Abstract
Hydrogen sulfide (H2S) is an endogenous, gaseous signaling molecule that plays a critical role in cardiac and vascular biology. H2S regulates vascular tone and oxidant defenses and exerts cytoprotective effects in the heart and circulation. Recent studies indicate that H2S modulates various components of metabolic syndrome, including obesity and glucose metabolism. This review will discuss studies exhibiting H2S -derived cardioprotective signaling in heart failure with reduced ejection fraction (HFrEF). We will also discuss the role of H2S in metabolic syndrome and heart failure with preserved ejection fraction (HFpEF).
1. H2S Therapy in Heart Failure with Reduced Ejection Fraction
Heart failure with reduced ejection fraction (HFrEF) is one of the leading causes of mortality in the United States, with a 5-year mortality rate of 75% [1]. Heart failure patients have reduced circulating H2S levels, and H2S levels progressively decline as heart failure symptoms worsen [2]. These diminished H2S levels have been recapitulated in several preclinical animal models of HFrEF [3]. In a mouse model of HFrEF, whereby mice underwent a transverse aortic constriction (TAC) procedure and subsequently developed HFrEF, there was > 60% reduction in circulating and cardiac hydrogen sulfide concentrations, highlighting the potential role of H2S as a cardioprotective signaling molecule [4]. Endogenously produced hydrogen sulfide exerts a myriad of cytoprotective actions in vivo, specifically in its role as an antioxidant through promoting Nrf2 and NRF-1 signaling [5,6,7] and augmenting nitric oxide-mediated signaling [8]. These signaling mechanisms provide a component of the antioxidant and anti-apoptotic effects that are observed with the administration of H2S donors in a broad range of disease models. In a cystathionine γ-lyase (CSE) knockout mouse model with attenuated circulating plasma H2S, cardiac injury was exacerbated following myocardial ischemia–reperfusion injury compared to wild-type mice. This deleterious phenotype was ameliorated following administration of a hydrogen sulfide donor. Interestingly, in an endothelial nitric oxide synthase (eNOS) phospho-mutant mouse, exogenous H2S failed to provide a protective effect in acute myocardial infarction, indicating the importance of eNOS-NO signaling in H2S-mediated protection [8]. These findings demonstrate hydrogen sulfide’s ability to activate or rescue eNOS functionality and may indicate a therapeutic niche for H2S in diseased states with high reactive oxygen species, such as heart failure, where eNOS is uncoupled into non-functioning monomers [8].
The mechanisms by which H2S activates eNOS involve promoting the recoupling of eNOS monomers, increasing the phosphorylation of eNOS at ser1177 while reducing the phosphorylation at thr495, and inhibiting endogenous eNOS inhibitors such as proline-rich tyrosine kinase 2 (PYK2) via s-sulfhydration [9]. Not only has H2S been shown to affect signaling trough sulfhydration, but it has also been investigated as a pan-inhibitor of phosphodiesterase, leading to a potential added benefit of increasing cGMP signaling in cardiovascular-diseased states [10]. This H2S mediated inhibition has been shown with decreases in both homodimers of PDE 5A and 5′-GMP content following incubation of rat vessels with NaHS or GYY4137 [11]. In addition, when human internal mammary arteries were exposed to NaHS, there was an observed increase in eNOS phosphorylation and subsequent decreased PDE5A level compared to that of arteries exposed to a Krebs control [12].
The tandem effects of H2S and NO, in addition to their metabolites and downstream signaling modulators, have been implicated in a range of cardioprotective effects [9,10,11,12,13]. Calvert et al. investigated the importance of CSE-mediated H2S generation in the setting of HFrEF. Myocardial CSE overexpression led to improved systolic function in a mouse model of ischemia-induced heart failure [6]. What remains unclear is whether the reduced H2S bioavailability observed in HFrEF is due to decreased enzymatic production through endogenous H2S-producing enzymes CSE, Cystathionine β-synthase (CBS) and 3-Mercaptopyruvate sulfurtransferase (3-MST) or due to increased metabolism of H2S, or both mechanisms.
Both polysulfides and persulfides have been shown to sulfhydrate cysteine residues of proteins in a much more potent manner compared to H2S [14]. Many novel polysulfide or persulfide compounds have been studied in the setting of various diseases [15,16,17,18,19]. However, due to the chemical nature of these compounds, they are also the source of H2S as a separate mechanism of action. Hence, it is very difficult to delineate their effects directly as a polysulfide or through its donation of H2S through extended chains of sulfur atoms. It is believed that these molecules themselves are in fact interchangeable in solution [20]. Nevertheless, several novel H2S-donating therapeutic compounds are known polysulfides [20,21,22,23,24].
SG-1002 is an alpha-sulfur polysulfide compound that also serves as a pro-H2S donor. In recent findings, wild-type mice and CSE-KO mice underwent TAC and then received either SG-1002 (P.O. in chow at 20 mg/kg/day) or vehicle. CSE-KO mice developed worsened cardiac remodeling and function compared to control mice. Interestingly, CSE-KO mice that received SG-1002 exhibited improved cardiac remodeling and function [6]. A subsequent phase 1 clinical trial was performed using SG-1002 for a 21-day period in healthy and heart failure subjects. The drug was well tolerated and SG-1002 effectively increased circulating H2S and nitrite levels [17]. To date, there are no other heart failure clinical trials that have been completed utilizing a H2S donor.
Sodium sulfide (Na2S) was one of the first H2S donors studied and has a very rapid release profile. Mice administered sodium sulfide 100 μg/kg/d through daily venous injections beginning 4 weeks after ischemia-induced heart failure had increased Nrf2 signaling along with preserved cardiac structure and function compared to vehicle control mice. Following the induction of ischemic heart failure in Nfr2-KO mice, sodium sulfide administration failed to alter cardiac function or structure compared to control. This indicates that H2S-mediated cardioprotection in ischemic heart failure is Nrf2-dependent [25]. It has also been reported that sodium sulfide leads to transient arterial pressure decreases when administered at 15 mg/kg i.v. [26]. The hemodynamic effects of H2S cannot be overlooked as a possible protective mechanism of action. Sodium sulfide has also been shown to upregulate Heme-oxygenase-1 in a volume overload mouse model of heart failure [27]. Reactive oxygen species in conjunction with cellular apoptosis are also decreased in HFrEF mice following Na2S administration [28]. These H2S-dependent antioxidant and anti-apoptotic effects have been reported in rodents in models of doxorubicin or anthracycline-induced heart failure [29].
Diallyl trisulfide (DATS) is a garlic-derived polysulfide that has a H2S-releasing profile that is more sustained than traditional H2S donors such as sodium sulfide. In a 12-week study of TAC-induced heart failure, DATS therapy (200 µg/day) led to improved LV ejection fraction compared to vehicle. Mice receiving DATS also had increased cardiac microvessel density, increased VEGF signaling, and attenuated myocardial fibrosis [16]. In addition, under certain conditions, DATS may react with its existing cysteine sulfhydyl residues forming trisulfide metabolites and elicit cytoprotective effects independent of H2S formation [30].
