
17-? Estradiol Rescued Immature Rat Brain against Glutamate-Induced Oxidative Stress and Neurodegeneration via Regulating Nrf2/HO-1 and MAP-Kinase Signaling Pathway


Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
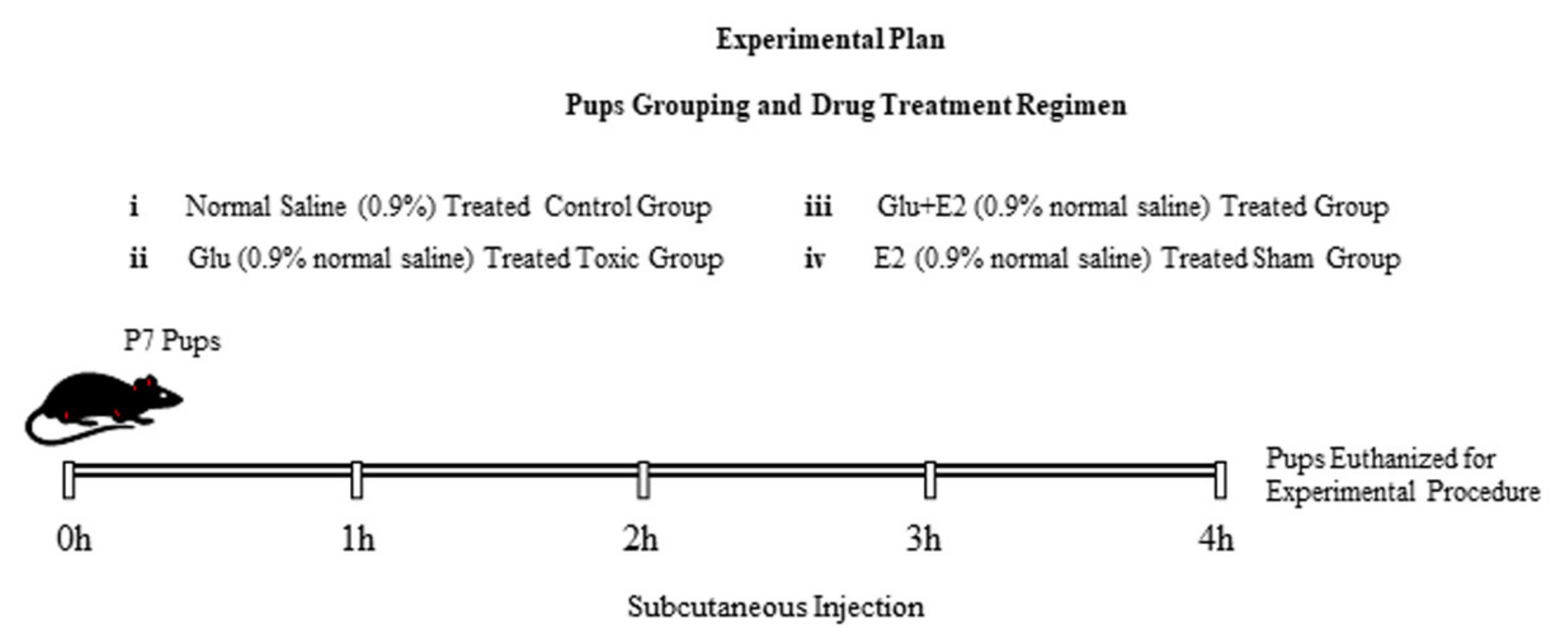
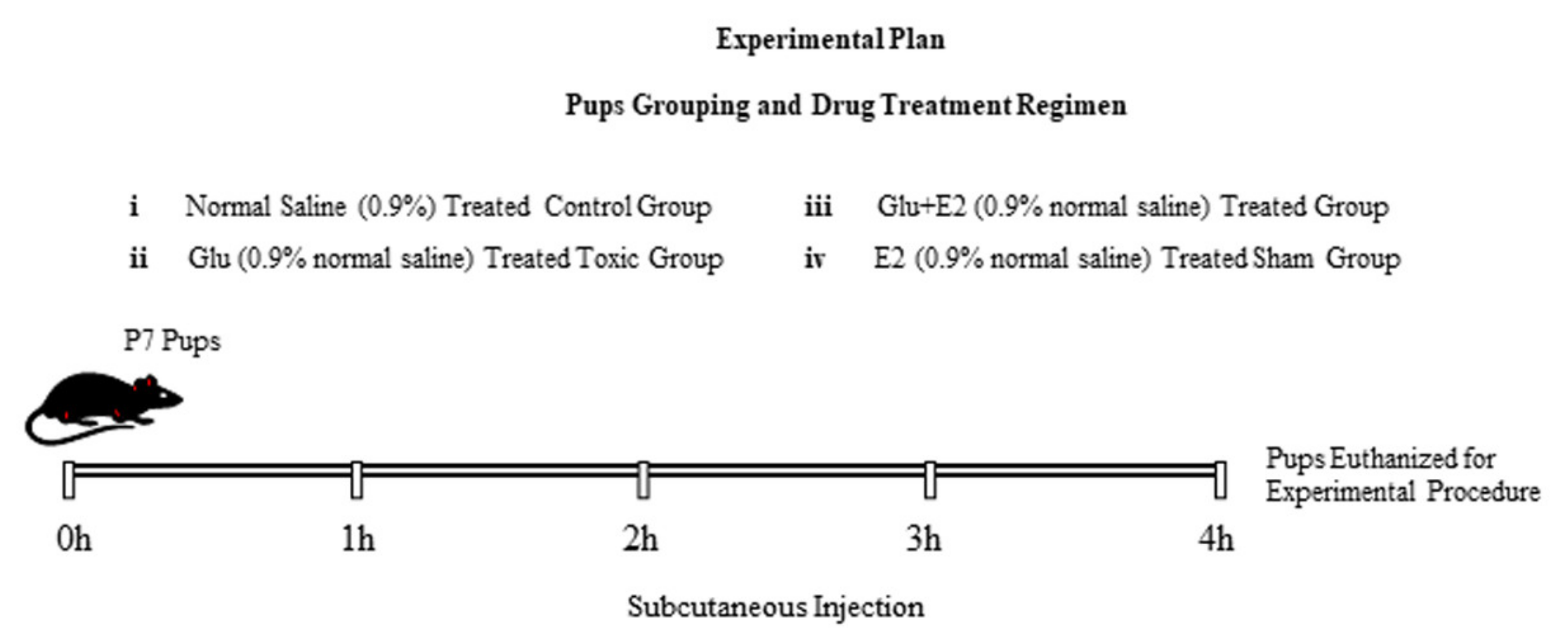
2.2. Animals and Drug Treatment
- (1)
- Control group (treated subcutaneously with 0.9% saline as a vehicle)
- (2)
- Glu group (treated subcutaneously with 10 mg/kg of glutamate)
- (3)
- Glu + E2 group (co-treated subcutaneously with 10 mg/kg of glutamate plus 10 mg/kg of 17β-estradiol).
- (4)
- E2 group (treated subcutaneously with 10 mg/kg of 17β-estradiol)
2.3. Brain Tissue Collection/Sample Preparation
2.4. Western Blot Analysis
2.5. Immunofluorescence
2.6. Antibodies
2.7. GSH Assays
2.8. Reactive Oxygen Species (ROS) Assay
2.9. Lipid Peroxidation (LPO) Assay
2.10. Statistical Analyses
3. Results
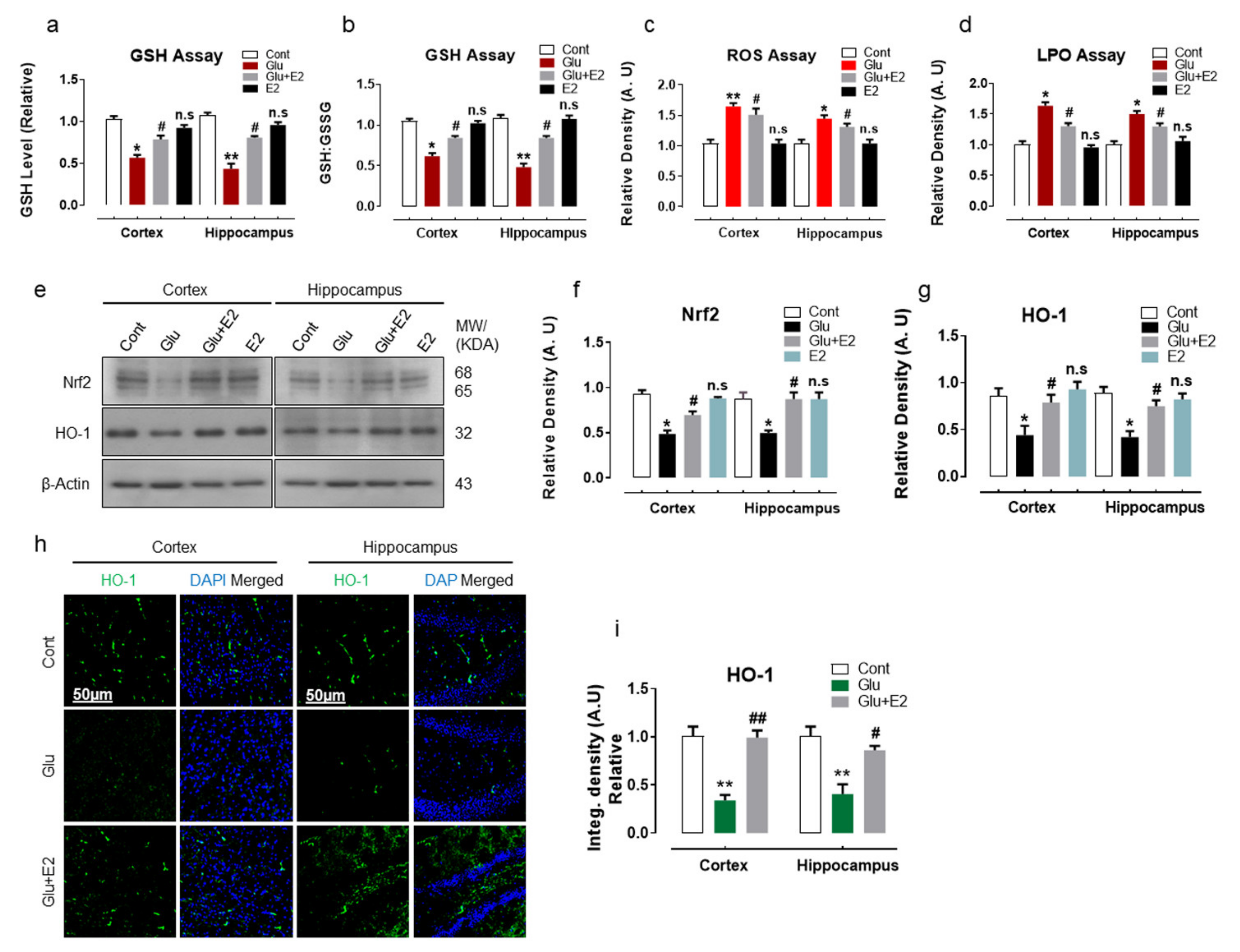
3.1. 17β-Estradiol Reduced Glutamate-Induced Oxidative Stress by Activating Nrf2/HO-1 Pathway and Enhanced Cellular Glutathione Stores in Postnatal Rat Brain
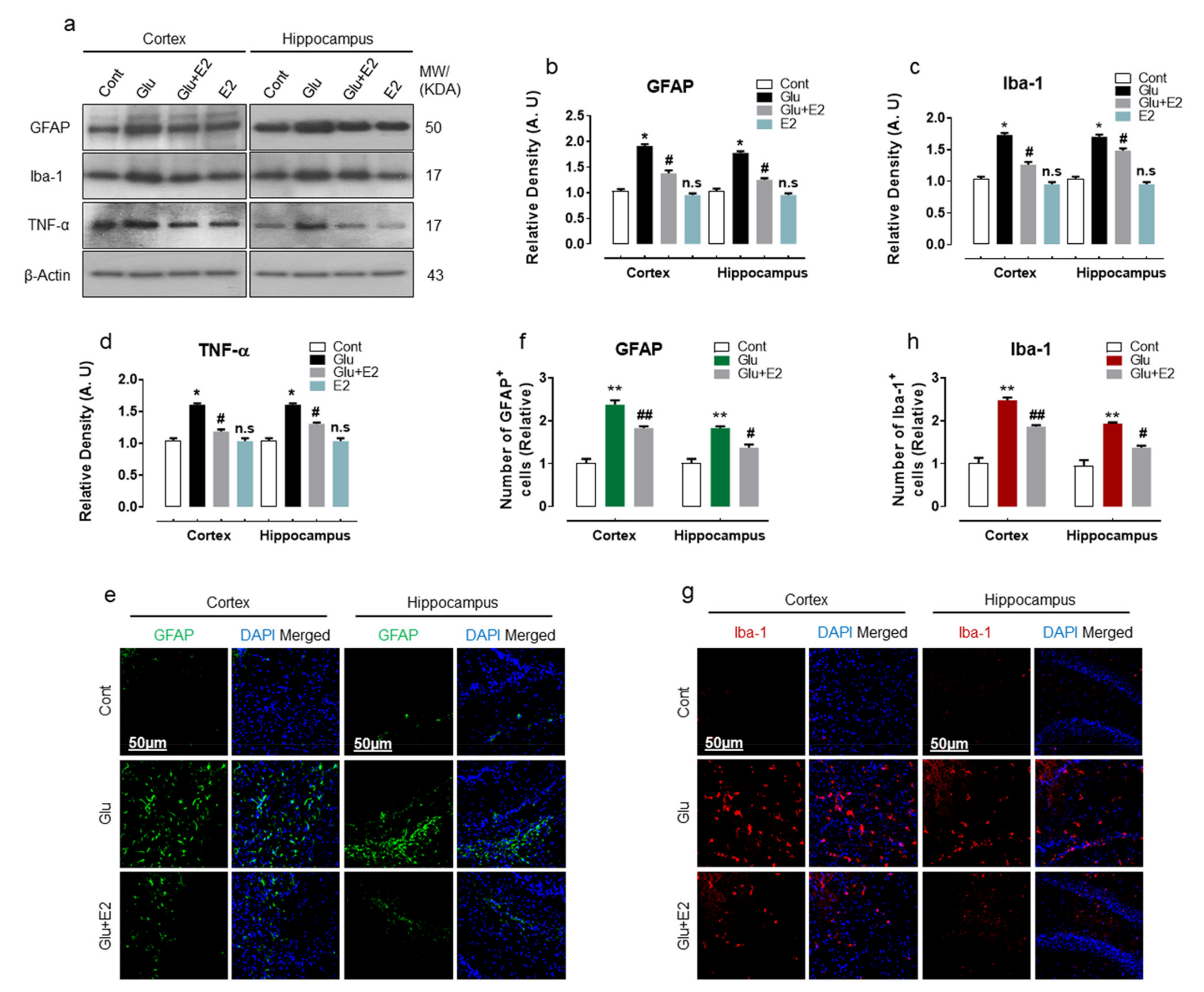
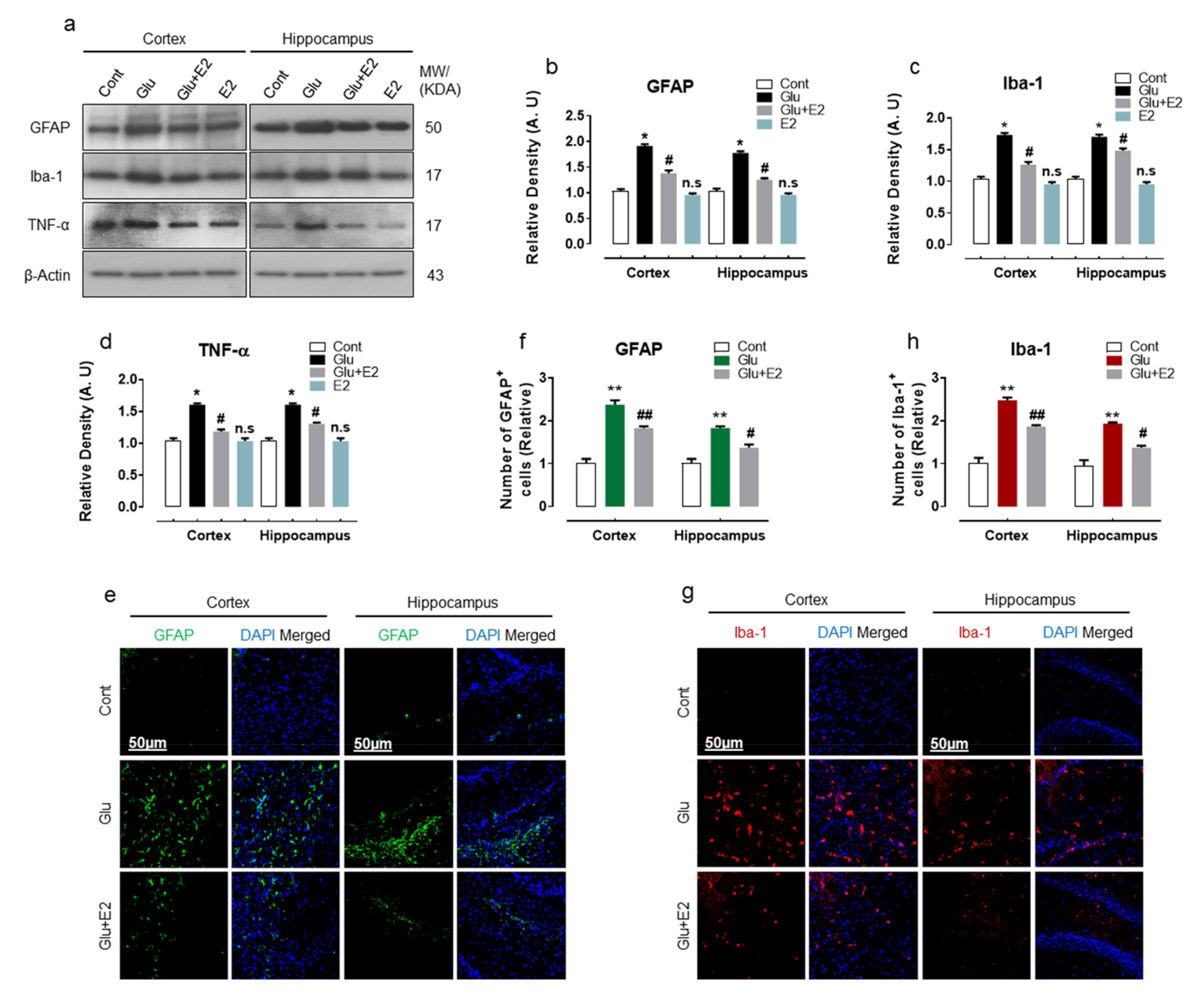
3.2. 17β-Estradiol Alleviates Glutamate Induced Neuroinflammation in Developing Rat Brain
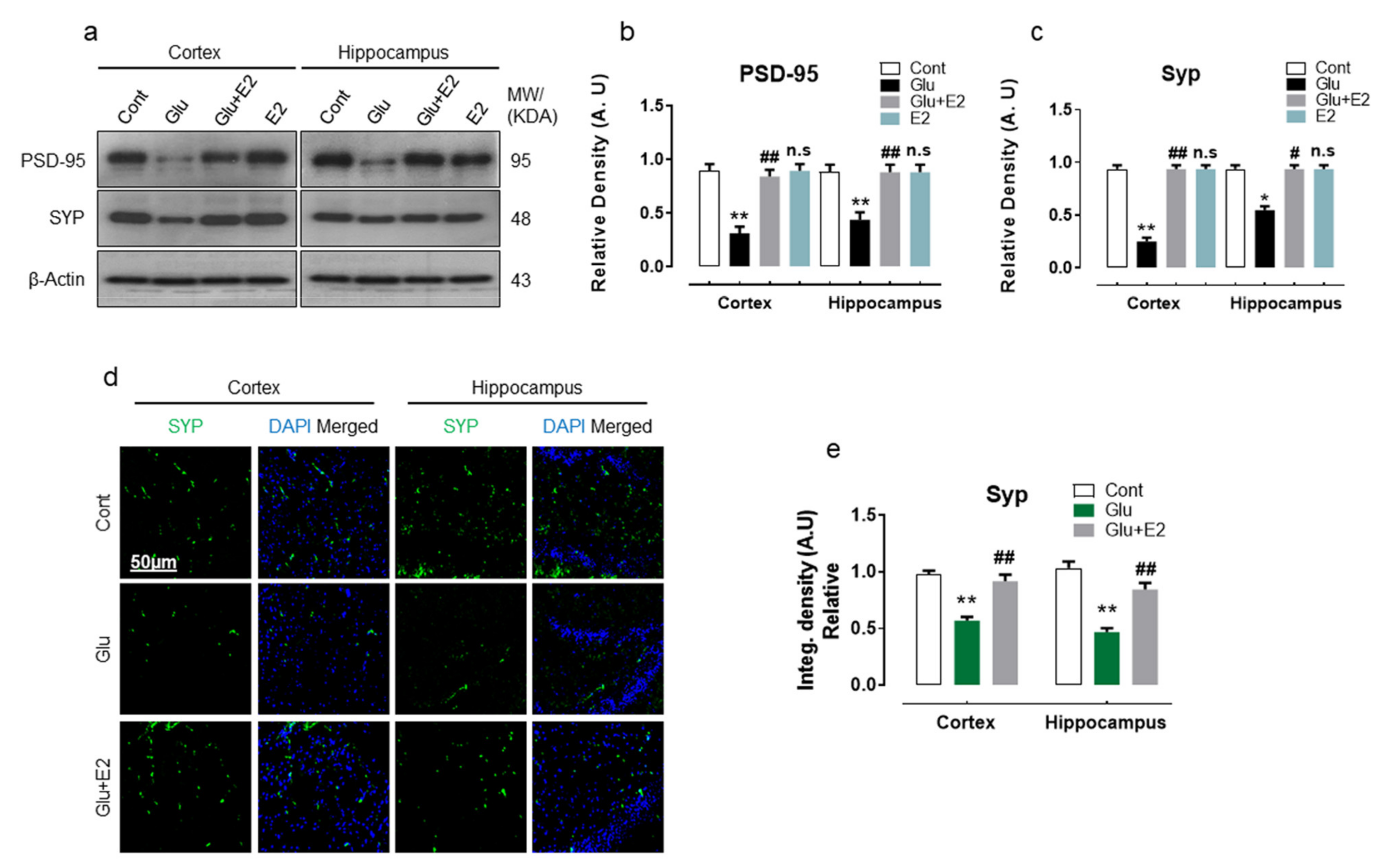
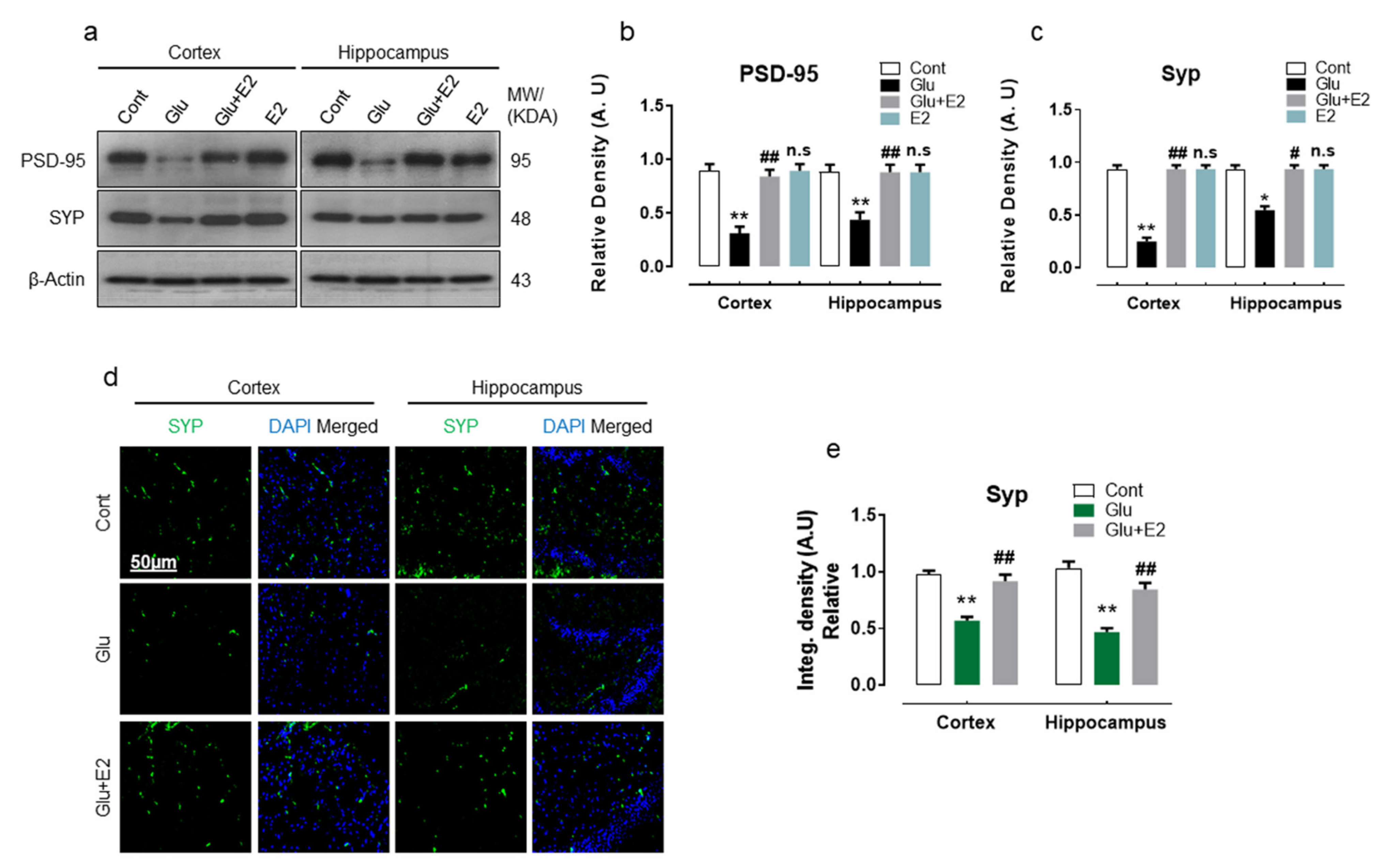
3.3. 17β-Estradiol Abrogates Glutamate-Induced Synaptic Dysfunction in Postnatal Rat Brain
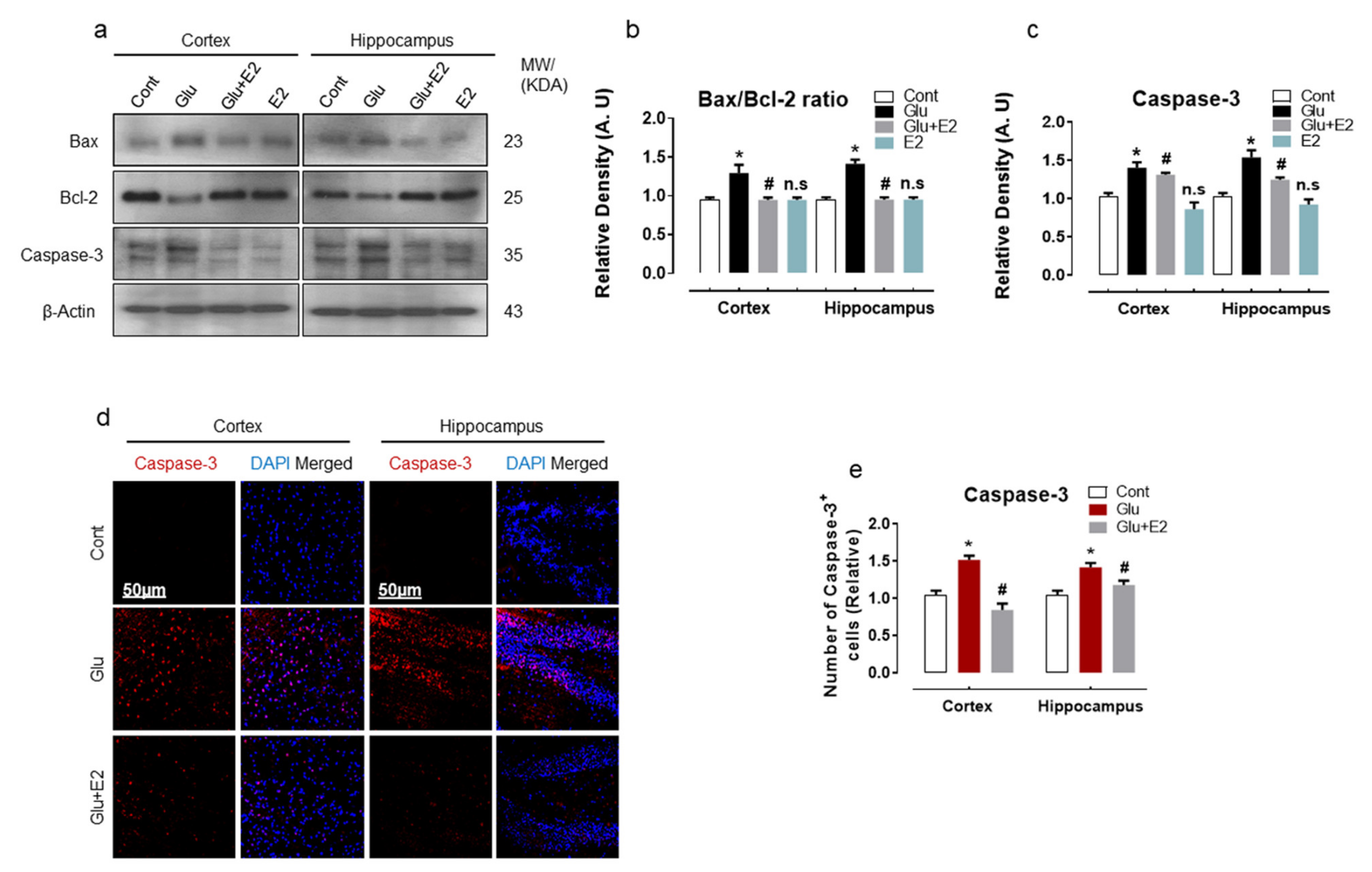
3.4. 17β-Estradiol Treatment Overcame Glutamate-Induced Neurodegeneration in Postnatal Rat Brain
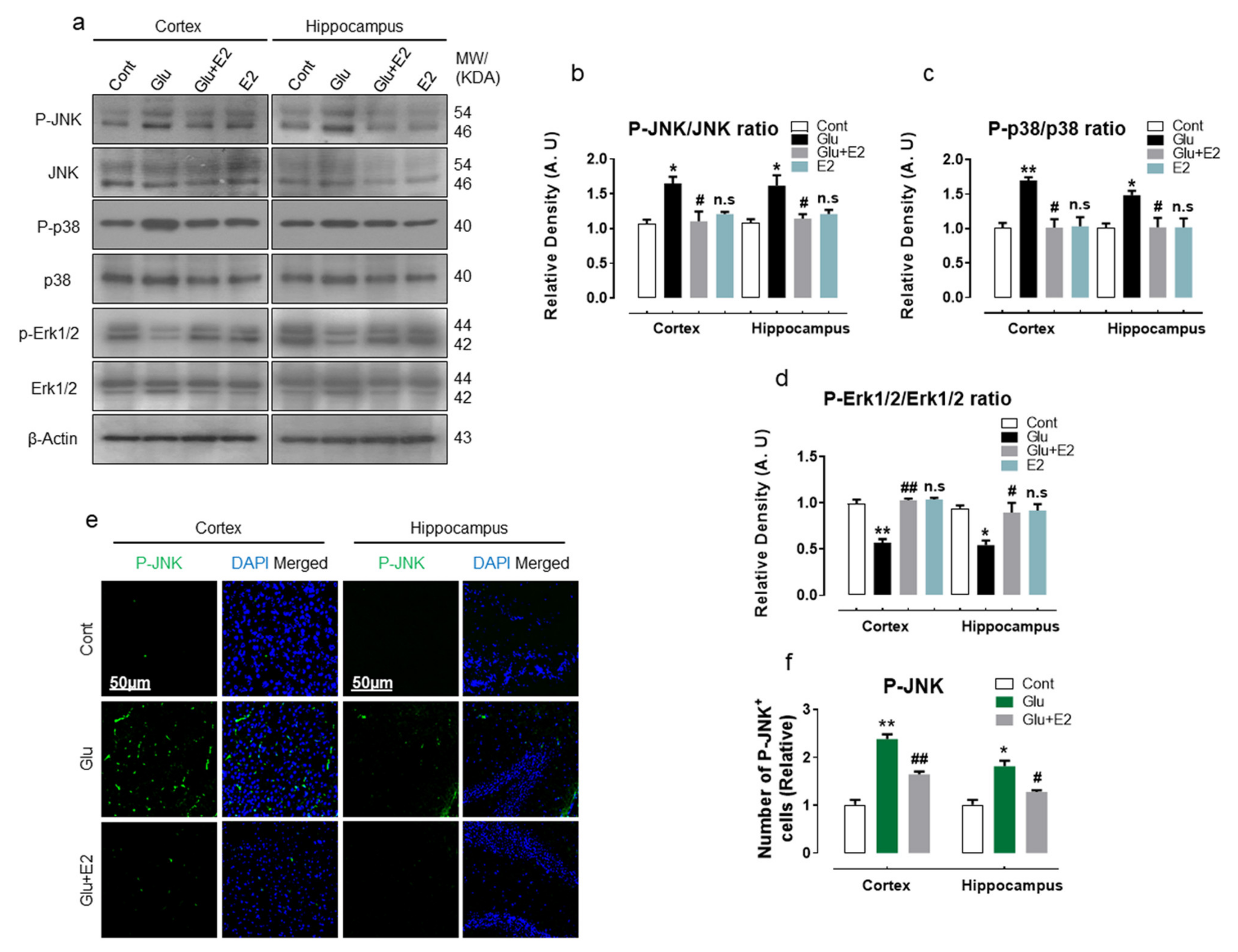
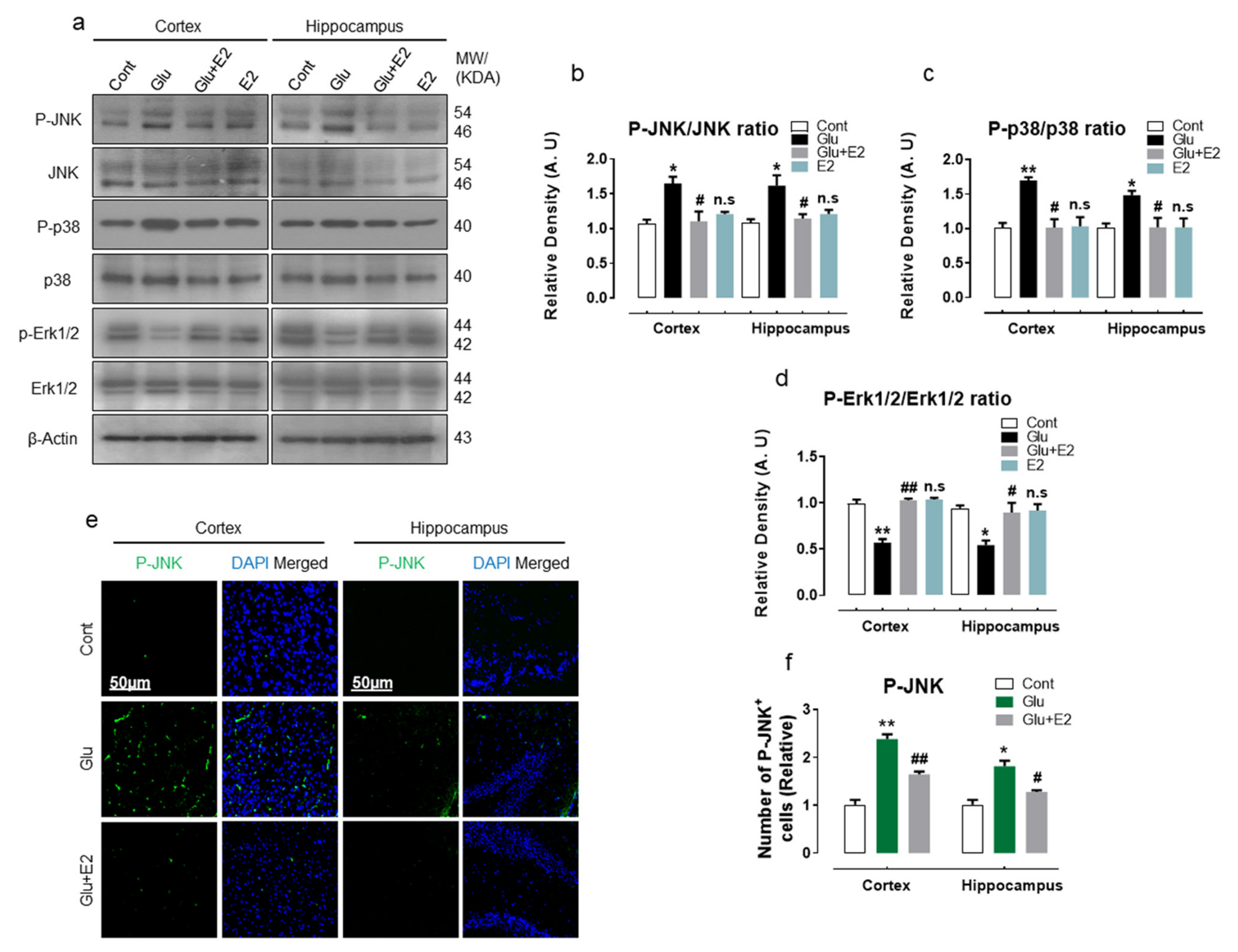
3.5. 17β-Estradiol Protect Developing Rat Brain against Glutamate-Induced Excitotoxicity through Regulating P-JNK/P38 and Erk1/2 MAPK Signaling Pathways
4. Discussion
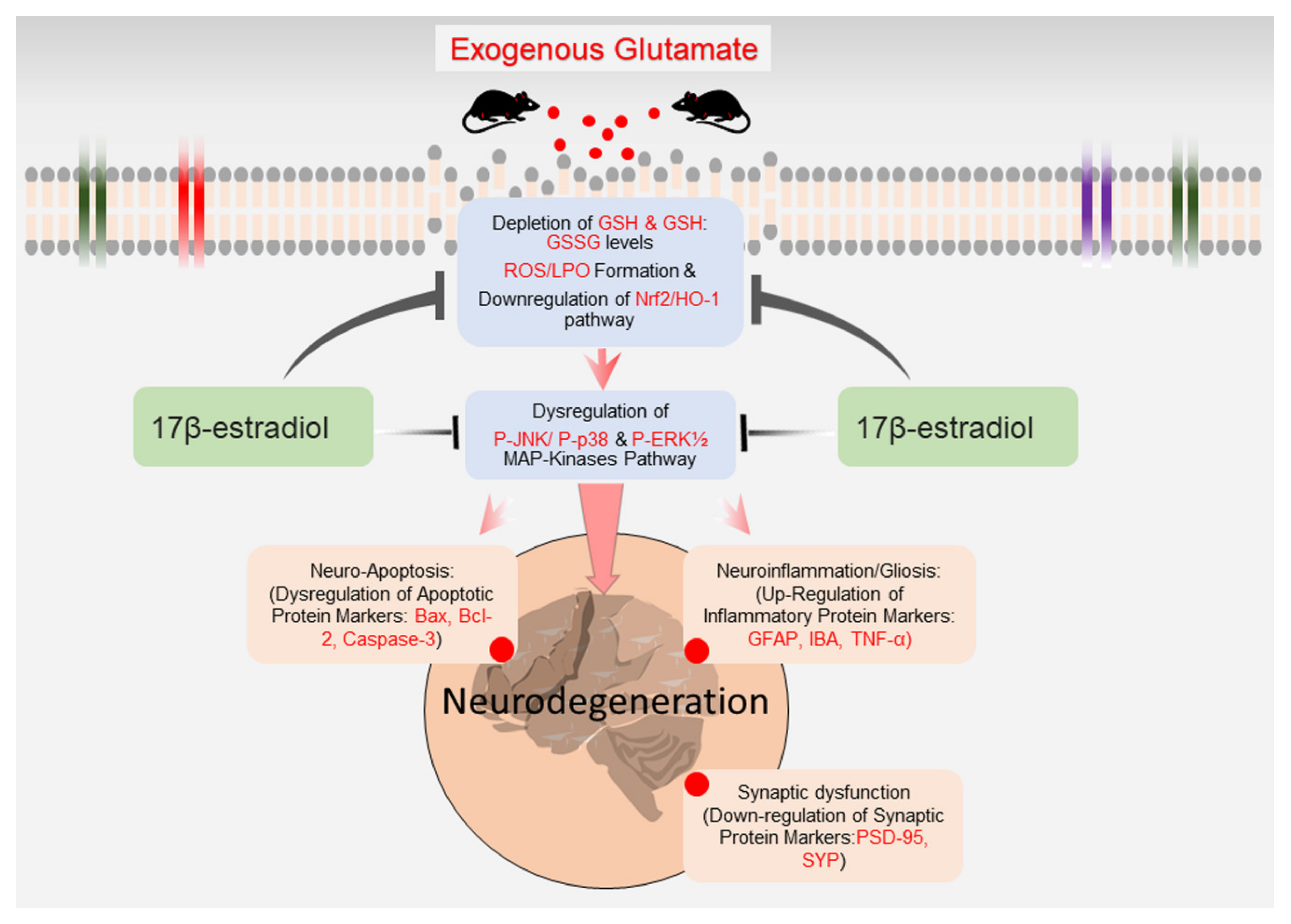
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| OS | Oxidative stress |
| ROS | Reactive oxygen species |
| E2 | Estradiol |
| Glu | Glutamate |
| LPO | Lipid peroxidation |
| Nrf2 | Nuclear factor erythroid 2-related factor 2 |
| HO-1 | Heme oxygenase-1 |
| p-JNK | c-Jun n-terminal kinase |
| P-p38 | phosphorylated p38 |
| P-Erk1/2 | Extracellular signal-regulated kinase 1 and 2 |
| GFAP | Glial fibrillary acidic protein |
| Iba-1 | Ionized calcium-binding adaptor molecule 1 |
| TNF-α | Tumor necrosis factor alpha |
| Bax | Bcl-2-associated X protein |
| Caspas3 | cysteine-aspartic acid protease 3 |
| Bcl-2 | B-cell lymphoma 2 |
References
- Lewerenz, J.; Maher, P. Chronic Glutamate Toxicity in Neurodegenerative Diseases—What is the Evidence? Front. Neurosci. 2015, 9, 469. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Reddy, P.H. Role of Glutamate and NMDA Receptors in Alzheimer’s Disease. J. Alzheimers Dis. 2017, 57, 1041–1048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossler, O.G.; Bauer, I.; Chung, H.Y.; Thiel, G. Glutamate-induced cell death of immortalized murine hippocampal neurons: Neuroprotective activity of heme oxygenase-1, heat shock protein 70, and sodium selenite. Neurosci. Lett. 2004, 362, 253–257. [Google Scholar] [CrossRef]
- Nishizawa, Y. Glutamate release and neuronal damage in ischemia. Life Sci. 2001, 69, 369–381. [Google Scholar] [CrossRef]
- Kritis, A.A.; Stamoula, E.G.; Paniskaki, K.A.; Vavilis, T.D. Researching glutamate-induced cytotoxicity in different cell lines: A comparative/collective analysis/study. Front. Cell Neurosci. 2015, 9, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, D.; Alavi, M.V.; Kim, K.Y.; Kang, T.; Scott, R.T.; Noh, Y.H.; Lindsey, J.D.; Wissinger, B.; Ellisman, M.H.; Weinreb, R.N.; et al. A new vicious cycle involving glutamate excitotoxicity, oxidative stress and mitochondrial dynamics. Cell Death Dis. 2011, 2, e240. [Google Scholar] [CrossRef]
- Trist, D.G. Excitatory amino acid agonists and antagonists: Pharmacology and therapeutic applications. Pharm. Acta Helv. 2000, 74, 221–229. [Google Scholar] [CrossRef]
- Mathisen, G.A.; Fonnum, F.; Paulsen, R.E. Contributing mechanisms for cysteine excitotoxicity in cultured cerebellar granule cells. Neurochem. Res. 1996, 21, 293–298. [Google Scholar] [CrossRef]
- Emerit, J.; Edeas, M.; Bricaire, F. Neurodegenerative diseases and oxidative stress. Biomed. Pharmcother. 2004, 58, 39–46. [Google Scholar] [CrossRef]
- Salganik, R.I. The benefits and hazards of antioxidants: Controlling apoptosis and other protective mechanisms in cancer patients and the human population. J. Am. Coll. Nutr. 2001, 20, 464S–472S, discussion 473S–475S. [Google Scholar] [CrossRef]
