
NADPH Oxidases (NOX): An Overview from Discovery, Molecular Mechanisms to Physiology and Pathology


Abstract
1. Introduction
2. NOX Family of NADPH Oxidases: Discovery of the Phagocytic Enzyme and History of NOX
3. Components of the Phagocytic NADPH Oxidase Complex
3.1. NOX2: NADPH Oxidase Prototype
3.2. The Components of the NADPH Oxidase Complex
3.2.1. The Flavocytochrome b558
NOX2
Electron Transfer
The p22phox Membrane Partner
3.3. Cytosolic Components
3.3.1. Cytosolic Factors
3.3.2. G Proteins
4. The Activation Mechanisms and Assembly of Phagocytic NADPH Oxidase
4.1. Activation
4.2. NOX Priming
4.3. Phosphorylation of Subunits of the NADPH Oxidase Complex
4.4. Activation by Lipids and Arachidonic Acid
5. NOX Homologs and Isoforms
5.1. NOX1-NOX3
5.2. NOX4
5.3. NOX5
5.4. DUOX1/DUOX2
6. NOX: From Bioinformatics to Structural Biology
6.1. Phylogenetic Analysis of Eukaryotic NOX
6.2. Experimental Discovery of Prokaryotic NOX
6.3. Structural Characterization of NOX Proteins
7. Involvement of NOX in Physiological Processes
7.1. Involvement in Host Defense and Inflammation
7.1.1. Phagocytosis: An ROS-Dependent Pathogen Clearance Process
7.1.2. Inactivation of Virulence Factors
7.1.3. Limitation of the Inflammatory Response
7.1.4. NET Activation
7.1.5. DUOX and Other NOXs in Host Defense
7.2. Role of NOX in Redox Signaling
7.2.1. Regulation of Signaling Pathways
Inhibition of Phosphatases
Activation of Kinases
7.2.2. Regulation of Calcium Ions
Membrane Calcium Channels
Release of Intracellular Calcium Ions
Calcium Pumps
- At low concentrations of ROS, the S-glutathionylation of cysteine residues by interaction of glutathione with peroxynitrite radicals leads to the formation of reversible disulfide bridges that stimulate Ca2+ pumps.
- At a higher ROS concentration, the excessive level of oxidative stress ends in irreversible thiol oxidation, resulting in the inactivation of enzymes [297].
7.2.3. Regulation of Cell Growth and Death
Cell Death
Cell Growth
7.2.4. Role in Biosynthesis Mechanisms
7.2.5. Role in Angiogenesis
The Proliferation Stage
The Sprouting Stage
The Migration and Tubule Formation Stages
8. Pathologies Related to NOX Deregulation
8.1. Chronic Granulomatous Disease
8.2. Central Nervous System Diseases
8.2.1. Parkinson’s Disease
8.2.2. Alzheimer’s Disease
8.3. Cancers
8.3.1. Tumor Development
8.3.2. Proliferation, Invasion and Metastasis
8.3.3. Tumor-Mediated Angiogenesis
8.4. Cardiovascular Pathologies
8.4.1. Hypertension
8.4.2. Atherosclerosis
8.4.3. Diabetes
9. Conclusions
Funding
Conflicts of Interest
References
- Warburg, O. Beobachtungen uber die Oxydationsprozeesse im Seeigelei. Z. Physiol. Chem. 1908, 57, 1–16. [Google Scholar] [CrossRef]
- Baldridge, C.W.; Gerard, R.W. The Extra Respiration of Phagocytosis. Am. J. Physiol. 1932, 103, 235–236. [Google Scholar] [CrossRef]
- Sbarra, A.; Karnovsky, M. The biochemical basis of phagocytosis. I. Metabolic changes during the ingestion of particles by polymorphonuclear leukocytes. J. Biol. Chem. 1959, 234, 1355–1362. [Google Scholar] [CrossRef]
- Iyer, G.Y.N.; Islam, M.F.; Quastel, J.H. Biochemical Aspects of Phagocytosis. Nature 1961, 192, 535–541. [Google Scholar] [CrossRef]
- Rossi, F.; Zatti, M. Biochemical Aspects of Phagocytosis in Polymorphonuclear Leucocytes. NADH and NADPH Oxidation by the Granules of Resting and Phagocytizing Cells. Experientia 1964, 20, 21–23. [Google Scholar] [CrossRef]
- Babior, B.; Curnutte, J.; Kipnes, B. Pyridine nucleotide-dependent superoxide production by a cell-free system from human granulocytes. J. Clin. Investig. 1975, 56, 1035–1042. [Google Scholar] [CrossRef]
- Clark, R.; Leidal, K.; Pearson, D.; Nauseef, W. NADPH oxidase of human neutrophils. Subcellular localization and characterization of an arachidonate-activatable superoxide generating system. J. Biol. Chem. 1987, 262, 4065–4074. [Google Scholar] [CrossRef]
- Klebanoff, S. Myeloperoxidase: Contribution to the microbicidal activity of intact leukocytes. Science 1970, 169, 1095–1097. [Google Scholar] [CrossRef] [PubMed]
- Babior, B.M.; Kipnes, R.S.; Curnutte, J.T. Biological defense mechanisms. The production by leukocytes of superoxide, a potential bactericidal agent. J. Clin. Investig. 1973, 52, 741–744. [Google Scholar] [CrossRef] [PubMed]
- Abuaita, B.; Schultz, T.; O’Riordan, M. Mitochondria-Derived Vesicles Deliver Antimicrobial Reactive Oxygen Species to Control Phagosome-Localized Staphylococcus aureus. Cell Host Microbe 2018, 24, 625–636. [Google Scholar] [CrossRef] [PubMed]
- Berendes, H.; Bridges, R.; Good, R.A. A fatal granulomatosus of childhood: The clinical study of a new syndrome. Minn. Med. 1957, 40, 309–312. [Google Scholar]
- Quie, P.G.; White, J.G.; Holmes, B.; Good, R.A. In Vitro Bactericidal Capacity of Human Polymorphonuclear Leukocytes: Diminished Activity in Chronic Granulomatous Disease of Childhood. J. Clin. Investig. 1967, 46, 668–679. [Google Scholar] [CrossRef] [PubMed]
- Johnston, R.B.; Baehner, R.L. Chronic Granulamatous Disease: Correlation Between Pathogenesis and Clinical Findings. Pediatrics 1971, 48, 730–739. [Google Scholar] [PubMed]
- Baehner, R.L.; Nathan, D.G. Leukocyte oxidase: Defective activity in chronic granulomatous disease. Science 1967, 155, 835–836. [Google Scholar] [CrossRef]
- Holmes, B.; Page, A.R.; Good, R.A. Studies of the metabolic activity of leukocytes from patients with a genetic abnormality of phagocytic function. J. Clin. Investig. 1967, 46, 1422–1432. [Google Scholar] [CrossRef]
- Cross, A.R.; Segal, A.W. The NADPH oxidase of professional phagocytes-prototype of the NOX electron transport chain systems. Biochim. Biophys. Acta 2004, 1657, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Bellavite, P.; Cross, A.; Serra, M.; Davoli, A.; Jones, O.; Rossi, F. The cytochrome b and flavin content and properties of the O2−-forming NADPH oxidase solubilized from activated neutrophils. Biochim. Biophys. Acta 1983, 746, 40–47. [Google Scholar] [CrossRef]
- Gabig, T.; Kipnes, R.; Babior, B. Solubilization of the 02--forming Activity Responsible for the Respiratory Burst in Human Neutrophils. J. Biol. Chem. 1978, 253, 6663–6665. [Google Scholar] [CrossRef]
- Bellavite, P. The superoxide-forming enzymatic system of phagocytes. Free Radic. Biol. Med. 1988, 4, 225–261. [Google Scholar] [CrossRef]
- Hattori, H. Studies on the labile, stable Nadi oxidase and peroxidase staining reactions in the isolated particles of horse granulocyte. Nagoya J. Med. Sci. 1961, 23, 362–378. [Google Scholar]
- Segal, B.; Veys, P.; Malech, H.; Cowan, M. Chronic granulomatous disease: Lessons from a rare disorder. Biol. Blood Marrow Transplant. 2011, 17, S123–S131. [Google Scholar] [CrossRef]
- Winkelstein, J.; Marino, M.; Johnston, R., Jr.; Boyle, J.; Curnutte, J.; Gallin, J.; Malech, H.; Holland, S.; Ochs, H.; Quie, P.R.; et al. Chronic granulomatous disease: Report on a national registry of 368 patients. Medicine 2000, 79, 155–169. [Google Scholar] [CrossRef]
- Segal, A.W.; Jones, O.T. Novel cytochrome b system in phagocytic vacuoles of human granulocytes. Nature 1978, 276, 515–517. [Google Scholar] [CrossRef]
- Segal, A.W.; Jones, O.T.G. The Subcellular Distribution and some Properties of the Cytochrome b Component of the Microbicidal Oxidase System of Human Neutrophils. Biochem. J. 1979, 182, 181–188. [Google Scholar] [CrossRef]
- Yu, L.; Quinn, M.T.; Cross, A.R.; Dinauer, M.C. Gp91phox is the heme binding subunit of the superoxide-generating NADPH oxidase. Proc. Natl. Acad. Sci. USA 1998, 95, 7993–7998. [Google Scholar] [CrossRef]
- Quinn, M.; Mullen, M.; Jesaitis, A. Human Neutrophil Cytochrome b Contains Multiple Hemes. J. Biol. Chem. 1992, 267, 7303–7309. [Google Scholar] [CrossRef]
- Cross, A.; Rae, J.; Curnutte, J. Cytochrome b−245 of the Neutrophil Superoxide-generating System Contains Two Nonidentical Hemes. J. Biol. Chem. 1995, 270, 17075–17077. [Google Scholar] [CrossRef]
- Segal, A.W.; West, I.; Wientjes, F.; Nugent, J.H.A.; Chavan, A.J.; Haley, B.; Scrace, G. Cytochrome b-245 is a flavocytochrome containing FAD and the NADPH-binding site of the microbicidal oxidase of phagocytes. Biochem. J. 1992, 284, 781–788. [Google Scholar] [CrossRef]
- Cross, A.; Parkinson, J.; Jones, O. The superoxide-generating oxidase of leucocytes. NADPH-dependent reduction of flavin and cytochrome b in solubilized preparations. Biochem. J. 1984, 223, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Dinauer, M.C.; Orkin, S.H.; Brown, R.; Jesaitis, A.J.; Parkos, C.A. The glycoprotein encoded by the X-linked chronic granulomatous disease locus is a component of the neutrophil cytochrome b complex. Nature 1987, 327, 717–720. [Google Scholar] [CrossRef] [PubMed]
- Parkos, C.; Allen, R.A.; Cochrane, C.; Jesaitis, A. Purified cytochrome b from human granulocyte plasma membrane is comprised of two polypeptides with relative molecular weights of 91,000 and 22,000. J. Clin. Investig. 1987, 80, 732–742. [Google Scholar] [CrossRef] [PubMed]
- Müller, D. Oxidation von Glukose mit Extrakten aus Aspegillus niger. Biochem. Z. 1928, 199, 136–170. [Google Scholar]
- Rossi, F.; Zatti, M. Changes in the Metabolic Pattern of Polymorpho-Nuclear Leucocytes during Phagocytosis. Br. J. Exp. Pathol. 1964, 45, 548–559. [Google Scholar]
- Klebanoff, S.J. Antimicrobial mechanisms in neutrophilic polymorphonuclear leukocytes. Semin. Hematol. 1975, 12, 117–142. [Google Scholar]
- Babior, B.M. Oxygen-dependent microbial killing by phagocytes (first of two parts). N. Engl. J. Med. 1978, 298, 659–668. [Google Scholar] [CrossRef]
- Bromberg, Y.; Pick, E. Unsaturated fatty acids stimulate NADPH-dependent superoxide production by cell-free system derived from macrophages. Cell. Immunol. 1984, 88, 213–221. [Google Scholar] [CrossRef]
- Royer-Pokora, B.; Kunkel, L.M.; Monaco, A.P.; Goff, S.C.; Newburger, P.E.; Baehner, R.L.; Cole, F.S.; Curnutte, J.T.; Orkin, S.H. Cloning the gene for an inherited human disorder-chronic granulomatous disease-on the basis of its chromosomal location. Nature 1986, 322, 32–38. [Google Scholar] [CrossRef]
- Teahan, C.; Rowe, P.; Parker, P.; Totty, N.; Segal, A.W. The X-linked chronic granulomatous disease gene codes for the beta-chain of cytochrome b-245. Nature 1987, 327, 720–721. [Google Scholar] [CrossRef] [PubMed]
- Nunoi, H.; Rotrosen, D.; Gallin, J.; Malech, H. Two forms of autosomal chronic granulomatous disease lack distinct neutrophil cytosol factors. Science 1988, 242, 1298–1301. [Google Scholar] [CrossRef]
- Volpp, B.; Nauseef, W.; Clark, R. Two cytosolic neutrophil oxidase components absent in autosomal chronic granulomatous disease. Science 1988, 242, 1295–1297. [Google Scholar] [CrossRef]
- Abo, A.; Pick, E.; Hall, A.; Totty, N.; Teahan, C.; Segal, A. Activation of the NADPH oxidase involves the small GTP-binding protein p21rac1. Nature 1991, 353, 668–670. [Google Scholar] [CrossRef]
- Knaus, U.; Heyworth, P.; Evans, T.; Curnutte, J.; Bokoch, G. Regulation of the phagocyte oxygen radical production by the GTP-binding protein Rac 2. Science 1991, 254, 1512–1515. [Google Scholar] [CrossRef]
- Wientjes, F.; Hsuan, J.; Totty, N.; Segal, A. p40phox, a third cytosolic componet of the activation complex of the NADPH oxidase that contain src homology 3 domains. Biochem. J. 1993, 296, 557–561. [Google Scholar] [CrossRef]
- Someya, A.; Nagaoka, I.; Yamashita, T. Purification of the 260 kDa cytosolic complex involved in the superoxide production of guinea pig neutrophils. FEBS Lett. 1993, 330, 215–218. [Google Scholar] [CrossRef]
- Groom, Q.J.; Torres, M.A.; Fordham-Skelton, A.P.; Hammond-Kosack, K.E.; Robinson, N.J.; Jones, J.D.G. RbohA, a rice homologue of the mammalian gp91phox respiratory burst oxidase gene. Plant J. 1996, 10, 515–522. [Google Scholar] [CrossRef]
- Suh, Y.A.; Arnold, R.S.; Lassegue, B.; Shi, J.; Xu, X.; Sorescu, D.; Chung, A.B.; Griendling, K.K.; Lambeth, J.D. Cell transformation by the superoxide-generating oxidase Mox1. Nature 1999, 401, 79–82. [Google Scholar] [CrossRef]
- Dupuy, C.; Ohayon, R.; Valent, A.; Noël-Hudson, M.-S.; Dème, D.; Virion, A. Purification of a Novel Flavoprotein Involved in the Thyroid NADPH Oxidase. J. Biol. Chem. 1999, 274, 37265–37269. [Google Scholar] [CrossRef]
- de Deken, X.; Wang, D.; Many, M.-C.; Costagliola, S.; Libert, F.; Vassart, G.; Dumont, J.E.; Miot, F. Cloning of Two Human Thyroid cDNAs Encoding New Members of the NADPH Oxidase Family. J. Biol. Chem. 2000, 275, 23227–23233. [Google Scholar] [CrossRef]
- Lapouge, K.; Smith, S.; Walker, P.; Gamblin, S.; Smerdon, S.; Rittinger, K. Structure of the TPR Domain of p67phox in Complex with Rac·GTP. Mol. Cell 2000, 6, 899–907. [Google Scholar] [CrossRef]
- Grizot, S.; Fauré, J.; Fieschi, F.; Vignais, P.; Dagher, M.; Pebay-Peyroula, E. Crystal structure of the Rac1-RhoGDI complex involved in nadph oxidase activation. Biochemistry 2001, 40, 10007–10013. [Google Scholar] [CrossRef]
- Bravo, J.; Karathanassis, D.; Pacold, C.M.; Pacold, M.E.; Ellson, C.D.; Anderson, K.E.; Butler, P.J.; Lavenir, I.; Perisic, O.; Hawkins, P.T.; et al. The crystal structure of the PX domain from p40(phox) bound to phosphatidylinositol 3-phosphate. Mol. Cell 2001, 8, 829–839. [Google Scholar] [CrossRef]
- Hiroaki, H.; Ago, T.; Sumimoto, I.T.H.; Kohda, D. Solution structure of the PX domain, a target of the SH3 domain. Nat. Struct. Biol. 2001, 8, 526–530. [Google Scholar] [CrossRef] [PubMed]
- Karathanassis, D.; Stahelin, R.V.; Bravo, J.; Perisic, O.; Pacold, C.M.; Cho, W.; Williams, R.L. Binding of the PX domain of p47(phox) to phosphatidylinositol 3,4-bisphosphate and phosphatidic acid is masked by an intramolecular interaction. EMBO J. 2002, 21, 5057–5068. [Google Scholar] [CrossRef] [PubMed]
