
Auraptene Enhances Junction Assembly in Cerebrovascular Endothelial Cells by Promoting Resilience to Mitochondrial Stress through Activation of Antioxidant Enzymes and mtUPR

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Oxygen-Glucose Deprivation (OGD)
2.2. Sulforhodamine B Assay (SRB) for Measurement of Cell Viability
2.3. Extraction of Protein and Western Blot
2.4. Isolation of RNA and Real-Time PCR
2.5. Flow Cytometry
2.6. Analysis of Oxygen Consumption Rate (OCR)
2.7. Immunofluorescence Staining
2.8. Statistical Analysis
3. Results
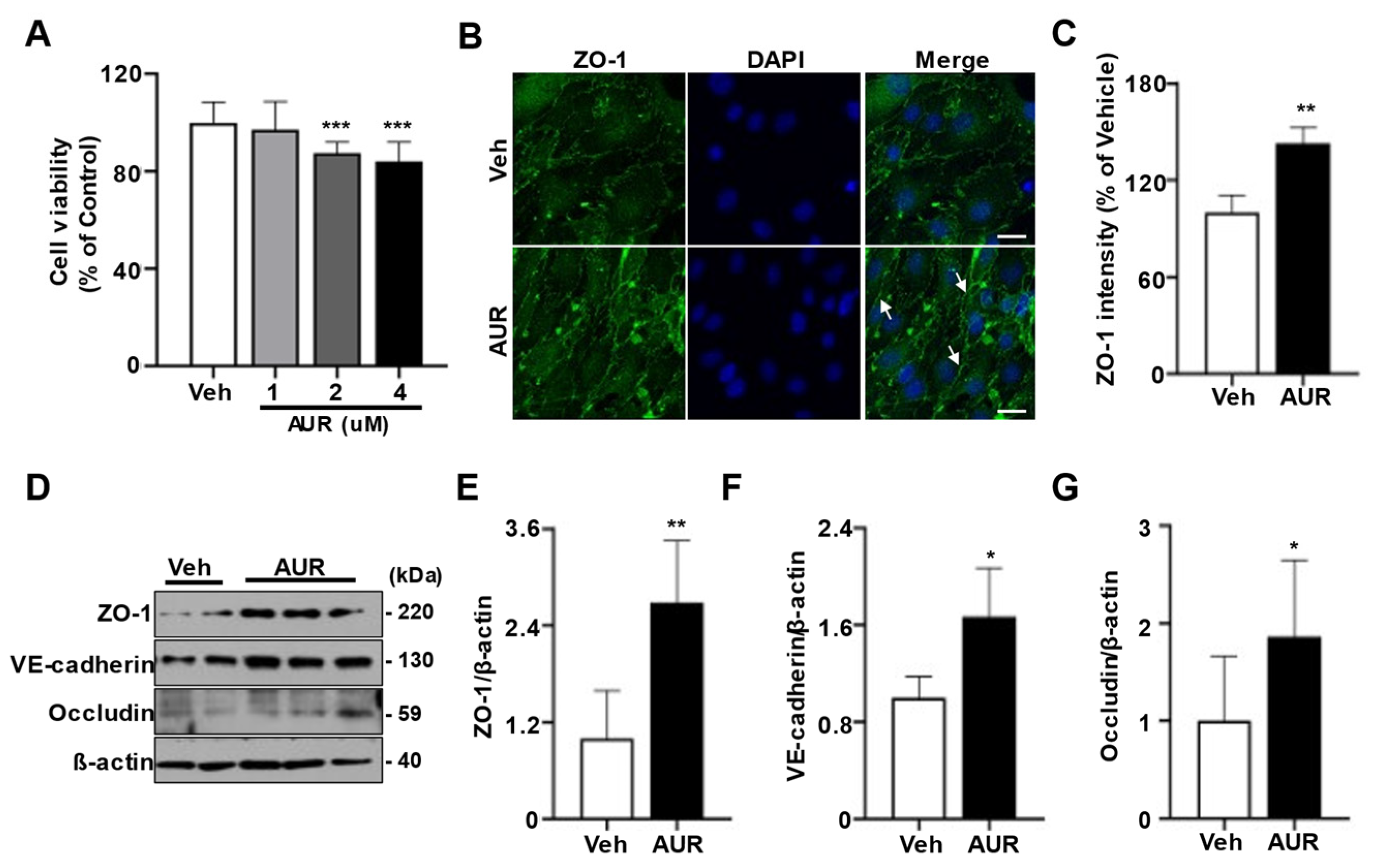
3.1. AUR Treatment Increases Junctional Proteins in bEnd.3 Cells
3.2. AUR Treatment Increases the Levels of mRNAs Encoding Antioxidant Enzymes in bEnd.3 Cells
3.3. AUR Reduces Mitochondrial Membrane Potential Maintaining Mitochondrial Respiration
3.4. AUR Promotes Resilience to Stress by Induction of mtUPR in bEnd.3 Cells
3.5. Pretreatment with AUR Protects against OGD Reduction in Junctional Protein Expression
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Daneman, R.; Prat, A. The blood-brain barrier. Cold Spring Harb. Perspect. Biol. 2015, 7, a020412. [Google Scholar] [CrossRef]
- Obermeier, B.; Daneman, R.; Ransohoff, R.M. Development, maintenance and disruption of the blood-brain barrier. Nat. Med. 2013, 19, 1584–1596. [Google Scholar] [CrossRef] [PubMed]
- Alquisiras-Burgos, I.; Peralta-Arrieta, I.; Alonso-Palomares, L.A.; Zacapala-Gómez, A.E.; Salmerón-Bárcenas, E.G.; Aguilera, P. Neurological Complications Associated with the Blood-Brain Barrier Damage Induced by the In-flammatory Response during SARS-CoV-2 Infection. Mol. Neurobiol. 2021, 58, 520–535. [Google Scholar] [CrossRef]
- Zlokovic, B.V. The Blood-Brain Barrier in Health and Chronic Neurodegenerative Disorders. Neuron 2008, 57, 178–201. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, G.A. Neurological Diseases in Relation to the Blood–Brain Barrier. Br. J. Pharmacol. 2012, 32, 1139–1151. [Google Scholar] [CrossRef] [PubMed]
- Minagar, A.; Alexander, J.S. Blood-brain barrier disruption in multiple sclerosis. Mult. Scler. J. 2003, 9, 540–549. [Google Scholar] [CrossRef]
- Huang, X.; Hussain, B.; Chang, J. Peripheral inflammation and blood–brain barrier disruption: Effects and mechanisms. CNS Neurosci. Ther. 2021, 27, 36–47. [Google Scholar] [CrossRef]
- Huppert, J.; Closhen, D.; Croxford, A.; White, R.; Kulig, P.; Pietrowski, E.; Bechmann, I.; Becher, B.; Luhmann, H.J.; Waisman, A.; et al. Cellular mechanisms of IL-17-induced blood-brain barrier disruption. FASEB J. 2010, 24, 1023–1034. [Google Scholar] [CrossRef]
