
Oxidative Stress as A Mechanism for Functional Alterations in Cardiac Hypertrophy and Heart Failure


Abstract
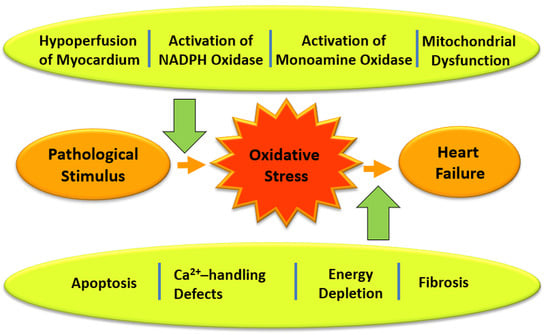
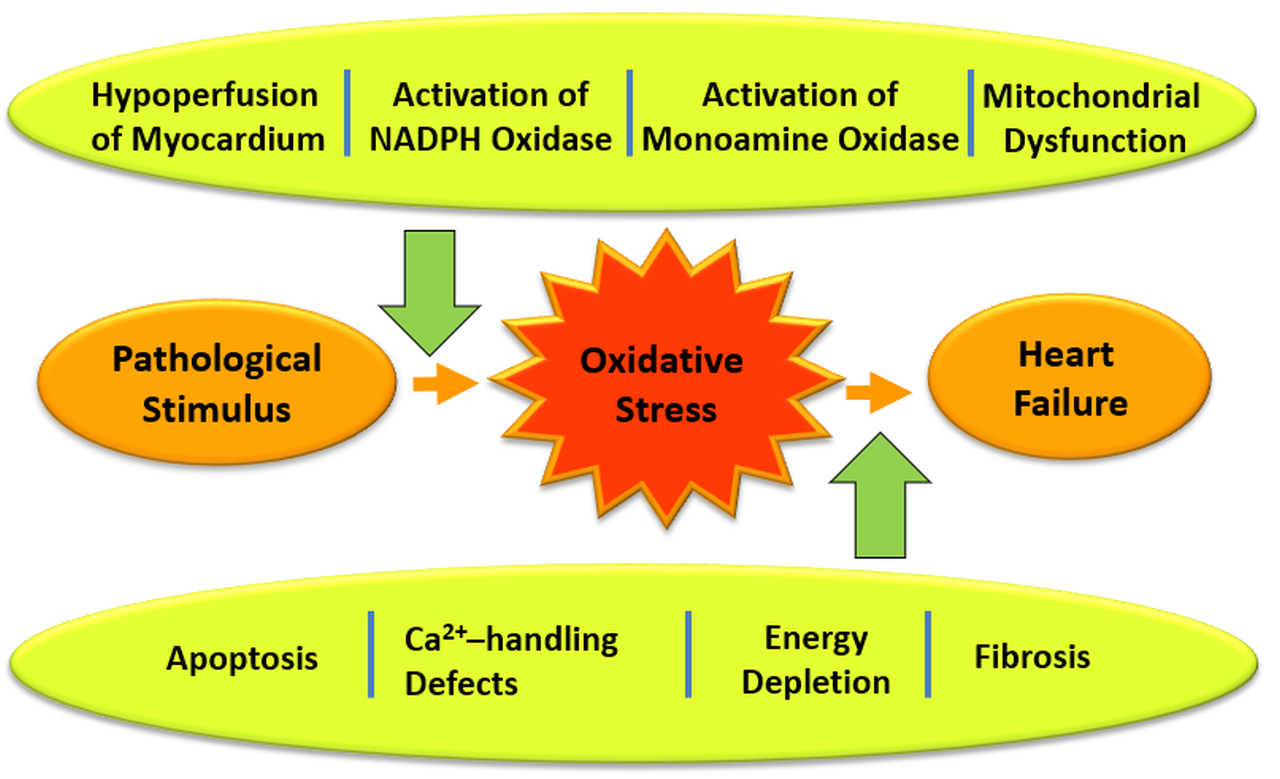{kind=link}
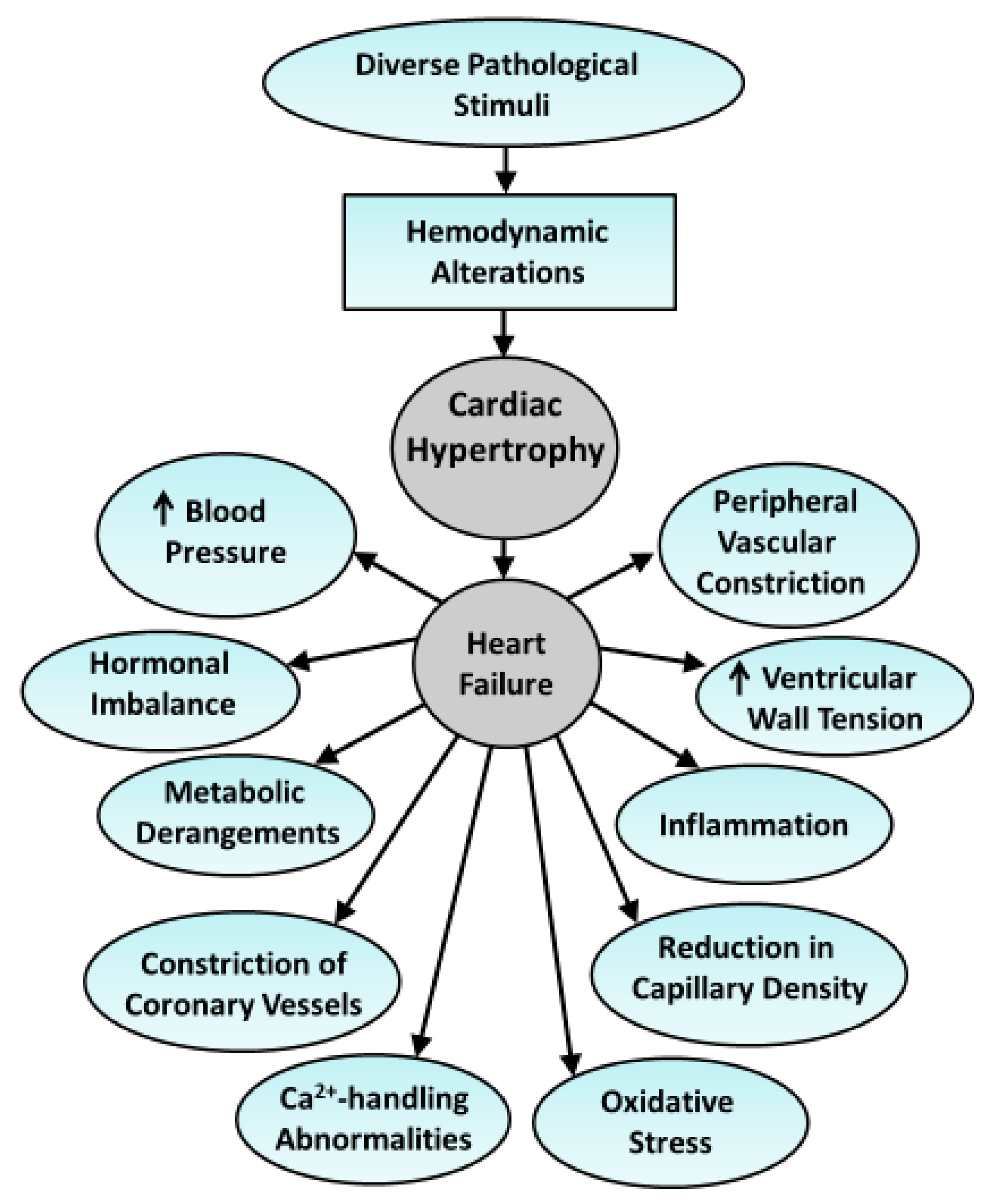{kind=link}
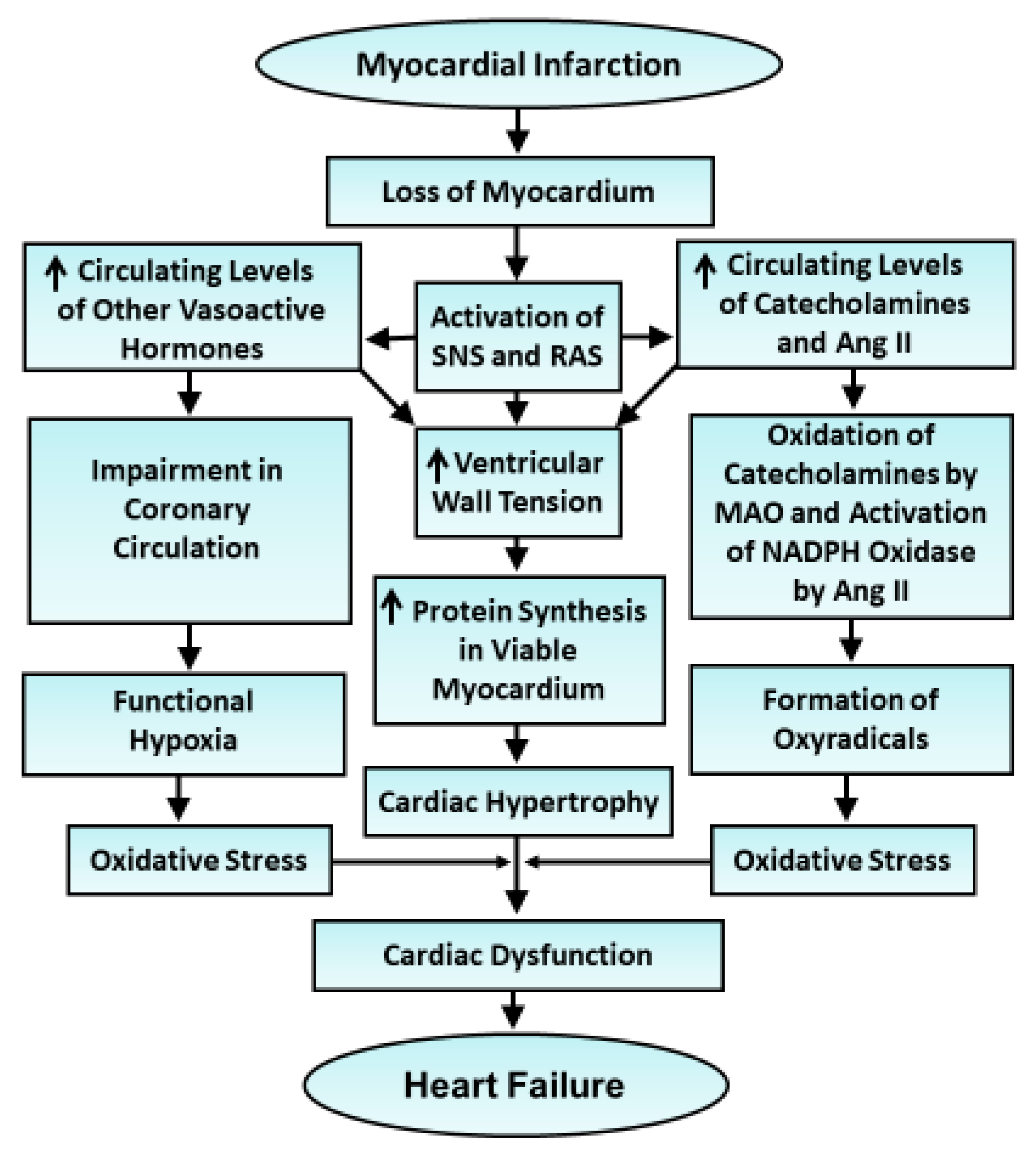{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Development of Cardiac Hypertrophy and Heart Failure
2.1. Development of Heart Failure Due to Myocardial Infarction
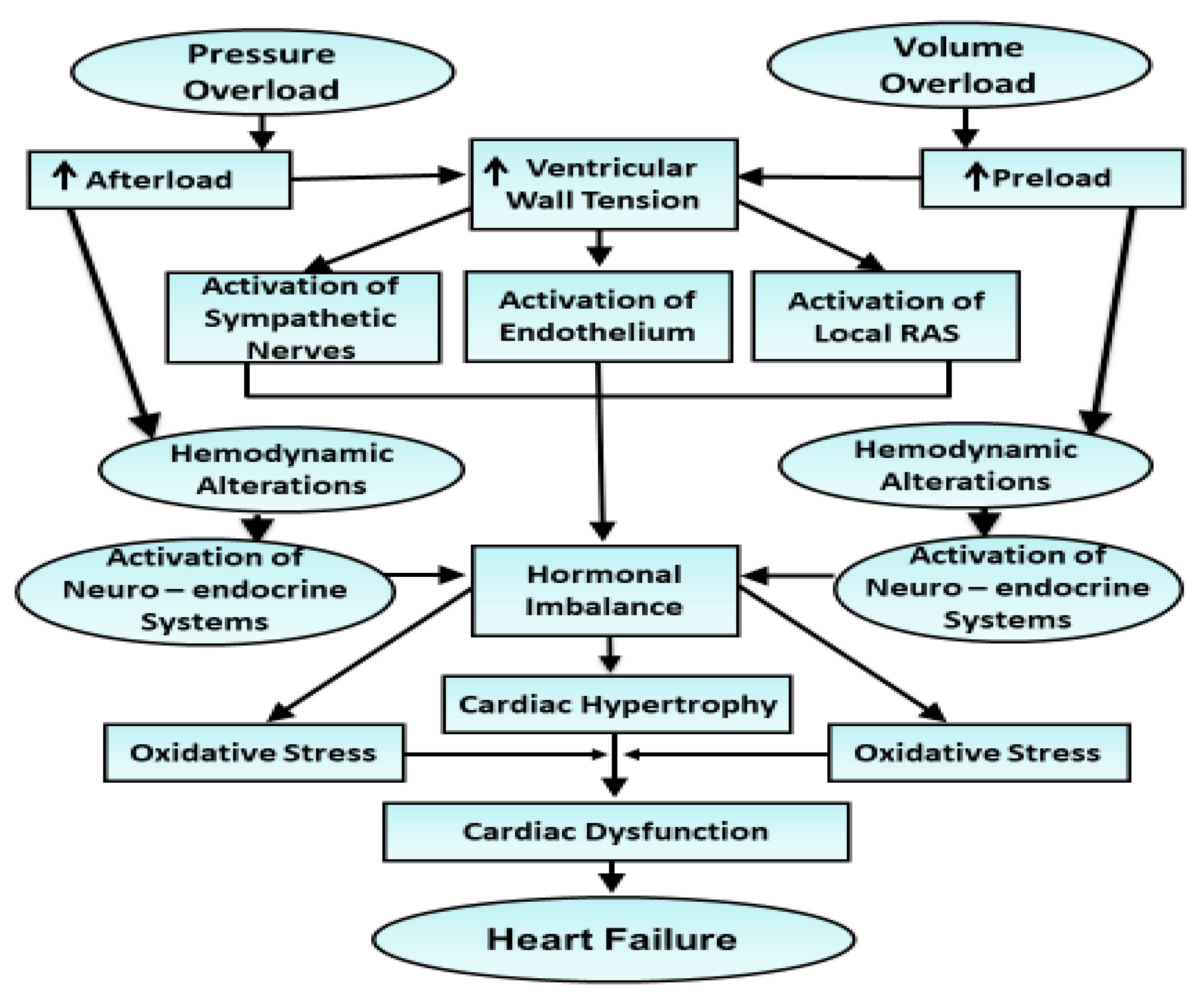
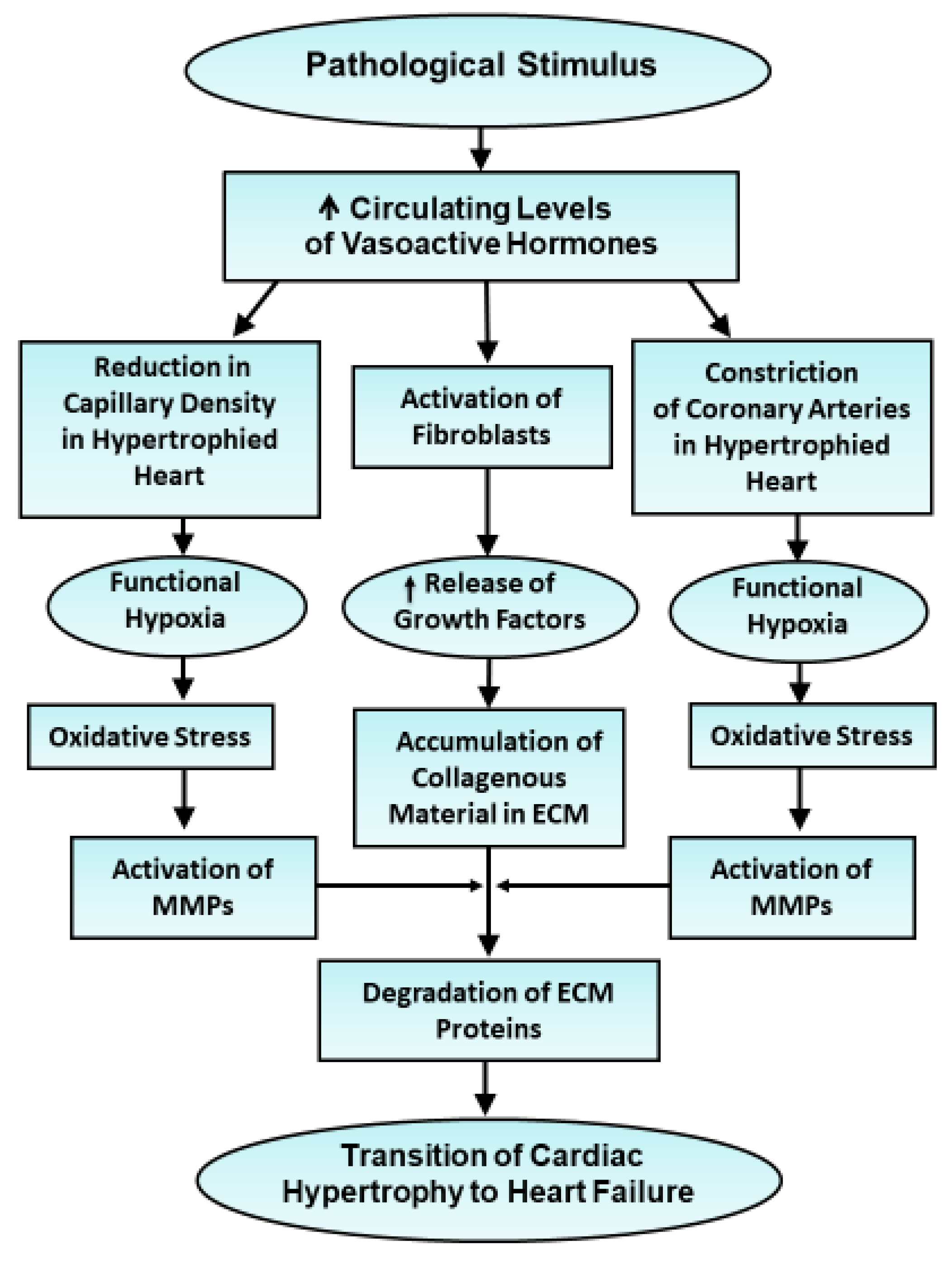
2.2. Development of Heart Failure Due to Pressure Overload and Volume Overload
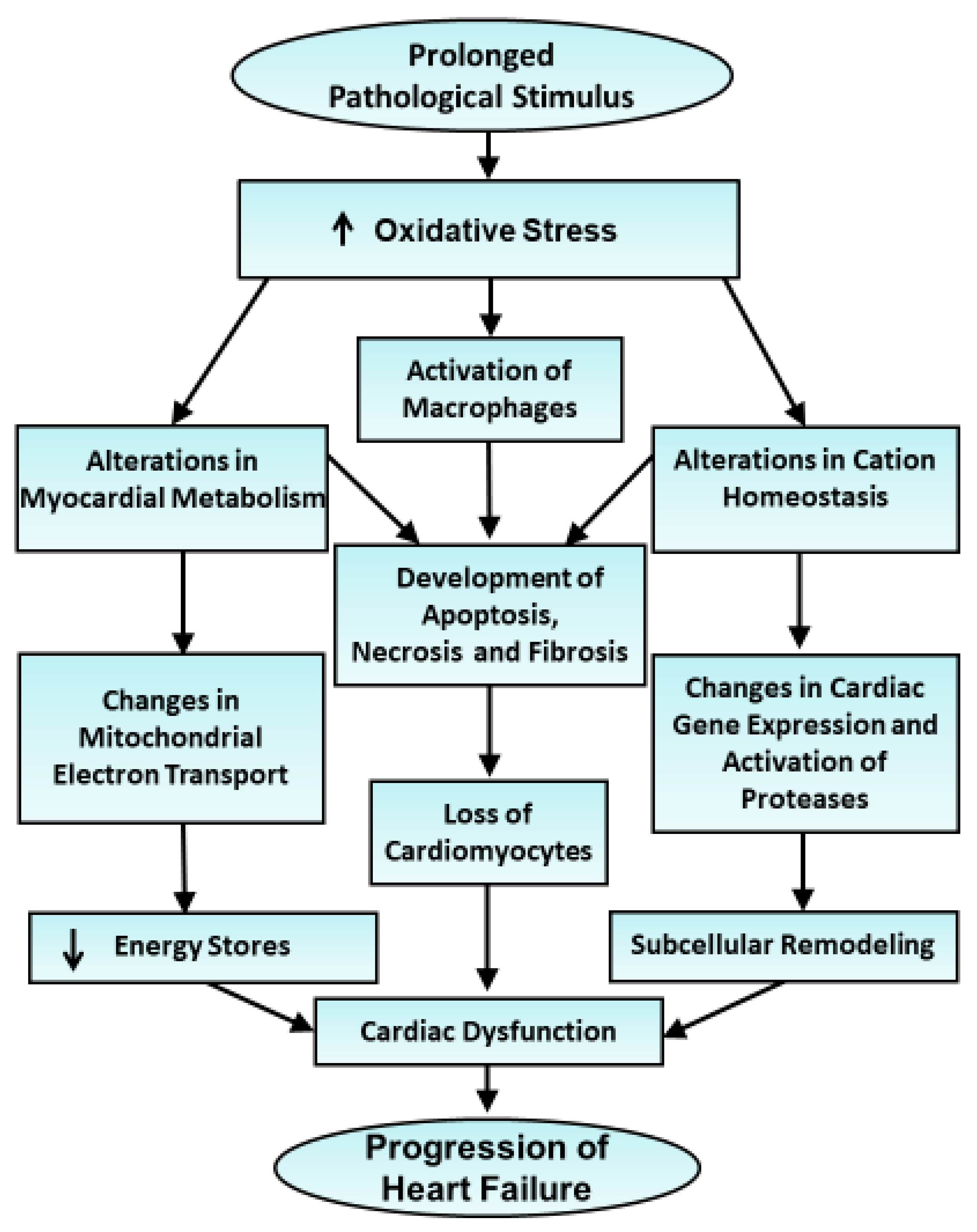
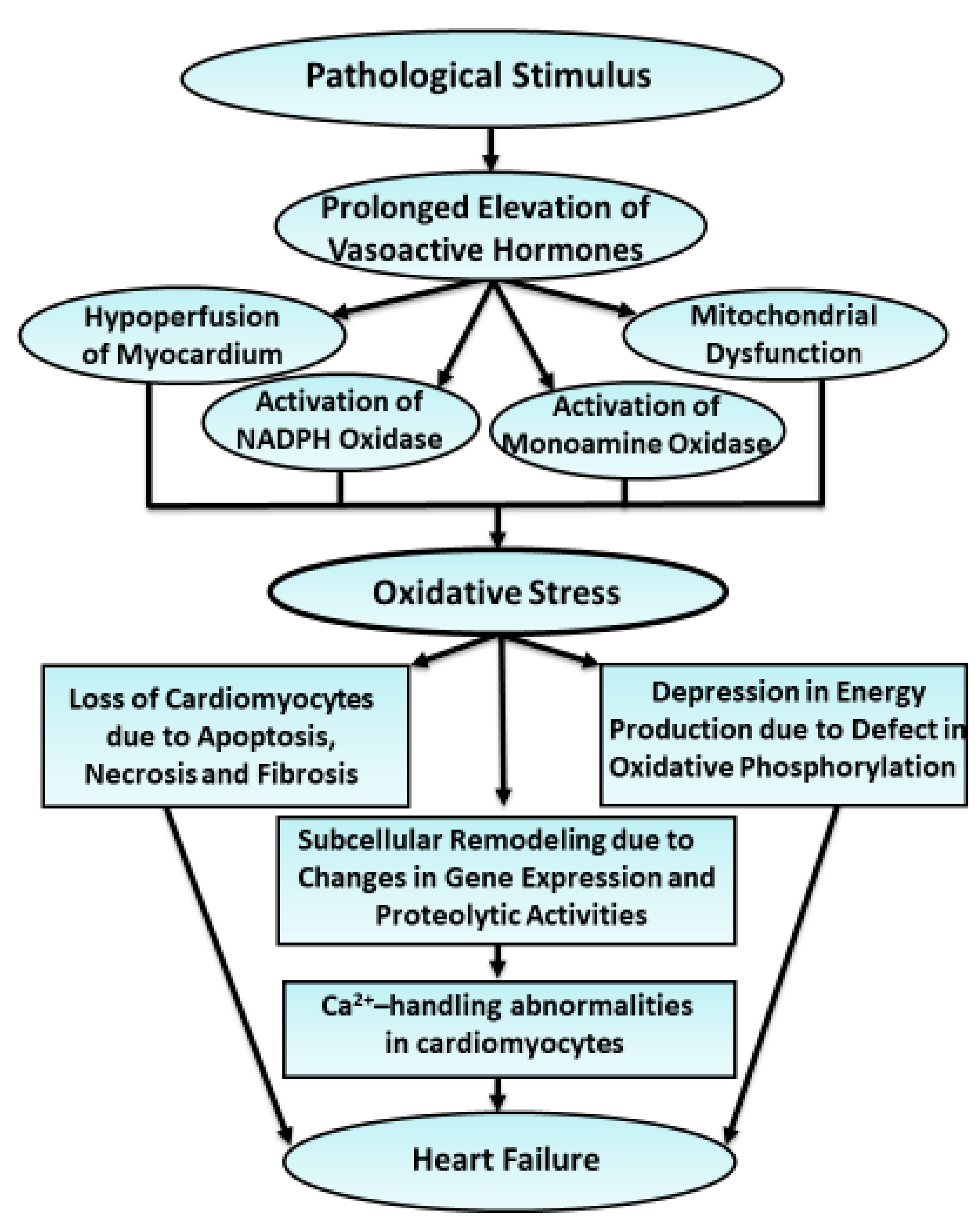
3. Generation of Oxyradicals, Redox Signaling and Consequences of Oxidative Stress in Failing Hearts
4. Evidence for the Implications of Oxidative Stress in Cardiac Dysfunction and Subcellular Remodeling
4.1. Alterations in Cardiac Function and Subcellular Activities in Ischemic Reperfused Hearts
4.2. Alterations in Cardiac Function and Subcellular Activities Due to Oxyradical Generating System or H2O2
4.3. Effects of Oxyradical Generating System and H2O2 on Subcellular Activities
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jessup, M.; Brozena, S. Heart failure. N. Engl. J. Med. 2003, 348, 2007–2018. [Google Scholar] [CrossRef]
- Parmley, W.W. Pathophysiology and current therapy of congestive heart failure. J. Am. Coll. Cardiol. 1989, 13, 771–785. [Google Scholar] [CrossRef]
- McMurray, J.J.; Stewart, S. Epidemiology, aetiology, and prognosis of heart failure. Heart 2000, 83, 596–602. [Google Scholar] [CrossRef]