- Kami, K.; Takeya, R.; Sumimoto, H.; Kohda, D. Diverse recognition of non-PxxP peptide ligands by the SH3 domains from p67phox, Grb2 and Pex13p. EMBO J. 2002, 21, 4268–4276. [Google Scholar] [CrossRef] [PubMed]
- Groemping, Y.; Lapouge, K.; Smerdon, S.; Rittinger, K. Molecular basis of phosphorylation-induced activation of the NADPH oxidase. Cell 2003, 113, 343–355. [Google Scholar] [CrossRef]
- Wilson, M.I.; Gill, D.J.; Perisic, O.; Quinn, M.T.; Williams, R.L. PB1 domain-mediated heterodimerization in NADPH oxidase and signaling complexes of atypical protein kinase C with Par6 and p62. Mol. Cell 2003, 12, 39–50. [Google Scholar] [CrossRef]
- Yuzawa, S.; Suzuki, N.N.; Fujioka, Y.; Ogura, K.; Sumimoto, H.; Inagaki, F. A molecular mechanism for autoinhibition of the tandem SH3 domains of p47phox, the regulatory subunit of the phagocyte NADPH oxidase. Genes Cells 2004, 9, 443–456. [Google Scholar] [CrossRef]
- Massenet, C.; Chenavas, S.; Cohen-Addad, C.; Dagher, M.C.; Brandolin, G.; Pebay-Peyroula, E.; Fieschi, F. Effects of p47phox C terminus phosphorylations on binding interactions with p40phox and p67phox. Structural and functional comparison of p40phox and p67phox SH3 domains. J. Biol. Chem. 2005, 280, 13752–13756. [Google Scholar] [CrossRef]
- Durand, D.; Cannella, D.; Dubosclard, V.; Pebay-Peyroula, E.; Vachette, P.; Fieschi, F. Small-Angle X-ray Scattering Reveals an Extended Organization for the Autoinhibitory Resting State of the p47phox Modular Protein. Biochemistry 2006, 45, 7185–7193. [Google Scholar] [CrossRef]
- Honbou, K.; Minakami, R.; Yuzawa, S.; Takeya, R.; Suzuki, N.; Kamakura, S.; Sumimoto, H.; Inagaki, F. Full-length p40phox structure suggests a basis for regulation mechanism of its membrane binding. EMBO J. 2007, 26, 1176–1186. [Google Scholar] [CrossRef] [PubMed]
- Marcoux, J.; Man, P.; Petit-Haertlein, I.; Vivès, C.; Forest, E.; Fieschi, F. p47phox Molecular Activation for Assembly of the Neutrophil NADPH Oxidase Complex. J. Biol. Chem. 2010, 285, 28980–28990. [Google Scholar] [CrossRef]
- Durand, D.; Vivès, C.; Cannella, D.; Pérez, J.; Pebay-Peyroula, E.; Vachette, P.; Fieschi, F. NADPH oxidase activator p67(phox) behaves in solution as a multidomain protein with semi-flexible linkers. J. Struct. Biol. 2010, 169, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Hajjar, C.; Cherrier, M.V.; Mirandela, G.D.; Petit-Hartlein, I.; Stasia, M.J.; Fontecilla-Camps, J.C.; Fieschi, F.; Dupuy, J. The NOX Family of Proteins Is Also Present in Bacteria. mBio 2017, 8, 1487–1517. [Google Scholar] [CrossRef] [PubMed]
- Magnani, F.; Nenci, S.; Fananas, E.M.; Ceccon, M.; Romero, E.; Fraaije, M.; Mattevi, A. Crystal structures and atomic model of NADPH oxidase. Proc. Natl. Acad. Sci. USA 2017, 114, 6764–6769. [Google Scholar] [CrossRef]
- Sun, J. Structures of Mouse DUOX1–DUOXA1 Provide Mechanistic Insights into Enzyme Activation and Regulation. Nat. Struct. Mol. Biol. 2020, 27, 1086–1093. [Google Scholar] [CrossRef]
- Harper, A.M.; Chaplin, M.F.; Segal, A.W. Cytochrome b-245 from human neutrophils is a glycoprotein. Biochem. J. 1985, 227, 783–788. [Google Scholar] [CrossRef]
- Pick, E. Cell-Free NADPH Oxidase Activation Assays: A Triumph of Reductionism. Methods Mol. Biol. 2020, 2087, 325–411. [Google Scholar]
- Wilde, M.; Carlson, K.; Manning, D.; Zigmond, S. Chemoattractant-stimulated GTPase activity is decreased on membranes from polymorphonuclear leukocytes incubated in chemoattractant. J. Biol. Chem. 1989, 264, 190–196. [Google Scholar] [CrossRef]
- Ambruso, D.; Knall, C.; Abell, A.; Penpinto, J.; Kurkchubasche, A.; Thurman, G.; Gonzalez-Aller, C.; Hiester, A.; deBoer, M.; Harbeck, R.; et al. Human neutrophil immunodeficiency syndrome is associated with an inhibitory Rac2 mutation. Proc. Natl. Acad. Sci. USA 2000, 97, 4654–4659. [Google Scholar] [CrossRef]
- Williams, D.; Tao, W.; Yang, F.; Kim, C.; Gu, Y.; Mansfield, P.; Levine, J.; Petryniak, B.; Derrow, C. Dominant negative mutation of the hematopoietic-specific Rho GTPase, Rac2, is associated with a human phagocyte immunodeficiency. Blood 2000, 96, 1646–1654. [Google Scholar] [PubMed]
- DeCoursey, T.E.; Cherny, V.V.; Morgan, D.; Katz, B.Z.; Dinauer, M.C. The gp91phox component of NADPH oxidase is not the voltage-gated proton channel in phagocytes, but it helps. J. Biol. Chem. 2001, 276, 36063–36066. [Google Scholar] [CrossRef]
- Lambeth, J.D. NOX enzymes and the biology of reactive oxygen. Nat. Rev. Immunol. 2004, 4, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Arbault, S.; Pantano, P.; Sojic, N.; Amatore, C.; Best-Belpomme, M.; Sarasin, A.; Vuillaume, M. Activation of the NADPH oxidase in human fibroblasts by mechanical intrusion of a single cell with an ultramicroelectrode. Carcinogenesis 1997, 18, 569–574. [Google Scholar] [CrossRef]
- Szatrowski, T.P.; Nathan, C.F. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res. 1991, 51, 794–798. [Google Scholar]
- Griendling, K.K.; Sorescu, D.; Ushio-Fukai, M. NAD(P)H Oxidase Role in Cardiovascular Biology and Disease. Circ Res. 2000, 86, 494–501. [Google Scholar] [CrossRef]
- Bánfi, B.; Maturana, A.; Jaconi, S.; Arnaudeau, S. A mammalian H+ channel generated through alternative splicing of the NADPH oxidase homolog NOH-1. Science 2000, 287, 138–142. [Google Scholar]
- Kikuchi, H.; Hikage, M.; Miyashita, H.; Fukumoto, M. NADPH oxidase subunit, gp91phox homologue, preferentially expressed in human colon epithelial cells. Gene 2000, 254, 237–243. [Google Scholar] [CrossRef]
- Geiszt, M.; Kopp, J.B.; Várnai, P.; Leto, T.L. Identification of Renox, an NAD(P)H oxidase in kidney. Proc. Natl. Acad. Sci. USA 2000, 97, 8010–8014. [Google Scholar] [CrossRef] [PubMed]
- Bánfi, B.; Molnar, G.; Maturana, A.; Steger, K.; Hegedûs, B.; Demaurex, N.; Krause, K. A Ca2+ activated NADPH Oxidase in Testis, Spleen, and Lymph Nodes. J. Biol. Chem. 2001, 276, 37594–37601. [Google Scholar] [CrossRef]
- Zhang, X.; Krause, K.H.; Xenarios, I.; Soldati, T.; Boeckmann, B. Evolution of the Ferric Reductase Domain (FRD) Superfamily: Modularity, Functional Diversification, and Signature Motifs. PLoS ONE 2013, 8, e58126. [Google Scholar] [CrossRef]
- Ambasta, R.; Kumar, P.; Griendling, K.; Schmidt, H.; Busse, R.; Brandes, R. Direct interaction of the novel Nox proteins with p22phox is required for the formation of a functionally active NADPH oxidase. J. Biol. Chem. 2004, 279, 45935–459341. [Google Scholar] [CrossRef]
- Kuroda, J.; Nakagawa, K.; Yamasaki, T.; Nakamura, K.; Takeya, R.; Kuribayashi, F.; Imajoh-Ohmi, S.; Igarashi, K.; Shibata, Y.; Sueishi, K.; et al. The superoxide-producing NAD(P)H oxidase Nox4 in the nucleus of human vascular endothelial cells. Genes Cells 2005, 10, 1139–1151. [Google Scholar] [CrossRef]
- Ago, T.; Kuroda, J.; Pain, J.; Fu, C.; Li, H.; Sadoshima, J. Upregulation of Nox4 by hypertrophic stimuli promotes apoptosis and mitochondrial dysfunction in cardiac myocytes. Circ. Res. 2010, 106, 1253–1264. [Google Scholar] [CrossRef]
- Gabig, T.G.; Babior, B.M. The O2(-)-forming oxidase responsible for the respiratory burst in human neutrophils. Properties of the solubilized enzyme. J. Biol. Chem. 1979, 254, 9070–9074. [Google Scholar] [CrossRef]
- Ray, P.D.; Huang, B.-W.; Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell. Signal. 2012, 24, 981–990. [Google Scholar] [CrossRef]
- Lambeth, J.D.; Neish, A.S. Nox Enzymes and New Thinking on Reactive Oxygen: A Double-Edged Sword Revisited. Annu. Rev. Pathol. 2014, 9, 119–145. [Google Scholar] [CrossRef]
- Nathan, C.; Cunningham-Bussel, A. Beyond oxidative stress: An immunologist’s guide to reactive oxygen species. Nat. Rev. Immunol. 2013, 13, 349–361. [Google Scholar] [CrossRef]
- Parvez, S.; Long, M.J.; Poganik, J.R.; Aye, Y. Redox Signaling by Reactive Electrophiles and Oxidants. Chem. Rev. 2018, 118, 8798–8888. [Google Scholar] [CrossRef]
- Bedard, K.; Krause, K.-H. The NOX Family of ROS-Generating NADPH Oxidases: Physiology and Pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Dang, P.M.C.; Cross, A.R.; Babior, B.M. Assembly of the neutrophil respiratory burst oxidase: A direct interaction between p67phox and cytochrome b558. Proc. Natl. Acad. Sci. USA 2001, 98, 3001–3005. [Google Scholar] [CrossRef]
- Babior, B.M. NADPH oxidase. Curr. Opin. Immunol. 2004, 16, 42–47. [Google Scholar] [CrossRef] [PubMed]
- Clark, R.; Volpp, B.; Leidal, K.; Nausee, W. Two cytosolic components of the human neutrophil respiratory burst oxidase translocate to the plasma membrane during cell activation. J. Clin. Investig. 1990, 85, 714–721. [Google Scholar] [CrossRef] [PubMed]
- Leto, T.L.; Geistz, M. Role of Nox Family NADPH Oxidases in Host Defense. Antioxid. Redox Signal. 2006, 8, 1549–1961. [Google Scholar] [CrossRef]
- Parkos, C.; Dinauer, M.; Jesaitis, A.; Orkin, S.; Curnutte, J. Absence of both the 91kD and 22kD subunits of human neutrophil cytochrome b in two genetic forms of chronic granulomatous disease. Blood 1989, 73, 1416–1420. [Google Scholar] [CrossRef]
- DeLeo, F.R.; Yu, L.; Burritt, J.B.; Loeterle, L.R.; Bond, C.W. Mapping sites of interaction of p47-phox and flavocytochrome b with random-sequence peptide phage display libraries. Proc. Natl. Acad. Sci. USA 1995, 92, 7110–7114. [Google Scholar] [CrossRef]
- Kawahara, T.; Quinn, M.T.; Lambeth, J.D. Molecular evolution of the reactive oxygen-generating NADPH oxidase (Nox/Duox) family of enzymes. BMC Evol. Biol. 2007, 7, 109. [Google Scholar] [CrossRef]
- Magnani, F.; Mattevi, A. Structure and mechanisms of ROS generation by NADPH Oxidases. Curr. Opin. Struct. Biol. 2019, 59, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Cross, A.R.; Curnutte, J.T. The Cytosolic Activating Factors p47phox and p67phox Have Distinct Roles in the Regulation of Electron Flow in NADPH Oxidase. J. Biol. Chem. 1995, 270, 6543–6548. [Google Scholar] [CrossRef]
- Nguyen, M.V.C.; Lardy, B.; Paclet, M.-H.; Rousset, F.; Berthier, S.; Baillet, A.; Grange, L.; Gaudin, P.; Morel, F. NADPH oxidases, Nox: New isoenzymes family. Med. Sci. 2015, 31, 43–52. [Google Scholar]
- Debeurme, F.; Picciocchi, A.; Dagher, M.; Grunwald, D.; Beaumel, S.; Fieschi, F.; Stasia, M. Regulation of NADPH oxidase activity in phagocytes: Relationship between FAD/NADPH binding and oxidase complex assembly. J. Biol. Chem. 2010, 285, 33197–33208. [Google Scholar] [CrossRef]
- Doussière, J.; Gaillard, J.; Vignais, P.V. Electron Transfer Across the O2-Generating Flavocytochrome b of Neutrophils. Evidence for a Transition from a Low-Spin State to a High-Spin State of the Heme Iron Component. Biochemistry 1996, 35, 13400–13410. [Google Scholar] [CrossRef]
- Vignais, P.V. The superoxide-generating NADPH oxidase: Structural aspects and activation mechanism. Cell. Mol. Life Sci. CMLS 2002, 59, 1428–1459. [Google Scholar] [CrossRef] [PubMed]
- Koshkin, V. Spatial and electrogenic properties of superoxide-producing cytochrome b-559 incorporated into liposomes. Biochim. Biophys. Acta 1995, 1229, 329–333. [Google Scholar] [CrossRef][Green Version]
- Cross, A.R.; Parkinson, J.F.; Jones, O.T.G. Mechanism of the superoxide-producing oxidase of neutrophils. Biochem. J. 1985, 226, 881–884. [Google Scholar] [CrossRef]
- Ceccon, M.; Fananas, E.M.; Massari, M.; Mattevi, A.; Magnani, F. Engineering stability in NADPH oxidases: A common strategy for enzyme production. Mol. Membr. Biol. 2017, 34, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Isogai, Y.; Iizuka, T.; Shiro, Y. The Mechanism of Electron Donation to Molecular Oxygen by Phagocytic Cytochrome b558. J. Biol. Chem. 1995, 270, 7853–7857. [Google Scholar] [CrossRef]
- DeLeo, F.; Burritt, J.; Yu, L.; Jesaitis, A.; Dinauer, M.; Nauseef, W. Processing and maturation of flavocytochrome b558 include incorporation of heme as a prerequisite for heterodimer assembly. J. Biol. Chem. 2000, 275, 13986–13993. [Google Scholar] [CrossRef] [PubMed]
- Leto, T.; Adams, A.; de Mendez, I. Assembly of the phagocyte NADPH oxidase: Binding of Src homology 3 domains to proline-rich targets. Proc. Natl. Acad. Sci. USA 1994, 91, 10650–10654. [Google Scholar] [CrossRef] [PubMed]
- Ogura, K.; Nobuhisa, I.; Yuzawa, S.; Takeya, R.; Torikai, S.; Saikawa, K.; Sumimoto, H.; Inagaki, F. NMR solution structure of the tandem Src homology 3 domains of p47phox complexed with a p22phox-derived proline-rich peptide. J. Biol. Chem. 2006, 281, 3660–3668. [Google Scholar] [CrossRef]
- Sumimoto, H. Structure, regulation and evolution of Nox-family NADPH oxidases that produce reactive oxygen species. FEBS J. 2008, 275, 3249–3277. [Google Scholar] [CrossRef]
- Taylor, R.; Burritt, J.; Baniulis, D.; Foubert, T.; Lord, C.; Dinauer, M.; Parkos, C.; Jesaitis, A. Site-Specific Inhibitors of NADPH Oxidase Activity and Structural Probes of Flavocytochrome b: Characterization of Six Monoclonal Antibodies to the p22phox Subunit. J. Immunol. 2004, 173, 7349–7357. [Google Scholar] [CrossRef] [PubMed]
- Dahan, I.; Issaeva, I.; Gorzalczany, Y.; Sigal, N.; Hirshberg, M.; Pick, E. Mapping of functional domains in the p22(phox) subunit of flavocytochrome b(559) participating in the assembly of the NADPH oxidase complex by peptide walking. J. Biol. Chem. 2002, 277, 8421–8432. [Google Scholar] [CrossRef]
- Rae, J.; Noack, D.; Heyworth, P.; Ellis, B.; Curnutte, J.; Cross, A. Molecular analysis of 9 new families with chronic granulomatous disease caused by mutations in CYBA, the gene encoding p22(phox). Blood 2000, 96, 1106–1112. [Google Scholar] [CrossRef]