- Olmez, I.; Ozyurt, H. Reactive oxygen species and ischemic cerebrovascular disease. Neurochem. Int. 2012, 60, 208–212. [Google Scholar] [CrossRef]
- Freeman, L.R.; Keller, J.N. Oxidative stress and cerebral endothelial cells: Regulation of the blood–brain-barrier and an-tioxidant based interventions. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2012, 1822, 822–829. [Google Scholar] [CrossRef] [PubMed]
- Sheu, S.-S.; Nauduri, D.; Anders, M. Targeting antioxidants to mitochondria: A new therapeutic direction. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2006, 1762, 256–265. [Google Scholar] [CrossRef] [PubMed]
- Ristow, M. Unraveling the Truth about Antioxidants: Mitohormesis explains ROS-induced health benefits. Nat. Med. 2014, 20, 709–711. [Google Scholar] [CrossRef] [PubMed]
- Yun, J.; Finkel, T. Mitohormesis. Cell Metab. 2014, 19, 757–766. [Google Scholar] [CrossRef]
- Münch, C. The different axes of the mammalian mitochondrial unfolded protein response. BMC Biol. 2018, 16, 81. [Google Scholar] [CrossRef] [PubMed]
- Rolland, S.G.; Schneid, S.; Schwarz, M.; Rackles, E.; Fischer, C.; Haeussler, S.; Regmi, S.; Yeroslaviz, A.; Habermann, B.; Mokranjac, D.; et al. Compromised Mitochondrial Protein Import Acts as a Signal for UPRmt. SSRN Electron. J. 2018, 28, 1659–1669.e5. [Google Scholar] [CrossRef] [PubMed]
- Santos-Parker, J.R.; Strahler, T.R.; Bassett, C.J.; Bispham, N.Z.; Chonchol, M.B.; Seals, D.R. Curcumin supplementation improves vascular endothelial function in healthy middle-aged and older adults by increasing nitric oxide bioavailability and reducing oxidative stress. Aging 2017, 9, 187–208. [Google Scholar] [CrossRef]
- Xiao, L.; Ding, M.; Fernandez, A.; Zhao, P.; Jin, L.; Li, X. Curcumin alleviates lumbar radiculopathy by reducing neuroinflammation, oxidative stress and nociceptive factors. Eur. Cells Mater. 2017, 33, 279–293. [Google Scholar] [CrossRef]
- Park, J.-H.; Choi, J.W.; Ju, E.J.; Pae, A.N.; Park, K.D. Antioxidant and Anti-Inflammatory Activities of a Natural Compound, Shizukahenriol, through Nrf2 Activation. Molecules 2015, 20, 15989–16003. [Google Scholar] [CrossRef]
- Zhao, X.; Chen, R.; Liu, M.; Feng, J.; Chen, J.; Hu, K. Remodeling the blood–brain barrier microenvironment by natural products for brain tumor therapy. Acta Pharm. Sin. B 2017, 7, 541–553. [Google Scholar] [CrossRef]
- Okuyama, S.; Minami, S.; Shimada, N.; Makihata, N.; Nakajima, M.; Furukawa, Y. Anti-inflammatory and neuroprotective effects of auraptene, a citrus coumarin, following cerebral global ischemia in mice. Eur. J. Pharmacol. 2013, 699, 118–123. [Google Scholar] [CrossRef]