Li et al. studied the cardioprotective effects of a novel H2S donor, JK-1, in a TAC-induced murine heart failure model. JK-1 releases H2S in a pH-dependent manner via a hydrolysis reaction and this release profile is ideally suited to pathological CV disease states such as heart failure. Delayed treatment of JK-1 following the onset of heart failure led to reduced left ventricular dilation and improved LV ejection fraction compared to a control group [31]. This study was also the first described evidence that H2S attenuates the activation of the renin–angiotensin–aldosterone system (RAAS) in the setting of heart failure. Downregulation of RAAS ultimately led to reduced renal fibrosis and improved renal function in the JK-1-treated group.
Wu et al. first described the novel compound, ZYZ-803, which activates both CSE and eNOS, leading to increased H2S and nitric oxide production simultaneously. ZYZ-803 has been tested in a mouse heart failure model induced by isoprenaline. Following administration of ZYZ-803, the mice had improved cardiac structural changes and increased LV ejection fraction compared to that of the vehicle group [32]. The drug appeared to effectively activate CSE and eNOS as plasma H2S and nitric oxide levels were increased at the study endpoint. Cardioprotective effects of ZYZ-803 were diminished with simultaneous administration of the eNOS inhibitor, L-NAME, and the CSE inhibitor, DL-propargylglycine (PAG). These results reveal that concurrent activation of these enzymes produces a greater effect than either one alone.
The slow-releasing H2S donor, GYY4137, has been shown to preserve left ventricular ejection fraction 7-days post-MI compared to that of control. Interestingly, despite improved cardiac function, these animals also had increased natriuretic peptide levels compared to control. The mechanism of H2S-mediated augmentation of natriuretic peptides remains unknown. In this same model, GYY4137 therapy led to increased NO-cGMP signaling post-MI [33]. In isolated cardiomyocytes infected with coxsackie virus, GYY4137 was also shown to produce anti-inflammatory actions through decreased NFKB and MAPK signaling [34].
Through both the investigation and success of these various H2S donors in relevant pre-clinical models such as HFrEF, we gain insight into the mechanisms of protective effects in pathological states seen in Figure 1. H2S therapy might be applied to similar states of not only pathological cardiac remodeling but a myriad of disease states involving, but not limited to, systemic inflammation, fibrosis, vascular diseases such as hypertension and peripheral artery disease [35,36,37,38], and overactive RAAS signaling.
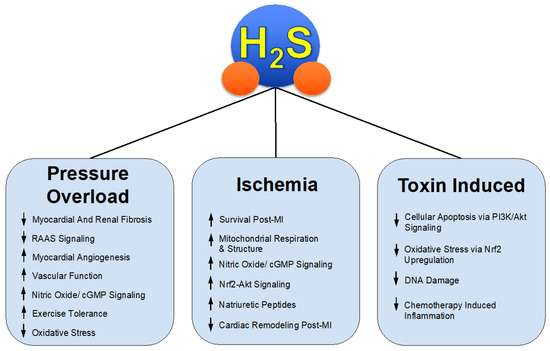
Figure 1.
H2S Mediated Protection in Heart Failure.
2. H2S Therapy in the Setting of Metabolic Syndrome
The prevalence of metabolic syndrome has increased at an alarming rate over the past several decades. It is estimated that approximately 1/3 of US adults, 12–37% of the Asian population, and 12–26% of the European population have metabolic syndrome [39,40]. These numbers equate to > 1 billion people worldwide. Those with metabolic syndrome have increased risk of cardiovascular morbidity and mortality. In fact, estimates suggest that 6–7% of all-cause mortality and 12–17% of cardiovascular disease can be attributed to metabolic syndrome [41]. In a survey of US adults, metabolic syndrome had an increased hazard ratio for coronary heart disease mortality of 2.02 and all-cause cardiovascular disease mortality of 1.82 [42]. We will now summarize the studies that have investigated the direct and indirect actions of H2S in the various components of metabolic syndrome summarized in Figure 2.
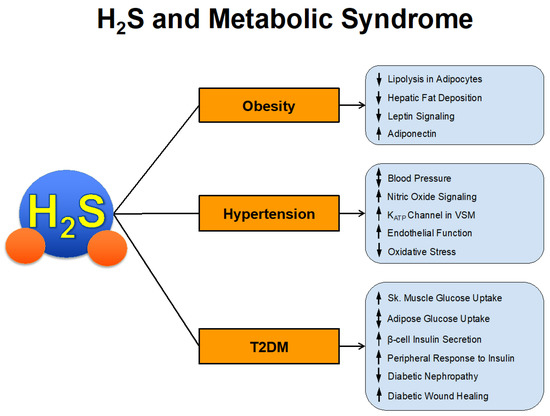
Figure 2.
H2S and Metabolic Syndrome.
2.1. H2S and Obesity
Obesity is a leading contributor to metabolic disease by numerous mechanisms. Obesity results in the release of increased nonesterified fatty acids (NEFAs), which, when acting on skeletal muscle, can promote insulin resistance and the development of fatty liver disease. Adipose tissue also synthesizes and secretes inflammatory cytokines, including TNF-alpha and IL-6, which are correlated with increased cardiovascular risk. Although adipose cells produce leptin, which is a hormone that helps to regulate the energy balance by inhibiting hunger and fat storage metabolism, there is leptin resistance in obese patients. Conversely, obese persons generally have lower levels of adiponectin, which is a hormone produced primarily in adipose tissue, with anti-inflammatory and anti-atherogenic properties.
It is unclear to what degree H2S regulates lipolysis or the development and pathology of obesity and metabolic syndrome. Some studies have suggested that H2S inhibits lipolysis, while others suggest that upregulation of H2S signaling stimulates adipose tissue lipolysis. Geng et al. reported that an H2S synthesis inhibitor, DL-propargylglycine (PAG), increased isoproterenol-stimulated lipolysis in rat adipocytes, while the H2S donor, GYY4137, reduced adipose tissue lipolysis [43]. In another study, sodium sulfide (Na2S) was administered via microdialysis probe into subcutaneous adipose tissue and lipolysis was estimated by measuring glycerol levels [44]. In a dose-dependent manner, Na2S led to increased glycerol production, which was accompanied by cAMP release. This release was abolished by the protein kinase A (PKA) inhibitor, KT5720, indicating that H2S-induced lipolysis is PKA-dependent. H2S-mediated glycerol release was greater in rats that were fed a high-fat diet. They also found that an inhibitor of H2S release (PAG) resulted in decreased glycerol production in obese rats.
The three H2S-generating enzymes, CSE, CBS, and 3-MST, are all produced in the liver and contribute to hepatic H2S production and hepatic physiology. Malfunction of hepatic H2S metabolism is involved in the pathogenesis of many liver diseases and H2S regulates lipid metabolism [45]. Studies have suggested that exogenous H2S improves fatty liver disease by improving lipid metabolism. In high-fat-diet-induced obese mice, daily injection of the H2S donor, NaHS, for 4 weeks resulted in improved hepatic cellular structure, decreased liver weight and Oil-red-O staining, and decreased triglycerides and total cholesterol [46]. The endogenous role of H2S signaling appears to center around the interplay of the H2S-producing enzymes. Meng et al. report that hepatic expression of 3-MST is upregulated in patients with non-alcoholic fatty liver disease as well as in mice fed a high-fat diet [47]. However, this increase in 3-MST led to a paradoxical decrease in H2S production because 3-MST directly interacted with and negatively regulated CSE in the liver. Furthermore, inhibition of 3-MST significantly enhanced, rather than decreased, H2S production and reduced FFA-induced fat accumulation in L02 cells.