- Lloyd-Jones, D.M.; Larson, M.G.; Leip, E.P.; Beiser, A.; D’Agostino, R.B.; Kannel, W.B.; Murabito, J.M.; Vasan, R.S.; Benjamin, E.J. Daniel Levy Lifetime risk for developing congestive heart failure: The Framingham Heart Study. Circulation 2002, 106, 3068–3072. [Google Scholar] [CrossRef]
- Cohn, J.N.; Ferrari, R.; Sharpe, N. Cardiac remodeling–concepts and clinical implications: A consensus paper from an international forum on cardiac remodeling. Behalf of an International Forum on Cardiac Remodeling. J. Am. Coll. Cardiol. 2000, 35, 569–582. [Google Scholar] [CrossRef]
- Dhalla, N.S.; Afzal, N.; Beamish, R.E.; Naimark, B.; Takeda, N.; Nagano, M. Pathophysiology of cardiac dysfunction in congestive heart failure. Can. J. Cardiol. 1993, 9, 873–887. [Google Scholar] [PubMed]
- Dhalla, N.S.; Saini–Chohan, H.K.; Rodriguez–Leyva, D.; Elimban, V.; Dent, M.R.; Tappia, P.S. Subcellular remodelling may induce cardiac dysfunction in congestive heart failure. Cardiovasc. Res. 2009, 81, 429–438. [Google Scholar] [CrossRef]
- Packer, M. Neurohormonal interactions and adaptations in congestive heart failure. Circulation 1988, 77, 721–730. [Google Scholar] [CrossRef]
- Mudd, J.O.; Kass, D.A. Tackling heart failure in the twenty–first century. Nature 2008, 451, 919–928. [Google Scholar] [CrossRef] [PubMed]
- Dhalla, N.S.; Dent, M.R.; Tappia, P.S.; Sethi, R.; Barta, J.; Goyal, R.K. Subcellular remodeling as a viable target for the treatment of congestive heart failure. J. Cardiovasc. Pharmacol. Ther. 2006, 11, 31–45. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Burchfield, J.S.; Hill, J.A. Pathological ventricular remodeling: Mechanisms: Part 1 of 2. Circulation 2013, 128, 388–400. [Google Scholar] [CrossRef] [PubMed]