- Jang, Y.; Choo, H.; Lee, M.J.; Han, J.; Kim, S.J.; Ju, X.; Cui, J.; Lee, Y.L.; Ryu, M.J.; Oh, E.S.; et al. Auraptene Mitigates Parkinson’s Disease-Like Behavior by Protecting Inhibition of Mitochondrial Respiration and Scavenging Reactive Oxygen Species. Int. J. Mol. Sci. 2019, 20, 3409. [Google Scholar] [CrossRef]
- Askari, V.R.; Rahimi, V.B.; Zargarani, R.; Ghodsi, R.; Boskabady, M.; Boskabady, M.H. Anti-oxidant and anti-inflammatory effects of auraptene on phytohemagglutinin (PHA)-induced inflam-mation in human lymphocytes. Pharmacol. Rep. 2020, 73, 154–162. [Google Scholar] [CrossRef]
- Carvalho, C.; Moreira, P.I. Oxidative Stress: A Major Player in Cerebrovascular Alterations Associated to Neurodegen-erative Events. Front. Physiol. 2018, 9, 806. [Google Scholar] [CrossRef]
- Soltani, F.; Mosaffa, F.; Iranshahi, M.; Karimi, G.; Malekaneh, M.; Haghighi, F.; Behravan, J. Auraptene from Ferula szowitsiana protects human peripheral lymphocytes against oxidative stress. Phytotherapy Res. 2009, 24, 85–89. [Google Scholar] [CrossRef]
- Holland, A.M.; Hyatt, H.W.; Smuder, A.J.; Sollanek, K.J.; Morton, A.B.; Roberts, M.D.; Kavazis, A.N. Influence of endurance exercise training on antioxidant enzymes, tight junction proteins, and inflammatory markers in the rat ileum. BMC Res. Notes 2015, 8, 1–9. [Google Scholar] [CrossRef]
- Moon, J.Y.; Kim, H.; Cho, S.K. Auraptene, a Major Compound of Supercritical Fluid Extract of Phalsak (CitrusHassaku Hort ex Tanaka), Induces Apoptosis through the Suppression of mTOR Pathways in Human Gastric Cancer SNU-1 Cells. Evid. Based Complement. Altern. Med. 2015, 2015, 402385. [Google Scholar] [CrossRef] [PubMed]
- Zorova, L.D.; Popkov, V.A.; Plotnikov, E.Y.; Silachev, D.N.; Pevzner, I.B.; Jankauskas, S.S.; Babenko, V.A.; Zorov, S.D.; Balakireva, A.V.; Juhaszova, M.; et al. Mitochondrial membrane potential. Anal. Biochem. 2018, 552, 50–59. [Google Scholar] [CrossRef]
- Huang, M.L.-H.; Chiang, S.; Kalinowski, D.S.; Bae, D.-H.; Sahni, S.; Richardson, D.R. The Role of the Antioxidant Response in Mitochondrial Dysfunction in Degenerative Diseases: Cross-Talk between Antioxidant Defense, Autophagy, and Apoptosis. Oxidative Med. Cell. Longev. 2019, 2019, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Kenny, T.C.; Manfredi, G.; Germain, D. The Mitochondrial Unfolded Protein Response as a Non-Oncogene Addiction to Support Adaptation to Stress during Transformation in Cancer and Beyond. Front. Oncol. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Holloway, P.M.; Gavins, F.N.E. Modeling Ischemic Stroke In Vitro: Status Quo and Future Perspectives. Stroke 2016, 47, 561–569. [Google Scholar] [CrossRef]