- Meijles, D.N.; Howlin, B.J.; Li, J.-M. Consensus in silico computational modelling of the p22phox subunit of the NADPH oxidase. Comput. Biol. Chem. 2012, 39, 6–13. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Marchal, C.; Casbon, A.; Stull, N.; von Löhneysen, K.; Knaus, U.; Jesaitis, A.; McCormick, S.; Nauseef, W.; Dinauer, M. Deletion Mutagenesis of p22phox Subunit of Flavocytochrome b558: Identification of regions critical for gp91phox maturation and NADPH oxidase activity. J. Biol. Chem. 2006, 281, 30336–30346. [Google Scholar] [CrossRef]
- Nobuhisa, I.; Takeya, R.; Ogura, K.; Ueno, N.; Kohda, D.; Inagaki, F.; Sumimoto, H. Activation of the superoxide-producing phagocyte NADPH oxidase requires co-operation between the tandem SH3 domains of p47phox in recognitionof a polyproline type II helix and an adjacent α-helix of p22phox. Biochem. J. 2006, 396, 183–192. [Google Scholar] [CrossRef]
- de Mendez, I.; Adams, A.; Sokolic, R.; Malech, H.; Leto, T. Multiple SH3 domain interactions regulate NADPH oxidase assembly in whole cells. EMBO J. 1996, 15, 1211–1220. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Matsui, Y.; Ago, T.; Ota, K.; Sumimoto, H. Novel modular domain PB1 recognizes PC motif to mediate functional protein-protein interactions. EMBO J. 2001, 20, 3938–3946. [Google Scholar] [CrossRef]
- Noda, Y.; Kohjima, M.; Izaki, T.; Ota, K.; Yoshinaga, S.; Inagaki, F.; Ito, T.; Sumimoto, H. Molecular recognition in dimerization between PB1 domains. J. Biol. Chem. 2003, 278, 43516–43524. [Google Scholar] [CrossRef]
- Ago, T.; Nunoi, H.; Ito, T.; Sumimoto, H. Mechanism for Phosphorylation-induced Activation of the Phagocyte NADPH Oxidase Protein p47 phox. J. Biol. Chem. 1999, 274, 33644–33653. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, C.S.; Bouchab, L.; Tramier, M.; Durand, D.; Fieschi, F.; Dupré-Crochet, S.; Mérola, F.; Nüße, O.; Erard, M. Quantitative live-cell imaging and 3D modeling reveal critical functional features in the cytosolic complex of phagocyte NADPH oxidase. J. Biol. Chem. 2019, 294, 3824–3836. [Google Scholar] [CrossRef] [PubMed]
- Nisimoto, Y.; Motalebi, S.; Han, C.H.; Lambeth, J.D. The p67(phox) activation domain regulates electron flow from NADPH to flavin in flavocytochrome b(558). J. Biol. Chem. 1999, 274, 22999–23005. [Google Scholar] [CrossRef]
- Suzuki-Inoue, K.; Yatomi, Y.; Asazuma, N.; Kainoh, M.; Tanaka, T.; Satoh, K.; Ozaki, Y. Rac, a small guanosine triphosphate–binding protein, and p21-activated kinase are activated during platelet spreading on collagen-coated surfaces: Roles of integrin α2β1. Blood 2001, 98, 3708–3716. [Google Scholar] [CrossRef]
- Caron, E. Cellular functions of the Rap1 GTP-binding protein: A pattern emerges. J. Cell Sci. 2003, 116, 435–440. [Google Scholar] [CrossRef]
- Ueyama, T.; Eto, M.; Kami, K.; Tatsuno, T.; Kobayashi, T.T.; Shirai, Y.; Lennartz, M.; Takeya, R.; Sumimoto, H.; Saito, N. Isoform-Specific Membrane Targeting Mechanism of Rac during FcγR-Mediated Phagocytosis: Positive Charge-Dependent and Independent Targeting Mechanism of Rac to the Phagosome. J. Immunol. 2005, 175, 2381–2390. [Google Scholar] [CrossRef] [PubMed]
- Jaśkiewicz, A.; Pająk, B.; Orzechowski, A. The Many Faces of Rap1 GTPase. Int. J. Mol. Sci. 2018, 19, 2848. [Google Scholar] [CrossRef]
- Joseph, G.; Gorzalczany, Y.; Koshkin, V.; Pick, E. Inhibition of NADPH oxidase activation by synthetic peptides mapping within the carboxyl-terminal domain of small GTP-binding proteins. Lack of amino acid sequence specificity and importance of polybasic motif. J. Biol. Chem. 1994, 269, 29024–29031. [Google Scholar] [CrossRef]
- Wilson, J.M.; Prokop, J.W.; Lorimer, E.; Ntantie, E.; Williams, C.L. Differences in the Phosphorylation-Dependent Regulation of Prenylation of Rap1A and Rap1B. J. Mol. Biol. 2016, 428, 4929–4945. [Google Scholar] [CrossRef] [PubMed]
- Miyano, K.; Ueno, N.; Takeya, R.; Sumimoto, H. Direct Involvement of the Small GTPase Rac in Activation of the Superoxide-producing NADPH Oxidase Nox1. J. Biol. Chem. 2006, 281, 21857–21868. [Google Scholar] [CrossRef]
- Quinn, M.T.; Gauss, K.A. Structure and regulation of the neutrophil respiratory burst oxidase: Comparison with nonphagocyte oxidases. J. Leuk. Biol. 2004, 76, 760–781. [Google Scholar] [CrossRef]
- Bokoch, G.; Quilliam, L.; Jesaitis, B.B.A.; Quinn, M. Inhibition of Rap1A binding to cytochrome b558 of NADPH oxidase by phosphorylation of Rap1A. Science 1991, 254, 1794–1796. [Google Scholar] [CrossRef]
- Gabig, T.; Crean, C.; Mantel, P.; Rosli, R. Function of wild-type or mutant Rac2 and Rap1a GTPases in differentiated HL60 cell NADPH oxidase activation. Blood 1995, 85, 804–811. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yan, J.; De, P.; Chang, H.; Yamauchi, A.; Christopherson, K.; Paranavitana, N.; Peng, X.; Kim, C.; Munugalavadla, V.; et al. Rap1a null mice have altered myeloid cell functions suggesting distinct roles for the closely related Rap1a and 1b proteins. J. Immunol. 2007, 179, 8322–8331. [Google Scholar] [CrossRef]
- Nguyen, G.; Green, E.; Mecsa, S. Neutrophils to the ROScue: Mechanisms of NADPH Oxidase Activation and Bacterial Resistance. Front Cell Infect. Microbiol. 2017, 25, 373–397. [Google Scholar] [CrossRef]
- Yang, C.-S.; Lee, J.-S.; Rodgers, M.; Min, C.-K.; Lee, J.-Y.; Kim, H.J.; Lee, K.; Kim, C.; Oh, B.; Zandi, E.; et al. Autophagy Protein Rubicon Mediates Phagocytic NADPH Oxidase Activation in Response to Microbial Infection or TLR Stimulation. Cell Host Microbe 2012, 11, 264–276. [Google Scholar] [CrossRef] [PubMed]
- DeLeo, F.; Allen, L.; Apicella, W.; Nauseef, M. NADPH oxidase activation and assembly during phagocytosis. J. Immunol. 1999, 163, 6732–6740. [Google Scholar] [PubMed]
- Ueyama, T.; Tatsuno, T.; Kawasaki, T.T.S.; Shirai, Y.; Sumimoto, H.; Leto, T.; Saito, N. A regulated adaptor function of p40phox: Distinct p67phox membrane targeting by p40phox and by p47phox. Mol. Biol. Cell 2007, 18, 441–454. [Google Scholar] [CrossRef] [PubMed]
- Freeman, J.; Lambeth, J. NADPH oxidase activity is independent of p47phox in vitro. J. Biol. Chem. 1996, 271, 22578–22582. [Google Scholar] [CrossRef]
- Koshkin, V.; Lotan, O.; Pick, E. The cytosolic component p47(phox) is not a sine qua non participant in the activation of NADPH oxidase but is required for optimal superoxide production. J. Biol. Chem. 1991, 271, 30326–30329. [Google Scholar] [CrossRef]
- Morgan, M.J.; Liu, Z.-G. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef]
- Dickinson, B.C.; Chang, C.J. Chemistry and biology of reactive oxygen species in signaling. Nat. Chem. Biol. 2012, 7, 504–511. [Google Scholar] [CrossRef] [PubMed]
- Alloul, N.; Gorzalczany, Y.; Itan, M.; Sigal, N.; Pick, E. Activation of the Superoxide-Generating NADPH Oxidase by Chimeric Proteins Consisting of Segments of the Cytosolic Component p67phox and the Small GTPase Rac1. Biochemistry 2001, 40, 14557–14566. [Google Scholar] [CrossRef]
- Berdichevsky, Y.; Mizrahi, A.; Ugolev, Y.; Molshanski-Mor, S.; Pick, E. Tripartite Chimeraripartite chimeras comprising functional domains derived from the cytosolic NADPH oxidase components p47phox, p67phox, and Rac1 elicit activator-independent superoxide production by phagocyte membranes. J. Biol. Chem. 2007, 282, 22122–22139. [Google Scholar] [CrossRef] [PubMed]
- Miyano, K.; Fukuda, H.; Ebisu, K.; Tamura, M. Remarkable Stabilization of Neutrophil NADPH Oxidase Using RacQ61L and a p67phox−p47phox Fusion Protein. Biochemistry 2003, 42, 184–190. [Google Scholar] [CrossRef]
- Ebisu, K.; Nagasawa, T.; Watanabe, K.; Kakinuma, K.; Miyano, K.; Tamura, M. Fused p47phox and p67phox truncations efficiently reconstitute NADPH oxidase with higher activity and stability than the individual components. J. Biol. Chem. 2001, 276, 24498–24505. [Google Scholar] [CrossRef] [PubMed]
- Valenta, H.; Dupré-Crochet, S.; Bizouarn, T.; Baciou, L.; Nüsse, O.; Deniset-Besseau, A.; Erard, M. Consequences of the constitutive NOX2 activity in living cells: Cytosol acidification, apoptosis, and localized lipid peroxidation. BioRxiv Prepr. 2021. [Google Scholar] [CrossRef]
- Si, J.; Behar, J.; Wands, J.; Beer, D.G.; Lambeth, D.; Chin, Y.E.; Cao, W. STAT5 Mediates Platelet-Activating Factor (PAF)-Induced NADPH Oxidase NOX5-S Expression In Barrett’s Esophageal Adenocarcinoma Cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 294, G174–G183. [Google Scholar] [CrossRef]
- Bylund, J.; Samuelsson, M.; Collins, L.; Karlsson, A. NADPH-oxidase activation in murine neutrophils via formyl peptide receptors. Exp. Cell. Res. 2003, 282, 70–77. [Google Scholar] [CrossRef]
- Kim, Y.-S.; Morgan, M.J.; Choksi, S.; Liu, Z.-G. TNF-induced activation of the Nox1 NADPH oxidase and its role in the induction of necrotic cell death. Mol. Cell 2007, 26, 675–687. [Google Scholar] [CrossRef] [PubMed]
- Boussetta, T.; Gougerot-Pocidalo, M.; Hayem, G.; Ciappelloni, S.; Raad, H.; Derkawi, R.A.; Bournier, O.; Kroviarski, Y.; Zhou, X.; Malter, J.; et al. The prolyl isomerase Pin1 acts as a novel molecular switch for TNF-alpha-induced priming of the NADPH oxidase in human neutrophils. Blood 2010, 116, 5795–5802. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Bedouhene, S.; Hurtado-Nedelec, M.; Pintard, C.; Pham, M.D.; Yu, S.; El-Benna, J. The Prolyl Isomerase Pin1 Controls Lipopolysaccharide-Induced Priming of NADPH Oxidase in Human Neutrophils. Front. Immunol. 2019, 10, 2567. [Google Scholar] [CrossRef] [PubMed]
- El Benna, J.; Faust, L.; Babior, B. The phosphorylation of the respiratory burst oxidase component p47(phox) during neutrophil activation. Phosphorylation of sites recognized by protein kinase C and by proline-directed kinases. J. Biol. Chem. 1994, 269, 23431–23436. [Google Scholar] [CrossRef]
- Inanami, O.; Johnson, J.; McAdara, J.; el Benna, J.; Faust, L.; Newburger, P.; Babior, B. Activation of the leukocyte NADPH oxidase by phorbol ester requires the phosphorylation of p47(PHOX) on serine 303 or 304. J. Biol. Chem. 1998, 273, 9539–9543. [Google Scholar] [CrossRef] [PubMed]
- Marcoux, J.; Man, P.; Castellan, M.; Vivès, C.; Forest, E.; Fieschi, F. Conformational changes in p47phox upon activation highlighted by mass spectrometry coupled to hydrogen/deuterium exchange and limited proteolysis. FEBS Lett. 2009, 583, 835–840. [Google Scholar] [CrossRef] [PubMed]
- Dang, P.M.C.; Stensballe, A.; Boussetta, T.; Raad, H.; Dewas, C.; Kroviarski, Y.; El-Benna, J. A specific p47phox-serine phosphorylated by convergent MAPKs mediates neutrophil NADPH oxidase priming at inflammatory sites. J. Clin. Investig. 2006, 116, 2033–2043. [Google Scholar] [CrossRef]
- Fontayne, A.; Dang, P.; Gougerot-Pocidalo, M.; El-Benna, J. Phosphorylation of p47phox sites by PKC alpha, beta II, delta, and zeta: Effect on binding to p22phox and on NADPH oxidase activation. Biochemistry 2002, 41, 7743–7750. [Google Scholar] [CrossRef]
- Inanami, O.; Johnson, J.L.; McAdara, J.K.; el Benna, J.; Faust, L.R.P. Phosphorylation of threonine 154 in p40phox is an important physiological signal for activation of the neutrophil NADPH oxidase. Blood 2010, 116, 6026–6036. [Google Scholar]
- DerMardirossian, C.; Schnelzer, A.; Bokoch, G.M. Phosphorylation of RhoGDI by Pak1 Mediates Dissociation of Rac GTPase. Mol. Cell 2004, 25, 117–127. [Google Scholar] [CrossRef]
- Pick, E. Role of the Rho GTPase Rac in the activation of the phagocyte NADPH oxidase: Outsourcing a key task. Small GTPases 2014, 5, e27952. [Google Scholar] [CrossRef]
- Nisimoto, Y.; Ogawa, H.; Miyano, K.; Tamura, M. Activation of the flavoprotein domain of gp91phox upon interaction with N-terminal p67phox (1-210) and the Rac complex. Biochemistry 2004, 43, 9567–9575. [Google Scholar] [CrossRef] [PubMed]
- Regier, D.S.; Greene, D.G.; Sergeant, S.; Jesaitis, A.J.; McPhail, L.C. Phosphorylation of p22phox is mediated by phospholipase D-dependant and independent mechanisms. Correlation of NADPH oxidase activity and p22phox phopshorylation. J. Biol. Chem. 2000, 275, 28406–28412. [Google Scholar] [CrossRef]
- Waite, K.A.; Wallin, R.; Qualliotine-Mann, D.; McPhail, L.C. Phosphatidic Acid-mediated Phosphorylation of the NADPH Oxidase Component p47-phox: Evidence That Phosphatidic Acid May Activate a Novel Protein Kinase. J. Biol. Chem. 1997, 272, 15569–15578. [Google Scholar] [CrossRef]
- Regier, D.S.; Waite, K.A.; McPhail, L.C. A novel protein kinase target for the lipid second messenger phosphatidic acid. J. Biol. Chem. 1999, 274, 36601–36608. [Google Scholar] [CrossRef]
- Beaumel, S.; Picciocchi, A.; Debeurme, F.; Vivès, C.; Hesse, A.M.; Ferro, M.; Grunwald, D.; Stieglitz, H.; Thepchatri, P.; Smith, S.M.E.; et al. Down-regulation of NOX2 activity in phagocytes mediated by ATM-kinase dependent phosphorylation. Free Radic. Biol. Med. 2017, 113, 1–15. [Google Scholar] [CrossRef]
- Raad, H.; Mouawia, H.; Hassan, H.; El-Seblani, M.; Arabi-Derkawi, R.; Boussetta, T.; Gougerot-Pocidalo, M.A.; Dang, P.M.C.; El-Benna, J. The protein kinase A negatively regulates reactive oxygen species production by phosphorylating gp91phox/NOX2 in human neutrophils. Free Radic. Biol. Med. 2020, 160, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Kanai, F.; Liu, H.; Field, S.J.; Akbary, H.; Matsuo, T.; Brown, G.E.; Cantley, L.C.; Yaffe, M.B. The PX domains of p47phox and p40phox bind to lipid products of PI(3)K. Nat. Cell Biol. 2001, 3, 675–678. [Google Scholar] [CrossRef] [PubMed]
- MLee, H.; Bell, R.M. Mechanism of protein kinase C activation by phosphatidylinositol 4,5-bisphosphate. Biochemistry 1991, 30, 1041–1049. [Google Scholar]
- Handlogten, M.; Huang, C.; Shiraishi, N.; Awata, H.; Miller, R.T. The Ca21-sensing Receptor Activates Cytosolic Phospholipase A2 via a Gqa-dependent ERK-independent Pathway. J. Biol. Chem. 2001, 276, 13941–13948. [Google Scholar] [CrossRef] [PubMed]