Adipose tissue is also an endocrine organ and the critical signaling hormones produced by adipose are leptin and adiponectin. Leptin resistance characterized by elevated circulating leptin levels is a hallmark of obesity. The effects of exogenous H2S in leptin signaling have been previously investigated. Wu et al. reported that pre-treatment of human umbilical vein endothelial cells with the H2S donor, sodium hydrosulfide, prior to glucose challenge resulted in decreased leptin as well as decreased leptin receptor expression [48]. Moreover, Zhuang et al. report that exogenous NaHS prevents high-glucose-induced injury by inhibiting the leptin-p38 MAPK signaling pathway in H9c2 cells. This group also reports that H2S attenuates leptin and leptin receptor expression [49]. On the other hand, circulating levels of adiponectin are negatively correlated with obesity and have been shown to be protective in cardiovascular disease [50]. Interestingly, there appears to be a positive correlation between circulating H2S and adiponectin levels in humans [24]. High glucose concentration has also been shown to decrease the expression of the H2S-producing enzyme, CSE, and lead to the downregulation of adiponectin [51]. Conversely, overexpression of CSE or exogenous treatment with NaHS resulted in increased adiponectin in adipocytes, indicating that H2S and adiponectin are not merely correlating bystanders but that H2S directly causes increased adiponectin production.
2.2. H2S and Type 2 Diabetes Mellitus
Type 2 diabetes mellitus (T2DM) carries a significant and increasing disease burden worldwide. Complications can be broadly categorized into microvascular and macrovascular complications. Microvascular complications, largely due to the accumulation of advanced glycosylation end-products and vascular injury, include retinopathy, nephropathy, neuropathy, and poor wound healing. Macrovascular complications of diabetes mellitus include increased risk of stroke, heart disease, and peripheral vascular disease. Interestingly, alterations in H2S signaling seem to play a role in glucose handling and the pathogenesis of T2DM.
Multiple groups have reported that patients with T2DM have lower circulating H2S levels than their age-matched non-diabetic counterparts [24,52]. Interestingly, H2S appears to decrease pancreatic beta-cell secretion of insulin but increase peripheral response to insulin-mediated glucose uptake peripherally [53]. In the liver, H2S likely mobilizes glucose stores by increasing gluconeogenesis and glycogenolysis. The interplay between H2S and NO in glucose regulation is somewhat unclear since studies report some discordant actions of NO compared to H2S. Similarly to H2S, NO promotes peripheral glucose uptake by skeletal muscle; however, its actions in the pancreas and liver differ. NO increases pancreatic insulin secretion by beta-cells. In the liver, NO inhibits glucose mobilization and promotes glucose storage by decreasing gluconeogenesis and glycolysis [54]. As we discussed earlier, H2S promotes NO signaling; thus, studies identifying which molecule plays a predominant role in glucose metabolism are warranted.
Several pre−clinical studies provide evidence that H2S attenuates many of the complications of T2DM. Wound healing is impaired in diabetic patients, largely due to impaired microvascular function and localized inflammation. Wang et al. found that localized daily treatment with bisulfide ointment for 21 days improved wound healing in diabetic rats [55]. They found that in these animals, H2S promoted angiogenic signaling while reducing markers of oxidative stress and inflammation. Diabetic nephropathy, and ultimately chronic kidney disease, develops because glycosylation of the vascular basement membrane leads to hyaline arteriolosclerosis. The efferent arteriole is preferentially affected, which leads to increased glomerular filtration pressure. Hyperfiltration leads to microalbuminuria and eventual progression to nephrotic syndrome. In a streptozotocin-induced diabetic rat model, Zhou et al. report that daily injections of NaHS for 12 weeks led to improved renal function, attenuated glomerular basement membrane thickening, and blunted interstitial fibrosis and mesangial matrix deposition [56]. Using the same rat model of streptozotocin-induced diabetes, Sun et al. demonstrated that the sulfhydration of SIRT1 by Na2S4, a polysulfide compound, leads to ameliorated diabetic nephropathy [19]. As in many of the studies examining H2S in various models of metabolic syndrome, there are no identifiable clinical trials that are examining or have examined the effects of an H2S donor in patients with T2DM. We believe that hydrogen sulfide, with its diverse mechanisms of action in rigorously performed in vitro and in vivo animal models, is an ideal candidate for the treatment of complications from T2DM.
2.3. H2S and Blood Pressure Regulation
H2S regulates vascular function and hemodynamics, but the precise role of H2S in vascular function remains unclear. Some studies report vasodilatory actions of H2S while others report vasoconstriction. It is possible that these discrepancies can be explained by dose-dependent effects, which vascular beds are under investigation, or the oxygen tension within that vascular bed. CSE KO mice, which are consequently deficient in H2S, are significantly hypertensive and have impaired endothelium-mediated vasorelaxation [57]. Exogenous H2S has been shown to act as a vasodilator at lower oxygen pressures (30 mmHg) and as a vasoconstrictor at elevated partial pressure of oxygen (150 mmHg) [58]. This discordance may suggest that H2S would have more dilatory effects on the venous side of the circulatory system with lower oxygen partial pressures. H2S appears to also play a role in the pulmonary vasculature, particularly in the setting of hypoxic pulmonary hypertension. Chunyu et al. reported that CSE expression in the lungs and plasma H2S levels are both decreased in hypoxia-induced pulmonary hypertension and suggest that exogenous H2S could oppose this rise in pulmonary arterial pressures [59].
There does not appear to be a single mechanism by which H2S modulates vascular tone; however, several have been proposed. It is likely that multiple signal transduction pathways are activated in the endothelium and vascular smooth muscle cells. Studies by Naik et al. suggest that H2S activates endothelial TRPV4 channels which allow for Ca2+ influx and subsequent vasodilation. Another mechanism of H2S-regulated blood pressure reduction may be its interplay with the potent vasodilator, nitric oxide (NO). Studies have shown that exogenous hydrogen sulfide activates eNOS and increases nitric oxide signaling [16]. Additionally, suppression of hydrogen sulfide production via CSE gene suppression leads to inhibited eNOS activity and diminished NO signaling. Others suggest that H2S-induced vasorelaxation is dependent on the activation of ATP-sensitive K+ channels in vascular smooth muscle [60,61]. In these studies, concentration-dependent H2S-induced vasodilation was inhibited by the KATP channel blocker glibenclamide [62]. However, another mechanism of H2S-mediated vascular regulation is its action as a powerful antioxidant. Oxidative stress causes endothelial dysfunction and has detrimental effects on vascular function and tone. Several models of elevated oxidative stress, including neuronal ischemia–reperfusion injury, myocardial ischemia–reperfusion injury, heart failure, and limb ischemia, report that H2S donors attenuate local and systemic oxidative stress and improve cellular health [6,14,63]. H2S scavenges O2− and reduces vascular NADPH oxidase-derived superoxide anion production [64]. H2S also inhibits H2O2-mediated mitochondrial dysfunction by preserving the activity of superoxide dismutase, catalase, glutathione peroxidase, and glutathione-S-transferase while promoting mitochondrial biogenesis via sulfhydrating PP2A and activating AMPK [65,66].