- Wallez, Y.; Huber, P. Endothelial adherens and tight junctions in vascular homeostasis, inflammation and angiogenesis. Biochim. Biophys. Acta (BBA) Biomembr. 2008, 1778, 794–809. [Google Scholar] [CrossRef]
- Qi, Z.; Yuan, S.; Liu, K.J. Occludin regulation of blood–brain barrier and potential therapeutic target in ischemic stroke. Brain Circ. 2020, 6, 152–162. [Google Scholar] [CrossRef]
- Svetlana, M.S.; Richard, F.K.; Anuska, V.A. Brain Endothelial Cell-Cell Junctions: How to Open the Blood Brain Barrier. Curr. Neuropharmacol. 2008, 6, 179–192. [Google Scholar]
- Corada, M.; Mariotti, M.; Thurston, G.; Smith, K.; Kunkel, R.; Brockhaus, M.; Lampugnani, M.G.; Martin-Padura, I.; Stoppacciaro, A.; Ruco, L.; et al. Vascular endothelial–cadherin is an important determinant of microvascular integrity in vivo. Proc. Natl. Acad. Sci. USA 1999, 96, 9815. [Google Scholar] [CrossRef]
- Huber, J.D.; Egleton, R.D.; Davis, T.P. Molecular physiology and pathophysiology of tight junctions in the blood–brain barrier. Trends Neurosci. 2001, 24, 719–725. [Google Scholar] [CrossRef]
- Lee, M.J.; Jang, Y.; Han, J.; Kim, S.J.; Ju, X.; Lee, Y.L.; Cui, J.; Zhu, J.; Ryu, M.J.; Choi, S.-Y.; et al. Endothelial-specific Crif1 deletion induces BBB maturation and disruption via the alteration of actin dynamics by impaired mitochondrial respiration. Br. J. Pharmacol. 2020, 40, 1546–1561. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.K.; Hyun, S.-W.; Jung, Y.-S. Yuzu and Hesperidin Ameliorate Blood-Brain Barrier Disruption during Hypoxia via Antioxidant Activity. Antioxidants 2020, 9, 843. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-S.; Namkoong, K.; Kim, D.-H.; Kim, K.-J.; Cheong, Y.-H.; Kim, S.-S.; Lee, W.-B.; Kim, K.-Y. Hydrogen peroxide-induced alterations of tight junction proteins in bovine brain microvascular endothelial cells. Microvasc. Res. 2004, 68, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Mo, W.; Cao, W.; Wu, X.; Jia, G.; Zhao, H.; Chen, X.; Wu, C.; Wang, J. Effects of spermine on ileal physical barrier, antioxidant capacity, metabolic profile and large intestinal bacteria in piglets. RSC Adv. 2020, 10, 26709–26716. [Google Scholar] [CrossRef]
- Cunha, F.M.; Da Silva, C.C.C.; Cerqueira, F.M.; Kowaltowski, A.J. Mild mitochondrial uncoupling as a therapeutic strategy. Curr. Drug Targets 2011, 12, 783–789. [Google Scholar] [CrossRef]
- Vyssokikh, M.Y.; Holtze, S.; Averina, O.A.; Lyamzaev, K.; Panteleeva, A.A.; Marey, M.V.; Zinovkin, R.A.; Severin, F.F.; Skulachev, M.V.; Fasel, N.; et al. Mild depolarization of the inner mitochondrial membrane is a crucial component of an anti-aging program. Proc. Natl. Acad. Sci. USA 2020, 117, 6491–6501. [Google Scholar] [CrossRef] [PubMed]