- Sellmayer, A.; Obermeier, H.; Danesch, U.; Aepfelbacher, M.; Weber, P.C. Arachidonic acid increases activation of NADPH oxidase in monocytic U937 cells by accelerated translocation of p47-phox and co-stimulation of protein kinase C. Cell. Signal. 1996, 8, 397–402. [Google Scholar] [CrossRef]
- Bizouarn, T.; Karimi, G.; Masoud, R.; Souabni, H.; Machillot, P.; Serfaty, X. Exploring the arachidonic acid-induced structural changes in phagocyte NADPH oxidase p47phox and p67phox via thiol accessibility and SRCD spectroscopy. FEBS J. 2016, 283, 2896–2910. [Google Scholar] [CrossRef] [PubMed]
- Doussière, J.; Gaillard, J.; Vignais, P. The heme component of the neutrophil NADPH oxidase complex is a target for aryliodonium compounds. Biochemistry 1999, 38, 3694–3703. [Google Scholar] [CrossRef] [PubMed]
- Shiose, A.; Sumimoto, H. Arachidonic acid and phosphorylation synergistically induce a conformational change of p47phox to activate the phagocyte NADPH oxidase. J. Biol. Chem. 2000, 275, 13793–13801. [Google Scholar] [CrossRef]
- Swain, S.D.; Helgerson, L.; Davis, A.R.; Nelson, L.K.; Quinn, M.T. Analysis of activation-induced conformational changes in p47phox using tryptophan fluorescence spectroscopy. J. Biol. Chem. 1997, 272, 29502–29510. [Google Scholar] [CrossRef] [PubMed]
- Bizouarn, T.; Souabni, H.; Serfaty, X.; Bouraoui, A.; Masoud, R.; Karimi, G.; Houée-Levin, C.; Baciou, L. A Close-Up View of the Impact of Arachidonic Acid on the Phagocyte NADPH Oxidase. Methods Mol. Biol. 2019, 1982, 75–101. [Google Scholar] [PubMed]
- Kim, C.; Dinauer, M.C. Impaired NADPH oxidase activity in Rac2-deficient murine neutrophils does not result from defective translocation of p47phox and p67phox and can be rescued by exogenous arachidonic acid. J. Leukoc. Biol. 2006, 79, 223–234. [Google Scholar] [CrossRef]
- Hata, K.; Ito, T.; Takeshige, K.; Sumimoto, H. Anionic amphiphile-independent activation of the phagocyte NADPH oxidase in a cell-free system by p47phox and p67phox, both in C terminally truncated forms. Implication for regulatory Src homology 3 domain-mediated interactions. J. Biol. Chem. 1998, 13, 4232–4236. [Google Scholar] [CrossRef]
- Davis, A.R.; Mascolo, P.L.; Bunger, P.L.; Sipes, K.M.; Quinn, M.T. Cloning and sequencing of the bovine flavocytochrome b subunit proteins, gp91-phox and p22-phox: Comparison with other known flavocytochrome b sequences. J. Leukoc. Biol. 1998, 64, 114–123. [Google Scholar] [CrossRef]
- Segal, A.W. The function of the NADPH oxidase of phagocytes and its relationship to other NOXs in plants, invertebrates, and mammals. Int. J. Biochem. Cell Biol. 2008, 40, 604–618. [Google Scholar] [CrossRef] [PubMed]
- Takemoto, D.; Tanaka, A.; Scott, B. NADPH oxidases in fungi: Diverse roles of reactive oxygen species in fungal cellular differentiation. Fungal Genet. Biol. 2007, 44, 1065–1076. [Google Scholar] [CrossRef]
- Bedard, K.; Lardy, B.; Krause, K. NOX family NADPH oxidases: Not just in mammals. Biochimie 2007, 89, 1107–1112. [Google Scholar] [CrossRef] [PubMed]
- Krause, K.H. Tissue distribution and putative physiological function of NOX family NADPH oxydases. J. Infect. Dis. 2004, 57, 28–29. [Google Scholar]
- Rybak, L.P.; Mukherjea, D.; Jajoo, S.; Kaur, T.; Ramkumar, V. siRNA-mediated knock-down of NOX3: Therapy for hearing loss? Cell. Mol. Life Sci. CMLS 2012, 69, 2429–2434. [Google Scholar] [CrossRef]
- Cooney, S.J.; Bermudez-Sabogal, S.L.; Byrnes, K.R. Cellular and temporal expression of NADPH oxidase (NOX) isotypes after brain injury. J. Neuroinflamm. 2013, 10, 917. [Google Scholar] [CrossRef]
- Zhang, X.; Shan, P.; Jiang, G.; Cohn, L.; Lee, P. Toll-like receptor four deficiency causes pulmonary emphysema. J. Clin. Investig. 2006, 116, 3050–3059. [Google Scholar] [CrossRef]
- Ruwanpura, S.M.; McLeod, L.; Lilja, A.R.; Brooks, G.; Dousha, L.F.; Seow, H.J.; Bozinovski, S.; Vlahos, R.; Hertzog, P.J.; Anderson, G.P.; et al. Non-essential role for TLR2 and its signaling adaptor Mal/TIRAP in preserving normal lung architecture in mice. PLoS ONE 2013, 8, e78095. [Google Scholar] [CrossRef]
- Nakano, Y.; Banfi, B.; Jesaitis, A.J.; Dinauer, M.C.; Allen, L.A.; Nauseef, W.M. Critical roles for p22phox in the structural maturation and subcellular targeting of Nox3. Biochem. J. 2007, 403, 97–108. [Google Scholar] [CrossRef]
- Miyano, K.; Sumimoto, H. N-linked glycosylation of the superoxide-producing NADPH oxidase Nox1. Biochem. Biophys. Res. Commun. 2014, 443, 1060–1065. [Google Scholar] [CrossRef] [PubMed]
- Takeya, R.; Ueno, N.; Kami, K.; Taura, M. Novel Human Homologues of p47phox and p67phox Participate in Activation of Superoxide-producing NADPH Oxidases. J. Biol. Chem. 2003, 278, 25234–25246. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.; Ritsick, D.; Lambeth, J.D. Nox3 Regulation by NOXO1, p47phox, and p67phox. J. Biol. Chem. 2004, 279, 34250–34255. [Google Scholar] [CrossRef] [PubMed]
- Nisimoto, Y.; Jackson, H.M.; Ogawa, H.; Kawahara, T.; Lambeth, J.D. Constitutive NADPH-Dependent Electron Transferase Activity of the Nox4 Dehydrogenase Domain. Biochemistry 2010, 49, 2433–2442. [Google Scholar] [CrossRef] [PubMed]
- Prior, K.K.; Leisegang, M.S.; Josipovic, I.; Löwe, O.; Shah, A.M.; Weissmann, N.; Schröder, K.; Brandes, R.P. CRISPR/Cas9-mediated knockout of p22phox leads to loss of Nox1 and Nox4, but not Nox5 activity. Redox Biol. 2016, 9, 287–295. [Google Scholar] [CrossRef] [PubMed]
- Shiose, A.; Kuroda, J.; Tsuruya, K.; Hirai, M.; Hirakata, H.; Naito, S.; Hattori, M.; Sakaki, Y.; Sumimoto, H. A novel superoxide-producing NAD(P)H oxidase in kidney. J. Biol. Chem. 2001, 276, 1417–1423. [Google Scholar] [CrossRef] [PubMed]
- Goyal, P.; Weissmann, N.; Rose, F.; Grimminger, F.; Schäfers, H.J.; Seeger, W.; Hänze, J. Identification of novel Nox4 splice variants with impact on ROS levels in A549 cells. Biochem. Biophys. Res. Commun. 2005, 329, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Lyle, A.N.; Deshpande, N.N.; Taniyama, Y.; Seidel-Rogol, B. Poldip2, a novel regulator of Nox4 and cytoskeletal integrity in vascular smooth muscle cells. Circ. Res. 2009, 105, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Kirber, M.T.; Xiao, H.; Yang, Y.; Keaney, J. Regulation of ROS signal transduction by NADPH oxidase 4 localization. J. Cell Biol. 2008, 181, 1129–1139. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.F.; Qiao, M.; Schöder, K.; Zhao, Q.; Asmis, R. Nox4 is a Novel Inducible Source of Reactive Oxygen Species in Monocytes and Macrophages and Mediates Oxidize. Circ. Res. 2010, 106, 1489–1497. [Google Scholar] [CrossRef]
- Nisimoto, Y.; Diebold, B.A.; Cosentino-Gomes, D.; Lambeth, J.D. Nox4: A hydrogen peroxide-generating oxygen sensor. Biochemistry 2014, 53, 5111–5120. [Google Scholar] [CrossRef]
- Takac, I.; Schröder, K.; Zhang, L.; Lardy, B.; Anilkumar, N.; Lambeth, J.D. The E-loop Is Involved in Hydrogen Peroxide Formation by the NADPH Oxidase Nox4. J. Biol. Chem. 2011, 286, 13304–13313. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, T.; Jackson, H.M.; Smith, S.M.; Simpson, P.D.; Lambeth, J.D. Nox5 forms a functional oligomer mediated by self-association of its dehydrogenase domain. Biochemistry 2011, 50, 2013–2025. [Google Scholar] [CrossRef]
- Kiyohara, T.; Miyano, K.; Kamakura, S.; Hayase, J.; Chishiki, K.; Kohda, A.; Sumimoto, H. Differential cell surface recruitment of the superoxide-producing NADPH oxidases Nox1, Nox2 and Nox5: The role of the small GTPase Sar1. Genes Cells 2018, 23, 480–493. [Google Scholar] [CrossRef]
- Chin, D.; Means, A. Calmodulin: A prototypical calcium sensor. Trends Cell Biol. 2000, 10, 322–328. [Google Scholar] [CrossRef]
- Wei, C.-C.; Reynolds, N.; Palka, C.; Wetherell, K.; Boyle, T.; Yang, Y.-P. Characterization of the 1st and 2nd EF-hands of NADPH oxidase 5 by fluorescence, isothermal titration calorimetry, and circular dichroism. Chem. Cent. J. 2012, 6, 1–11. [Google Scholar] [CrossRef]
- Tirone, F.; Cox, J.A. NADPH oxidase 5 (NOX5) interacts with and is regulated by calmodulin. FEBS Lett. 2007, 581, 1202–1208. [Google Scholar] [CrossRef]
- Meitzler, J.L.; de Montellano, P.R.O. Caenorhabditis elegans and human dual oxidase 1 (DUOX1) peroxidase domains: Insights into heme binding and catalytic activity. J. Biol. Chem. 2009, 284, 18634–18643. [Google Scholar] [CrossRef] [PubMed]
- de Deken, X.; Wang, D.; Dumont, J.; Miot, F. Characterization of ThOX proteins as components of the thyroid H2O2-generating system. Exp. Cell Res. 2002, 273, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Morand, S.; Chaaraoui, M.; Kaniewski, J.; Dème, D.; Ohayon, R.; Noel-Hudson, M.S.; Virion, A.; Dupuy, C. Effect of iodide on nicotinamide adenine dinucleotide phosphate oxidase activity and Duox2 protein expression in isolated porcine thyroid follicles. Endocrinology 2003, 144, 1241–1248. [Google Scholar] [CrossRef]
- Grasberger, H.; De Deken, X.; Mayo, O.B.; Raad, H.; Weiss, M.; Liao, X.H.; Refetoff, S. Mice deficient in dual oxidase maturation factors are severely hypothyroid. Mol. Endocrinol. 2012, 26, 481–492. [Google Scholar] [CrossRef]
- Ueyama, T.; Sakuma, M.; Ninoyu, Y.; Hamada, T.; Dupuy, C.; Geiszt, M.; Leto, T.L.; Saito, N. The extracellular A-loop of dual oxidases affects the specificity of reactive oxygen species release. J. Biol. Chem. 2015, 290, 6495–6506. [Google Scholar] [CrossRef] [PubMed]
- Milenkovic, M.; de Deken, X.; Jin, L.; de Felice, M.; di Lauro, R.; Dumont, J.E.; Corvilain, B.; Miot, F. Duox expression and related H2O2 measurement in mouse thyroid: Onset in embryonic development and regulation by TSH in adult. J. Endocrinol. 2007, 192, 615–626. [Google Scholar] [CrossRef]
- von Rozycki, T.; Yen, M.R.; Lende, E.E.; Saier, M.H.J. The YedZ family: Possible heme binding proteins that can be fused to transporters and electron carriers. J. Mol. Microbiol. Biotechnol. 2004, 8, 129–140. [Google Scholar] [CrossRef]
- Sanchez-Pulido, L.; Rojas, A.M.; Valencia, A.; Martinez, C.; Andrade, M.A. ACRATA: A novel electron transfer domain associated to apoptosis and cancer. BMC Cancer 2004, 4, 98. [Google Scholar] [CrossRef]
- Hervé, C.; Tonon, T.; Collén, J.; Corre, E.; Boyen, C. NADPH oxidases in Eukaryotes: Red algae provide new hints! Curr. Genet. 2006, 49, 190–204. [Google Scholar] [CrossRef] [PubMed]
- Keller, T.; Damude, H.G.; Werner, D.; Doerner, P.; Dixon, R.A.; Lamb, C. A plant homolog of the neutrophil NADPH oxidase gp91phox subunit gene encodes a plasma membrane protein with Ca2+ binding motifs. Plant Cell 1998, 10, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Lardy, B.; Bof, M.; Aubry, L.; Paclet, M.H.; Morel, F.; Satre, M.; Klein, G. NADPH oxidase homologs are required for normal cell differentiation and morphogenesis in Dictyostelium discoideum. Biochim. Biophys. Acta 2005, 1744, 199–212. [Google Scholar] [CrossRef]
- Lara-Ortíz, T.; Riveros-Rosas, H.; Aguirre, J. Reactive oxygen species generated by microbial NADPH oxidase NoxA regulate sexual development in Aspergillus nidulans. Mol. Microbiol. 2003, 50, 1241–1255. [Google Scholar] [CrossRef]
- Aguirre, J.; Lambeth, J.D. Nox enzymes from fungus to fly to fish and what they tell us about Nox function in mammals. Free Radic. Biol. Med. 2010, 49, 1342–1353. [Google Scholar] [CrossRef]
- Segal, A.W. Biochemistry and molecular biology of chronic granulomatous disease. J. Inherit. Metab. Dis. 1992, 15, 683–686. [Google Scholar] [CrossRef]
- Finegold, A.A.; Shatwell, K.P.; Segal, A.W.; Klausner, R.D.; Dancis, A. Intramembrane bis-heme motif for transmembrane electron transport conserved in a yeast iron reductase and the human NADPH oxidase. J. Biol. Chem. 1996, 271, 31021–31024. [Google Scholar] [CrossRef]
- Cramer, W.Z.H. Consequences of the structure of the cytochrome b6f complex for its charge transfer pathways. Biochim. Biophys. Acta 2006, 1757, 339–345. [Google Scholar] [CrossRef]
- Baniulis, D.; Zhang, H.; Zakharova, T.; Hasan, S.; Cramer, W. Purification and crystallization of the cyanobacterial cytochrome b6f complex. Methods Mol. Biol. 2011, 684, 65–77. [Google Scholar]
- Juillan-Binard, C.; Picciocchi, A.; Andrieu, J.-P.; Dupuy, J.; Petit-Hartlein, I.; Caux-Thang, C.; Vivès, C.; Nivière, V.; Fieschi, F. A Two-component NADPH Oxidase (NOX)-like System in Bacteria Is Involved in the Electron Transfer Chain to the Methionine Sulfoxide Reductase MsrP. J. Biol. Chem. 2017, 10, 2485–2494. [Google Scholar] [CrossRef]
- Gennaris, A.; Ezraty, B.; Henry, C.; Agrebi, R.; Vergnes, A.; Oheix, E.; Bos, J.; Leverrier, P.; Espinosa, L.; Szewczyk, J.; et al. Repairing oxidized proteins in the bacterial envelope using respiratory chain electrons. Nature 2015, 528, 409–412. [Google Scholar] [CrossRef]
- Taylor, W.; Jones, D.; Segal, A. A structural model for the nucleotide binding domains of the flavocytochrome b-245 beta-chain. Protein Sci. 1993, 2, 1675–1685. [Google Scholar] [CrossRef]
- Burritt, J.; Foubert, T.; Baniulis, D.; Lord, C.; Taylor, R.; Mills, J.; Baughan, T.; Roos, D.; Parkos, C.; Jesaitis, A. Functional epitope on human neutrophil flavocytochrome b558. J. Immunol. 2003, 170, 6082–6089. [Google Scholar] [CrossRef] [PubMed]
- Streeter, J.; Schickling, B.; Jiang, S.; Stanic, B.; Thiel, W.; Gakhar, L.; Houtman, J.; Miller, F.J. Phosphorylation of Nox1 regulates association with NoxA1 activation domain. Circ. Res. 2014, 115, 911–918. [Google Scholar] [CrossRef]
- Vermot, A.; Petit-Hartlein, I.; Breyton, C.; le Roy, A.; Thépaut, M.; Vivès, C.; Moulin, M.; Härtlein, M.; Grudinin, S.; Smith, S.M.E.; et al. Interdomain Flexibility within NADPH Oxidase Suggested by SANS Using LMNG Stealth Carrier. Biophys. J. 2020, 119, 605–618. [Google Scholar] [CrossRef]
- Pick, E.; Bromberg, Y.; Shpungin, S.; Gadba, R. Activation of the superoxide forming NADPH oxidase in a cell-free system by sodium dodecyl sulfate. Characterization of the membrane-associated component. J. Biol. Chem. 1987, 262, 16476–16483. [Google Scholar] [CrossRef]