- Gilgun-Sherki, Y.; Melamed, E.; Offen, D. Oxidative stress induced-neurodegenerative diseases: The need for antioxidants that penetrate the blood brain barrier. Neuropharmacology 2001, 40, 959–975. [Google Scholar] [CrossRef]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Harvey, C.J.; Thimmulappa, R.K.; Singh, A.; Blake, D.J.; Ling, G.; Wakabayashi, N.; Fujii, J.; Myers, A.; Biswal, S. Nrf2-regulated glutathione recycling independent of biosynthesis is critical for cell survival during oxidative stress. Free Radic. Biol. Med. 2009, 46, 443–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnham, K.J.; Masters, C.L.; Bush, A.I. Neurodegenerative diseases and oxidative stress. Nat. Rev. Drug Discov. 2004, 3, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Son, Y.; Cheong, Y.K.; Kim, N.H.; Chung, H.T.; Kang, D.G.; Pae, H.O. Mitogen-Activated Protein Kinases and Reactive Oxygen Species: How Can ROS Activate MAPK Pathways? J. Signal. Transduct. 2011, 2011, 792639. [Google Scholar] [CrossRef] [PubMed]
- Ortuno-Sahagun, D.; Gonzalez, R.M.; Verdaguer, E.; Huerta, V.C.; Torres-Mendoza, B.M.; Lemus, L.; Rivera-Cervantes, M.C.; Camins, A.; Zarate, C.B. Glutamate excitotoxicity activates the MAPK/ERK signaling pathway and induces the survival of rat hippocampal neurons in vivo. J. Mol. Neurosci. 2014, 52, 366–377. [Google Scholar] [CrossRef] [PubMed]
- Kaminska, B.; Gozdz, A.; Zawadzka, M.; Ellert-Miklaszewska, A.; Lipko, M. MAPK signal transduction underlying brain inflammation and gliosis as therapeutic target. Anat. Rec. 2009, 292, 1902–1913. [Google Scholar] [CrossRef]
- Lee, J.K.; Kim, N.J. Recent Advances in the Inhibition of p38 MAPK as a Potential Strategy for the Treatment of Alzheimer’s Disease. Molecules 2017, 22, 1287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molz, S.; Decker, H.; Dal-Cim, T.; Cremonez, C.; Cordova, F.M.; Leal, R.B.; Tasca, C.I. Glutamate-induced toxicity in hippocampal slices involves apoptotic features and p38 MAPK signaling. Neurochem. Res. 2008, 33, 27–36. [Google Scholar] [CrossRef]
- Turgeon, J.L.; Carr, M.C.; Maki, P.M.; Mendelsohn, M.E.; Wise, P.M. Complex actions of sex steroids in adipose tissue, the cardiovascular system, and brain: Insights from basic science and clinical studies. Endocr. Rev. 2006, 27, 575–605. [Google Scholar] [CrossRef] [Green Version]
- Fink, G.; Sumner, B.E.; Rosie, R.; Grace, O.; Quinn, J.P. Estrogen control of central neurotransmission: Effect on mood, mental state, and memory. Cell Mol. Neurobiol. 1996, 16, 325–344. [Google Scholar] [CrossRef]
- Mauvais-Jarvis, F.; Clegg, D.J.; Hevener, A.L. The role of estrogens in control of energy balance and glucose homeostasis. Endocr. Rev. 2013, 34, 309–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gruber, C.J.; Tschugguel, W.; Schneeberger, C.; Huber, J.C. Production and actions of estrogens. N. Engl. J. Med. 2002, 346, 340–352. [Google Scholar] [CrossRef] [PubMed]
- Rao, M.L.; Kolsch, H. Effects of estrogen on brain development and neuroprotection—Implications for negative symptoms in schizophrenia. Psychoneuroendocrinology 2003, 28 (Suppl. 2), 83–96. [Google Scholar] [CrossRef]
- Baulieu, E.E. Neurosteroids: A novel function of the brain. Psychoneuroendocrinology 1998, 23, 963–987. [Google Scholar] [CrossRef]
- Cui, J.; Shen, Y.; Li, R. Estrogen synthesis and signaling pathways during aging: From periphery to brain. Trends Mol. Med. 2013, 19, 197–209. [Google Scholar] [CrossRef] [Green Version]
- Vrtacnik, P.; Ostanek, B.; Mencej-Bedrac, S.; Marc, J. The many faces of estrogen signaling. Biochem. Med. 2014, 24, 329–342. [Google Scholar] [CrossRef] [Green Version]
- Green, P.S.; Simpkins, J.W. Neuroprotective effects of estrogens: Potential mechanisms of action. Int. J. Dev. Neurosci. 2000, 18, 347–358. [Google Scholar] [CrossRef]
- Henderson, V.W. Estrogen-containing hormone therapy and Alzheimer’s disease risk: Understanding discrepant inferences from observational and experimental research. Neuroscience 2006, 138, 1031–1039. [Google Scholar] [CrossRef]
- Wise, P.M. Estrogens and neuroprotection. Trends Endocrinol. Metab. 2002, 13, 229–230. [Google Scholar] [CrossRef]
- Yang, S.H.; Liu, R.; Perez, E.J.; Wang, X.; Simpkins, J.W. Estrogens as protectants of the neurovascular unit against ischemic stroke. Curr. Drug Targets Cns Neurol. Disord. 2005, 4, 169–177. [Google Scholar] [CrossRef]
- Raghava, N.; Das, B.C.; Ray, S.K. Neuroprotective effects of estrogen in CNS injuries: Insights from animal models. Neurosci. Neuroecon. 2017, 6, 15–29. [Google Scholar] [CrossRef] [Green Version]
- Bjornstrom, L.; Sjoberg, M. Mechanisms of estrogen receptor signaling: Convergence of genomic and nongenomic actions on target genes. Mol. Endocrinol. 2005, 19, 833–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haas, M.J.; Raheja, P.; Jaimungal, S.; Sheikh-Ali, M.; Mooradian, A.D. Estrogen-dependent inhibition of dextrose-induced endoplasmic reticulum stress and superoxide generation in endothelial cells. Free Radic. Biol. Med. 2012, 52, 2161–2167. [Google Scholar] [CrossRef] [PubMed]