- Lyon, R.C.; Zanella, F.; Omens, J.H.; Sheikh, F. Mechanotransduction in cardiac hypertrophy and failure. Circ. Res. 2015, 116, 1462–1476. [Google Scholar] [CrossRef] [PubMed]
- Wikman–Coffelt, J.; Parmley, W.W.; Mason, D.T. The cardiac hypertrophy process: Analyses of factors determining pathological vs. physiological development. Circ. Res. 1979, 45, 697–707. [Google Scholar] [CrossRef] [PubMed]
- Dhalla, N.S.; Heyliger, C.E.; Beamish, R.E.; Innes, I.R. Pathophysiological aspects of myocardial hypertrophy. Can. J. Cardiol. 1987, 3, 183–196. [Google Scholar] [PubMed]
- Shimizu, I.; Minamino, T. Physiological and pathological cardiac hypertrophy. J. Mol. Cell. Cardiol. 2016, 97, 245–262. [Google Scholar] [CrossRef]
- Maillet, M.; van Berlo, J.H.; Molkentin, J.D. Molecular basis of physiological heart growth: Fundamental concepts and new players. Nat. Rev. Mol. Cell. Biol. 2013, 14, 38–48. [Google Scholar] [CrossRef]
- Bernardo, B.C.; Weeks, K.L.; Pretorius, L.; McMullen, J.R. Molecular distinction between physiological and pathological cardiac hypertrophy: Experimental findings and therapeutic strategies. Pharmacol. Ther. 2010, 128, 191–227. [Google Scholar] [CrossRef]
- Molkentin, J.D.; Dorn, G.W., II. Cytoplasmic signaling pathways that regulate cardiac hypertrophy. Annuv. Rev. Physiol. 2001, 63, 391–426. [Google Scholar] [CrossRef]
- Heineke, J.; Molkentin, J.D. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat. Rev. Mol. Cell Biol. 2006, 7, 589–600. [Google Scholar] [CrossRef]