An important question is whether chronic H2S therapy could have consistent and sustained actions on blood pressure control. Many of the studies reported in the literature describe the acute vasodilatory effects of a H2S donor bolus. Numerous studies also discuss the development of hypertension in models of H2S deficiency. However, we have yet to find conclusive studies reporting that H2S can safely and effectively reduce blood pressure in the setting of chronic systemic hypertension. A likely reason for this is the release profile of most of the H2S donors. Most of these compounds release H2S in the order of seconds to minutes, which would unlikely be clinically therapeutic in a chronic disease state where effective treatment requires blood pressure reduction throughout the entire day [2]. However, as discussed, a few of the proposed blood-pressure-lowering effects, specifically the role of H2S as an antioxidant and an eNOS activator, may lead to more sustained anti-hypertensive effects than the abrupt H2S release profile of many of the developed compounds. Further studies in models of chronic hypertension are required to answer these questions.
3. The Potential of H2S Therapy in HFpEF
Heart failure with preserved ejection fraction (HFpEF) is a complex heterogeneous multi-organ disease that has been at the forefront of heart failure research over the past decade. HFpEF is the most common of all heart failure (HF) diagnoses, expected to amass > 60% of all clinical HF cases by 2030 [67]. In contrast to HFrEF, the list of disappointing HFpEF clinical trials is expanding, with no FDA-approved pharmacotherapies showing significant decreases in patient hospitalization or mortality rates. These failures contribute to HFpEF being widely regarded as the largest unmet clinical need in cardiovascular medicine [1,68].
HFpEF therapy development is further complicated by its heterogeneous phenotype and non-unified diagnostic criteria [1,69]. Until recently, HFpEF was broadly defined as a patient experiencing symptoms of HF while sustaining normal left ventricular ejection fraction (> 50%) [70]. Given the generality of the HFpEF diagnosis, the current treatment regimen lacks precision and is largely limited to symptom management targeting the accompanying comorbidities.
In-depth investigation into HFpEF clinical presentations has led to the identification and stratification in three distinct phenogroups that include: (1) elderly patients with moderate diastolic dysfunction, hypertension, and relatively preserved brain natriuretic peptide (BNP), (2) obese, diabetic patients with significantly impaired left ventricular relaxation, and (3) older patients with chronic kidney disease, pulmonary hypertension, and right ventricular dysfunction. Stratification into these phenotypes revealed correlative comorbidities and mortality rate among groups [71,72]. The identification of multiple phenogroups within the HFpEF population has raised the possibility that a single mechanistic treatment approach likely will not yield optimal outcomes in this heterogeneous syndrome.
Phenogroup 2 HFpEF patients suffer from severe cardiometabolic disease and have the highest incidence of obesity, diabetes mellitus, and obstructive sleep apnea. Moreover, > 50% of this HFpEF subgroup contains patients with dyslipidemia, type 2 diabetes mellitus, hypertension, and/or obesity [71]. The combination of these comorbidities leads to metabolic syndrome, an energetically dysregulated state known to cause deleterious cardiometabolic effects [41,73]. As described previously, H2S has been shown to positively modulate obesity, dyslipidemia, glucose control, and insulin resistance. Therefore, H2S therapy may improve outcomes in phenogroup 2 HFpEF patients by addressing the deleterious accompanying comorbidities. It is also possible that H2S may have direct myocardial cardioprotective actions on the heart, as it does in HFrEF. Specifically, H2S has been shown to decrease cardiac fibrosis, which is critically important in treating a disease state whose hallmark is a non-compliant ventricle [3]. Given that H2S has been previously researched as a treatment strategy for these specific HFpEF-inducing comorbidities, as well as its involvement in HFrEF progression, we identify this specific phenotype as the most interesting candidate to investigate the use of H2S as an effective therapeutic approach [74].
Cardiometabolic HFpEF poses an interesting challenge in HFpEF research in that systemic metabolic dysregulation, vascular dysfunction, inflammation, and oxidative stress predominate this disease. The accumulation of these systemic insults, though initially compensated by increasing the work of the heart, eventually results in overwhelming systemic stress, leading to significant left ventricular diastolic dysfunction. The predominance of metabolic and inflammatory disturbances in cardiometabolic-HFpEF makes it a most interesting target for H2S therapy, especially regarding the potential effect that H2S will have on attenuating the pro-inflammatory metabolic state.
Small animal modeling of cardiometabolic-HFpEF has been thoroughly described with the ZSF1 obese rat [75,76]. The ZSF1 obese rat presents with severe HFpEF signs and symptoms as early as 20 weeks of age [76]. The rapid development of the disease also makes the ZSF1 obese rats ideal for assessing the progression of HFpEF from asymptomatic to severe presentations. Plasma sulfane sulfur is a metabolic breakdown product of H2S that is a direct indication of H2S levels, allowing for a less volatile marker of H2S in the plasma [8]. Sulfane sulfur levels both prior to onset of HFpEF symptoms and in late-stage HFpEF are shown in Figure 3. With the presence of the metabolic disturbances at the 14-week timepoint, there are extremely low levels of plasma sulfane sulfur present. Of significant importance, however, is that the progression of HFpEF symptoms depleted the remaining sulfane sulfur levels. These observations highlight a novel finding in the setting of cardiometabolic-HFpEF, suggesting that therapeutic supplementation with H2S donors could potentially benefit both metabolic and cardiovascular components of this disease.
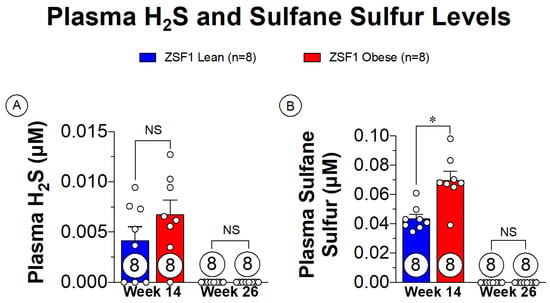
Figure 3.
Plasma free H2S and sulfane sulfure levels in ZSF1 obese rats prior at multiple time points in disease progression. (A) Plasma H2S levels at 14 weeks of age (prior to HFpEF onset) and at 26 weeks of age (late-stage HFpEF). (B) Plasma sulfane sulfur levels at 14 and 26 weeks of age. NS: Not Significant, * p < 0.05.
4. Conclusions
Attempts to treat the systemic pathology associated with HFpEF using therapeutics previously approved for HFrEF have failed to provide positive results despite their clear-cut benefits in HFrEF. This is likely due to the heterogeneity of the HFpEF patient population and the highly complex and diverse pathology of this disease. Stratification of HFpEF patient populations and the use of combination therapies in future clinical studies may reveal a viable therapeutic solution. We propose that the previously described actions of H2S, including its global involvement with metabolic syndrome, RAAS, sympathetic output, blood pressure, and cytoprotectant effects, make it a worthy candidate for future investigation as a treatment for cardiometabolic-HFpEF and perhaps additional HFpEF phenotypes.