- Richardson, T.E.; Yu, A.E.; Wen, Y.; Yang, S.H.; Simpkins, J.W. Estrogen prevents oxidative damage to the mitochondria in Friedreich’s ataxia skin fibroblasts. PLoS ONE 2012, 7, e34600. [Google Scholar] [CrossRef]
- Behl, C.; Holsboer, F. The female sex hormone oestrogen as a neuroprotectant. Trends Pharmacol. Sci. 1999, 20, 441–444. [Google Scholar] [CrossRef]
- Mize, A.L.; Shapiro, R.A.; Dorsa, D.M. Estrogen receptor-mediated neuroprotection from oxidative stress requires activation of the mitogen-activated protein kinase pathway. Endocrinology 2003, 144, 306–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fitzpatrick, J.L.; Mize, A.L.; Wade, C.B.; Harris, J.A.; Shapiro, R.A.; Dorsa, D.M. Estrogen-mediated neuroprotection against beta-amyloid toxicity requires expression of estrogen receptor alpha or beta and activation of the MAPK pathway. J. Neurochem. 2002, 82, 674–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, M.; Ullah, R.; Rehman, S.U.; Shah, S.A.; Saeed, K.; Muhammad, T.; Park, H.Y.; Jo, M.H.; Choe, K.; Rutten, B.P.F.; et al. 17beta-Estradiol Modulates SIRT1 and Halts Oxidative Stress-Mediated Cognitive Impairment in a Male Aging Mouse Model. Cells 2019, 8, 928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishihara, Y.; Itoh, K.; Ishida, A.; Yamazaki, T. Selective estrogen-receptor modulators suppress microglial activation and neuronal cell death via an estrogen receptor-dependent pathway. J. Steroid Biochem. Mol. Biol. 2015, 145, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Vegeto, E.; Pollio, G.; Ciana, P.; Maggi, A. Estrogen blocks inducible nitric oxide synthase accumulation in LPS-activated microglia cells. Exp. Gerontol. 2000, 35, 1309–1316. [Google Scholar] [CrossRef]
- Suuronen, T.; Nuutinen, T.; Huuskonen, J.; Ojala, J.; Thornell, A.; Salminen, A. Anti-inflammatory effect of selective estrogen receptor modulators (SERMs) in microglial cells. Inflamm. Res. 2005, 54, 194–203. [Google Scholar] [CrossRef]
- Zhang, L.; Nair, A.; Krady, K.; Corpe, C.; Bonneau, R.H.; Simpson, I.A.; Vannucci, S.J. Estrogen stimulates microglia and brain recovery from hypoxia-ischemia in normoglycemic but not diabetic female mice. J. Clin. Investig. 2004, 113, 85–95. [Google Scholar] [CrossRef]
- Habib, P.; Dreymueller, D.; Ludwig, A.; Beyer, C.; Dang, J. Sex steroid hormone-mediated functional regulation of microglia-like BV-2 cells during hypoxia. J. Steroid Biochem. Mol. Biol. 2013, 138, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Henderson, V.W.; Paganini-Hill, A.; Emanuel, C.K.; Dunn, M.E.; Buckwalter, J.G. Estrogen replacement therapy in older women. Comparisons between Alzheimer’s disease cases and nondemented control subjects. Arch. Neurol. 1994, 51, 896–900. [Google Scholar] [CrossRef]
- Kawas, C.; Resnick, S.; Morrison, A.; Brookmeyer, R.; Corrada, M.; Zonderman, A.; Bacal, C.; Lingle, D.D.; Metter, E. A prospective study of estrogen replacement therapy and the risk of developing Alzheimer’s disease: The Baltimore Longitudinal Study of Aging. Neurology 1997, 48, 1517–1521. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.A.; Yoon, G.H.; Chung, S.S.; Abid, M.N.; Kim, T.H.; Lee, H.Y.; Kim, M.O. Novel osmotin inhibits SREBP2 via the AdipoR1/AMPK/SIRT1 pathway to improve Alzheimer’s disease neuropathological deficits. Mol. Psychiatry 2017, 22, 407–416. [Google Scholar] [CrossRef]
- Fukui, M.; Song, J.H.; Choi, J.; Choi, H.J.; Zhu, B.T. Mechanism of glutamate-induced neurotoxicity in HT22 mouse hippocampal cells. Eur. J. Pharmacol. 2009, 617, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Guo, C.; Kong, J. Oxidative stress in neurodegenerative diseases. Neural Regen. Res. 2012, 7, 376–385. [Google Scholar] [CrossRef] [PubMed]
- Uttara, B.; Singh, A.V.; Zamboni, P.; Mahajan, R.T. Oxidative stress and neurodegenerative diseases: A review of upstream and downstream antioxidant therapeutic options. Curr. Neuropharmacol. 2009, 7, 65–74. [Google Scholar] [CrossRef] [Green Version]
- Spampinato, S.F.; Copani, A.; Nicoletti, F.; Sortino, M.A.; Caraci, F. Metabotropic Glutamate Receptors in Glial Cells: A New Potential Target for Neuroprotection? Front. Mol. Neurosci. 2018, 11, 414. [Google Scholar] [CrossRef] [Green Version]
- Haroon, E.; Miller, A.H.; Sanacora, G. Inflammation, Glutamate, and Glia: A Trio of Trouble in Mood Disorders. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2017, 42, 193–215. [Google Scholar] [CrossRef] [PubMed]