- Heger, J.; Schulz, R.; Euler, G. Molecular switches under TGFβ signaling during progression from cardiac hypertrophy to heart failure. Br. J. Pharmacol. 2016, 173, 3–14. [Google Scholar] [CrossRef]
- Schirone, L.; Forte, M.; Palmerio, S.; Yee, D.; Nocella, C.; Angelini, F.; Pagano, F.; Schiavon, S.; Bordin, A.; Carrizzo, A.; et al. A review of the molecular mechanisms underlying the development and progression of cardiac remodeling. Oxid. Med. Cell. Longev. 2017, 2017, 3920195. [Google Scholar] [CrossRef]
- Nakamura, M.; Sadoshima, J. Mechanisms of physiological and pathological cardiac hypertrophy. Nat. Rev. Cardiol. 2018, 15, 387–407. [Google Scholar] [CrossRef] [PubMed]
- Sygitowicz, G.; Maciejak-Jastrzebska, A.; Sitkiewicz, D. MicroRNAs in the development of left ventricular remodeling and postmyocardial infarction heart failure. Pol. Arch. Intern. Med. 2020, 130, 59–65. [Google Scholar] [PubMed]
- Mishra, S.; Kass, K.A. Cellular and molecular pathobiology of heart failure with preserved ejection fraction. Nat. Rev. Cardiol. 2021, 18, 400–423. [Google Scholar] [CrossRef] [PubMed]
- Sethi, R.; Saini, H.K.; Guo, X.; Wang, X.; Elimban, V.; Dhalla, N.S. Dependence of changes in beta–adrenoceptor signal transduction on type and stage of cardiac hypertrophy. J. Appl. Physiol. 2007, 102, 978–984. [Google Scholar] [CrossRef] [PubMed]
- Oldfield, C.J.; Duhamel, T.A.; Dhalla, N.S. Mechanisms for the transition from physiological to pathological cardiac hypertrophy. Can. J. Physiol. Pharmacol. 2019. [Google Scholar] [CrossRef]
- Nicholls, D.P.; Onuoha, G.N.; McDowell, G.; Elborn, J.S.; Riley, M.S.; Nugent, A.-M.; Steele, I.C.; Shaw, C.; Buchanan, K.D. Neuroendocrine changes in chronic cardiac failure. Basic Res. Cardiol. 1996, 91 (Suppl. 1), 13–20. [Google Scholar]