- Shpungin, S.; Dotan, I.; Abo, A.; Pick, E. Activation of the superoxide forming NADPH oxidase in a cell-free system by sodium dodecyl sulfate. Absolute lipid dependence of the solubilized enzyme. J. Biol. Chem. 1989, 264, 9195–9203. [Google Scholar] [CrossRef]
- Heyneman, R.A.; Vercauteren, R.E. Activation of a NADPH oxidase from horse polymorphonuclear leukocytes in a cell-free system. J. Leukoc. Biol. 1984, 36, 751–759. [Google Scholar] [CrossRef] [PubMed]
- McPhail, L.C.; Shirley, P.S.; Clayton, C.C.; Snyderman, R. Activation of the respiratory burst enzyme from human neutrophils in a cell-free system. Evidence for a soluble cofactor. J. Clin. Investig. 1985, 75, 1735–1739. [Google Scholar] [CrossRef][Green Version]
- Curnutte, J.T. Activation of human neutrophil nicotinamide adenine dinucleotide phosphate, reduced (triphosphopyridine nucleotide, reduced) oxidase by arachidonic acid in a cell-free system. J. Clin. Investig. 1985, 75, 1740–1743. [Google Scholar] [CrossRef]
- Levine, A.P.; Segal, A.W. The NADPH Oxidase and Microbial Killing by Neutrophils, With a Particular Emphasis on the Proposed Antimicrobial Role of Myeloperoxidase within the Phagocytic Vacuole. Microbiol. Spectr. 2016, 4, 1–14. [Google Scholar]
- Rada, B.; Leto, T.L. Oxidative innate immune defenses by Nox/Duox family NADPH Oxidases. Contrib. Microbiol. 2008, 15, 164–187. [Google Scholar] [PubMed]
- Fu, H.; Bylund, J.; Karlsson, A.; Pellmé, S.; Dahlgren, C. The mechanism for activation of the neutrophil NADPH-oxidase by the peptides formyl-Met-Leu-Phe and Trp-Lys-Tyr-Met-Val-Met differs from that for interleukin-8. Immunology 2004, 112, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Uhing, R.J.; Snyderman, R. Chemoattractant stimulus-response coupling. In Inflammation Basic Principles and Clinical Correlates; Gallin, J.I., Snyderman, R., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 1999; pp. 607–626. [Google Scholar]
- DeCoursey, T.E. The Intimate and Controversial Relationship between Voltage Gated Proton Channels and the Phagocyte NADPH Oxidase. Immunol. Rev. 2016, 273, 194–218. [Google Scholar] [CrossRef]
- Henderson, L.M.; Chappell, J.B.; Jones, O.T. The superoxide-generating NADPH oxidase of human neutrophils is electrogenic and associated with an H+ channel. Biochem. J. 1987, 246, 325–329. [Google Scholar] [CrossRef]
- Schrenzel, J.; Serrander, L.; Bánfi, B.; Nüße, O.; Fouyouzi, R.; Lew, D.P.; Demaurex, N.; Krause, K.H. Electron currents generated by the human phagocyte NADPH oxidase. Nature 1998, 392, 734–737. [Google Scholar] [CrossRef]
- Nunes, P.; Demaurex, N.; Dinauer, M.C. Regulation of the NADPH Oxidase and Associated Ion Fluxes during Phagocytosis. Traffic 2013, 14, 1118–1131. [Google Scholar] [CrossRef] [PubMed]
- Capasso, M.; DeCoursey, T.E.; Dyer, M.J.S. pH regulation and beyond: Unanticipated functions for the voltage-gated proton channel, HVCN1. Trends Cell Biol. 2011, 21, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Morgan, D.; Capasso, M.; Musset, B.; Cherny, V.V.; Ríos, E.; Dyer, M.J.S.; DeCoursey, T.E. Voltage-gated proton channels maintain pH in human neutrophils during phagocytosis. Proc. Natl. Acad. Sci. USA 2009, 106, 18022–18027. [Google Scholar] [CrossRef]
- Nanda, A.; Romanek, R.; Curnutte, J.T.; Grinstein, S. Assessment of the contribution of the cytochrome b moiety of the NADPH oxidase to the transmembrane H+ conductance of leukocytes. J. Biol. Chem. 1994, 269, 27280–27285. [Google Scholar] [CrossRef]
- Morgan, D.; Cherny, V.V.; Price, M.O.; Dinauer, M.C.; DeCoursey, T.E. Absence of proton channels in COS-7 cells expressing functional NADPH oxidase components. J. Gen. Physiol. 2002, 119, 571–580. [Google Scholar] [CrossRef]
- Ramsey, I.; Moran, M.; Chong, J.; Clapham, D. A voltage-gated proton-selective channel lacking the pore domain. Nature 2006, 440, 1213–1216. [Google Scholar] [CrossRef]
- Sasaki, M.; Takagi, M.; Okamura, Y. A Volt. Sens.-Domain Protein Is A Volt.-Gated Proton Channel. Science 2006, 392, 589–592. [Google Scholar] [CrossRef]
- Ramsey, I.S.; Ruchti, E.; Kaczmarek, J.S.; Clapham, D.E. Hv1 proton channels are required for high-level NADPH oxidase-dependent superoxide production during the phagocyte respiratory burst. Proc. Natl. Acad. Sci. USA 2009, 106, 7642–7647. [Google Scholar] [CrossRef]
- Okamura, Y.; Sasaki, M. Phagocytosis and membrane potential. Seikagaku 2007, 79, 454–458. [Google Scholar]
- Femling, J.K.; Cherny, V.V.; Morgan, D.; Rada, B.; Davis, A.P.; Czirják, G.; Enyedi, P.; England, S.K.; Moreland, J.G.; Ligeti, E.; et al. The antibacterial activity of human neutrophils and eosinophils requires proton channels but not BK channels. J. Gen. Physiol. 2006, 127, 659–672. [Google Scholar] [CrossRef]
- Klebanoff, S.J.; Kettle, A.J.; Rosen, H.; Winterbourn, C.C.; Nauseef, W.M. Myeloperoxidase: A front-line defender against phagocytosed microorganisms. J. Leukoc. Biol. 2013, 93, 185–198. [Google Scholar] [CrossRef]
- Rothfork, J.M.; Timmins, G.S.; Harris, M.N.; Chen, X.; Lusis, A.J.; Otto, M.; Cheung, A.L.; Gresham, H.D. Inactivation of a bacterial virulence pheromone by phagocyte-derived oxidants: New role for the NADPH oxidase in host defense. Proc. Natl. Acad. Sci. USA 2004, 101, 13867–13872. [Google Scholar] [CrossRef]
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive oxygen species in inflammation and tissue injury. Antioxid. Redox Signal. 2014, 20, 1126–1167. [Google Scholar] [CrossRef]
- Valavanidis, A.; Vlachogianni, T.; Fiotakis, K.; Loridas, S. Pulmonary oxidative stress, inflammation and cancer: Respirable particulate matter, fibrous dusts and ozone as major causes of lung carcinogenesis through reactive oxygen species mechanisms. Int. J. Environ. Res. Public Health 2013, 10, 3886–3907. [Google Scholar] [CrossRef]
- Shaeib, F.; Khan, S.N.; Thakur, M.; Kohan-Ghadr, H.R.; Drewlo, S.; Saed, G.M.; Pennathur, S.; Abu-Soud, H.M. The Impact of Myeloperoxidase and Activated Macrophages on Metaphase II Mouse Oocyte Quality. PLoS ONE 2016, 11, e0151160. [Google Scholar] [CrossRef]
- Davies, M.J. The oxidative environment and protein damage. Biochim. Biophys. Acta 2005, 1703, 93–109. [Google Scholar] [CrossRef]
- Hazen, S.L.; Hsu, F.F.; Gaut, J.P.; Crowley, J.R.; Heinecke, W.J. Modification of proteins and lipids by myeloperoxidase. Methods Enzym. 1999, 300, 88–105. [Google Scholar]
- Bagaitkar, J.; Pech, N.K.; Ivanov, S.; Austin, A.; Zeng, M.Y.; Pallat, S.; Huang, G.; Randolph, G.J.; Dinauer, M.C. NADPH oxidase controls neutrophilic response to steril inflammation in mice by regulating the IL-1α/G-CSF axis. Blood 2015, 126, 2724–2733. [Google Scholar] [CrossRef]
- Dupont, M.; Ouachée, A.; Royer, J.; Dupuy, C. NADPH oxidase: Double agent during inflammation? Med. Sci. 2016, 32, 833–835. [Google Scholar]
- Takei, H.; Haraki, A.; Watanabe, H.; Ichinose, A.; Sando, F. Rapid killing of human neutrophils by the potent activator phorbol 12-myristate 13-acetat (PMA) accompanied by changes different from typical apoptosis or necrosis. J. Leukoc. Biol. 1996, 59, 229–240. [Google Scholar] [CrossRef]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil Extracellular Traps Kill Bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef]
- Kumar, S.V.; Kulkarni, O.P.; Mulay, S.R.; Darisipudi, M.N.; Romoli, S.; Thomasova, D.; Scherbaum, C.R.; Hohenstein, B.; Hugo, C.; Müller, S.; et al. Neutrophil Extracellular Trap-Related Extracellular Histones Cause Vascular Necrosis in Severe GN. J. Am. Soc. Nephrol. 2015, 26, 2399–2413. [Google Scholar] [CrossRef]
- Parker, H.; Winterbourn, C. Reactive oxydants and myeloperoxidase and their involvment in neutrophil extracellular traps. Front. Immunol. 2013, 3, 424. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, T.; Abed, U.; Goosman, C.; Hurwitz, R.; Schulze, I.; Wahn, V.; Weinrauch, Y.; Brinkmann, V.; Zychlinsky, A. Novel cell death program leads to neutrophil extracellular traps. J. Cell. Biol. 2007, 176, 231–241. [Google Scholar] [CrossRef]
- Parker, H.; Dragunow, M.; Hampton, M.B.; Kettle, A.J.; Winterbourn, C.C. Requirements for NADPH oxidase and myeloperoxidase in neutrophil extracellular trap formation differ depending on the stimulus. J. Leukoc. Biol. 2012, 92, 841–849. [Google Scholar] [CrossRef]
- Röhm, M.; Grimm, M.J.; D’Auria, A.C.; Almyroudis, N.G.; Segal, B.H.; Urban, C.F. NADPH Oxidase Promotes Neutrophil Extracellular Trap Formation in Pulmonary Aspergillosis. Infect. Immun. 2014, 82, 1766–1777. [Google Scholar] [CrossRef]
- Bianchi, M.; Hakkim, A.; Brinkmann, V.; Siler, U.; Seger, R.A.; Zychlinsky, A.; Reichenbach, J. Restoration of NET formation by gene therapy in CGD controls aspergillosis. Blood 2009, 114, 2619–2622. [Google Scholar] [CrossRef]
- Chen, K.; Nishi, H.; Travers, R.; Tsuboi, N.; Martinod, K.; Wagner, D.D.; Stan, R.; Croce, K.; Mayadas, T.N. Endocytosis of soluble immune complexes leads to their clearance by FcγRIIIB but induces neutrophil extracellular traps via FcγRIIA in vivo. Blood 2012, 120, 4421–4431. [Google Scholar] [CrossRef]
- Byrd, A.S.; O’Brien, X.M.; Johnson, C.M.; Lavigne, L.M.; Reichner, J.S. An extracellular matrix-based mechanism of rapid neutrophil extracellular trap formation in response to Candida albicans. J. Immunol. 2013, 190, 4136–4148. [Google Scholar] [CrossRef]
- Tatsiy, O.; McDonald, P.P. Physiological Stimuli Induce PAD4-Dependent, ROS-Independent NETosis, With Early and Late Events Controlled by Discrete Signaling Pathways. Front. Immunol. 2018, 9, 2036. [Google Scholar] [CrossRef] [PubMed]
- Grandvaux, N.; Soucy-Faulkner, A.; Fink, K. Innate host defense: Nox and Duox on phox’s tail. Biochimie 2007, 89, 1113–1122. [Google Scholar] [CrossRef]
- Geiszt, M.; Witta, J.; Baffi, J.; Lekstrom, K.; Leto, T.L. Dual oxidases represent novel hydrogen peroxide sources supporting mucosal surface host defense. FASEB J. 2003, 17, 1502–1504. [Google Scholar] [CrossRef]
- Ha, E.M.; Oh, C.T.; Bae, Y.S.; Lee, W.J. A direct role for dual oxidase in Drosophila gut immunity. Science 2005, 310, 847–850. [Google Scholar] [CrossRef]
- Geiszt, M.; Lekstrom, K.; Brenner, S.; Hewitt, S.M.; Dana, R.; Malech, H.L.; Leto, T.L. NAD(P)H oxidase 1, a product of differentiated colon epithelial cells, can partially replace glycoprotein 91phox in the regulated production of superoxide by phagocytes. J. Immunol. 2003, 171, 299–306. [Google Scholar] [CrossRef]
- Kuwano, Y.; Kawahara, T.; Yamamoto, H.; Teshima-Kondo, S.; Tominaga, K.; Masuda, K.; Kishi, K.; Morita, K.; Rokutan, K. Interferon-gamma activates transcription of NADPH oxidase 1 gene and upregulates production of superoxide anion by human large intestinal epithelial cells. Am. J. Physiol. Cell Physiol. 2006, 290, C433. [Google Scholar] [CrossRef] [PubMed]
- Park, H.S.; Chun, J.N.; Jung, H.Y.; Choi, C.; Bae, Y.S. Role of NADPH oxidase 4 in lipopolysaccharide-induced proinflammatory responses by human aortic endothelial cells. Cardiovas. Res. 2006, 72, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Park, H.S.; Jung, H.Y.; Park, E.Y.; Kim, J.; Lee, W.J.; Bae, Y.S. Cutting edge: Direct interaction of TLR4 with NAD(P)H oxidase 4 isozyme is essential for lipopolysaccharide-induced production of reactive oxygen species and activation of NF-kappa B. J. Immunol. 2004, 173, 3589–3593. [Google Scholar] [CrossRef]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.A.; Waypa, G.B.; Schumacker, P.T. Redox signaling during hypoxia in mammalian cells. Redox Biol. 2017, 13, 228–234. [Google Scholar] [CrossRef]
- Winterbourn, C.C. Hydrogen peroxide reactivity and specificity in thiol-based cell signalling. Biochem. Soc. Trans. 2020, 48, 745–754. [Google Scholar] [CrossRef] [PubMed]
- Knaus, U.G. Oxidants in Physiological Processes. Handb. Exp. Pharm. 2021, 264, 27–47. [Google Scholar]
- Heo, S.; Kim, S.; Kang, D. The Role of Hydrogen Peroxide and Peroxiredoxins throughout the Cell Cycle. Antioxidants 2020, 9, 280. [Google Scholar] [CrossRef]
- Winterbourn, C.C. Reconciling the chemistry and biology of reactive oxygen species. Nat. Chem. Biol. 2008, 4, 278–286. [Google Scholar] [CrossRef]
- Hunter, T. Signaling: 2000 and Beyond. Cell 2000, 100, 113–127. [Google Scholar] [CrossRef]
- Barford, D. The role of cysteine residues as redox-sensitive regulatory switches. Curr. Opin. Struct. Biol. 2004, 14, 679–686. [Google Scholar] [CrossRef]
- Salmeen, A.; Barford, D. Functions and Mechanisms of Redox Regulation of Cysteine-Based Phosphatases. Antioxid. Redox Signal. 2005, 7, 560–577. [Google Scholar] [CrossRef]
- Denu, J.M.; Tanner, K.G. Specific and Reversible Inactivation of Protein Tyrosine Phosphatases by Hydrogen Peroxide: Evidence for a Sulfenic Acid Intermediate and Implications for Redox Regulation. Biochemistry 1998, 37, 5633–5642. [Google Scholar] [CrossRef]
- Goldstein, B.J.; Kalyankar, M.; Wu, X. Redox Paradox: Insulin Action Is Facilitated by Insulin-Stimulated Reactive Oxygen Species With Multiple Potential Signaling Targets. Diabetes 2005, 54, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Kwon, J.; Qu, C.-K.; Maeng, J.-S.; Falahati, R.; Lee, C.; Williams, M.S. Receptor-stimulated oxidation of SHP-2 promotes T-cell adhesion through SLP-76–ADAP. EMBO J. 2005, 24, 2331–2341. [Google Scholar] [CrossRef] [PubMed]
- Han, M.-J.; Kim, B.-Y.; Yoon, S.-O.; Chung, A.-S. Cell Proliferation Induced by Reactive Oxygen Species Is Mediated via Mitogen-activated Protein Kinase in Chinese Hamster Lung Fibroblast (V79) Cells. Mol. Cells 2003, 15, 94–101. [Google Scholar] [PubMed]