- Simoes, A.P.; Silva, C.G.; Marques, J.M.; Pochmann, D.; Porciuncula, L.O.; Ferreira, S.; Oses, J.P.; Beleza, R.O.; Real, J.I.; Kofalvi, A.; et al. Glutamate-induced and NMDA receptor-mediated neurodegeneration entails P2Y1 receptor activation. Cell Death Dis. 2018, 9, 297. [Google Scholar] [CrossRef] [PubMed]
- Sattler, R.; Xiong, Z.; Lu, W.Y.; Hafner, M.; MacDonald, J.F.; Tymianski, M. Specific coupling of NMDA receptor activation to nitric oxide neurotoxicity by PSD-95 protein. Science 1999, 284, 1845–1848. [Google Scholar] [CrossRef]
- Lepeta, K.; Lourenco, M.V.; Schweitzer, B.C.; Martino Adami, P.V.; Banerjee, P.; Catuara-Solarz, S.; de La Fuente Revenga, M.; Guillem, A.M.; Haidar, M.; Ijomone, O.M.; et al. Synaptopathies: Synaptic dysfunction in neurological disorders—A review from students to students. J. Neurochem. 2016, 138, 785–805. [Google Scholar] [CrossRef] [PubMed]
- Abramov, A.Y.; Duchen, M.R. Mechanisms underlying the loss of mitochondrial membrane potential in glutamate excitotoxicity. Biochim. Biophys. Acta 2008, 1777, 953–964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hara, M.R.; Snyder, S.H. Cell signaling and neuronal death. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 117–141. [Google Scholar] [CrossRef]
- Vieira, M.; Fernandes, J.; Burgeiro, A.; Thomas, G.M.; Huganir, R.L.; Duarte, C.B.; Carvalho, A.L.; Santos, A.E. Excitotoxicity through Ca2+-permeable AMPA receptors requires Ca2+-dependent JNK activation. Neurobiol. Dis. 2010, 40, 645–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawasaki, H.; Morooka, T.; Shimohama, S.; Kimura, J.; Hirano, T.; Gotoh, Y.; Nishida, E. Activation and involvement of p38 mitogen-activated protein kinase in glutamate-induced apoptosis in rat cerebellar granule cells. J. Biol. Chem. 1997, 272, 18518–18521. [Google Scholar] [CrossRef] [Green Version]
- Savolainen, K.M.; Loikkanen, J.; Naarala, J. Amplification of glutamate-induced oxidative stress. Toxicol. Lett. 1995, 82–83, 399–405. [Google Scholar] [CrossRef]
- Nakatsu, Y.; Kotake, Y.; Komasaka, K.; Hakozaki, H.; Taguchi, R.; Kume, T.; Akaike, A.; Ohta, S. Glutamate excitotoxicity is involved in cell death caused by tributyltin in cultured rat cortical neurons. Toxicol. Sci. Off. J. Soc. Toxicol. 2006, 89, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Kim, G.H.; Kim, J.E.; Rhie, S.J.; Yoon, S. The Role of Oxidative Stress in Neurodegenerative Diseases. Exp. Neurobiol. 2015, 24, 325–340. [Google Scholar] [CrossRef] [PubMed]
- Coyle, J.T.; Puttfarcken, P. Oxidative stress, glutamate, and neurodegenerative disorders. Science 1993, 262, 689–695. [Google Scholar] [CrossRef] [PubMed]
- Pereira, C.M.; Oliveira, C.R. Glutamate toxicity on a PC12 cell line involves glutathione (GSH) depletion and oxidative stress. Free Radic. Biol. Med. 1997, 23, 637–647. [Google Scholar] [CrossRef]
- Penugonda, S.; Mare, S.; Goldstein, G.; Banks, W.A.; Ercal, N. Effects of N-acetylcysteine amide (NACA), a novel thiol antioxidant against glutamate-induced cytotoxicity in neuronal cell line PC12. Brain Res. 2005, 1056, 132–138. [Google Scholar] [CrossRef] [Green Version]
- Miyamoto, M.; Murphy, T.H.; Schnaar, R.L.; Coyle, J.T. Antioxidants protect against glutamate-induced cytotoxicity in a neuronal cell line. J. Pharmacol. Exp. Ther. 1989, 250, 1132–1140. [Google Scholar]
- Pereira, C.F.; Oliveira, C.R. Oxidative glutamate toxicity involves mitochondrial dysfunction and perturbation of intracellular Ca2+ homeostasis. Neurosci. Res. 2000, 37, 227–236. [Google Scholar] [CrossRef] [Green Version]
- Murphy, T.H.; Schnaar, R.L.; Coyle, J.T. Immature cortical neurons are uniquely sensitive to glutamate toxicity by inhibition of cystine uptake. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 1990, 4, 1624–1633. [Google Scholar] [CrossRef]
- Brandes, M.S.; Gray, N.E. NRF2 as a Therapeutic Target in Neurodegenerative Diseases. ASN Neuro 2020, 12, 1759091419899782. [Google Scholar] [CrossRef]
- Loboda, A.; Damulewicz, M.; Pyza, E.; Jozkowicz, A.; Dulak, J. Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: An evolutionarily conserved mechanism. Cell. Mol. Life Sci. CMLS 2016, 73, 3221–3247. [Google Scholar] [CrossRef] [Green Version]
- Satoh, T.; Okamoto, S.I.; Cui, J.; Watanabe, Y.; Furuta, K.; Suzuki, M.; Tohyama, K.; Lipton, S.A. Activation of the Keap1/Nrf2 pathway for neuroprotection by electrophilic [correction of electrophillic] phase II inducers. Proc. Natl. Acad. Sci. USA 2006, 103, 768–773. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Chen, J.; Sun, S.; Zhao, J.; Dong, X.; Wang, J. Effects of Estradiol on Autophagy and Nrf-2/ARE Signals after Cerebral Ischemia. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2017, 41, 2027–2036. [Google Scholar] [CrossRef]