Through previous animal modeling in HFrEF, H2S has been shown to provide cytoprotective effects through NRF2- and eNOS-dependent pathways, alleviating systemic oxidative stress and improving vascular function found in pathological inflammatory states. In investigating models of obesity and metabolic syndrome, hydrogen sulfide has been shown to not only beneficially impact circulating lipids but improve glucose signaling in a diabetic state. With regard to hypertension, H2S has been shown to improve endothelial function and nitric oxide signaling, with mixed reports regarding overall blood pressure regulation. In the HFpEF ZSF1 rat model (Figure 3), we have now shown that circulating plasma sulfane sulfur levels are severely diminished during the progression of this disease, providing some new evidence that loss of H2S may be involved in the pathogenesis of HFpEF. Additional studies are required to determine if reductions in H2S in fact contribute to HFpEF pathology.
While this review emphasizes the therapeutic role of H2S supplementation in heart failure and metabolic syndrome, it must also be mentioned the potentially toxic role of H2S in excess. H2S, along with other signaling molecules, must remain in a homeostatic concentration in the body, stressing the importance and potential danger of supplementing H2S with supraphysiologic levels. It is also worth noting that the potential role of contamination of NaHS in solutions, although not investigated thoroughly, is an ongoing issue in the H2S field [77].
Although H2S has been shown to be beneficial in these models of HFrEF and metabolic syndrome, further studies are required to investigate its true role in the setting of HFpEF pathology. It may be that several therapeutics are necessary to combat the heterogeneity of this disease, where H2S may contribute in a supportive role.
Author Contributions
K.B.L., D.J.P., H.A.H., J.E.D., D.J.L., Z.L.: writing—original draft preparation. All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded in part by the National Institutes of Health (NIH), grants R01HL146098, 1R01 HL146514, 7R01 HL070241, and 1R01 HL151398, to D.J.L.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Shah, K.S.; Xu, H.; Matsouaka, R.A.; Bhatt, D.L.; Heidenreich, P.A.; Hernandez, A.F.; Devore, A.D.; Yancy, C.W.; Fonarow, G.C. Heart failure with preserved, borderline, and reduced ejection fraction: 5-year outcomes. J. Am. Coll. Cardiol. 2017, 70, 2476–2486. [Google Scholar] [CrossRef]
- Polhemus, D.J.; Calvert, J.W.; Butler, J.; Lefer, D.J. The Cardioprotective Actions of Hydrogen Sulfide in Acute Myocardial Infarction and Heart Failure. Scientifica 2014, 2014, 1–8. [Google Scholar] [CrossRef]
- Li, Z.; Organ, C.L.; Kang, J.; Polhemus, D.J.; Trivedi, R.K.; Sharp, T.E.; Jenkins, J.S.; Tao, Y.-X.; Xian, M.; Lefer, D.J. Hydrogen Sulfide Attenuates Renin Angiotensin and Aldosterone Pathological Signaling to Preserve Kidney Function and Improve Exercise Tolerance in Heart Failure. JACC Basic Transl. Sci. 2018, 3, 796–809. [Google Scholar] [CrossRef] [PubMed]
- Kondo, K.; Bhushan, S.; King, A.L.; Prabhu, S.D.; Hamid, T.; Koenig, S.; Murohara, T.; Predmore, B.L.; Gojon, G.; Wang, R.; et al. H2S Protects Against Pressure Overload–Induced Heart Failure via Upregulation of Endothelial Nitric Oxide Synthase. Circulation 2013, 127, 1116–1127. [Google Scholar] [CrossRef]
- Ling, K.; Zhou, W.; Guo, Y.; Hu, G.; Chu, J.; Xie, F.; Li, Y.; Wang, W. H2S attenuates oxidative stress via nrf2/nf-κb signaling to regulate restenosis after percutaneous transluminal angioplasty. Exp. Biol. Med. 2020, 246, 1–14. [Google Scholar] [CrossRef]
- Calvert, J.W.; Elston, M.; Nicholson, C.K.; Gundewar, S.; Jha, S.; Elrod, J.W.; Ramachandran, A.; Lefer, D.J. Genetic and Pharmacologic Hydrogen Sulfide Therapy Attenuates Ischemia-Induced Heart Failure in Mice. Circulation 2010, 122, 11–19. [Google Scholar] [CrossRef]
- Peake, B.F.; Nicholson, C.K.; Lambert, J.P.; Hood, R.L.; Amin, H.; Amin, S.; Calvert, J.W. Hydrogen sulfide preconditions the db/db diabetic mouse heart against ischemia-reperfusion injury by activating Nrf2 signaling in an Erk-dependent manner. Am. J. Physiol. Circ. Physiol. 2013, 304, H1215–H1224. [Google Scholar] [CrossRef]
- King, A.L.; Polhemus, D.J.; Bhushan, S.; Otsuka, H.; Kondo, K.; Nicholson, C.K.; Bradley, J.M.; Islam, K.N.; Calvert, J.W.; Tao, Y.X.; et al. Hydrogen sulfide cytoprotective signaling is endothelial nitric oxide synthase-nitric oxide dependent. Proc. Natl. Acad. Sci. USA 2014, 111, 3182–3187. [Google Scholar] [CrossRef] [PubMed]
- Andreadou, I.; Schulz, R.; Papapetropoulos, A.; Turan, B.; Ytrehus, K.; Ferdinandy, P.; Daiber, A.; Di Lisa, F. The role of mitochondrial reactive oxygen species, NO and H2S in ischaemia/reperfusion injury and cardioprotection. J. Cell. Mol. Med. 2020, 24, 6510–6522. [Google Scholar] [CrossRef] [PubMed]
- Bucci, M.; Papapetropoulos, A.; Vellecco, V.; Zhou, Z.; Zaid, A.; Giannogonas, P.; Cantalupo, A.; Dhayade, S.; Karalis, K.P.; Wang, R.; et al. Cgmp-dependent protein kinase contributes to hydrogen sulfide-stimulated vasorelaxation. PLoS ONE 2012, 7, e53319. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Huang, Y.; Yu, W.; Chen, S.; Yao, Q.; Zhang, C.; Bu, D.; Tang, C.; Du, J.; Jin, H. Sulfhydration-associated phosphodiesterase 5A dimerization mediates vasorelaxant effect of hydrogen sulfide. Oncotarget 2017, 8, 31888–31900. [Google Scholar] [CrossRef]