- Rouleau, J.L. The neurohormonal hypothesis and the treatment of heart failure. Can. J. Cardiol. 1996, 12 (Suppl. l), 3F–8F. [Google Scholar]
- Packer, M. The neurohormonal hypothesis: A theory to explain the mechanism of disease progression in heart failure. J. Am. Coll. Cardiol. 1992, 20, 248–254. [Google Scholar] [CrossRef]
- Francis, G.S.; Benedict, C.; Johnstone, D.E.; Kirlin, P.C.; Nicklas, J.; Liang, C.S.; Kubo, S.H.; Rudin-Toretsky, E.; Yusuf, S. Comparison of neuroendocrine activation in patients with left ventricular dysfunction with and without congestive heart failure. A substudy of the studies of left ventricular dysfunction (SOLVD). Circulation 1990, 82, 1724–1729. [Google Scholar] [CrossRef]
- Weber, K.T.; Brilla, C.G. Pathological hypertrophy and cardiac interstitium. Fibrosis and renin–angiotensin–aldosterone system. Circulation 1991, 83, 1849–1865. [Google Scholar] [CrossRef] [PubMed]
- Janicki, J.S.; Brower, G.L.; Gardner, J.D.; Chancey, A.L.; Stewart, J.A., Jr. The dynamic interaction between matrix metalloproteinase activity and adverse myocardial remodeling. Heart Fail. Rev. 2004, 9, 33–42. [Google Scholar] [CrossRef]
- Felker, G.M.; Thompson, R.E.; Hare, J.M.; Hruban, R.H.; Clemetson, D.E.; Howard, D.; Baughman, K.L.; Kasper, E.K. Underlying causes and long–term survival in patients with initially unexplained cardiomyopathy. N. Engl. J. Med. 2000, 342, 1077–1084. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.M.; Sanderson, J.E.; Shum, I.O.; Chan, S.; Yeung, L.Y.; Hung, Y.T.; Cockram, C.S.; Woo, K.S. Diastolic dysfunction and natriuretic peptides in systolic heart failure. Higher ANP and BNP levels are associated with the restrictive filling pattern. Eur. Heart J. 1996, 17, 1694–1702. [Google Scholar] [CrossRef]