- Wu, Q.; Zhang, X.S.; Wang, H.D.; Zhang, X.; Yu, Q.; Li, W.; Zhou, M.L.; Wang, X.L. Astaxanthin activates nuclear factor erythroid-related factor 2 and the antioxidant responsive element (Nrf2-ARE) pathway in the brain after subarachnoid hemorrhage in rats and attenuates early brain injury. Mar. Drugs 2014, 12, 6125–6141. [Google Scholar] [CrossRef]
- Saeed, K.; Shah, S.A.; Ullah, R.; Alam, S.I.; Park, J.S.; Saleem, S.; Jo, M.H.; Kim, M.W.; Hahm, J.R.; Kim, M.O. Quinovic Acid Impedes Cholesterol Dyshomeostasis, Oxidative Stress, and Neurodegeneration in an Amyloid-beta-Induced Mouse Model. Oxid. Med. Cell Longev. 2020, 2020, 9523758. [Google Scholar] [CrossRef]
- Zhang, R.; Zhang, J.; Fang, L.; Li, X.; Zhao, Y.; Shi, W.; An, L. Neuroprotective effects of sulforaphane on cholinergic neurons in mice with Alzheimer’s disease-like lesions. Int. J. Mol. Sci. 2014, 15, 14396–14410. [Google Scholar] [CrossRef]
- Yang, L.; Calingasan, N.Y.; Thomas, B.; Chaturvedi, R.K.; Kiaei, M.; Wille, E.J.; Liby, K.T.; Williams, C.; Royce, D.; Risingsong, R.; et al. Neuroprotective effects of the triterpenoid, CDDO methyl amide, a potent inducer of Nrf2-mediated transcription. PLoS ONE 2009, 4, e5757. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.; Shah, S.A.; Kim, M.O. 17beta-Estradiol via SIRT1/Acetyl-p53/NF-kB Signaling Pathway Rescued Postnatal Rat Brain Against Acute Ethanol Intoxication. Mol. Neurobiol. 2018, 55, 3067–3078. [Google Scholar] [CrossRef]
- Wu, J.; Williams, D.; Walter, G.A.; Thompson, W.E.; Sidell, N. Estrogen increases Nrf2 activity through activation of the PI3K pathway in MCF-7 breast cancer cells. Exp. Cell Res. 2014, 328, 351–360. [Google Scholar] [CrossRef]
- Song, C.H.; Kim, N.; Kim, D.H.; Lee, H.N.; Surh, Y.J. 17-beta estradiol exerts anti-inflammatory effects through activation of Nrf2 in mouse embryonic fibroblasts. PLoS ONE 2019, 14, e0221650. [Google Scholar] [CrossRef]
- Stakhiv, T.M.; Mesia-Vela, S.; Kauffman, F.C. Phase II antioxidant enzyme activities in brain of male and female ACI rats treated chronically with estradiol. Brain Res. 2006, 1104, 80–91. [Google Scholar] [CrossRef]
- Saitoh, M.; Nishitoh, H.; Fujii, M.; Takeda, K.; Tobiume, K.; Sawada, Y.; Kawabata, M.; Miyazono, K.; Ichijo, H. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J. 1998, 17, 2596–2606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pearson, G.; Robinson, F.; Beers Gibson, T.; Xu, B.E.; Karandikar, M.; Berman, K.; Cobb, M.H. Mitogen-activated protein (MAP) kinase pathways: Regulation and physiological functions. Endocr. Rev. 2001, 22, 153–183. [Google Scholar] [CrossRef] [Green Version]
- Reynolds, I.J.; Hastings, T.G. Glutamate induces the production of reactive oxygen species in cultured forebrain neurons following NMDA receptor activation. J. Neurosci. Off. J. Soc. Neurosci. 1995, 15, 3318–3327. [Google Scholar] [CrossRef] [Green Version]
- Song, J.H.; Lee, H.J.; Kang, K.S. Procyanidin C1 Activates the Nrf2/HO-1 Signaling Pathway to Prevent Glutamate-Induced Apoptotic HT22 Cell Death. Int. J. Mol. Sci. 2019, 20, 142. [Google Scholar] [CrossRef] [Green Version]
- Xia, Z.; Dickens, M.; Raingeaud, J.; Davis, R.J.; Greenberg, M.E. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science 1995, 270, 1326–1331. [Google Scholar] [CrossRef]
- Friedman, A.; Perrimon, N. A functional RNAi screen for regulators of receptor tyrosine kinase and ERK signalling. Nature 2006, 444, 230–234. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Bode, A.M. Dialogue between ERKs and JNKs: Friendly or antagonistic? Mol. Interv. 2003, 3, 306–308. [Google Scholar] [CrossRef]
- Black, E.J.; Walker, M.; Clark, W.; MacLaren, A.; Gillespie, D.A. Cell transformation by v-Jun deactivates ERK MAP kinase signalling. Oncogene 2002, 21, 6540–6548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Y.H.; Godlewski, J.; Zhu, J.; Sathyanarayana, P.; Leaner, V.; Birrer, M.J.; Rana, A.; Tzivion, G. Cross-talk between JNK/SAPK and ERK/MAPK pathways: Sustained activation of JNK blocks ERK activation by mitogenic factors. J. Biol. Chem. 2003, 278, 26715–26721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Junttila, M.R.; Li, S.P.; Westermarck, J. Phosphatase-mediated crosstalk between MAPK signaling pathways in the regulation of cell survival. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2008, 22, 954–965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westermarck, J.; Li, S.P.; Kallunki, T.; Han, J.; Kahari, V.M. p38 mitogen-activated protein kinase-dependent activation of protein phosphatases 1 and 2A inhibits MEK1 and MEK2 activity and collagenase 1 (MMP-1) gene expression. Mol. Cell. Biol. 2001, 21, 2373–2383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Q.; Hofmann, P.A. Protein phosphatase 2A-mediated cross-talk between p38 MAPK and ERK in apoptosis of cardiac myocytes. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H2204–H2212. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Hofmann, P.A. Modulation of protein phosphatase 2a by adenosine A1 receptors in cardiomyocytes: Role for p38 MAPK. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H97–H103. [Google Scholar] [CrossRef] [Green Version]