- Yuan, C.; Hou, H.T.; Chen, H.X.; Wang, J.; Wang, Z.Q.; Chen, T.N.; Novakovic, A.; Marinko, M.; Yang, Q.; Liu, Z.G.; et al. Hydrogen sulfide-mediated endothelial function and the interaction with enos and pde5a activity in human internal mammary arteries. J. Int. Med. Res. 2019, 47, 3778–3791. [Google Scholar] [CrossRef] [PubMed]
- Yong, Q.-C.; Hu, L.-F.; Wang, S.; Huang, D.; Bian, J.-S. Hydrogen sulfide interacts with nitric oxide in the heart: Possible involvement of nitroxyl. Cardiovasc. Res. 2010, 88, 482–491. [Google Scholar] [CrossRef]
- Kimura, Y.; Goto, Y.-I.; Kimura, H. Hydrogen Sulfide Increases Glutathione Production and Suppresses Oxidative Stress in Mitochondria. Antioxid. Redox Signal. 2010, 12, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Yu, B.; Li, Z.; Yuan, Z.; Organ, C.L.; Trivedi, R.K.; Wang, S.; Lefer, D.J.; Wang, B. An esterase-sensitive prodrug approach for controllable delivery of persulfide species. Angew. Chem. 2017, 70112, 11749–11753. [Google Scholar] [CrossRef] [PubMed]
- Polhemus, D.J.; Kondo, K.; Bhushan, S.; Bir, S.C.; Kevil, C.G.; Murohara, T.; Lefer, D.J.; Calvert, J.W. Hydrogen Sulfide Attenuates Cardiac Dysfunction After Heart Failure Via Induction of Angiogenesis. Circ. Heart Fail. 2013, 6, 1077–1086. [Google Scholar] [CrossRef]
- Polhemus, D.J.; Li, Z.; Pattillo, C.B.; Gojon, G.; Giordano, T.; Krum, H. A Novel Hydrogen Sulfide Prodrug, SG 1002, Promotes Hydrogen Sulfide and Nitric Oxide Bioavailability in Heart Failure Patients. Cardiovasc. Ther. 2015, 33, 216–226. [Google Scholar] [CrossRef] [PubMed]
- Jeremic, J.N.; Jakovljevic, V.L.; Zivkovic, V.I.; Srejovic, I.M.; Bradic, J.V.; Bolevich, S.; Nikolic Turnic, T.R.; Mitrovic, S.L.; Jovicic, N.U.; Tyagi, S.C.; et al. The cardioprotective effects of diallyl trisulfide on diabetic rats with ex vivo induced ischemia/reperfusion injury. Mol. Cell. Biochem. 2019, 460, 151–164. [Google Scholar] [CrossRef]
- Sun, H.J.; Xiong, S.P.; Cao, X.; Cao, L.; Zhu, M.Y.; Wu, Z.Y.; Bian, J.S. Polysulfide-mediated sulfhydration of sirt1 prevents diabetic nephropathy by suppressing phosphorylation and acetylation of p65 nf-κb and stat3. Redox Biol. 2021, 38, 101813. [Google Scholar] [CrossRef] [PubMed]
- Greiner, R.; Pálinkás, Z.; Bäsell, K.; Becher, D.; Antelmann, H.; Nagy, P.; Dick, T.P. Polysulfides link H2S to protein thiol oxidation. Antioxid. Redox Signal. 2013, 19, 1749–1765. [Google Scholar] [CrossRef]
- Cortese-Krott, M.M.; Kuhnle, G.G.C.; Dyson, A.; Fernandez, B.O.; Grman, M.; Dumond, J.F.; Barrow, M.P.; McLeod, G.; Nakagawa, H.; Ondrias, K.; et al. Key bioactive reaction products of the NO/H2S interaction are S/N-hybrid species, polysulfides, and nitroxyl. Proc. Natl. Acad. Sci. USA 2015, 112, E4651–E4660. [Google Scholar] [CrossRef]
- Gojon, G.; Morales, G.A. SG1002 and Catenated Divalent Organic Sulfur Compounds as Promising Hydrogen Sulfide Prodrugs. Antioxid. Redox Signal. 2020, 33, 1010–1045. [Google Scholar] [CrossRef] [PubMed]
- Moustafa, A.; Habara, Y. Cross talk between polysulfide and nitric oxide in rat peritoneal mast cells. Am. J. Physiol. Cell Physiol. 2016, 310, C894–C902. [Google Scholar] [CrossRef]
- Jain, S.K.; Bull, R.; Rains, J.L.; Bass, P.F.; Levine, S.N.; Reddy, S.; McVie, R.; Bocchini, J.A. Low Levels of Hydrogen Sulfide in the Blood of Diabetes Patients and Streptozotocin-Treated Rats Causes Vascular Inflammation? Antioxid. Redox Signal. 2010, 12, 1333–1337. [Google Scholar] [CrossRef]
- Shimizu, Y.; Nicholson, C.K.; Lambert, J.P.; Barr, L.A.; Kuek, N.; Herszenhaut, D.; Tan, L.; Murohara, T.; Hansen, J.M.; Husain, A.; et al. Sodium Sulfide Attenuates Ischemic-Induced Heart Failure by Enhancing Proteasomal Function in an Nrf2-Dependent Manner. Circ. Heart Fail. 2016, 9, e002368. [Google Scholar] [CrossRef] [PubMed]
- Swan, K.W.; Song, B.M.; Chen, A.L.; Chen, T.J.; Chan, R.A.; Guidry, B.T.; Katakam, P.V.G.; Kerut, E.K.; Giles, T.D.; Kadowitz, P.J. Analysis of decreases in systemic arterial pressure and heart rate in response to the hydrogen sulfide donor sodium sulfide. Am. J. Physiol. Circ. Physiol. 2017, 313, H732–H743. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.-Y.; Li, X.-H.; Zhang, T.; Fu, J.; Cui, X.-D. Hydrogen sulfide upregulates heme oxygenase-1 expression in rats with volume overload-induced heart failure. Biomed. Rep. 2013, 1, 454–458. [Google Scholar] [CrossRef] [PubMed]
- Mishra, P.K.; Tyagi, N.; Sen, U.; Givvimani, S.; Tyagi, S.C. H2S ameliorates oxidative and proteolytic stresses and protects the heart against adverse remodeling in chronic heart failure. Am. J. Physiol. Circ. Physiol. 2010, 298, H451–H456. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Zhang, W.; Zhang, M.; Jin, M.; Xu, W.; Zhou, X. Gas signaling molecule hydrogen sulfide attenuates doxorubicin-induced dilated cardiomyopathy. Oncotarget 2017, 8, 95425–95431. [Google Scholar] [CrossRef] [PubMed]
- Szabo, C.; Papapetropoulos, A. International Union of Basic and Clinical Pharmacology. CII: Pharmacological Modulation of H2S Levels: H2S Donors and H2S Biosynthesis Inhibitors. Pharm. Rev. 2017, 69, 497–564. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Organ, C.L.; Zibilich, C.M.; Kang, J.; Xian, M.; Lefer, D.J. A novel hydrogen sulfide donor, jk1, preserves left ventricular ejection fraction and improves hemodynamics in the setting of heart failure. Circulation 2017, 136, A16459. [Google Scholar]