- Clerico, A.; Iervasi, G.; Del Chicca, M.G.; Emdin, M.; Maffei, S.; Nannipieri, M.; Sabatino, L.; Forini, F.; Manfredi, C.; Donato, L. Circulating levels of cardiac natriuretic peptides (ANP and BNP) measured by highly sensitive and specific immunoradiometric assays in normal subjects and in patients with different degrees of heart failure. J. Endocrinol. Investig. 1998, 21, 170–179. [Google Scholar] [CrossRef]
- Ruetten, H.; Dimmeler, S.; Gehring, D.; Ihling, C.; Zeiher, A.M. Concentric left ventricular remodeling in endothelial nitric oxide synthase knockout mice by chronic pressure overload. Cardiovasc. Res. 2005, 66, 444–453. [Google Scholar] [CrossRef]
- Dhalla, N.S.; Elimban, V.; Shah, A.K.; Nusier, M. Mechanisms of cardiac dysfunction in heart failure due to myocardial infarction. J. Integr. Cardiol. 2019, 2, 2–7. [Google Scholar] [CrossRef]
- Wang, J.; Liu, X.; Arneja, A.S.; Dhalla, N.S. Alterations in protein kinase A and protein kinase C levels in heart failure due to genetic cardiomyopathy. Can. J. Cardiol. 1999, 15, 683–690. [Google Scholar] [PubMed]
- English, J.M.; Cobb, M.H. Pharmacological inhibitors of MAPK pathways. Trends Pharmacol. Sci. 2002, 23, 40–45. [Google Scholar] [CrossRef]
- Zhang, W.; Elimban, V.; Nijjar, M.S.; Gupta, S.K.; Dhalla, N.S. Role of mitogen-activated protein kinase in cardiac hypertrophy and heart failure. Exp. Clin. Cardiol. 2003, 8, 173–183. [Google Scholar]