- De Felice, F.G.; Houzel, J.C.; Garcia-Abreu, J.; Louzada, P.R., Jr.; Afonso, R.C.; Meirelles, M.N.; Lent, R.; Neto, V.M.; Ferreira, S.T. Inhibition of Alzheimer’s disease β-amyloid aggregation, neurotoxicity, and in vivo deposition by ni-trophenols: Implications for Alzheimer’s therapy. FASEB J. 2001, 15, 1297–1299. [Google Scholar] [CrossRef] [PubMed]
- Kalinovich, A.V.; Shabalina, I.G. Novel Mitochondrial Cationic Uncoupler C4R1 Is an Effective Treatment for Combat-ing Obesity in Mice. Biochemistry 2015, 80, 620–628. [Google Scholar] [PubMed]
- Yi, H.-S.; Chang, J.Y.; Shong, M. The mitochondrial unfolded protein response and mitohormesis: A perspective on met-abolic diseases. J. Mol. Endocrinol. 2018, 61, R91–R105. [Google Scholar] [CrossRef] [PubMed]
- Narasimhan, P.; Liu, J.; Song, Y.S.; Massengale, J.L.; Chan, P.H. VEGF Stimulates the ERK 1/2 Signaling Pathway and Apoptosis in Cerebral Endothelial Cells after Ischemic Conditions. Stroke 2009, 40, 1467–1473. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, M.J.; Jang, Y.; Zhu, J.; Namgung, E.; Go, D.; Seo, C.; Ju, X.; Cui, J.; Lee, Y.L.; Kang, H.; et al. Auraptene Enhances Junction Assembly in Cerebrovascular Endothelial Cells by Promoting Resilience to Mitochondrial Stress through Activation of Antioxidant Enzymes and mtUPR. Antioxidants 2021, 10, 475. https://doi.org/10.3390/antiox10030475
Lee MJ, Jang Y, Zhu J, Namgung E, Go D, Seo C, Ju X, Cui J, Lee YL, Kang H, et al. Auraptene Enhances Junction Assembly in Cerebrovascular Endothelial Cells by Promoting Resilience to Mitochondrial Stress through Activation of Antioxidant Enzymes and mtUPR. Antioxidants. 2021; 10(3):475. https://doi.org/10.3390/antiox10030475
Chicago/Turabian StyleLee, Min Joung, Yunseon Jang, Jiebo Zhu, Eunji Namgung, Dahyun Go, Changjun Seo, Xianshu Ju, Jianchen Cui, Yu Lim Lee, Hyoeun Kang, and et al. 2021. "Auraptene Enhances Junction Assembly in Cerebrovascular Endothelial Cells by Promoting Resilience to Mitochondrial Stress through Activation of Antioxidant Enzymes and mtUPR" Antioxidants 10, no. 3: 475. https://doi.org/10.3390/antiox10030475
APA StyleLee, M. J., Jang, Y., Zhu, J., Namgung, E., Go, D., Seo, C., Ju, X., Cui, J., Lee, Y. L., Kang, H., Kim, H., Chung, W., & Heo, J. Y. (2021). Auraptene Enhances Junction Assembly in Cerebrovascular Endothelial Cells by Promoting Resilience to Mitochondrial Stress through Activation of Antioxidant Enzymes and mtUPR. Antioxidants, 10(3), 475. https://doi.org/10.3390/antiox10030475