- Wu, D.; Hu, Q.; Xiong, Y.; Zhu, D.; Mao, Y.; Zhu, Y.Z. Novel H2S-NO hybrid molecule (ZYZ-803) promoted synergistic effects against heart failure. Redox. Biol. 2018, 15, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Lilyanna, S.; Peh, M.T.; Liew, O.W.; Wang, P.; Moore, P.K.; Richards, A.M.; Martinez, E.C. GYY4137 attenuates remodeling, preserves cardiac function and modulates the natriuretic peptide response to ischemia. J. Mol. Cell. Cardiol. 2015, 87, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Peng, H.; Du, Q.; Lin, W.; Liu, Y. GYY4137, a hydrogen sulfide-releasing molecule, inhibits the inflammatory response by suppressing the activation of nuclear factor-kappa B and mitogen-activated protein kinases in Coxsackie virus B3-infected rat cardiomyocytes. Mol. Med. Rep. 2014, 11, 1837–1844. [Google Scholar] [CrossRef]
- Al-Magableh, M.R.; Kemp-Harper, B.K.; Hart, J.L. Hydrogen sulfide treatment reduces blood pressure and oxidative stress in angiotensin II-induced hypertensive mice. Hypertens. Res. 2015, 38, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Kolluru, G.K.; Bir, S.C.; Yuan, S.; Shen, X.; Pardue, S.; Wang, R.; Kevil, C.G. Cystathionine γ-lyase regulates arteriogenesis through NO-dependent monocyte recruitment. Cardiovasc. Res. 2015, 107, 590–600. [Google Scholar] [CrossRef]
- Li, Z.; Polhemus, D.J.; Lefer, D.J. Evolution of Hydrogen Sulfide Therapeutics to Treat Cardiovascular Disease. Circ. Res. 2018, 123, 590–600. [Google Scholar] [CrossRef]
- Zhang, F.; Ma, B.; Liang, G.; Chen, Y.; Zhang, H. Effect of hydrogen sulfide on restenosis of peripheral arteries after angioplasty. Mol. Med. Rep. 2012, 5, 1497–1502. [Google Scholar] [CrossRef]
- Saklayen, M.G. The Global Epidemic of the Metabolic Syndrome. Curr. Hypertens. Rep. 2018, 20, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Sigit, F.S.; Tahapary, D.L.; Trompet, S.; Sartono, E.; Van Dijk, K.W.; Rosendaal, F.R.; De Mutsert, R. The prevalence of metabolic syndrome and its association with body fat distribution in middle-aged individuals from Indonesia and the Netherlands: A cross-sectional analysis of two population-based studies. Diabetol. Metab. Syndr. 2020, 12, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ford, E.S. Risks for All-Cause Mortality, Cardiovascular Disease, and Diabetes Associated with the Metabolic Syndrome: A summary of the evidence. Diabetes Care 2005, 28, 1769–1778. [Google Scholar] [CrossRef] [PubMed]
- Malik, S.; Wong, N.D.; Franklin, S.S.; Kamath, T.V.; L’Italien, G.J.; Pio, J.R.; Williams, G.R. Impact of the Metabolic Syndrome on Mortality from Coronary Heart Disease, Cardiovascular Disease, and All Causes in United States Adults. Circulation 2004, 110, 1245–1250. [Google Scholar] [CrossRef] [PubMed]
- Geng, B.; Cai, B.; Liao, F.; Zheng, Y.; Zeng, Q.; Fan, X.; Gong, Y.; Yang, J.; Cui, Q.H.; Tang, C.; et al. Increase or Decrease Hydrogen Sulfide Exert Opposite Lipolysis, but Reduce Global Insulin Resistance in High Fatty Diet Induced Obese Mice. PLoS ONE 2013, 8, e73892. [Google Scholar] [CrossRef] [PubMed]
- Bełtowski, J.; Jamroz-Wiśniewska, A. Hydrogen Sulfide in the Adipose Tissue—Physiology, Pathology and a Target for Pharmacotherapy. Molecules 2016, 22, 63. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.; Cao, W.; Wu, L.; Wang, R. Hydrogen sulfide and the liver. Nitric. Oxide 2014, 41, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Zheng, N.; Ziqiang, S.; Cheng, H.; Sun, Z.; Gao, B.; Zhang, Y.; Pang, W.; Huangfu, C.; Ji, S.; et al. Exogenous hydrogen sulfide mitigates the fatty liver in obese mice through improving lipid metabolism and antioxidant potential. Med. Gas. Res. 2015, 5, 1–8. [Google Scholar] [CrossRef]
- Li, M.; Xu, C.; Shi, J.; Ding, J.; Wan, X.; Chen, D.; Gao, J.; Li, C.; Zhang, J.; Lin, Y.; et al. Fatty acids promote fatty liver disease via the dysregulation of 3-mercaptopyruvate sulfurtransferase/hydrogen sulfide pathway. Gut 2017, 67, 2169–2180. [Google Scholar] [CrossRef]
- Wu, D.-B.; Chen, J.-F.; Xu, Q.; Lin, J.-Q.; Liao, J.-Q.; Wu, W. Exogenous hydrogen sulfide inhibits high-glucose-induced injuries via regulating leptin/leptin receptor signaling pathway in human umbilical vein endothelial cells. Nan Fang Yi Ke Da Xue Xue Bao J. South. Med. Univ. 2016, 36, 1055–1061. [Google Scholar]
- Zhuang, X.-D.; Hu, X.; Long, M.; Dong, X.-B.; Liu, D.-H.; Liao, X.-X. Exogenous hydrogen sulfide alleviates high glucose-induced cardiotoxicity via inhibition of leptin signaling in H9c2 cells. Mol. Cell. Biochem. 2014, 391, 147–155. [Google Scholar] [CrossRef]
- Yang, W.-S.; Lee, W.-J.; Funahashi, T.; Tanaka, S.; Matsuzawa, Y.; Chao, C.-L.; Chen, C.-L.; Tai, T.-Y.; Chuang, L.-M. Plasma Adiponectin Levels in Overweight and Obese Asians. Obes. Res. 2002, 10, 1104–1110. [Google Scholar] [CrossRef]
- Pan, Z.; Wang, H.; Liu, Y.; Yu, C.; Zhang, Y.; Chen, J.; Wang, X.; Guan, Q. Involvement of CSE/ H2S in high glucose induced aberrant secretion of adipokines in 3T3-L1 adipocytes. Lipids Health Dis. 2014, 13, 155. [Google Scholar] [CrossRef]
- Suzuki, K.; Sagara, M.; Aoki, C.; Tanaka, S.; Aso, Y. Clinical Implication of Plasma Hydrogen Sulfide Levels in Japanese Patients with Type 2 Diabetes. Intern. Med. 2017, 56, 17–21. [Google Scholar] [CrossRef] [PubMed]
- Xue, R.; Hao, D.-D.; Sun, J.-P.; Li, W.-W.; Zhao, M.-M.; Li, X.-H.; Chen, Y.; Zhu, J.-H.; Ding, Y.-J.; Liu, J.; et al. Hydrogen Sulfide Treatment Promotes Glucose Uptake by Increasing Insulin Receptor Sensitivity and Ameliorates Kidney Lesions in Type 2 Diabetes. Antioxid. Redox Signal. 2013, 19, 5–23. [Google Scholar] [CrossRef]
- Gheibi, S.; Jeddi, S.; Kashfi, K.; Ghasemi, A. Regulation of vascular tone homeostasis by NO and H2S: Implications in hypertension. Biochem. Pharm. 2018, 149, 42–59. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Li, W.; Chen, Q.; Jiang, Y.; Lu, X.; Zhao, X. Hydrogen sulfide accelerates wound healing in diabetic rats. Int. J. Clin. Exp. Pathol. 2015, 8, 5097–5104. [Google Scholar] [PubMed]