- Kirchhefer, U.; Schmitz, W.; Scholz, H.; Neumann, J. Activity of cAMP-dependent protein kinase and Ca2+/calmodulin-dependent protein kinase in failing and nonfailing human hearts. Cardiovasc. Res. 1999, 42, 254–261. [Google Scholar] [CrossRef]
- Weber, K.T. Cardiac interstitium in health and disease. J. Am. Coll. Cardiol. 1989, 13, 1637–1652. [Google Scholar] [CrossRef]
- Ju, H.; Zhane, S.; Jassal, D.S.; Dixon, I.M.C. Effect of AT1 receptor blockade on cardiac collagen remodeling after myocardial infarction. Cardiovasc. Res. 1997, 35, 223–232. [Google Scholar] [CrossRef]
- Briest, W.; Holzl, A.; Rabler, B.; Deten, A.; Beba, H.A.; Zimmer, H. Significance of matrix metalloproteinases in norepinephrine-induced remodeling of rat hearts. Cardiovasc. Res. 2003, 57, 379–387. [Google Scholar] [CrossRef]
- Zak, R. Cell proliferation during cardiac growth. Am. J. Cardiol. 1973, 31, 211–219. [Google Scholar] [CrossRef]
- Westermann, D.; Kasner, M.; Steendijk, P.; Spillmann, F.; Riad, A.; Weitmann, K.; Hoffmann, W.; Poller, W.; Pauschinger, M.; Spillmann, F.; et al. Role of left ventricular stiffness in heart failure with normal ejection fraction. Circulation 2008, 117, 2051–2060. [Google Scholar] [CrossRef]
- Borbely, A.; van der Velden, J.; Papp, Z.; Bronzwaer, J.G.; Edes, I.; Stienen, G.; Paulus, W.J. Cardiomyocyte stiffness in diastolic heart failure. Circulation 2005, 111, 774–781. [Google Scholar] [CrossRef] [PubMed]
- Bartekova, M.; Radosinska, J.; Jelemensky, M.; Dhalla, N.S. Role of cytokines and inflammation in heart function during health and disease. Heart Fail. Rev. 2018, 23, 733–758. [Google Scholar] [CrossRef]