- Fürst, R.; Brueckl, C.; Kuebler, W.M.; Zahle, S.; Krötz, F.; Görlach, A.; Vollmar, A.M.; Kiemer, A.K. Atrial Natriuretic Peptide Induces Mitogen-Activated Protein Kinase Phosphatase-1 in Human Endothelial Cells via Rac1 and NAD(P)H Oxidase/Nox2-Activation. Circ. Res. 2005, 7, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Stouffs, M.; Serrander, L.; Banfi, B. The NADPH Oxidase NOX4 Drives Cardiac Differentiation: Role in Regulating Cardiac Transcription Factors and MAP Kinase Activation. Mol. Biol. Cell. 2006, 17, 3978–3988. [Google Scholar] [CrossRef] [PubMed]
- Görlach, A.; Bertram, K.; Hudecova, S.; Krizanova, O. Calcium and ROS: A mutual interplay. Redox Biol. 2015, 6, 260–271. [Google Scholar] [CrossRef]
- Santos, C.X.C.; Anilkumar, N.; Zhang, M.; Brewer, A.C.; Shah, A.M. Redox signaling in cardiac myocytes. Free Radic. Biol. Med. 2011, 50, 777–793. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Deng, X.; Hewavitharana, T.; Soboloff, J.; Gill, D.L. STIM, ORAI and TRPC Channels in the Control of Calcium Entry Signals in Smooth Muscle. Clin. Exp. Pharm. Physiol. 2008, 35, 1127–1133. [Google Scholar] [CrossRef] [PubMed]
- Grupe, M.; Myers, G.; Penner, R.; Fleig, A. Activation of store-operated ICRAC by hydrogen peroxide. Cell Calcium 2010, 48, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.H.; Lee, C.H.; Ahn, Y.; Kim, H.; Kim, H.; Ahn, C.Y.; Yang, K.S.; Lee, S.R. Redox regulation of PTEN and protein tyrosine phosphatases in H2O2-mediated cell signaling. FEBS Lett. 2004, 560, 7–13. [Google Scholar] [CrossRef]
- Lei, H.; Kazlauskas, A. Growth factors outside of the platelet-derived growth factor (PDGF) family employ reactive oxygen species/Src family kinases to activate PDGF receptor alpha and thereby promote proliferation and survival of cells. J. Biol. Chem. 2009, 284, 6329–6336. [Google Scholar] [CrossRef]
- Granados, M.P.; Salido, G.M.; González, A.; Pariente, J.A. Dose-dependent effect of hydrogen peroxide on calcium mobilization in mouse pancreatic acinar cells. Biochem. Cell Biol. 2006, 84, 39–48. [Google Scholar] [CrossRef]
- Wang, X.; Takeda, S.; Mochizuki, S.; Jindal, R.; Dhalla, N.S. Mechanisms of Hydrogen Peroxide-Induced Increase in Intracellular Calcium in Cardiomyocytes. J. Cardiovasc. Pharm. 1999, 4, 41–48. [Google Scholar] [CrossRef]
- Liu, G.; Pessah, I.N. Molecular Interaction between Ryanodine Receptor andMolecular Interaction between Ryanodine Receptor and Glycoprotein Triadin Involves Redox Cycling of Functionally Important Hyperreactive Sulfhydryls. J. Biol. Chem. 1994, 269, 33028–33034. [Google Scholar] [CrossRef]
- Favero, T.G.; Zable, A.C.; Abramson, J.J. Hydrogen peroxide stimuthe Ca2+ release channel from skeletal muscle sarcoplasmic reticulum. J. Biol. Chem. 1995, 270, 25557–25563. [Google Scholar] [CrossRef]
- Suzuki, Y.; Cleemann, L.; Abernethy, D.; Morad, M. Glutathione is a cofactor for H2O2-mediated stimulation of Ca2+induced Ca2+ release in cardiac myocytes. Free Radic. Biol. Med. 1998, 24, 318–325. [Google Scholar] [CrossRef]
- Kawakami, M.; Okabe, E. Superoxide anion radical-triggered Ca2+ release from cardiac sarcoplasmic reticulum through ryanodine receptor Ca2+ channel. Mol. Pharmacol. 1998, 53, 497–503. [Google Scholar] [CrossRef] [PubMed]
- Cheranov, S.Y.; Jaggar, J.H. TNF-alpha dilates cerebral arteries via NAD(P)H oxidase-dependent Ca2+ spark activation. Am. J. Physiol. Cell. Physiol. 2006, 290, C964–C971. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo, C.; Sanchez, G.; Barrientos, G.; Aracena-Parks, P. A transverse tubule NOX activity stimulates calcium release from isolated triads via RYR1 S-glutathionylation. J. Biol. Chem. 2006, 281, 26473–26482. [Google Scholar] [CrossRef]
- Yi, X.Y.; Li, V.X.; Zhang, F.; Yi, F.; Matson, D.R.; Jiang, M.T.; Li, P.L. Characteristics and actions of NAD(P)H oxidase on the sarcoplasmic reticulum of coronary artery smooth muscle. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H1136–H1144. [Google Scholar] [CrossRef]
- Xie, Y.; Yang, Y.; Galice, S.; Bers, D.M.; Sato, D. Size Matters: Ryanodine Receptor Cluster Size Heterogeneity Potentiates Calcium Waves. Biophys. J. 2019, 116, 530–539. [Google Scholar] [CrossRef]
- Hu, Q.; Zu-Xi, Y.; Ferrans, V.J.; Takeda, K.; Irani, K.; Ziegelstein, R.C. Critical Role of NADPH Oxidase-derived Reactive Oxygen Species in Generating Ca2+ Oscillations in Human Aortic Endothelial Cells Stimulated by Histamine. J. Biol. Chem. 2002, 277, 32546–32551. [Google Scholar] [CrossRef]
- Adachi, T.; Weisbrod, R.M.; Pimentel, D.R.; Ying, J.; Sharov, V.S.; Schöneich, C.; Cohen, R.A. S-Glutathiolation by peroxynitrite activates SERCA during arterial relaxation by nitric oxide. Nat. Med. 2004, 10, 1200–1207. [Google Scholar] [CrossRef]
- Redondo, P.C.; Salido, G.M.; Rosado, J.A.; Pariente, J.A. Effect of hydrogen peroxide on Ca2+ mobilisation in human platelets through sulphydryl oxidation dependent and independent mechanisms. Biochem. Pharm. 2004, 67, 491–502. [Google Scholar] [CrossRef]
- Irani, K. Oxidant Signaling in Vascular Cell Growth, Death, and Survival: A Review of the Roles of Reactive Oxygen Species in Smooth Muscle and Endothelial Cell Mitogenic and Apoptotic Signaling. Circ. Res. 2000, 87, 179–183. [Google Scholar] [CrossRef]
- Hampton, M.B.; Fadeel, B.; Orrenius, S. Redox Regulation of the Caspases during Apoptosis. Ann. N. Y. Acad. Sci. 1997, 854, 328–335. [Google Scholar] [CrossRef]
- Hampton, M.B.; Orrenius, S. Dual regulation of caspase activity by hydrogen peroxide: Implications for apoptosis. FEBS Lett. 1997, 414, 552–555. [Google Scholar] [CrossRef]
- Deshpande, S.S.; Angkeow, P.; Huang, J. Rac1 inhibits TNF-alpha-induced endothelial cell apoptosis: Dual regulation by reactive oxygen species. FASEB J. 2000, 14, 1705–1714. [Google Scholar] [CrossRef]
- Mochizuki, T.; Furuta, S.; Mitsushita, J.; Shang, W.H.; Ito, M.; Yokoo, Y.; Yamaura, M.; Ishizone, S.; Nakayama, J.; Konagai, A.K.; et al. Inhibition of NADPH oxidase 4 activates apoptosis via the AKT/apoptosis signal-regulating kinase 1 pathway in pancreatic cancer PANC-1 cells. Oncogene 2006, 25, 3699–3707. [Google Scholar] [CrossRef]
- Clement, M.V.; Stamenkovic, I. Superoxide anion is a natural inhibitor of FAS-mediated cell death. EMBO J. 1996, 15, 216–225. [Google Scholar] [CrossRef]
- Adachi, T.; Togashi, H.; Suzuki, A.; Kasai, S.; Ito, J.; Sugahara, K.; Kawata, S. NAD(P)H oxidase plays a crucial role in PDGF-induced proliferation of hepatic stellate cells. Hepathology 2005, 41, 1272–1281. [Google Scholar] [CrossRef]
- Irani, K.; Xia, Y.; Zweier, J.L.; Sollott, S.J.; Der, C.J.; Fearon, E.R.; Sundaresan, M.; Finkel, T.; Goldschmidt-Clermont, P.J. Mitogenic signaling mediated by oxidants in Ras-transformed fibroblasts. Science 1997, 275, 1649–1652. [Google Scholar] [CrossRef]
- Laurent, A.; Nicco, C.; Chéreau, C.; Goulvestre, C. Controlling Tumor Growth by Modulating Endogenous Production of Reactive Oxygen Species. Cancer Res. 2005, 65, 948–956. [Google Scholar]
- Arnold, R.S.; Shi, J.; Murad, E.; Whalen, A.M.; Sun, C.Q.; Polavarapu, R.; Parthasarathy, S.; Petros, J.A.; Lambeth, J.D. Hydrogen peroxide mediates the cell growth and transformation caused by the mitogenic oxidase Nox1. Proc. Natl. Acad. Sci. USA 2001, 98, 5550–5555. [Google Scholar] [CrossRef]
- Fu, X.; Beer, D.G.; Behar, J.; Wands, J.; Lambeth, D.; Cao, W. cAMP-response element-binding protein mediates acid-induced NADPH oxidase NOX5-S expression in Barrett esophageal adenocarcinoma cells. J. Biol. Chem. 2006, 281, 20368–20382. [Google Scholar] [CrossRef]
- Kitamoto, K.; Miura, Y.; Karman, S.; Ota, K.; Konishi, H.; Hosokawa, Y.; Sato, K. Inhibition of NADPH oxidase 2 induces apoptosis in osteosarcoma: The role of reactive oxygen species in cell proliferation. Oncol. Lett. 2018, 15, 7955–7962. [Google Scholar] [CrossRef]
- Brault, J.; Vigne, B.; Meunier, M.; Beaumel, S.; Mollin, M.; Park, S.; Stasia, M.J. NOX4 is the main NADPH oxidase involved in the early stages of hematopoietic differentiation from human induced pluripotent stem cells. Free Radic. Biol. Med. 2020, 146, 107–118. [Google Scholar] [CrossRef]
- Szanto, I.; Pusztaszeri, M.; Mavromati, M. H2O2 Metabolism in Normal Thyroid Cells and in Thyroid Tumorigenesis: Focus on NADPH Oxidases. Antioxidants 2019, 8, 126. [Google Scholar] [CrossRef] [PubMed]
- Moreno, J.C.; Bikker, H.; Kempers, M.J.; van Trotsenburg, A.S.; Baas, F.; de Vijlder, J.J.; Vulsma, T.; Ris-Stalpers, C. Inactivating Mutations in the Gene for Thyroid Oxidase(THOX2) and Congenital Hypothroidism. N. Engl. J. Med. 2002, 347, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Bánfi, B.; Malgrange, B.; Knisz, J.; Steger, K.; Dubois-Dauphin, M.; Krause, K.H. NOX3: A superoxide-generating NADPH oxidase of the inner ear. J. Biol. Chem. 2004, 279, 46065–46072. [Google Scholar] [CrossRef] [PubMed]
- Rousset, F.; Carnesecchi, S.; Senn, P.; Krause, K.H. NOX3-Targeted therapies for inner ear pathologies. Curr. Pharm. Des. 2015, 21, 5977–5987. [Google Scholar] [CrossRef] [PubMed]
- Mohri, H.; Ninoyu, Y.; Sakaguchi, H.; Hirano, S.; Saito, N.; Ueyama, T. Nox3-derived superoxide in cochleae induces sensorineural hearing loss Mechanisms of Nox3-dependent hearing loss. J. Neurosci. 2021. [Google Scholar] [CrossRef]
- Malec, V.; Gottschald, O.R.; Li, S.; Rose, F.; Seeger, W.; Hänze, J. HIF-1α signaling is augmented during intermittent hypoxia by induction of the Nrf2 pathway in NOX1-expressing adenocarcinoma A549 cells. Free Radic. Biol. Med. 2010, 48, 1626–1635. [Google Scholar] [CrossRef]
- Kim, Y.M.; Kim, S.J.; Tatsunami, R.; Yamamura, H.; Fukai, T.; Ushio-Fukai, M. ROS-induced ROS release orchestrated by Nox4, Nox2, and mitochondria in VEGF signaling and angiogenesis. Am. J. Physiol. Cell Physiol. 2017, 312, C749–C764. [Google Scholar] [CrossRef]
- Wang, H.; Yang, Z.; Jiang, Y.; Hartnett, M.E. Endothelial NADPH oxidase 4 mediates vascular endothelial growth factor receptor 2-induced intravitreal neovascularization in a rat model of retinopathy of prematurity. Mol. Vis. 2014, 20, 231–241. [Google Scholar]
- di Bartolo, B.A.; Cartland, S.P.; Prado-Lourenco, L.; Griffith, T.S.; Gentile, C.; Ravindran, J.; Azahri, N.S.; Thai, T.; Yeung, A.W.; Thomas, S.R.; et al. Tumor necrosis factor-related apoptosis-inducing ligand (trail) promotes angiogenesis and ischemia-induced neneovascularization via NADPH oxidase 4 (NOX4) and nitric oxide-dependent mechanisms. J. Am. Heart Assoc. 2015, 4, e002527. [Google Scholar] [CrossRef]
- Peshavariya, H.M.; Chan, E.C.; Liu, G.S.; Jiang, F.; Dusting, G.J. Transforming growth factor-β1 requires NADPH oxidase 4 for angiogenesis in vitro and in vivo. J. Cell. Mol. Med. 2014, 18, 1172–1183. [Google Scholar] [CrossRef]
- Chen, L.; Xiao, J.; Kuroda, J.; Ago, T.; Sadoshima, J.; Cohen, R.A.; Tong, X. Both hydrogen peroxide and transforming growth factor β1 contribute to endothelial Nox4 mediated angiogenesis in endothelial Nox4 transgenic mouse lines. Biochim. Biophys. Acta. 2014, 1842, 2489–2499. [Google Scholar] [CrossRef]
- Shafique, E.; Torina, A.; Reichert, K.; Colantuono, B.; Nur, N.; Zeeshan, K.; Ravichandran, V.; Liu, Y.; Feng, J.; Zeeshan, K.; et al. Mitochondrial redox plays a critical role in the paradoxical effects of NAPDH oxidase-derived ROS on coronary endothelium. Cardiovasc. Res. 2017, 113, 234–246. [Google Scholar] [CrossRef] [PubMed]
- Evangelista, A.; Thompson, M.; Bolotina, V.; Tong, X.; Cohen, R. Nox4- and Nox2-dependent oxidant production is required for VEGF-induced SERCA cysteine-674 s-glutathiolation and endothelial cell migration. Free Rad. Biol. Med. 2012, 53, 2327–2334. [Google Scholar] [CrossRef] [PubMed]
- Craige, S.M.; Chen, K.; Pei, Y.; Li, C.; Huang, X.; Chen, C.; Shibata, R.; Sato, K.; Walsh, K.; Keaney, J.F. NADPH oxidase 4 promotes endothelial angiogenesis through endothelial nitric oxide synthase activation. Circulation 2011, 124, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.X.; Zeng, H.; Tuo, Q.H.; Yu, H.; Meyrick, B.; Aschner, J.L. NADPH oxidase modulates myocardial Akt, ERK1/2 activation, and angiogenesis after hypoxia-reoxygenation. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, 1664–1674. [Google Scholar] [CrossRef]
- Garrido-Urbani, S.; Jemelin, S.; Deffert, C.; Carnesecchi, S.; Basset, O.; Szyndralewiez, C.; Heitz, F.; Page, P.; Montet, X.; Michalik, L.; et al. Targeting vascular NADPH oxidase 1 blocks tumor angiogenesis through a PPARα mediated mechanism. PLoS ONE 2011, 6, 14665. [Google Scholar] [CrossRef]
- Krock, B.L.; Skuli, N.N.; Simon, M.C. Hypoxia-Induced Angiogenesis Good and Evil. Genes Cancer 2011, 2, 1117–1133. [Google Scholar] [CrossRef]
- Pendyala, S.; Gorshkova, I.A.; Usatyuk, P.V.; He, D.; Pennathur, A.; Lambeth, J.D.; Thannickal, V.J.; Natarajan, V. Role of Nox4 and Nox2 in Hyperoxia-Induced Reactive Oxygen Species Generation and Migration of Human Lung Endothelial Cells. Antioxid. Redox Signal 2009, 11, 747–764. [Google Scholar] [CrossRef]
- Menden, H.; Welak, S.; Cossette, S.; Ramchandran, R.; Sampath, V. Lipopolysaccharide (LPS)-mediated angiopoietin-2-dependent autocrine angiogenesis is regulated by NADPH oxidase 2 (Nox2) in human pulmonary microvascular endothelial cells. J. Biol. Chem. 2015, 290, 5449–5461. [Google Scholar] [CrossRef]
- Ho, T.K.; Tsui, J.; Xu, S.; Leoni, P.; Abraham, D.J.; Baker, D.M. Angiogenic effects of stromal cell-derived factor-1 (SDF-1/CXCL12) variants in vitro and the in vivo expressions of CXCL12 variants and CXCR4 in human critical leg ischemia. J. Vasc. Surg. 2010, 51, 689–699. [Google Scholar] [CrossRef]