- Giordano, G.; Klintworth, H.M.; Kavanagh, T.J.; Costa, L.G. Apoptosis induced by domoic acid in mouse cerebellar granule neurons involves activation of p38 and JNK MAP kinases. Neurochem. Int. 2008, 52, 1100–1105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavanaugh, J.E. Role of extracellular signal regulated kinase 5 in neuronal survival. Eur. J. Biochem. 2004, 271, 2056–2059. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, Y.; Takemoto, T.; Ishida, A.; Yamazaki, T. Protective actions of 17beta-estradiol and progesterone on oxidative neuronal injury induced by organometallic compounds. Oxidative Med. Cell. Longev. 2015, 2015, 343706. [Google Scholar] [CrossRef] [Green Version]
- Dominguez, R.; Liu, R.; Baudry, M. 17-Beta-estradiol-mediated activation of extracellular-signal regulated kinase, phosphatidylinositol 3-kinase/protein kinase B-Akt and N-methyl-D-aspartate receptor phosphorylation in cortical synaptoneurosomes. J. Neurochem. 2007, 101, 232–240. [Google Scholar] [CrossRef] [Green Version]
- Borras, C.; Gambini, J.; Gomez-Cabrera, M.C.; Sastre, J.; Pallardo, F.V.; Mann, G.E.; Vina, J. 17beta-oestradiol up-regulates longevity-related, antioxidant enzyme expression via the ERK1 and ERK2[MAPK]/NFkappaB cascade. Aging Cell 2005, 4, 113–118. [Google Scholar] [CrossRef]
- Singer, C.A.; Figueroa-Masot, X.A.; Batchelor, R.H.; Dorsa, D.M. The mitogen-activated protein kinase pathway mediates estrogen neuroprotection after glutamate toxicity in primary cortical neurons. J. Neurosci. Off. J. Soc. Neurosci. 1999, 19, 2455–2463. [Google Scholar] [CrossRef] [Green Version]
- Skaper, S.D.; Facci, L.; Zusso, M.; Giusti, P. An Inflammation-Centric View of Neurological Disease: Beyond the Neuron. Front. Cell. Neurosci. 2018, 12, 72. [Google Scholar] [CrossRef]
- Olmos, G.; Llado, J. Tumor necrosis factor alpha: A link between neuroinflammation and excitotoxicity. Mediat. Inflamm. 2014, 2014, 861231. [Google Scholar] [CrossRef] [PubMed]
- Ki, Y.W.; Park, J.H.; Lee, J.E.; Shin, I.C.; Koh, H.C. JNK and p38 MAPK regulate oxidative stress and the inflammatory response in chlorpyrifos-induced apoptosis. Toxicol. Lett. 2013, 218, 235–245. [Google Scholar] [CrossRef] [PubMed]
- Kyriakis, J.M.; Avruch, J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol. Rev. 2001, 81, 807–869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, J.; Dai, Q.; Han, K.; Hong, W.; Jia, D.; Mo, Y.; Lv, Y.; Tang, H.; Fu, H.; Geng, W. JNK-IN-8, a c-Jun N-terminal kinase inhibitor, improves functional recovery through suppressing neuroinflammation in ischemic stroke. J. Cell. Physiol. 2020, 235, 2792–2799. [Google Scholar] [CrossRef]
- Roy Choudhury, G.; Ryou, M.G.; Poteet, E.; Wen, Y.; He, R.; Sun, F.; Yuan, F.; Jin, K.; Yang, S.H. Involvement of p38 MAPK in reactive astrogliosis induced by ischemic stroke. Brain Res. 2014, 1551, 45–58. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Harada, C.; Namekata, K.; Matsuzawa, A.; Camps, M.; Ji, H.; Swinnen, D.; Jorand-Lebrun, C.; Muzerelle, M.; Vitte, P.A.; et al. Regulation of the severity of neuroinflammation and demyelination by TLR-ASK1-p38 pathway. EMBO Mol. Med. 2010, 2, 504–515. [Google Scholar] [CrossRef] [PubMed]
- Villa, A.; Vegeto, E.; Poletti, A.; Maggi, A. Estrogens, Neuroinflammation, and Neurodegeneration. Endocr. Rev. 2016, 37, 372–402. [Google Scholar] [CrossRef] [Green Version]
- Priyanka, H.P.; Nair, R.S. Neuroimmunomodulation by estrogen in health and disease. AIMS Neurosci. 2020, 7, 401–417. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, M.; Haque, A.; Banik, N.L.; Nagarkatti, P.; Nagarkatti, M.; Ray, S.K. Estrogen receptor agonists for attenuation of neuroinflammation and neurodegeneration. Brain Res. Bull. 2014, 109, 22–31. [Google Scholar] [CrossRef] [Green Version]
- Vegeto, E.; Bonincontro, C.; Pollio, G.; Sala, A.; Viappiani, S.; Nardi, F.; Brusadelli, A.; Viviani, B.; Ciana, P.; Maggi, A. Estrogen prevents the lipopolysaccharide-induced inflammatory response in microglia. J. Neurosci. Off. J. Soc. Neurosci. 2001, 21, 1809–1818. [Google Scholar] [CrossRef] [Green Version]
