiox10030408