- Pi, X.; Xie, L.; Portbury, A.L.; Kumar, S.; Lockyer, P.; Li, X.; Patterson, C. NADPH Oxidase-Generated Reactive Oxygen Species Are Required for Stromal Cell-Derived Factor-1 -Stimulated Angiogenesis. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2023–2032. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, S.; Brault, J.; Stasia, M.J.; Knaus, U.G. Genetic disorders coupled to ROS deficiency. Redox Biol. 2015, 6, 135–156. [Google Scholar] [CrossRef] [PubMed]
- Roos, D.; Kuhns, D.B.; Maddalena, A.; Roesler, J.; Lopez, J.A.; Ariga, T.; Avcin, T.; de Boer, M.; Bustamante, J.; Condino-Neto, A.; et al. Hematologically important mutations: X-linked chronic granulomatous disease (third update). Blood Cells Mol. Dis. 2010, 45, 246–265. [Google Scholar] [CrossRef]
- Beaumel, S.; Stasia, M.J. The X-CGD PLB-985 Cell Model for NOX2 Structure-Function Analysis. Methods Mol. Biol. 2019, 1982, 153–171. [Google Scholar]
- Stasia, M.J.; Li, X.J. Genetics and immunopathology of chronic granulomatous disease. Semin. Immunopathol. 2008, 30, 209–235. [Google Scholar] [CrossRef]
- Rae, J.; Newburger, P.E.; Dinauer, M.C.; Noack, D.; Hopkins, P.J.; Kuruto, R.; Curnutte, J.T. X-Linked Chronic Granulomatous Disease: Mutations in the CYBB Gene. Am. J. Hum. Genet. 1998, 62, 1320–1331. [Google Scholar] [CrossRef]
- Porter, C.; Parkar, M.; Verhoeven, A.; Levinsky, R.; Collins, M.; Kinnon, C. p22-phox-deficient chronic granulomatous disease: Reconstitution by retrovirus-mediated expression and identification of a biosynthetic intermediate of gp91-phox. Blood 1994, 84, 2767–2775. [Google Scholar] [CrossRef]
- Stasia, M.J.; Cathebras, P.; Lutz, M.F.; Durieu, I. Chronic-granulomatous disease. Rev. Med. Interne 2009, 30, 221–232. [Google Scholar] [CrossRef]
- de Boer, M.; Klein, A.; Hossle, J.-P.; Seger, R.; Corbeel, L.; Weening, R.S.; Roos, D. Cytochrome b558-negative, autosomal recessive chronic granulomatous disease: Two new mutations in the cytochrome b558 light chain of the NADPH oxidase (p22-phox). Am. J. Hum. Genet. 1992, 51, 1127–1135. [Google Scholar]
- Roos, D.; Kuhns, D.B.; Maddalena, A.; Bustamante, J.; Kannengiesser, C.; de Boer, M.; van Leeuwen, K.; Köker, M.Y.; Wolach, B.; Roesler, J.; et al. Hematologically important mutations: The autosomal recessive forms of chronic granulomatous disease (second update). Blood Cells Mol. Dis. 2010, 44, 291–299. [Google Scholar] [CrossRef]
- van den Berg, J.M.; van Koppen, E.; Ahlin, A.; Belohradsky, B.; Bernatowska, E.; Corbeel, L.; Español, T.; Fischer, A.; Kurenko-Deptuch, M.; Mouy, R.; et al. Chronic Granulomatous Disease: The European Experience. PLoS ONE 2009, 4, e5234. [Google Scholar] [CrossRef]
- Popa-Wagner, A.; Mitran, S.; Sivanesan, S.; Chang, E.; Buga, A.M. ROS and brain diseases: The good, the bad, and the ugly. Oxid. Med. Cell. Longev. 2013, 2013, 963520. [Google Scholar] [CrossRef]
- Sorce, S.; Krause, K.H. NOX enzymes in the central nervous system: From signaling to disease. Antioxid. Redox Signal 2009, 11, 2481–2504. [Google Scholar] [CrossRef]
- Chéret, C.; Gervais, A.; Lelli, A.; Colin, C.; Amar, L.; Ravassard, P.; Mallet, J.; Cumano, A.; Krause, K.H.; Mallat, M. Neurotoxic activation of microglia is promoted by a nox1-dependent NADPH oxidase. J. Neurosci. 2008, 12, 12039–12051. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Bedard, K.; Sorce, S.; Hinz, B.; Dubois-Dauphin, M.; Krause, K.H. NOX4 expression in human microglia leads to constitutive generation of reactive oxygen species and to constitutive IL-6 expression. J. Innate Immun. 2009, 1, 570–581. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Ago, T.; Kitazono, T.; Nabika, T. NADPH Oxidase-Related Pathophysiology in Experimental Models of Stroke. Int. J. Mol. Sci. 2017, 18, 2123. [Google Scholar] [CrossRef] [PubMed]
- Hernandes, M.S.; Café-Mendes, C.C.; Britto, L.R.G. NADPH oxidase and the degeneration of dopaminergic neurons in parkinsonian mice. Oxid. Med. Cell. Longev. 2013, 2013, 157857. [Google Scholar] [CrossRef]
- Przedborski, S.; Vila, M. The 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model: A tool to explore the pathogenesis of Parkinson’s disease. Ann. N. Y. Acad. Sci. 2003, 991, 189–198. [Google Scholar] [CrossRef]
- Meredith, G.E.; Rademacher, D.J. MPTP mouse models of Parkinson’s disease: An update. J. Parkinsons Dis. 2011, 1, 19–33. [Google Scholar] [CrossRef]
- George, J.L.; Mok, S.; Moses, D.; Wilkins, S.; Bush, A.I.; Cherny, R.A.; Finkelstein, D.I. Targeting the Progression of Parkinson’s Disease. Curr. Neuropharmacol. 2009, 7, 9–36. [Google Scholar] [CrossRef] [PubMed]
- Huh, S.H.; Chung, Y.C.; Piao, Y.; Jin, M.Y.; Son, H.J.; Yoon, N.S.; Hong, J.Y.; Pak, Y.K.; Kim, Y.S.; Hong, J.K.; et al. Ethyl pyruvate rescues nigrostriatal dopaminergic neurons by regulating glial activation in a mouse model of Parkinson’s disease. J. Immunol. 2011, 187, 960–969. [Google Scholar] [CrossRef] [PubMed]
- Dias, V.; Junn, E.; Mouradian, M.M. The role of oxidative stress in Parkinson’s disease. J. Parkinsons Dis. 2013, 3, 461–491. [Google Scholar] [CrossRef]
- Guo, J.; Zhao, X.; Li, Y.; Li, G.; Liu, X. Damage to dopaminergic neurons by oxidative stress in Parkinson’s disease. Int. J. Mol. Med. 2018, 41, 1817–1825. [Google Scholar] [CrossRef]
- Belarbi, K.; Cuvelier, E.; Destée, A.; Gressier, B.; Chartier-Harlin, M.-C. NADPH oxidases in Parkinson’s disease: A systematic review. Mol. Neurodegener. 2017, 12, 84. [Google Scholar] [CrossRef]
- Rodriguez-Pallares, J.; Rey, P.; Parga, J.; Muñoz, A.; Guerra, M.; Labandeira-Garcia, J. Brain angiotensin enhances dopaminergic cell death via microglial activation and NADPH-derived ROS. Neurobiol. Dis. 2008, 31, 58–73. [Google Scholar] [CrossRef]
- Kim, S.; Hwang, J.; Lee, W.H.; Hwang, D.Y.; Suk, K. Role of protein kinase in paraquat-induced glial cell death. J. Neurosci. Res. 2008, 86, 2062–2070. [Google Scholar] [CrossRef]
- Shimohama, S.; Tanino, H.; Kawakami, N.; Okamura, N.; Kodama, H.; Yamaguchi, T.; Hayakawa, T.; Nunomura, A.; Chiba, S.; Perry, G.; et al. Activation of NADPH Oxidase in Alzheimer’s Disease Brains. Biochem. Biophys. Res. Commun. 2000, 273, 5–9. [Google Scholar] [CrossRef]
- Park, L.; Zhou, P.; Pitstick, R.; Capone, C.; Anrather, J.; Norris, E.H.; Younkin, L.; Younkin, S.; Carlson, G.; McEwen, B.S.; et al. Nox2-derived radicals contribute to neurovascular and behavioral dysfunction in mice overexpressing the amyloid precursor protein. Proc. Natl. Acad. Sci. USA 2008, 105, 1347–1352. [Google Scholar] [CrossRef]
- Gamba, P.; Leonarduzzi, G.; Tamagno, E.; Guglielmotto, M.; Testa, G.; Sottero, B.; Gargiulo, S.; Biasi, F.; Mauro, A.; Viña, J.; et al. Interaction between 24-hydroxycholesterol, oxidative stress, and amyloid-b in amplifying neuronal damage in Alzheimer’s disease: Three partners in crime. Aging Cell 2011, 10, 403–417. [Google Scholar] [CrossRef]
- Bruce-Keller, A.J.; Gupta, S.; Knight, A.G.; Beckett, T.L.; McMullen, J.M.; Davis, P.R.; Murphy, M.P.; van Eldik, L.J.; Clair, D.S.; Keller, J.N. Cognitive Impairment in Humanized APPxPS1 Mice is Linked to Aβ1-42 and NOX Activation. Neurobiol. Dis. 2011, 44, 317–326. [Google Scholar] [CrossRef] [PubMed]
- Akhter, H.; Katre, A.; Li, L.; Liu, X.; Liu, R.M. Therapeutic potential and anti-amyloidosis mechanisms of tert-butylhydroquinone for Alzheimer’s disease. J. Alzheimers Dis. 2011, 26, 767–778. [Google Scholar] [CrossRef]
- Choi, D.H.; Lee, K.H.; Kim, J.H.; Seo, J.H.; Kim, H.Y.; Shin, C.Y.; Han, J.S.; Han, S.H.; Kim, Y.S.; Lee, J. NADPH Oxidase 1, a Novel Molecular Source of ROS in Hippocampal Neuronal Death in Vascular Dementia. Antioxid. Redox Signal. 2014, 21, 533–550. [Google Scholar] [CrossRef]
- de la Monte, S.M.; Wands, J.R. Molecular indices of oxidative stress and mitochondrial dysfunction occur early and often progress with severity of Alzheimer’s disease. J. Alzheimers Dis. 2006, 9, 167–181. [Google Scholar] [CrossRef]
- Hernandes, M.S.; Britto, L.R.G. NADPH Oxidase and Neurodegeneration. Curr. Neuropharmacol. 2012, 10, 321–327. [Google Scholar] [CrossRef]
- Meitzler, J.L.; Antony, S.; Wu, Y.; Juhasz, A.; Liu, H.; Jiang, G.; Lu, J.; Roy, K.; Doroshow, J.H. NADPH Oxidases: A Perspective on Reactive Oxygen Species Production in Tumor Biology. Antioxid. Redox Signal. 2014, 20, 2873–2888. [Google Scholar] [CrossRef]
- Juhasz, A.; Ge, Y.; Markel, S.; Chiu, A.; Matsumoto, L.; van Balgooy, J.; Roy, K.; Doroshow, J.H. Expression of NADPH oxidase homologues and accessory genes in human cancer cell lines, tumours and adjacent normal tissues. Free Radic. Res. 2009, 43, 523–532. [Google Scholar] [CrossRef] [PubMed]
- Day, R.M.; Suzuki, Y.J. Cell Proliferation, Reactive Oxygen and Cellular Glutathione. Dose Response 2005, 3, 425–442. [Google Scholar] [CrossRef]
- Wang, P.; Shi, Q.; Deng, W.-H.; Yu, J.; Zuo, T.; Mei, F.C.; Wang, W.X. Relationship between expression of NADPH oxidase 2 and invasion and prognosis of human gastric cancer. World J. Gastroenterol. 2015, 21, 6271–6279. [Google Scholar] [CrossRef]
- You, X.; Ma, M.; Hou, G.; Hu, Y.; Shi, X. Gene expression and prognosis of NOX family members in gastric cancer. Onco Targets Ther. 2018, 11, 3065–3074. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.G.; Xia, H.L.; Just, T.; Dai, Y.R. Hydroxyl radical-induced apoptosis in human tumor cells is associated with telomere shortening but not telomerase inhibition and caspase activation. FEBS Lett. 2001, 488, 123–132. [Google Scholar] [CrossRef]
- Matsura, T.; Kai, M.; Fujii, Y.; Ito, H.; Yamada, K. Hydrogen peroxide-induced apoptosis in HL-60 cells requires caspase-3 activation. Free Radic. Res. 1999, 30, 73–83. [Google Scholar] [CrossRef]
- Simizu, S.; Takada, M.; Umezawa, K.; Imoto, M. Requirement of Caspase-3(-like) Protease-mediated Hydrogen Peroxide Production for Apoptosis Induced by Various Anticancer Drugs. J. Biol. Chem. 1998, 273, 26900–26907. [Google Scholar] [CrossRef]
- Skonieczna, M.; Hejmo, T.; Poterala-Hejmo, A.; Cieslar-Pobuda, A.; Buldak, R.J. NADPH Oxidases: Insights into Selected Functions and Mechanisms of Action in Cancer and Stem Cells. Oxid. Med. Cell. Longev. 2017, 2017. [Google Scholar] [CrossRef]
- Limoli, C.L.; Giedzinski, E. Induction of chromosomal instability by chronic oxidative stress. Neoplasia 2003, 5, 339–346. [Google Scholar] [CrossRef]
- Hunt, C.R.; Sim, J.E.; Sullivan, S.J.; Featherstone, T.; Golden, W.; Von Kapp-Herr, C.; Hock, R.A.; Gomez, R.A.; Parsian, A.J.; Spitz, D.R. Genomic instability and catalase gene amplification induced by chronic exposure to oxidative stress. Cancer Res. 1998, 58, 3986–3992. [Google Scholar]
- Limoli, C.L.; Hartmann, A.; Shephard, L.; Yang, C.R.; Boothman, D.A.; Bartholomew, J.; Morgan, W.F. Apoptosis, reproductive failure, and oxidative stress in Chinese hamster ovary cells with compromised genomic integrity. Cancer Res. 1998, 58, 3712–3718. [Google Scholar]
- Clutton, S.M.; Townsend, K.M.S.; Walker, C.; Ansell, J.D.; Wright, E.G. Radiation-induced genomic instability and persisting oxidative stress in primary bone marrow cultures. Carcinogenesis 1996, 17, 1633–1639. [Google Scholar] [CrossRef]
- Graham, K.; Kulawiec, M.; Owens, K.; Li, X.; Desouki, M.; Chandra, D.; Singh, K. NADPH oxidase 4 is an oncoprotein localized to mitochondria. Cancer Biol. 2010, 10, 223–231. [Google Scholar] [CrossRef]
- Kartha, G.; Moshal, K.; Sen, U.; Joshua, I.; Tyagi, N.; Steed, M.; Tyagi, S. Renal mitochondrial damage and protein modification in type-2 diabetes. Acta Diabetol. 2008, 45, 75–81. [Google Scholar] [CrossRef]
- Spencer, N.Y.; Yan, Z.; Boudreau, R.L.; Zhang, Y.; Luo, M.; Li, Q.; Tian, X.; Shah, A.M.; Davisson, R.L.; Davidson, B.; et al. Control of Hepatic Nuclear Superoxide Production by Glucose 6-Phosphate Dehydrogenase and NADPH Oxidase-4. J. Biol. Chem. 2011, 286, 8977–8987. [Google Scholar] [CrossRef] [PubMed]
- Salmeen, A.; Park, B.; Meyer, T. The NADPH oxidases NOX4 and DUOX2 regulate cell cycle entry via a p53-dependent pathway. Oncogene 2010, 29, 4473–4484. [Google Scholar] [CrossRef]
- Eid, A.A.; Ford, B.M.; Block, K.; Kasinath, B.S.; Gorin, Y.; Ghosh-Choudhury, G.; Barnes, J.L.; Abboud, H.E. AMP-activated protein kinase (AMPK) negatively regulates Nox4-dependent activation of p53 and epithelial cell apoptosis in diabetes. J. Biol. Chem. 2010, 285, 37503–37512. [Google Scholar] [CrossRef]
- Boudreau, H.E.; Casterline, B.W.; Burke, D.J.; Leto, T.L. Wild-type and mutant p53 differentially regulate NADPH oxidase 4 in TGF-beta-mediated migration of human lung and breast epithelial cells. Br. J. Cancer 2014, 110, 2569–2582. [Google Scholar] [CrossRef]
- Vaziri, H.; Dessain, S.K.; Eaton, E.N.; Imai, S.I.; Frye, R.A.; Pandita, T.K.; Guarente, L.; Weinberg, R.A. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell 2001, 107, 149–159. [Google Scholar] [CrossRef]
- Laurent, E.; McCoy III, J.W.; Macina, R.A.; Liu, W.; Cheng, G.; Robine, S.; Papkoff, J.; Lambeth, J.D. Nox1 is over-expressed in human colon cancers and correlates with activating mutations in K-Ras. Int. J. Cancer 2008, 123, 100–107. [Google Scholar] [CrossRef]
- Thiery, J.P. Epithelial–mesenchymal transitions in development and pathologies. Curr. Opin Cell. Biol. 2003, 15, 740–746. [Google Scholar] [CrossRef]
- Leong, H.S.; Robertson, A.E.; Stoletov, K.; Chin, C.A.; Chien, A.E.; Hague, M.N.; Ablack, A.; Carmine-Simmen, K.; McPherson, V.A.; Postenka, C.O.; et al. Invadopodia Are Required for Cancer Cell Extravasation and Are a Therapeutic Target for Metastasis. Cell Rep. 2014, 8, 1558–1570. [Google Scholar] [CrossRef] [PubMed]
- Diaz, B.; Shani, G.; Pass, D.A.I.; Quintavalle, M.; Courtneidge, S.A. Tks5-dependent, Nox-mediated Generation of Reactive Oxygen Species is Necessary for Invadopodia Formation. Sci. Signal. 2009, 2, 1–15. [Google Scholar] [CrossRef]
- van Zijl, F.; Krupitza, G.; Mikulits, W. Initial steps of metastasis: Cell invasion and endothelial transmigration. Mutat. Res. 2011, 728, 23–34. [Google Scholar] [CrossRef]