- Vegeto, E.; Belcredito, S.; Etteri, S.; Ghisletti, S.; Brusadelli, A.; Meda, C.; Krust, A.; Dupont, S.; Ciana, P.; Chambon, P.; et al. Estrogen receptor-alpha mediates the brain antiinflammatory activity of estradiol. Proc. Natl. Acad. Sci. USA 2003, 100, 9614–9619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruce-Keller, A.J.; Keeling, J.L.; Keller, J.N.; Huang, F.F.; Camondola, S.; Mattson, M.P. Antiinflammatory effects of estrogen on microglial activation. Endocrinology 2000, 141, 3646–3656. [Google Scholar] [CrossRef]
- Lei, D.L.; Long, J.M.; Hengemihle, J.; O’Neill, J.; Manaye, K.F.; Ingram, D.K.; Mouton, P.R. Effects of estrogen and raloxifene on neuroglia number and morphology in the hippocampus of aged female mice. Neuroscience 2003, 121, 659–666. [Google Scholar] [CrossRef]
- Armada-Moreira, A.; Gomes, J.I.; Pina, C.C.; Savchak, O.K.; Goncalves-Ribeiro, J.; Rei, N.; Pinto, S.; Morais, T.P.; Martins, R.S.; Ribeiro, F.F.; et al. Going the Extra (Synaptic) Mile: Excitotoxicity as the Road Toward Neurodegenerative Diseases. Front. Cell. Neurosci. 2020, 14, 90. [Google Scholar] [CrossRef]
- Benarroch, E.E. Glutamatergic synaptic plasticity and dysfunction in Alzheimer disease: Emerging mechanisms. Neurology 2018, 91, 125–132. [Google Scholar] [CrossRef]
- Falcicchia, C.; Tozzi, F.; Arancio, O.; Watterson, D.M.; Origlia, N. Involvement of p38 MAPK in Synaptic Function and Dysfunction. Int. J. Mol. Sci. 2020, 21, 5624. [Google Scholar] [CrossRef]
- Munoz, L.; Ralay Ranaivo, H.; Roy, S.M.; Hu, W.; Craft, J.M.; McNamara, L.K.; Chico, L.W.; Van Eldik, L.J.; Watterson, D.M. A novel p38 alpha MAPK inhibitor suppresses brain proinflammatory cytokine up-regulation and attenuates synaptic dysfunction and behavioral deficits in an Alzheimer’s disease mouse model. J. Neuroinflamm. 2007, 4, 21. [Google Scholar] [CrossRef] [Green Version]
- Nagata, Y.; Todokoro, K. Requirement of activation of JNK and p38 for environmental stress-induced erythroid differentiation and apoptosis and of inhibition of ERK for apoptosis. Blood 1999, 94, 853–863. [Google Scholar] [CrossRef]
- Hammouda, M.B.; Ford, A.E.; Liu, Y.; Zhang, J.Y. The JNK Signaling Pathway in Inflammatory Skin Disorders and Cancer. Cells 2020, 9, 857. [Google Scholar] [CrossRef] [Green Version]
- Yarza, R.; Vela, S.; Solas, M.; Ramirez, M.J. c-Jun N-terminal Kinase (JNK) Signaling as a Therapeutic Target for Alzheimer’s Disease. Front. Pharmacol. 2015, 6, 321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piscopo, P.; Bellenghi, M.; Manzini, V.; Crestini, A.; Pontecorvi, G.; Corbo, M.; Ortona, E.; Care, A.; Confaloni, A. A Sex Perspective in Neurodegenerative Diseases: microRNAs as Possible Peripheral Biomarkers. Int. J. Mol. Sci. 2021, 22, 4423. [Google Scholar] [CrossRef]
- Ferretti, M.T.; Iulita, M.F.; Cavedo, E.; Chiesa, P.A.; Schumacher Dimech, A.; Santuccione Chadha, A.; Baracchi, F.; Girouard, H.; Misoch, S.; Giacobini, E.; et al. Sex differences in Alzheimer disease—The gateway to precision medicine. Nat. Rev. Neurol. 2018, 14, 457–469. [Google Scholar] [CrossRef] [PubMed]
- Luchetti, S.; van Eden, C.G.; Schuurman, K.; van Strien, M.E.; Swaab, D.F.; Huitinga, I. Gender differences in multiple sclerosis: Induction of estrogen signaling in male and progesterone signaling in female lesions. J. Neuropathol. Exp. Neurol. 2014, 73, 123–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez-Lee, C.; Kodama, L.; Gan, L. Sex Differences in Neurodegeneration: The Role of the Immune System in Humans. Biol. Psychiatry 2021. [Google Scholar] [CrossRef] [PubMed]
- Costa, G.; Sisalli, M.J.; Simola, N.; Della Notte, S.; Casu, M.A.; Serra, M.; Pinna, A.; Feliciello, A.; Annunziato, L.; Scorziello, A.; et al. Gender Differences in Neurodegeneration, Neuroinflammation and Na(+)-Ca(2+) Exchangers in the Female A53T Transgenic Mouse Model of Parkinson’s Disease. Front. Aging Neurosci. 2020, 12, 118. [Google Scholar] [CrossRef] [PubMed]
- Mosconi, L.; Berti, V.; Quinn, C.; McHugh, P.; Petrongolo, G.; Osorio, R.S.; Connaughty, C.; Pupi, A.; Vallabhajosula, S.; Isaacson, R.S.; et al. Correction: Perimenopause and emergence of an Alzheimer’s bioenergetic phenotype in brain and periphery. PLoS ONE 2018, 13, e0193314. [Google Scholar] [CrossRef] [Green Version]
- Mosconi, L.; Berti, V.; Quinn, C.; McHugh, P.; Petrongolo, G.; Varsavsky, I.; Osorio, R.S.; Pupi, A.; Vallabhajosula, S.; Isaacson, R.S.; et al. Sex differences in Alzheimer risk: Brain imaging of endocrine vs chronologic aging. Neurology 2017, 89, 1382–1390. [Google Scholar] [CrossRef]
- Scheyer, O.; Rahman, A.; Hristov, H.; Berkowitz, C.; Isaacson, R.S.; Diaz Brinton, R.; Mosconi, L. Female Sex and Alzheimer’s Risk: The Menopause Connection. J. Prev. Alzheimers Dis. 2018, 5, 225–230. [Google Scholar] [CrossRef]