- Zhou, X.; Feng, Y.; Zhan, Z.; Chen, J. Hydrogen Sulfide Alleviates Diabetic Nephropathy in a Streptozotocin-induced Diabetic Rat Model. J. Biol. Chem. 2014, 289, 28827–28834. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Wu, L.; Jiang, B.; Yang, W.; Qi, J.; Cao, K.; Meng, Q.; Mustafa, A.K.; Mu, W.; Zhang, S.; et al. H2S as a physiologic vasorelaxant: Hypertension in mice with deletion of cystathionine γ-lyase. Science 2008, 322, 587–590. [Google Scholar] [CrossRef] [PubMed]
- Koenitzer, J.R.; Isbell, T.S.; Patel, H.D.; Benavides, G.A.; Dickinson, D.A.; Patel, R.P.; Darley-Usmar, V.M.; Lancaster, J.R.; Doeller, J.E.; Kraus, D.W. Hydrogen sulfide mediates vasoactivity in an O2-dependent manner. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, 1953–1960. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Du, J.; Bu, D.; Yan, H.; Tang, X.; Tang, C. The regulatory effect of hydrogen sulfide on hypoxic pulmonary hypertension in rats. Biochem. Biophys. Res. Commun. 2003, 302, 810–816. [Google Scholar]
- Liu, Y.H.; Yan, C.D.; Bian, J.S. Hydrogen sulfide: A novel signaling molecule in the vascular system. J. Cardiovasc. Pharmacol. 2011, 58, 560–569. [Google Scholar] [CrossRef] [PubMed]
- Wang, R. Signaling pathways for the vascular effects of hydrogen sulfide. Curr. Opin. Nephrol. Hypertens 2011, 20, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Zhang, J.; Lu, Y.; Wang, R. The vasorelaxant effect of H2S as a novel endogenous gaseous KATP channel opener. Embo J. 2001, 20, 6008–6016. [Google Scholar] [CrossRef] [PubMed]
- Rushing, A.M.; Donnarumma, E.; Polhemus, D.J.; Au, K.R.; Victoria, S.E.; Schumacher, J.D.; Li, Z.; Jenkins, J.S.; Lefer, D.J.; Goodchild, T.T. Effects of a novel hydrogen sulfide prodrug in a porcine model of acute limb ischemia. J. Vasc. Surg. 2019, 69, 1924–1935. [Google Scholar] [CrossRef]
- Al-Magableh, M.R.; Kemp-Harper, B.K.; Ng, H.H.; Miller, A.A.; Hart, J.L. Hydrogen sulfide protects endothelial nitric oxide function under conditions of acute oxidative stress in vitro. Naunyn-Schmiedeberg’s Arch. Pharm. 2013, 387, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, Y.; Polavarapu, R.; Eskla, K.-L.; Nicholson, C.K.; Koczor, C.A.; Wang, R.; Lewis, W.; Shiva, S.; Lefer, D.J.; Calvert, J.W. Hydrogen sulfide regulates cardiac mitochondrial biogenesis via the activation of AMPK. J. Mol. Cell. Cardiol. 2018, 116, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.-D.; Wang, H.; Kho, S.-H.; Rinkiko, S.; Sheng, X.; Shen, H.-M.; Zhu, Y.-Z. Hydrogen Sulfide Protects HUVECs against Hydrogen Peroxide Induced Mitochondrial Dysfunction and Oxidative Stress. PLoS ONE 2013, 8, e53147. [Google Scholar] [CrossRef] [PubMed]
- Oktay, A.A.; Rich, J.D.; Shah, S.J. The Emerging Epidemic of Heart Failure with Preserved Ejection Fraction. Curr. Heart Fail. Rep. 2013, 10, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K.; Kass, D.A. Heart failure with preserved ejection fraction: Mechanisms, clinical features, and therapies. Circ. Res. 2014, 115, 79–96. [Google Scholar] [CrossRef] [PubMed]
- Caruana, L.; Petrie, M.C.; Davie, A.P.; McMurray, J.J.V. Do patients with suspected heart failure and preserved left ventricular systolic function suffer from “diastolic heart failure” or from misdiagnosis? A prospective descriptive study. BMJ 2000, 321, 215–218. [Google Scholar] [CrossRef]
- Samson, R.; Jaiswal, A.; Ennezat, P.V.; Cassidy, M.; Le Jemtel, T.H. Clinical Phenotypes in Heart Failure with Preserved Ejection Fraction. J. Am. Heart Assoc. 2016, 5, e002477. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.J.; Katz, D.H.; Selvaraj, S.; Burke, M.A.; Yancy, C.W.; Gheorghiade, M.; Bonow, R.O.; Huang, C.-C.; Deo, R.C. Phenomapping for Novel Classification of Heart Failure with Preserved Ejection Fraction. Circulation 2015, 131, 269–279. [Google Scholar] [CrossRef] [PubMed]
- Lewis, G.A.; Schelbert, E.B.; Williams, S.G.; Cunnington, C.; Ahmed, F.; McDonagh, T.A.; Miller, C.A. Biological Phenotypes of Heart Failure with Preserved Ejection Fraction. J. Am. Coll. Cardiol. 2017, 70, 2186–2200. [Google Scholar] [CrossRef] [PubMed]
- Savji, M.; Meijers, W.C.; Bartz, T.M.; Bhambhani, V.; Cushman, M.; Nayor, M.; Kizer, J.R.; Sarma, A.; Blaha, M.J.; Gansevoort, R.T.; et al. The association of obesity and cardiometabolic traits with incident hfpef and hfref. Physiol. Behav. 2019, 176, 139–148. [Google Scholar] [CrossRef]
- Bryan, N.S.; Lefer, D.J. Update on Gaseous Signaling Molecules Nitric Oxide and Hydrogen Sulfide: Strategies to Capture their Functional Activity for Human Therapeutics. Mol. Pharm. 2019, 96, 109–114. [Google Scholar] [CrossRef]
- Valero-Muñoz, M.; Backman, W.; Sam, F. Murine models of heart failure with preserved ejection fraction: A “fishing expedition”. JACC Basic Transl. Sci. 2017, 2, 770–789. [Google Scholar] [CrossRef] [PubMed]
- Hamdani, N.; Franssen, C.; Lourenço, A.; Falca o-Pires, I.; Fontoura, D.; Leite, S.; Plettig, L.; Lopez, B.; Ottenheijm, C.A.; Becher, P.M.; et al. Myocardial titin hypophosphorylation importantly contributes to heart failure with preserved ejection fraction in a rat metabolic risk model. Circ. Heart Fail. 2013, 6, 1239–1249. [Google Scholar] [CrossRef] [PubMed]
- Nagy, P.; Pálinkás, Z.; Nagy, A.; Budai, B.; Tóth, I.; Vasas, A. Chemical aspects of hydrogen sulfide measurements in physiological samples. Biochim. Biophys. Acta (BBA) Gen. Subj. 2014, 1840, 876–891. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).