- Paz, H.; Pathak, N.; Yang, J. Invading one step at a time: The role of invadopodia in tumor metastasis. Oncogene 2014, 33, 4193–4202. [Google Scholar] [CrossRef]
- Lock, P.; Abram, C.L.; Gibson, T.; Courtneidge, S.A. A new method for isolating tyrosine kinase substrates used to identify fish, an SH3 and PX domain-containing protein, and Src substrate. EMBO J. 1998, 17, 4346–4357. [Google Scholar] [CrossRef]
- Gianni, D.; Diaz, B.; Taulet, N.; Fowler, B.; Courtneidge, S.; Bokoch, G. Novel p47(phox)-related organizers regulate localized NADPH oxidase 1 (Nox1) activity. Sci. Signal. 2009, 2, ra54. [Google Scholar] [CrossRef]
- Seals, D.F.; Azucena, E.F.J.; Pass, I.; Tesfay, L.; Gordon, R.; Woodrow, M.; Resau, J.H.; Courtneidge, S.A. The adaptor protein Tks5/Fish is required for podosome formation and function, and for the protease-driven invasion of cancer cells. Cancer Cell 2005, 7, 155–165. [Google Scholar] [CrossRef]
- Revach, O.Y.; Sandler, O.; Samuels, Y.; Geiger, B. Cross-talk between receptor tyrosine kinases AXL and ERBB3 regulates invadopodia formation in melanoma cells. Cancer Res. 2019, 79, 2634–2648. [Google Scholar] [CrossRef]
- Liu, X.; Pei, C.; Yan, S.; Liu, G.; Liu, G.; Chen, W.; Cui, Y.; Liu, Y. NADPH oxidase 1-dependent ROS is crucial for TLR4 signaling to promote tumor metastasis of non-small cell lung cancer. Tumour Biol. 2015, 36, 1493–1502. [Google Scholar] [CrossRef] [PubMed]
- Pei, J.; Lou, Y.; Zhong, R.; Han, B. MMP9 activation triggered by epidermal growth factor induced FoxO1 nuclear exclusion in nonsmall cell lung cancer. Tumour Biol. 2014, 35, 6673–6678. [Google Scholar] [CrossRef]
- Choi, Y.H.; Burdick, M.D.; Strieter, B.A.; Mehrad, B.; Strieter, R.M. CXCR4, but not CXCR7, discriminates metastatic behavior in nonsmall cell lung cancer cells. Mol. Cancer Res. 2014, 12, 38–47. [Google Scholar] [CrossRef]
- O’Leary, D.P.; Bhatt, L.; Woolley, J.F.; Gough, D.R.; Wang, J.H.; Cotter, T.G.; Redmond, H.P. TLR-4 signalling accelerates colon cancer cell adhesion via NF-κB mediated transcriptional up-regulation of Nox-1. PLoS ONE 2012, 7, e44176. [Google Scholar] [CrossRef]
- Huang, H.S.; Liu, Z.M.; Chen, P.C.; Tseng, H.Y.; Yeh, B.W. TG-interacting factor-induced superoxide production from NADPH oxidase contributes to the migration/invasion of urothelial carcinoma. Free. Radic. Biol. Med. 2012, 53, 769–778. [Google Scholar] [CrossRef] [PubMed]
- Nishida, N.; Yano, H.; Nishida, T.; Kamura, T.; Kojiro, M. Angiogenesis in cancer. Vasc. Health Risk Manag. 2006, 2, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Helfinger, V.; Henke, N.; Harenkamp, S.; Walter, M.; Epah, J.; Penski, C.; Mittelbronn, M.; Schröder, K. The NADPH oxidase Nox4 mediates tumour angiogenesis. Acta Physiol. 2016, 216, 435–446. [Google Scholar] [CrossRef]
- Jung, S.N.; Yang, W.K.; Kim, J.; Kim, H.S.; Kim, E.J.; Yun, H.; Park, H.; Kim, S.S.; Choe, W.; Kang, I.; et al. Reactive oxygen species stabilize hypoxia-inducible factor-1 alpha protein and stimulate transcriptional activity via AMP-activated protein kinase in DU145 human prostate cancer cells. Carcinogenesis 2008, 29, 713–721. [Google Scholar] [CrossRef]
- Xia, C.; Meng, Q.; Liu, L.Z.; Rojanasakul, Y.; Wang, X.R.; Jiang, B.H. Reactive oxygen species regulate angiogenesis and tumor growth through vascular endothelial growth factor. Cancer Res. 2007, 67, 10823–10830. [Google Scholar] [CrossRef]
- Arbiser, J.L.; Petros, J.; Klafter, R.; Govindajaran, B.; McLaughlin, E.R.; Brown, L.F.; Cohen, C.; Moses, M.; Kilroy, S.; Arnold, R.S.; et al. Reactive oxygen generated by Nox1 triggers the angiogenic switch. Proc. Natl. Acad. Sci. USA 2002, 99, 715–720. [Google Scholar] [CrossRef]
- Karar, J.; Maity, A. PI3K/AKT/mTOR pathway in angiogenesis. Front. Mol. Neurosci. 2011, 4, 51. [Google Scholar] [CrossRef]
- Block, K.; Gorin, Y. Aiding and abetting roles of NOX oxidases in cellular transformation. Nat. Rev. Cancer 2012, 12, 627–637. [Google Scholar] [CrossRef]
- Govindarajan, B.; Sligh, J.; Vincent, B.; Li, M.; Canter, J.; Nickoloff, B.; Rodenburg, R.; Smeitink, J.; Oberley, L.; Zhang, Y.; et al. Overexpression of Akt converts radial growth melanoma to vertical growth melanoma. J. Clin. Investig. 2007, 117, 719–729. [Google Scholar] [CrossRef] [PubMed]
- Pozzi, A.; Ibanez, M.R.; Gatica, A.E.; Yang, S.; Wei, S.; Mei, S.; Falck, J.R.; Capdevila, J.H. Peroxisomal proliferator-activated receptor-α-dependent inhibition of endothelial cell proliferation and tumorigenesis. J. Biol. Chem. 2007, 282, 17685–17695. [Google Scholar] [CrossRef] [PubMed]
- Banskota, S.; Gautam, J.; Regmi, S.C.; Gurung, P.; Park, M.H.; Kim, S.J.; Nam, T.G.; Jeong, B.S.; Kim, J.A. BJ-1108, a 6-amino- 2,4,5-trimethylpyridin-3-ol analog, inhibits serotonininduced angiogenesis and tumor growth through PI3K/NOX pathway. PLoS ONE 2016, 11, e0148133. [Google Scholar] [CrossRef] [PubMed]
- Konior, A.; Schramm, A.; Czesnikiewicz-Guzik, M.; Guzik, T.J. NADPH Oxidases in Vascular Pathology. Antioxid. Redox Signal. 2014, 20, 2794–2814. [Google Scholar] [CrossRef]
- Zhang, Y.; Murugesan, P.; Huang, K.; Cai, H. NADPH oxidases and oxidase crosstalk in cardiovascular diseases: Novel therapeutic targets. Nat. Rev. Cardiol. 2020, 17, 170–194. [Google Scholar] [CrossRef]
- Burtenshaw, D.; Hakimjavadi, R.; Redmond, E.; Cahill, P. Nox Reactive Oxygen Species and Regulation of Vascular Cell Fate. Antioxidants 2017, 6, 90. [Google Scholar] [CrossRef] [PubMed]
- de Keulenaer, G.W.; Alexander, R.W.; Ushio-Fukai, M.; Ishizaka, N.; Griendling, K.K. Tumour necrosis factor alpha activates a p22phox-based NADH oxidase in vascular smooth muscle. Biochem. J. 1998, 329, 653–657. [Google Scholar] [CrossRef] [PubMed]
- Brandes, R.P.; Viedt, C.; Nguyen, K.; Beer, S.; Kreuzer, J.; Busse, R.; Gorlach, A. Thrombin-induced MCP-1 expression involves activation of the p22phox-containing NADPH oxidase in human vascular smooth muscle cells. Thromb. Haemost. 2001, 85, 1104–1110. [Google Scholar] [PubMed]
- Jansen, F.; Yang, X.; Franklin, B.S.; Hoelscher, M.; Schmitz, T.; Bedorf, J.; Nickenig, G.; Werner, N. High glucose condition increases NADPH oxidase activity in endothelial microparticles that promote vascular inflammation. Cardiovasc. Res. 2013, 98, 94–106. [Google Scholar] [CrossRef]
- Brandes, R.P. Vascular Functions of NADPH Oxidases. Hypertension 2010, 56, 17–21. [Google Scholar] [CrossRef] [PubMed]
- Rajagopalan, S.; Kurz, S.; Münzel, T.; Tarpey, M.; Freeman, B.A.; Griendling, K.K.; Harrison, D.G. Angiotensin II-mediated hypertension in the rat increases vascular superoxide production via membrane NADH/NADPH oxidase activation. Contribution to alterations of vasomotor tone. J. Clin. Investig. 1996, 97, 1916–1923. [Google Scholar] [CrossRef]
- Guzik, T.J.; Mussa, S.; Gastaldi, D.; Sadowski, J.; Ratnatunga, C.; Pillai, R.; Channon, K.M. Mechanisms of increased vascular superoxide production in human diabetes mellitus: Role of NAD(P)H oxidase and endothelial nitric oxide synthase. Circulation 2002, 105, 1656–1662. [Google Scholar] [CrossRef] [PubMed]
- Dikalova, A.; Clempus, R.; Lassègue, B.; Cheng, G.; McCoy, J.; Dikalov, S.; Martin, A.S.; Lyle, A.; Weber, D.S.; Weiss, D.; et al. Nox1 Overexpression Potentiates Angiotensin II-Induced Hypertension and Vascular Smooth Muscle Hypertrophy in Transgenic Mice. Circulation 2005, 112, 2668–2676. [Google Scholar] [CrossRef]
- Landmesser, U.; Cai, H.; Dikalov, S.; McCann, L.; Hwang, J.; Jo, H.; Holland, S.M.; Harrison, D.G. Role of p47phox in Vascular Oxidative Stress and Hypertension Caused by Angiotensin II. Hypertension 2002, 40, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Landmesser, U.; Dikalov, S.; Price, S.R. Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J. Clin. Investig. 2003, 111, 1201–1209. [Google Scholar] [CrossRef]
- Schröder, K.; Zhang, M.; Benkhoff, S.; Mieth, A.; Pliquett, R.; Kosowski, J.; Kruse, C.; Luedike, P.; Michaelis, U.R.; Weissmann, N.; et al. Nox4 is a protective reactive oxygen species generating vascular NADPH oxidase. Circ. Res. 2012, 110, 1217–1225. [Google Scholar] [CrossRef]
- Ray, R.; Murdoch, C.E.; Wang, M.; Santos, C.X. Endothelial Nox4 NADPH Oxidase Enhances Vasodilatation and Reduces Blood Pressure In Vivo. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1368–1376. [Google Scholar] [CrossRef]
- Quesada, I.M.; Lucero, A.; Amaya, C.; Meijles, D.N.; Cifuentes, M.E.; Pagano, P.J.; Castro, C. Selective inactivation of NADPH oxidase 2 causes regression of vascularization and the size and stability of atherosclerotic plaques. Atherosclerosis 2015, 242, 469–475. [Google Scholar] [CrossRef]
- Khatri, J.J.; Johnson, C.; Magid, R.; Lessner, S.M. Vascular Oxidant Stress Enhances Progression and Angiogenesis of Experimental Atheroma. Circulation 2004, 109, 520–525. [Google Scholar] [CrossRef]
- Meyer, J.W.; Schmitt, M.E. A central role for the endothelial NADPH oxidase in atherosclerosis. FEBS Lett. 2000, 472, 1–4. [Google Scholar] [CrossRef]
- Hink, U.; Li, H.; Mollnau, H.; Oelze, M.; Matheis, E.; Hartmann, M.; Skatchkov, M.; Thaiss, F.; Stahl, R.A.; Warnholtz, A.; et al. Mechanisms Underlying Endothelial Dysfunction in Diabetes Mellitus. Circ. Res. 2001, 8, E14–E22. [Google Scholar] [CrossRef]
- Guzik, T.J.; West, N.E.J.; Black, E.; McDonald, D.; Ratnatunga, C.; Pillai, R.; Channon, K.M. Vascular Superoxide Production by NAD(P)H Oxidase. Association With Endothelial Dysfunction and Clinical Risk Factors. Circ. Res. 2000, 86, e85–e90. [Google Scholar] [PubMed]
- Ding, H.; Hashem, M.; Triggle, C. Increased oxidative stress in the streptozotocin-induced diabetic apoE-deficient mouse: Changes in expression of NADPH oxidase subunits and eNOS. Eur. J. Pharm. 2007, 561, 121–128. [Google Scholar] [CrossRef]
- Liang, C.F.; Liu, J.T.C.; Wang, Y.; Xu, A.; Vanhoutte, P.M. Toll-Like Receptor 4 Mutation Protects Obese Mice Against Endothelial Dysfunction by Decreasing NADPH Oxidase Isoforms 1 and 4. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 777–784. [Google Scholar] [CrossRef]
- Gray, S.P.; di Marco, E.; Okabe, J.; Szyndralewiez, C.; Heitz, F.; Montezano, A.C.; de Haan, J.B.; Koulis, C.; El-Osta, A.; Andrews, K.L.; et al. NADPH Oxidase 1 Plays a Key Role in Diabetes Mellitus–Accelerated Atherosclerosis. Circulation 2013, 127, 1888–1902. [Google Scholar] [CrossRef]
- Augsburger, F.; Filippova, A.; Rasti, D.; Seredenina, T.; Lam, M.; Maghzal, G.; Mahiout, Z.; Jansen-Dürr, P.; Knaus, U.G.; Doroshow, J.; et al. Pharmacological characterization of the seven human NOX isoforms and their inhibitors. Redox Biol. 2019, 26, 101272. [Google Scholar] [CrossRef] [PubMed]
- Schröder, K.; Wandzioch, K.; Helmcke, I.; Brandes, R.P. Nox4 Acts as a Switch Between Differentiation and Proliferation in Preadipocytes. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Barton, M.; Haas, E.; Bhattacharya, I. Getting Radical About Obesity New Links Between Fat and Heart Disease. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 447–448. [Google Scholar] [CrossRef] [PubMed]
- Wautier, M.-P.; Chappey, O.; Corda, S.; Stern, D.M.; Schmidt, A.M.; Wautier, J.-L. Activation of NADPH oxidase by AGE links oxidant stress to altered gene expression via RAGE. Am. J. Physiol. Endocrinol. Metab. 2001, 280, E685–E694. [Google Scholar] [CrossRef]

























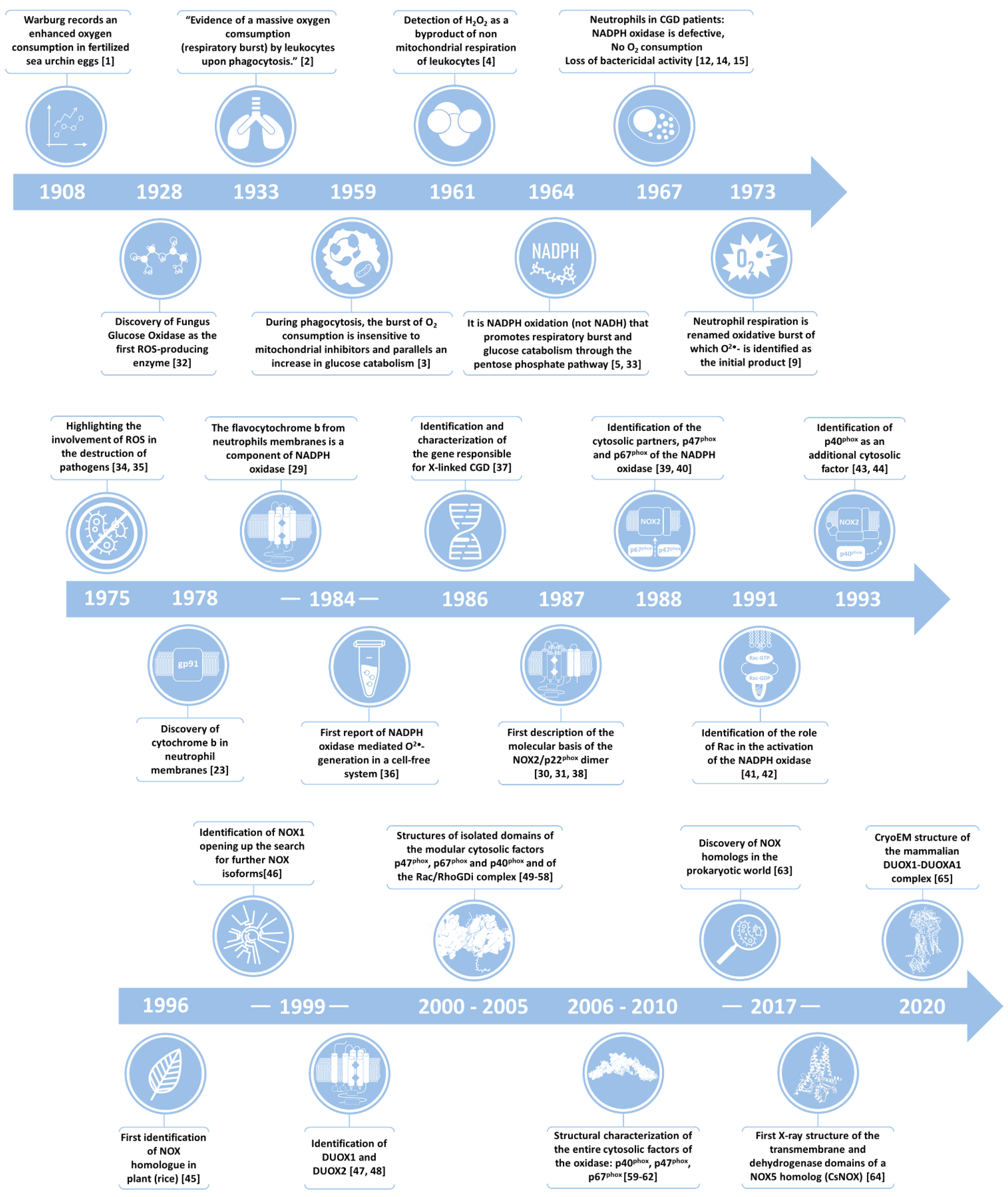{kind=link}
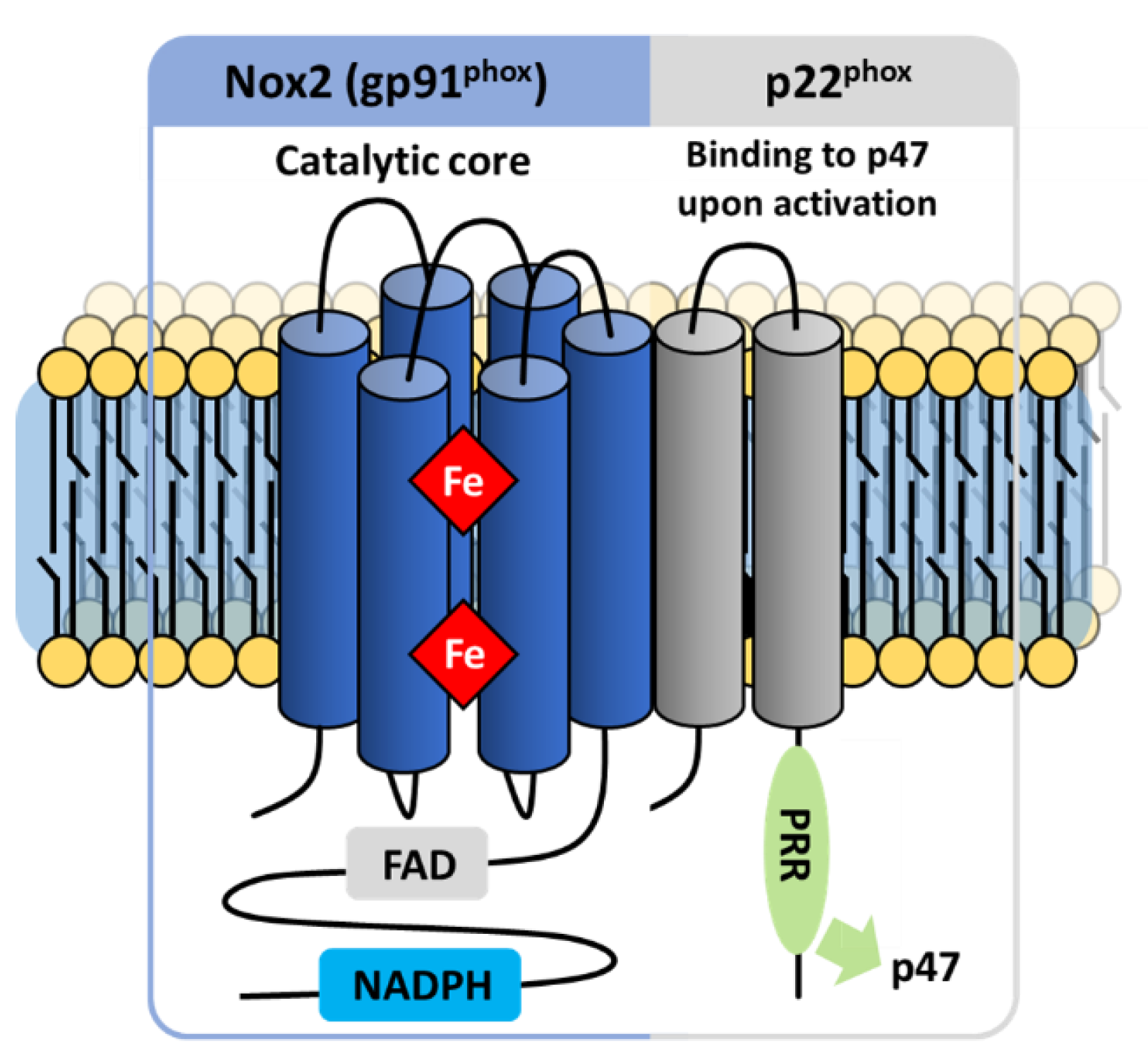{kind=link}
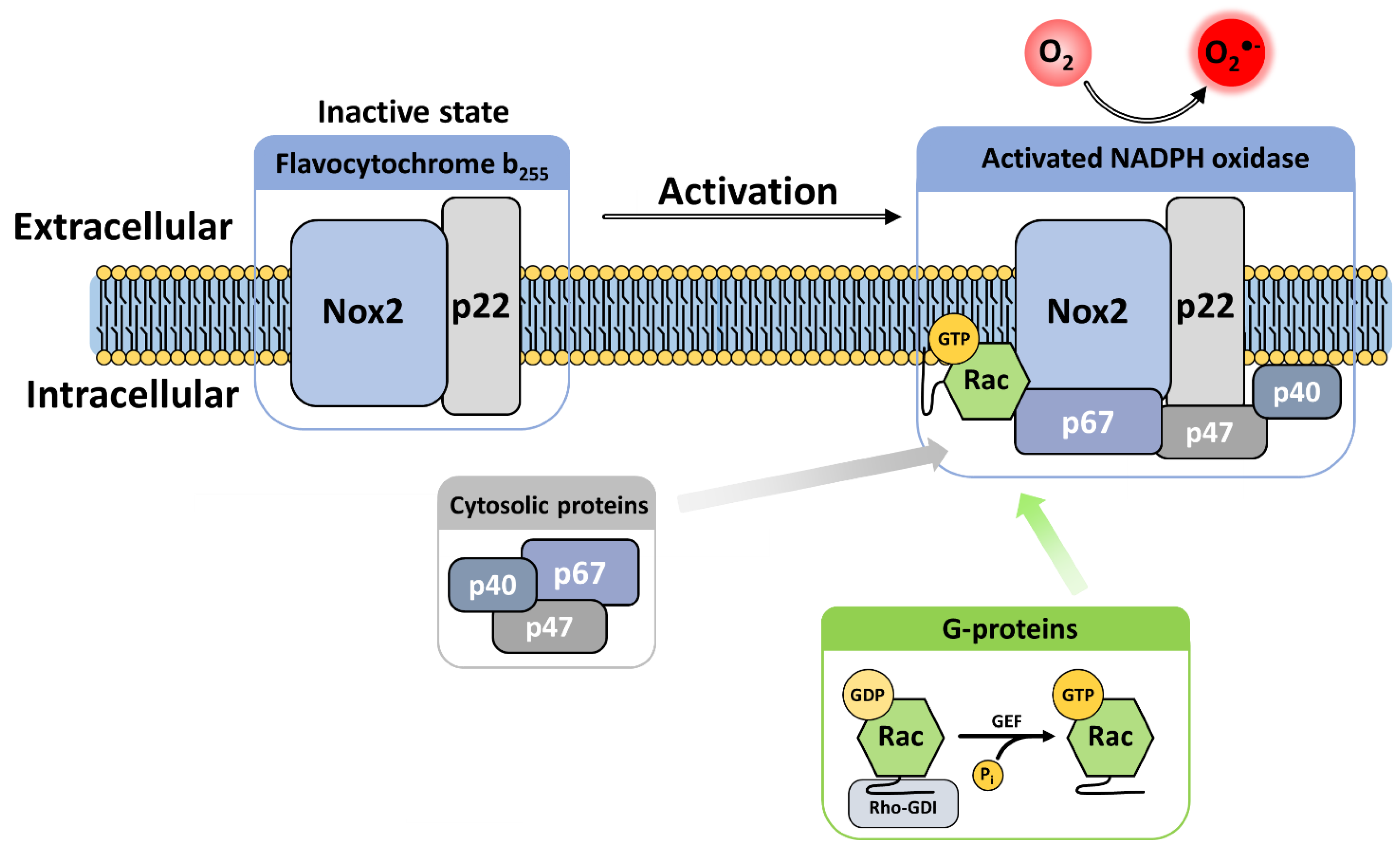{kind=link}
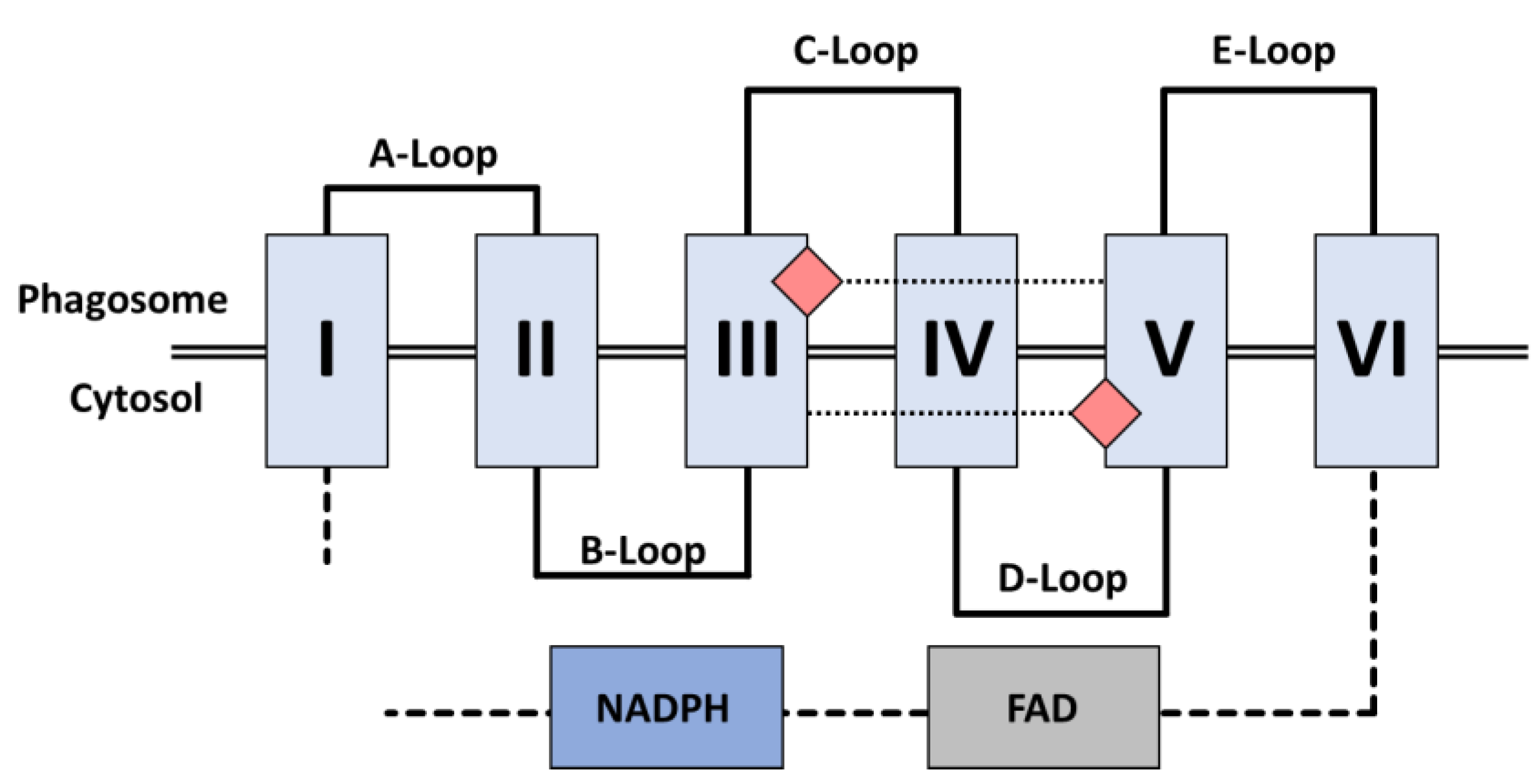{kind=link}
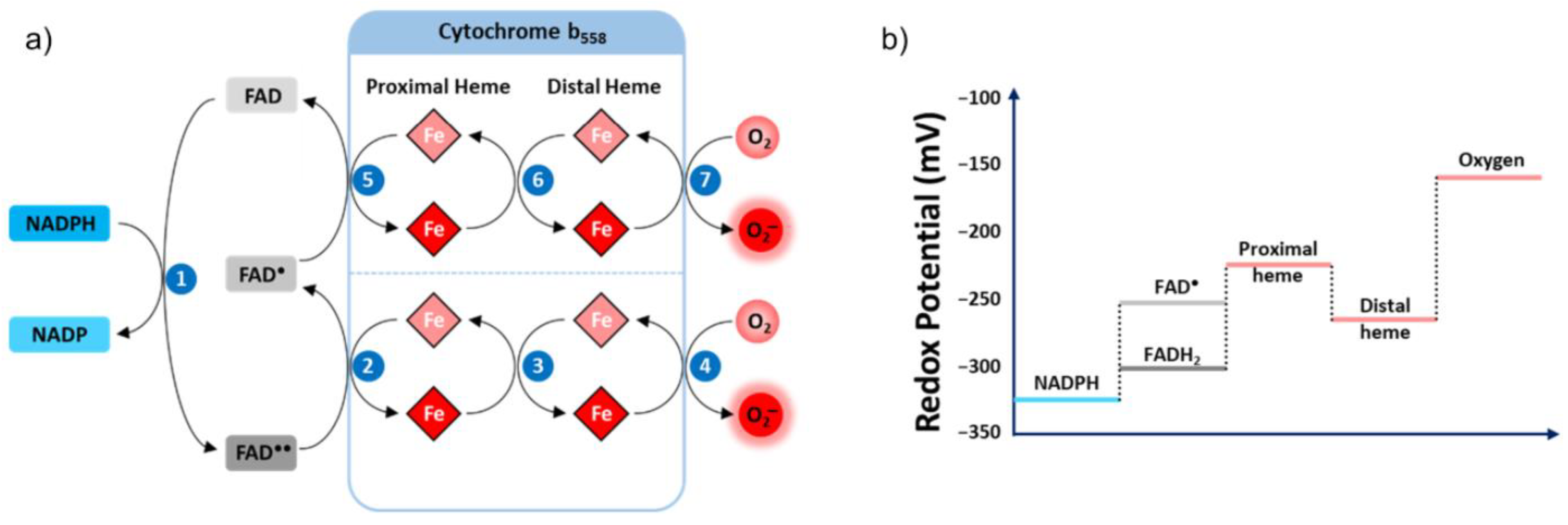{kind=link}
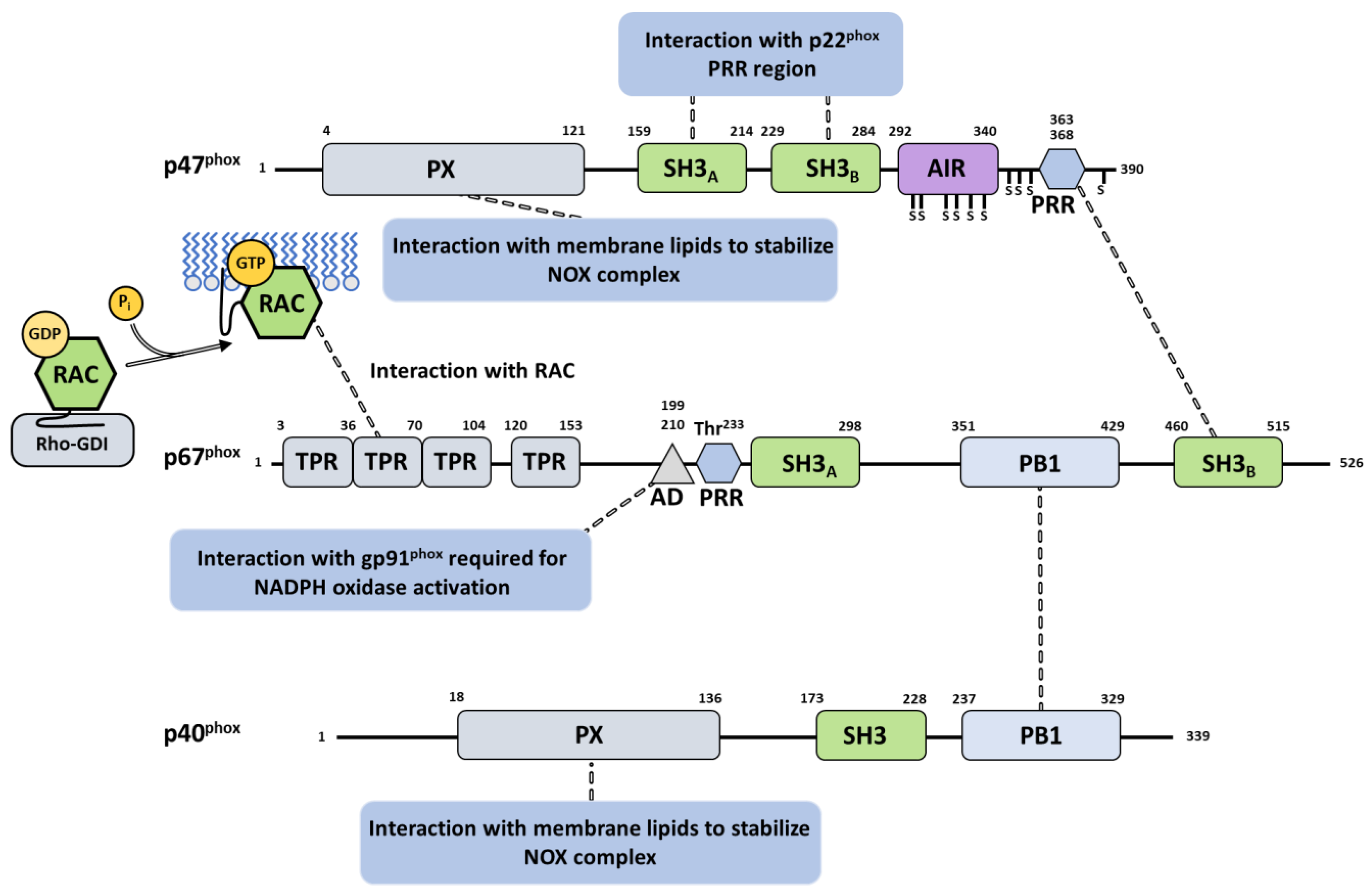{kind=link}
{kind=link}
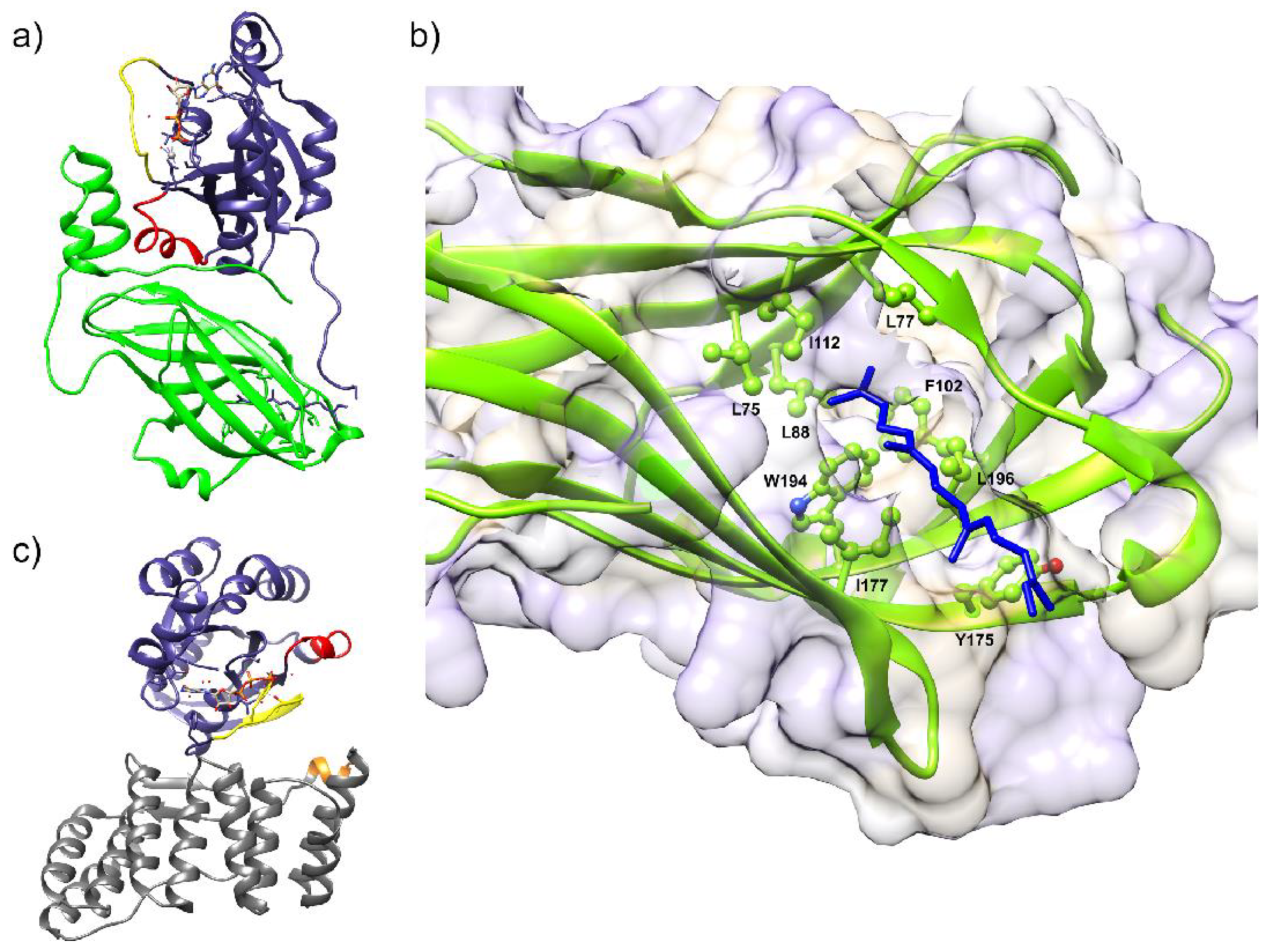{kind=link}
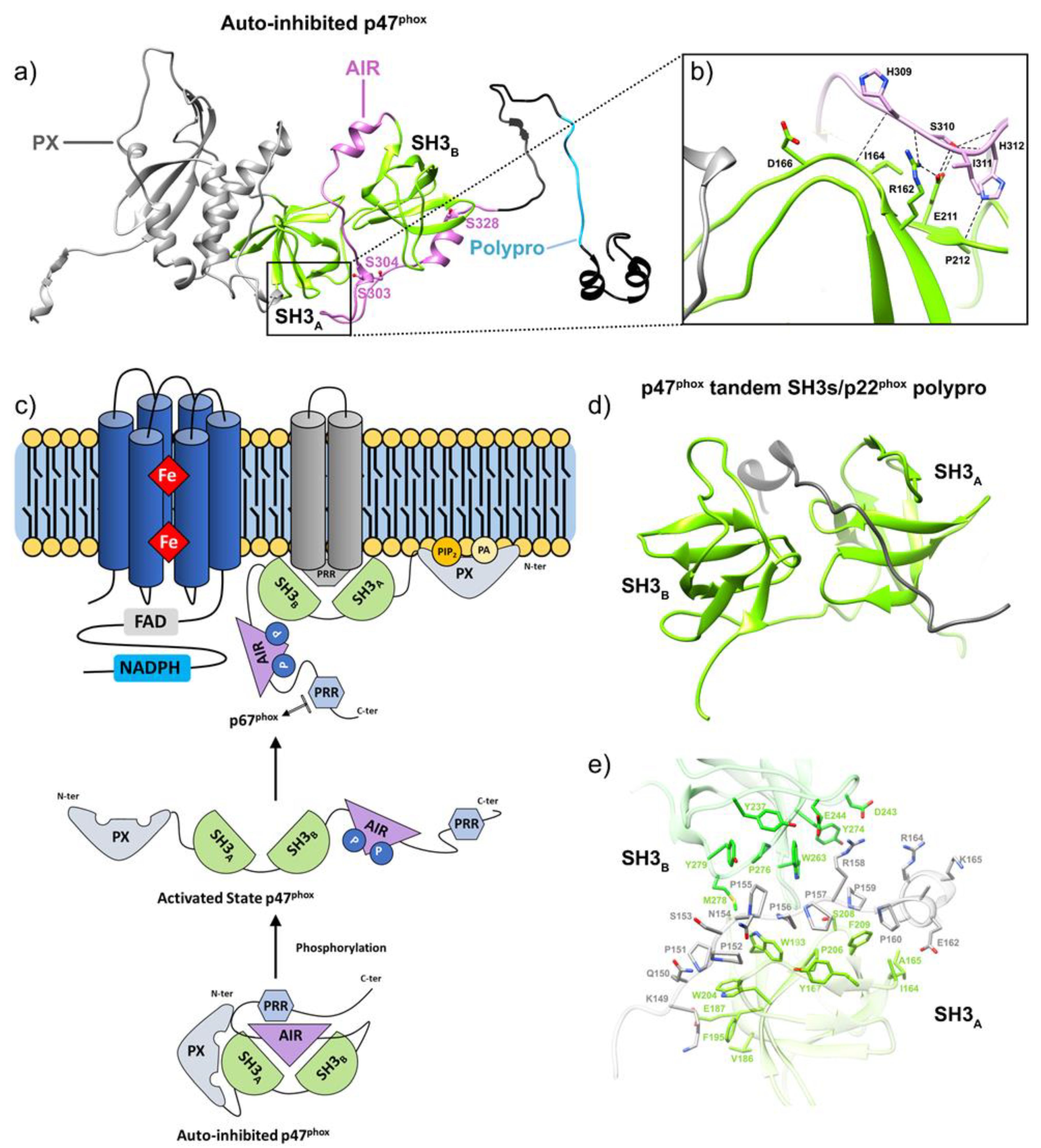{kind=link}
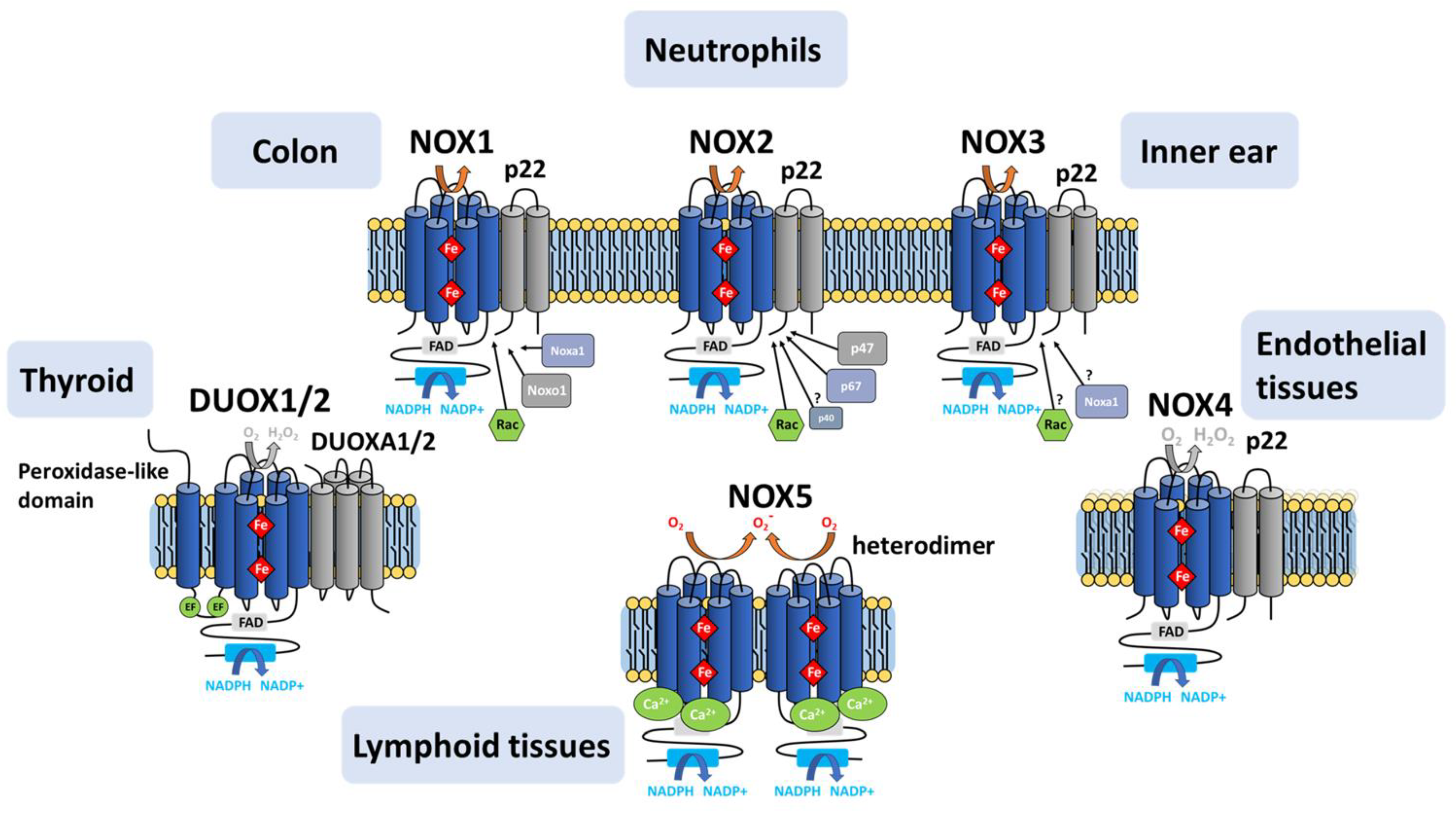{kind=link}
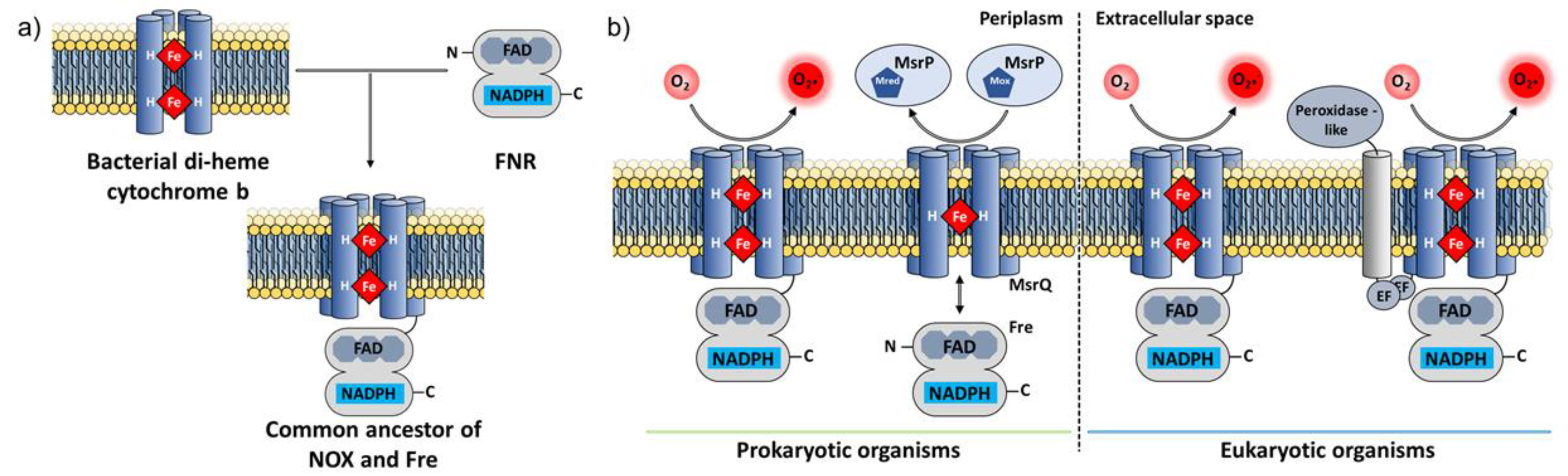{kind=link}
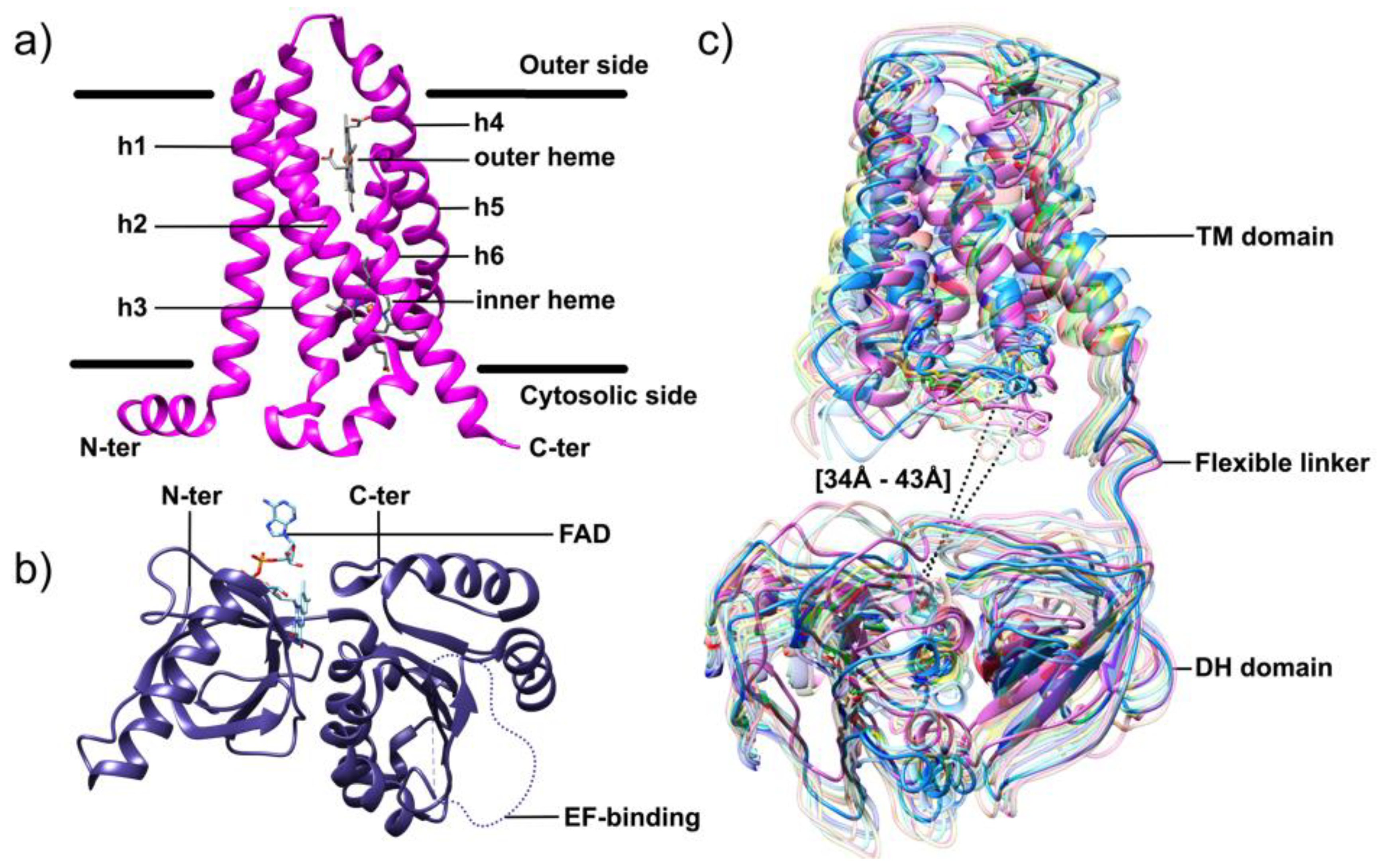{kind=link}
{kind=link}
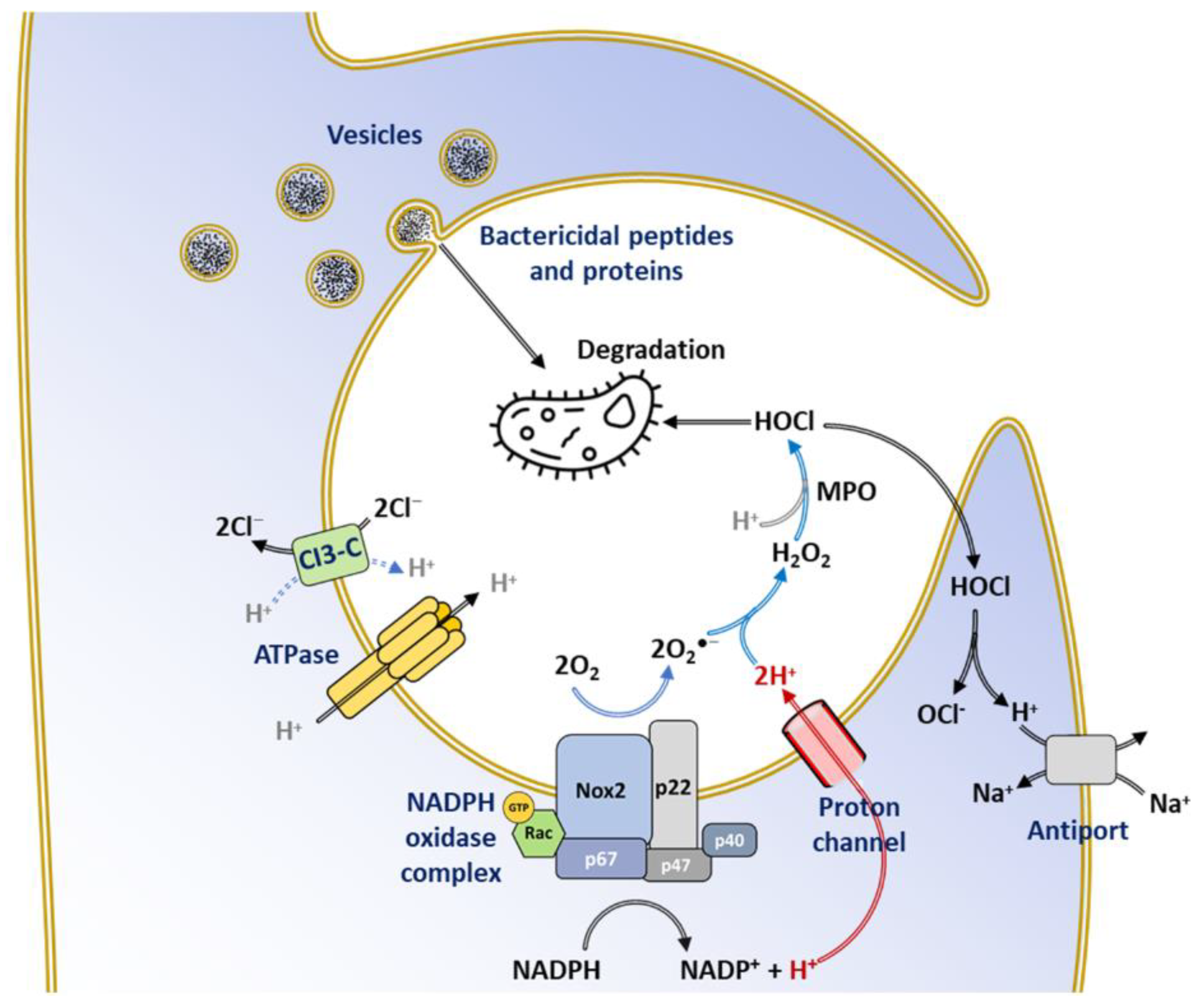{kind=link}
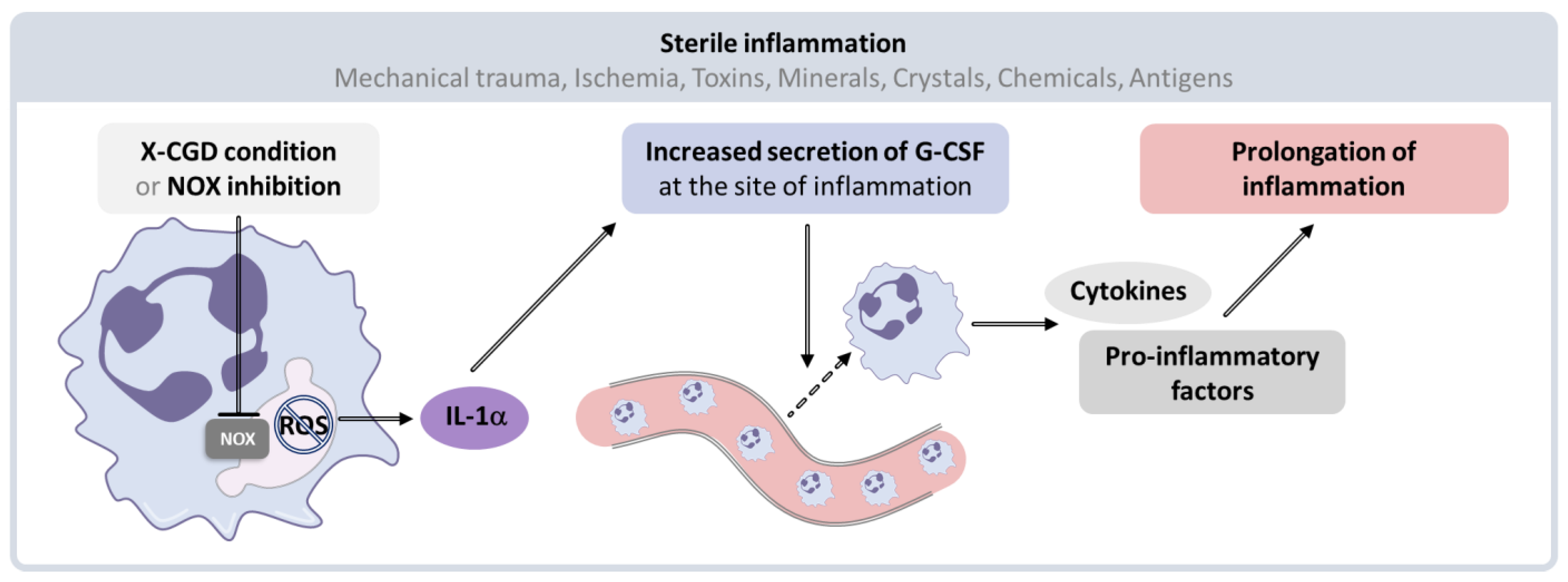{kind=link}
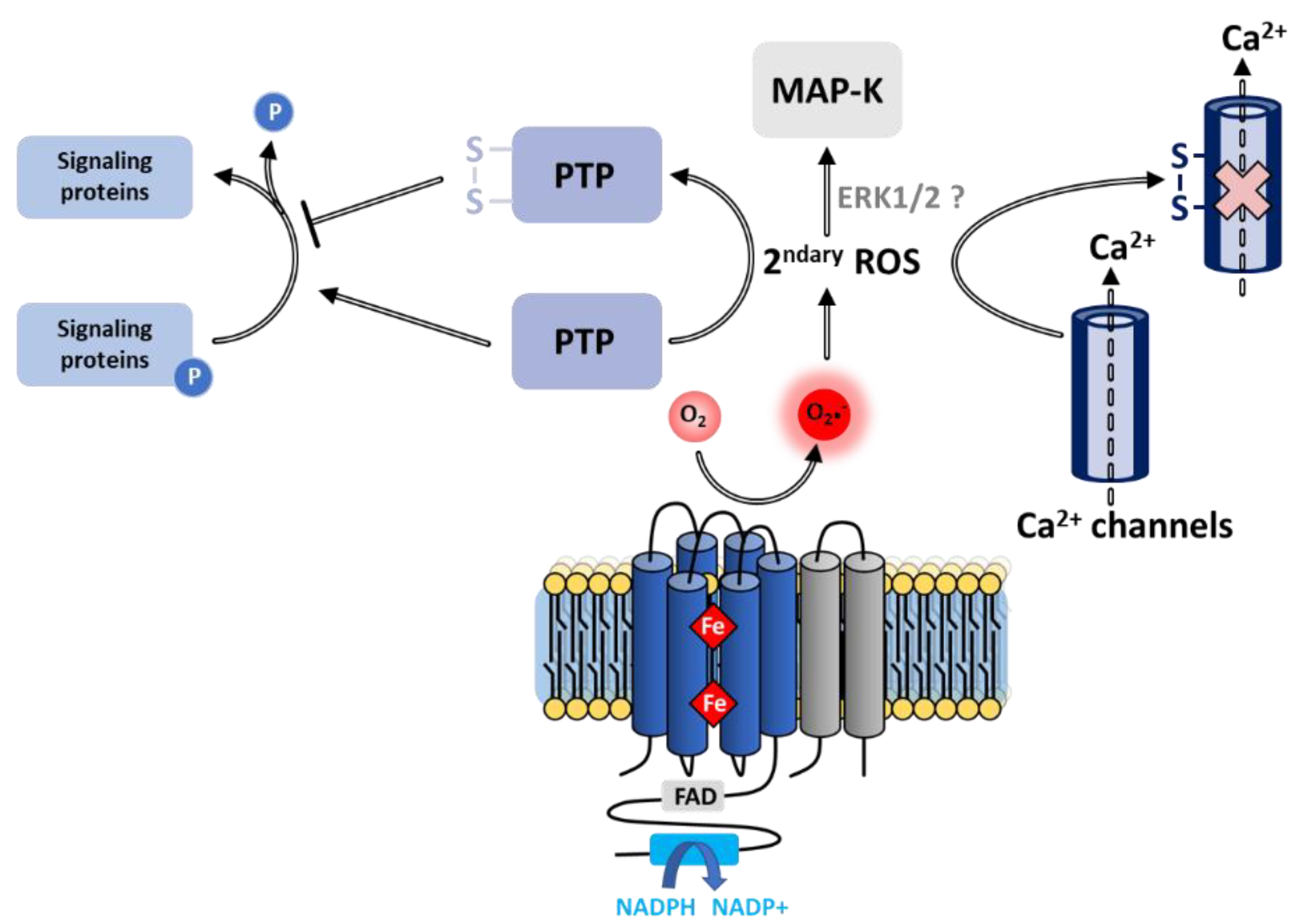{kind=link}
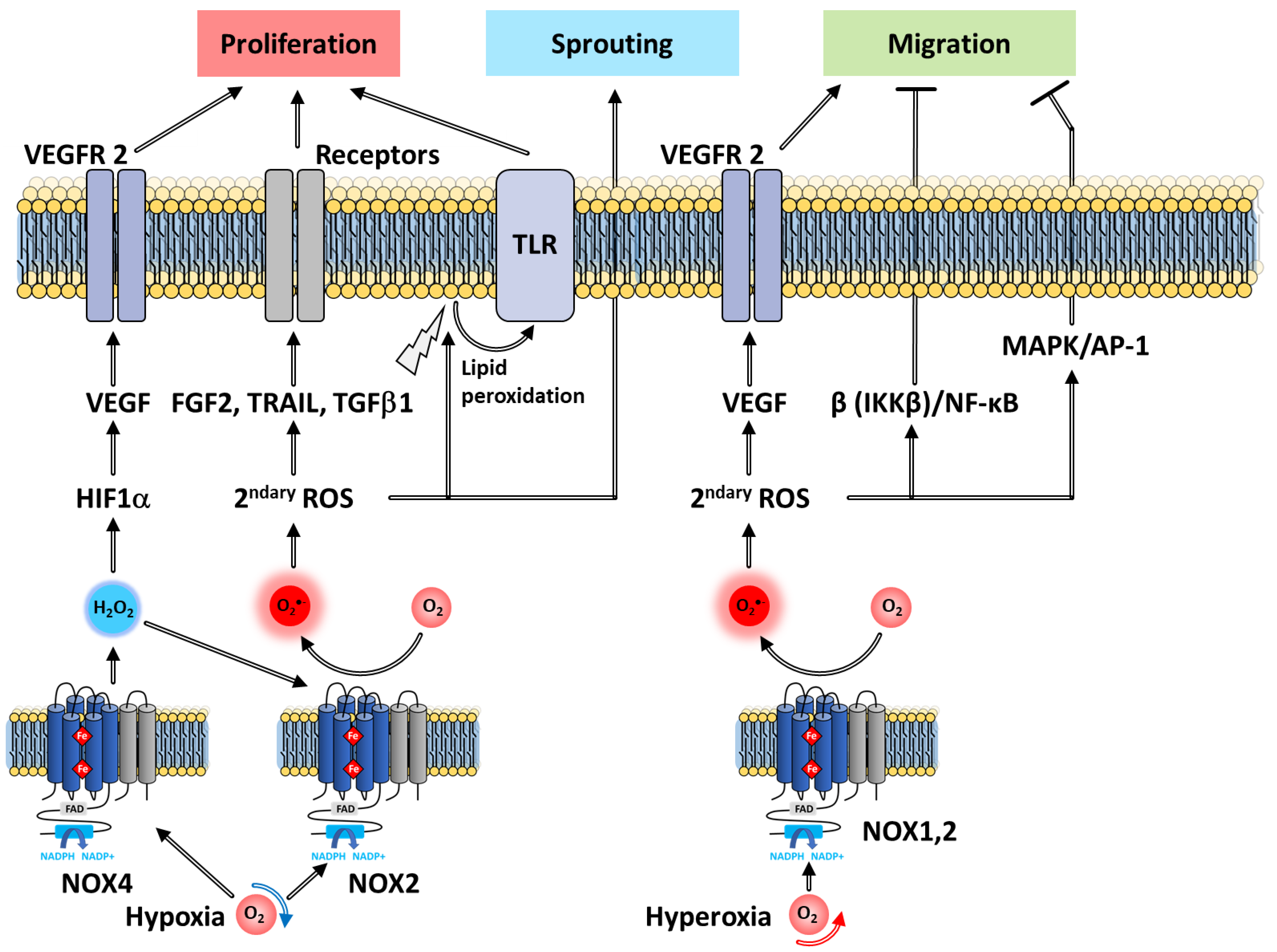{kind=link}
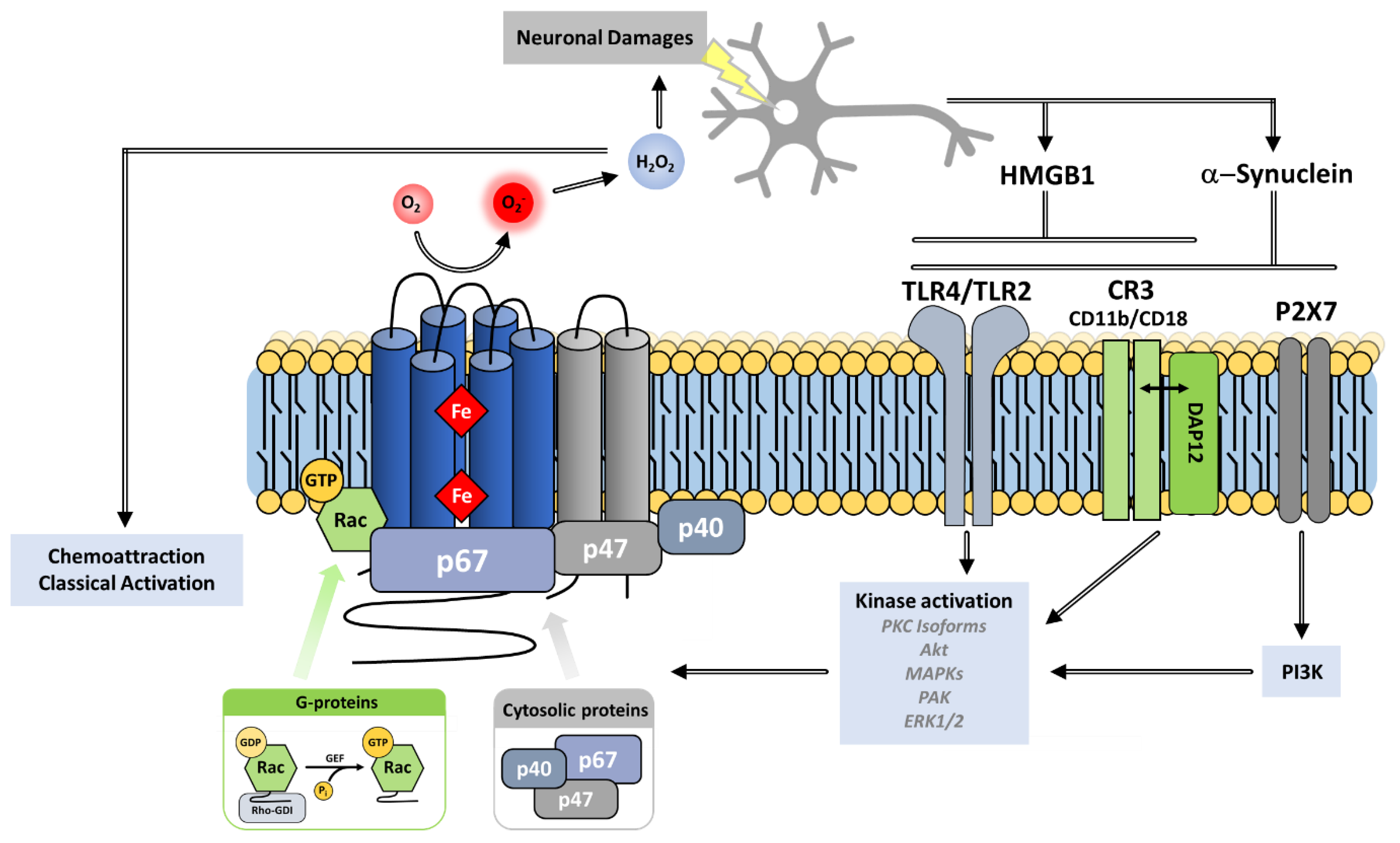{kind=link}
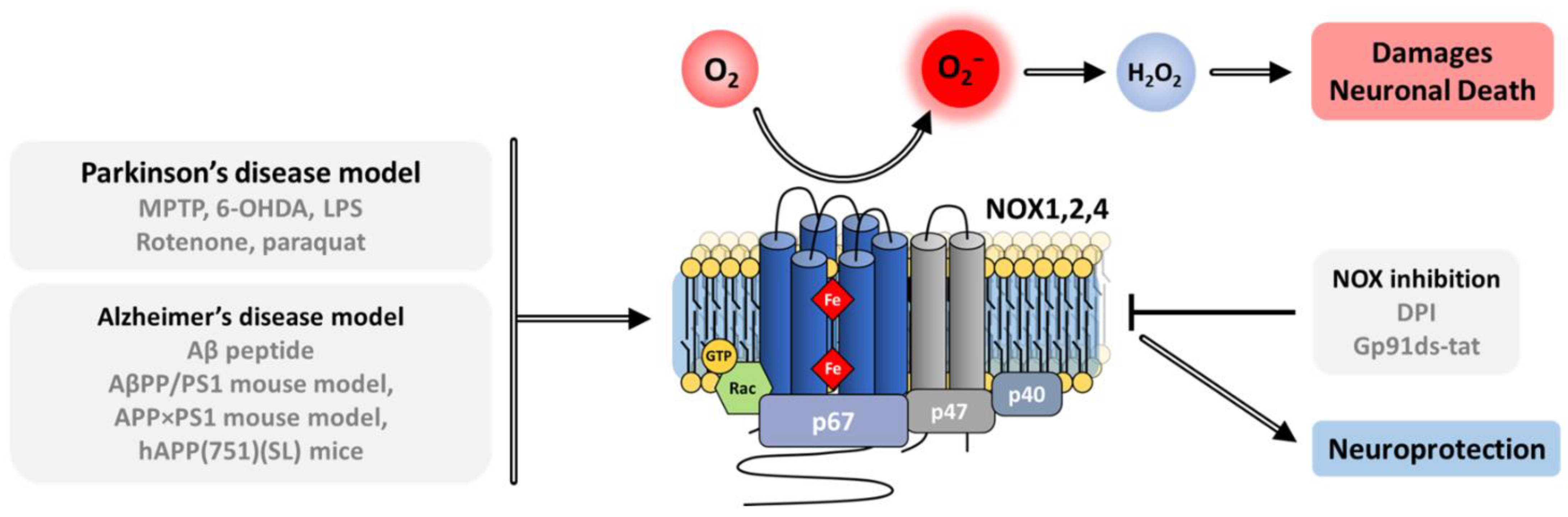{kind=link}
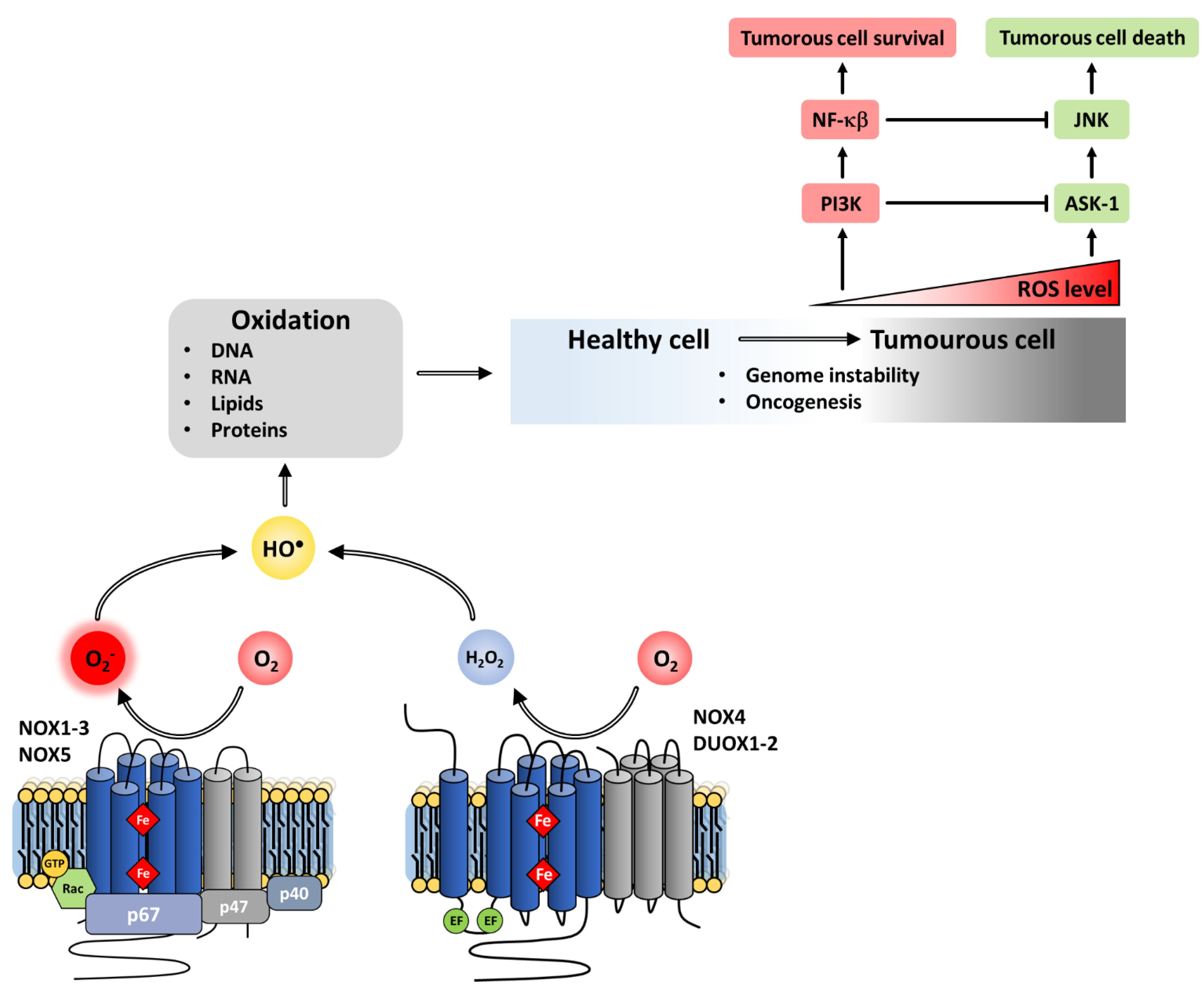{kind=link}
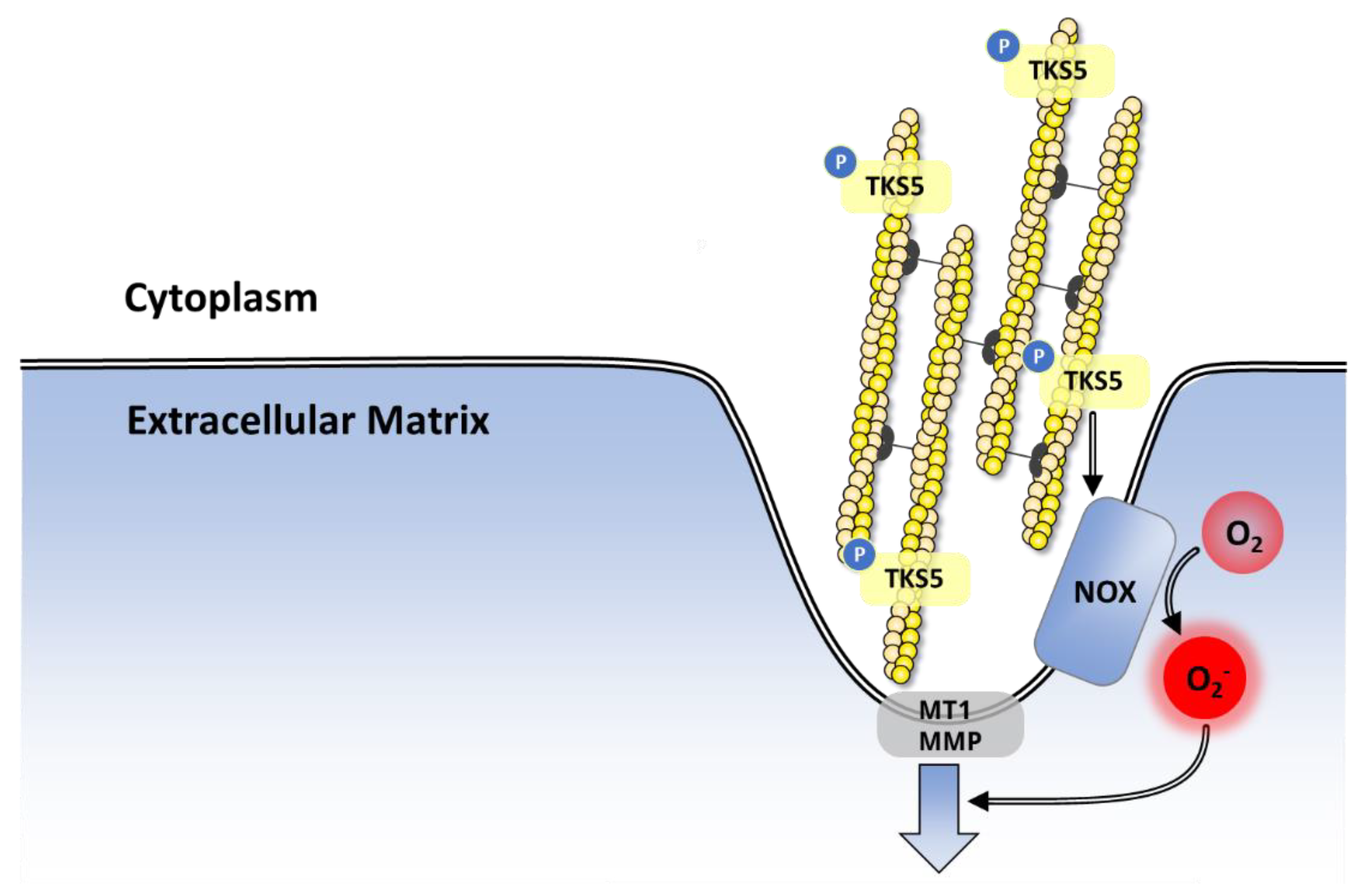{kind=link}
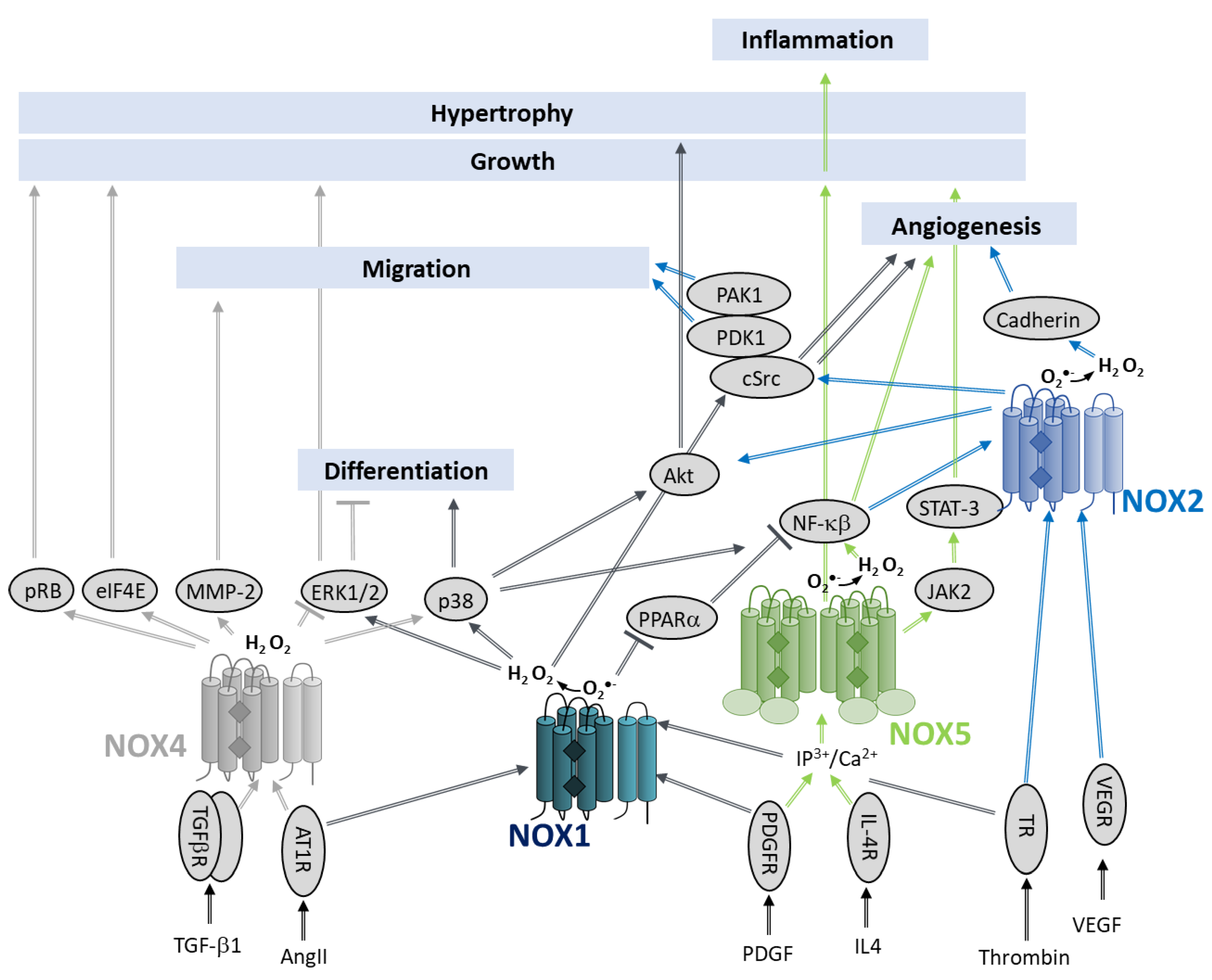{kind=link}
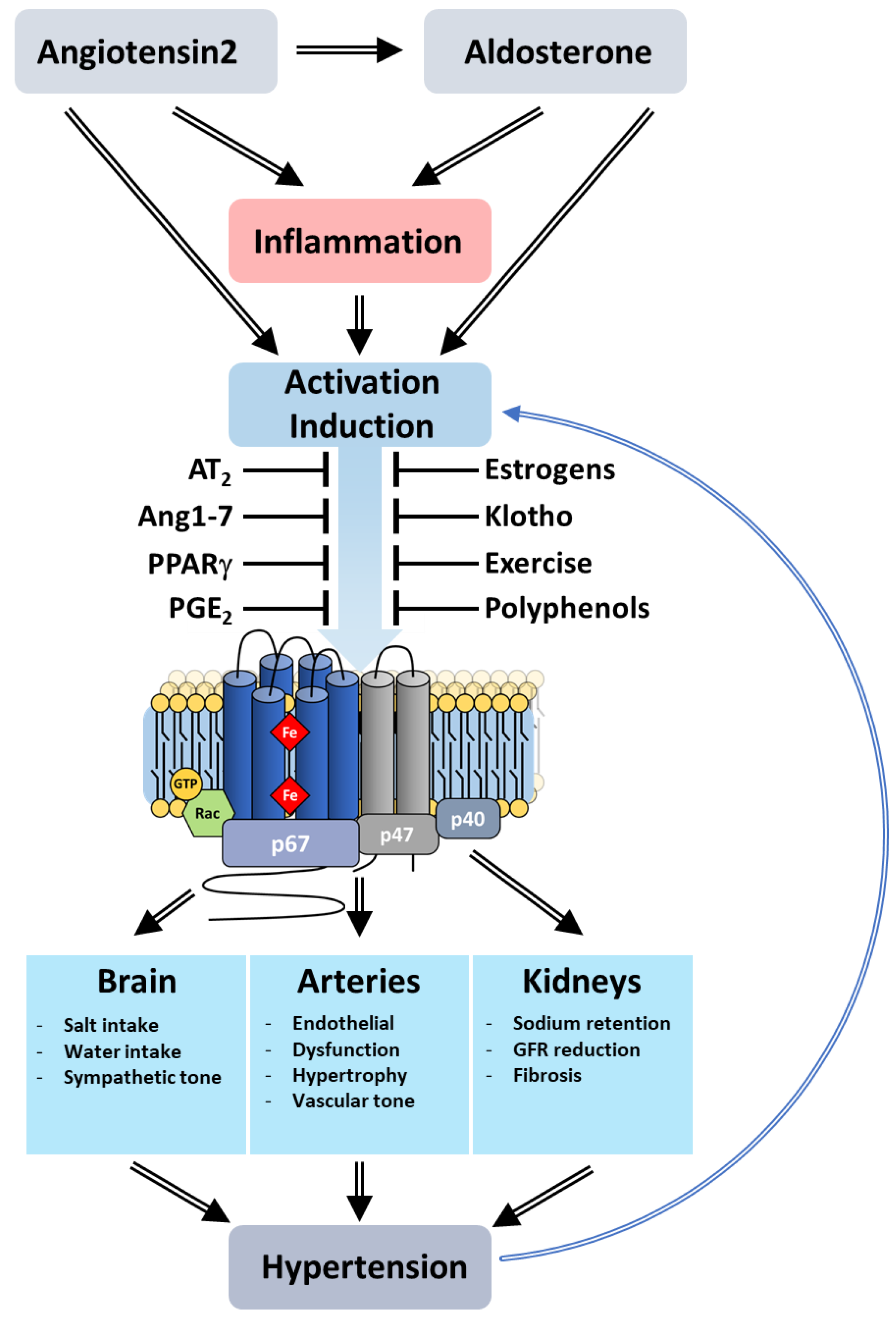{kind=link}
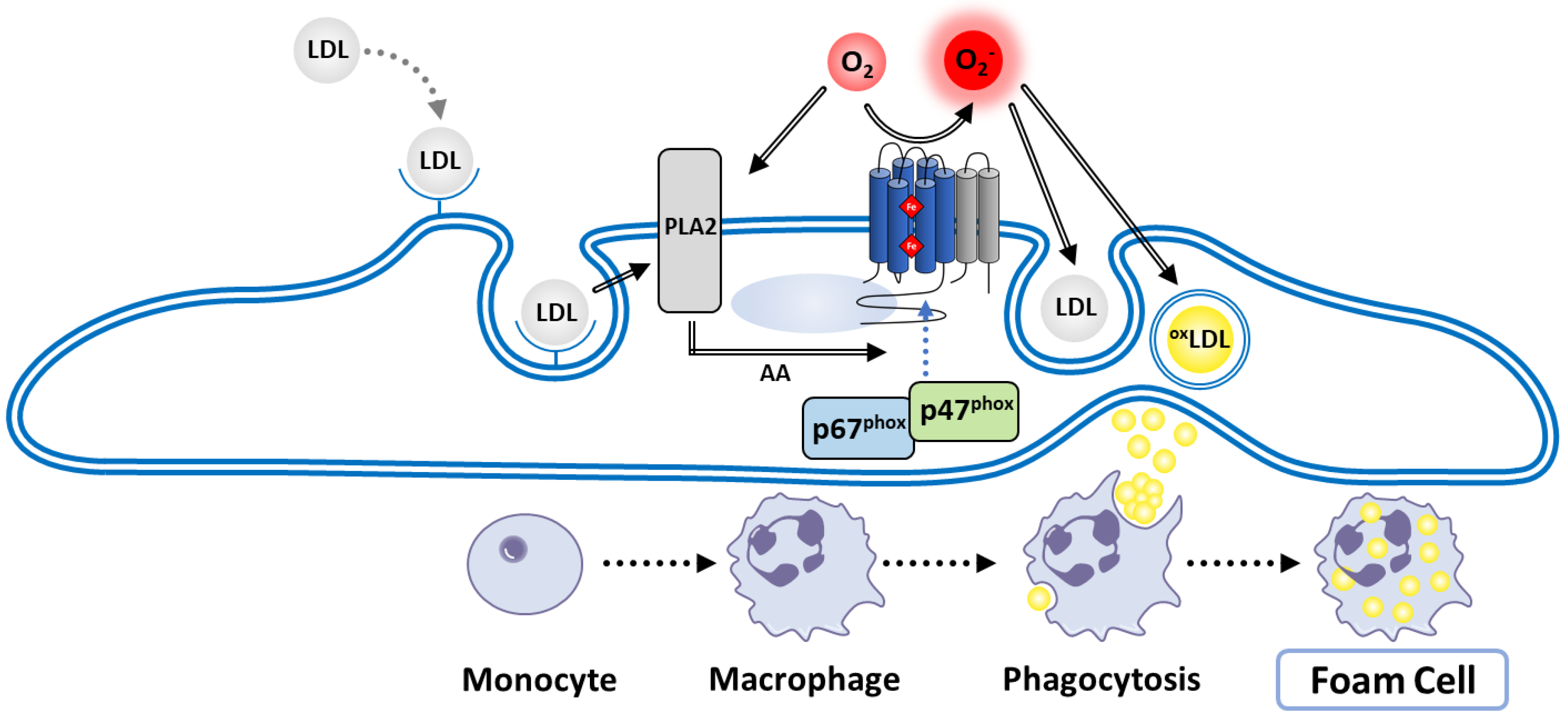{kind=link}
| NADPH Oxidase |
|---|
| O2 consumption |
| Cyano-resistant |
| Dependent on FAD and NADPH |
| Presence of heme groups |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vermot, A.; Petit-Härtlein, I.; Smith, S.M.E.; Fieschi, F. NADPH Oxidases (NOX): An Overview from Discovery, Molecular Mechanisms to Physiology and Pathology. Antioxidants 2021, 10, 890. https://doi.org/10.3390/antiox10060890
Vermot A, Petit-Härtlein I, Smith SME, Fieschi F. NADPH Oxidases (NOX): An Overview from Discovery, Molecular Mechanisms to Physiology and Pathology. Antioxidants. 2021; 10(6):890. https://doi.org/10.3390/antiox10060890
Chicago/Turabian StyleVermot, Annelise, Isabelle Petit-Härtlein, Susan M. E. Smith, and Franck Fieschi. 2021. "NADPH Oxidases (NOX): An Overview from Discovery, Molecular Mechanisms to Physiology and Pathology" Antioxidants 10, no. 6: 890. https://doi.org/10.3390/antiox10060890
APA StyleVermot, A., Petit-Härtlein, I., Smith, S. M. E., & Fieschi, F. (2021). NADPH Oxidases (NOX): An Overview from Discovery, Molecular Mechanisms to Physiology and Pathology. Antioxidants, 10(6), 890. https://doi.org/10.3390/antiox10060890