- Shi, C.; Xu, X.W.; Forster, E.L.; Tang, L.F.; Ge, Z.; Yew, D.T.; Xu, J. Possible role of mitochondrial dysfunction in central neurodegeneration of ovariectomized rats. Cell Biochem. Funct. 2008, 26, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Unal, D.; Halici, Z.; Altunkaynak, Z.; Keles, O.N.; Oral, E.; Unal, B. A new hypothesis about neuronal degeneration appeared after a rat model of menopause. Neurodegener. Dis. 2012, 9, 25–30. [Google Scholar] [CrossRef]
- Mohamd, E.M.; Ahmed, H.H.; Estefan, S.F.; Farrag, A.E.; Salah, R.S. Windows into estradiol effects in Alzheimer’s disease therapy. Eur. Rev. Med. Pharm. Sci. 2011, 15, 1131–1140. [Google Scholar]
- Zhang, X.; Wang, J.; Xing, Y.; Gong, L.; Li, H.; Wu, Z.; Li, Y.; Wang, J.; Wang, Y.; Dong, L.; et al. Effects of ginsenoside Rg1 or 17beta-estradiol on a cognitively impaired, ovariectomized rat model of Alzheimer’s disease. Neuroscience 2012, 220, 191–200. [Google Scholar] [CrossRef]
- Zhiping, H.; Imam, M.U.; Ismail, M.; Ismail, N.; Yida, Z.; Ideris, A.; Sarega, N.; Mahmud, R. Effects of edible bird’s nest on hippocampal and cortical neurodegeneration in ovariectomized rats. Food Funct. 2015, 6, 1701–1711. [Google Scholar] [CrossRef] [PubMed]
- Woolley, C.S.; McEwen, B.S. Estradiol mediates fluctuation in hippocampal synapse density during the estrous cycle in the adult rat. J. Neurosci. Off. J. Soc. Neurosci. 1992, 12, 2549–2554. [Google Scholar] [CrossRef]
- Akama, K.T.; McEwen, B.S. Estrogen stimulates postsynaptic density-95 rapid protein synthesis via the Akt/protein kinase B pathway. J. Neurosci. Off. J. Soc. Neurosci. 2003, 23, 2333–2339. [Google Scholar] [CrossRef] [Green Version]
- Waters, E.M.; Yildirim, M.; Janssen, W.G.; Lou, W.Y.; McEwen, B.S.; Morrison, J.H.; Milner, T.A. Estrogen and aging affect the synaptic distribution of estrogen receptor beta-immunoreactivity in the CA1 region of female rat hippocampus. Brain Res. 2011, 1379, 86–97. [Google Scholar] [CrossRef] [Green Version]
- Bailey, M.E.; Wang, A.C.; Hao, J.; Janssen, W.G.; Hara, Y.; Dumitriu, D.; Hof, P.R.; Morrison, J.H. Interactive effects of age and estrogen on cortical neurons: Implications for cognitive aging. Neuroscience 2011, 191, 148–158. [Google Scholar] [CrossRef] [Green Version]
- Hara, Y.; Waters, E.M.; McEwen, B.S.; Morrison, J.H. Estrogen Effects on Cognitive and Synaptic Health Over the Lifecourse. Physiol. Rev. 2015, 95, 785–807. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Catalog/Product # | Application (Conc.) | Host | Manufacturer |
|---|---|---|---|---|
| β-Actin | SC-47,778 | WB (1:1000) | Mouse | Santa Cruz Biotech |
| Nrf2 | SC-722 | WB/IF (1:1000) | = | = |
| HO1 | SC-136,961 | WB/IF (1:1000/1:100) | = | = |
| GFAP | SC-33,673 | WB/IF (1:1000/1:100) | = | = |
| Iba-1 | SC-32,725 | WB (1:1000) | = | = |
| Iba-1 | PA5-27,436 | IF (1:100) | Rabbit | Thermo Fisher |
| TNF-α | SC-52,746 | WB (1:1000) | Mouse | Santa Cruz Biotech |
| PSD-95 | SC-71,933 | WB (1:1000) | = | = |
| SYP | SC-17,750 | WB (1:1000) | = | = |
| Bax | 2772S | WB (1:1000) | Rabbit | Cell Signaling |
| Bcl-2 | SC-7382 | WB (1:1000) | Mouse | Santa Cruz Biotech |
| Caspase-3 | SC-7272 | WB (1:1000) | = | = |
| Caspase-3 | 9661S | IF (1:100) | Rabbit | Cell Signaling |
| P-JNK | SC-6254 | WB/IF (1:1000/1:100) | Mouse | Santa Cruz Biotech |
| JNK | SC-7345 | WB (1:1000) | = | = |
| P-p38 | #9212 | WB (1:10,000) | Rabbit | Cell Signaling |
| p38 | #9211s | WB (1:1000) | = | = |
| P-Erk1/2 | #9101 | WB (1:10,000) | = | = |
| Erk1/2 | 9102S | WB (1:10,000) | = | = |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khan, I.; Saeed, K.; Jo, M.G.; Kim, M.O. 17-? Estradiol Rescued Immature Rat Brain against Glutamate-Induced Oxidative Stress and Neurodegeneration via Regulating Nrf2/HO-1 and MAP-Kinase Signaling Pathway. Antioxidants 2021, 10, 892. https://doi.org/10.3390/antiox10060892
Khan I, Saeed K, Jo MG, Kim MO. 17-? Estradiol Rescued Immature Rat Brain against Glutamate-Induced Oxidative Stress and Neurodegeneration via Regulating Nrf2/HO-1 and MAP-Kinase Signaling Pathway. Antioxidants. 2021; 10(6):892. https://doi.org/10.3390/antiox10060892
Chicago/Turabian StyleKhan, Ibrahim, Kamran Saeed, Min Gi Jo, and Myeong Ok Kim. 2021. "17-? Estradiol Rescued Immature Rat Brain against Glutamate-Induced Oxidative Stress and Neurodegeneration via Regulating Nrf2/HO-1 and MAP-Kinase Signaling Pathway" Antioxidants 10, no. 6: 892. https://doi.org/10.3390/antiox10060892
APA StyleKhan, I., Saeed, K., Jo, M. G., & Kim, M. O. (2021). 17-? Estradiol Rescued Immature Rat Brain against Glutamate-Induced Oxidative Stress and Neurodegeneration via Regulating Nrf2/HO-1 and MAP-Kinase Signaling Pathway. Antioxidants, 10(6), 892. https://doi.org/10.3390/antiox10060892