- Sanganalmath, S.K.; Barta, J.; Takeda, N.; Kumamoto, H.; Dhalla, N.S. Antiplatelet therapy mitigates cardiac remodeling and dysfunction in congestive heart failure due to myocardial infarction. Can. J. Physiol. Pharmacol. 2008, 86, 180–189. [Google Scholar] [CrossRef]
- Sanganalmath, S.K.; Babick, A.; Barta, J.; Kumamoto, H.; Takeda, N.; Dhalla, N.S. Antiplatelet therapy attenuates subcellular remodelling in congestive heart failure. J. Cell. Mol. Med. 2008, 12, 1728–1738. [Google Scholar] [CrossRef]
- Vanhoutte, P.M. Endothelium and control of vascular function. Hypertension 1989, 13, 658–667. [Google Scholar] [CrossRef] [PubMed]
- Rehsia, N.S.; Dhalla, N.S. Potential of endothelin-1 and vasopressin antagonists for the treatment of congestive heart failure. Heart Fail. Rev. 2010, 15, 85–101. [Google Scholar] [CrossRef] [PubMed]
- Gupta, M.; Singal, P.K. Higher antioxidant capacity during a chronic stable heart hypertrophy. Circ. Res. 1989, 64, 398–406. [Google Scholar] [CrossRef] [PubMed]
- Singal, P.K.; Gupta, M.; Randhawa, A.K. Reduced myocardial injury due to exogenous oxidants in pressure induced heart hypertrophy. Basic Res. Cardiol. 1991, 86, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Dhalla, N.S.; Temsah, R.M.; Netticadan, T. Role of oxidative stress in cardiovascular diseases. J. Hypertens. 2000, 18, 655–673. [Google Scholar] [CrossRef]
- Belch, J.J.; Bridges, A.B.; Scott, N.; Chopra, M. Oxygen free radicals and congestive heart failure. Br. Heart J. 1991, 65, 245–248. [Google Scholar] [CrossRef]
- Diaz-Velez, C.R.; Garcia-Castinieras, S.; Mendoza Ramos, E.; Hernandez Lopez, E. Increased malondialdehyde in peripheral blood of patients with congestive heart failure. Am. Heart J. 1996, 131, 146–152. [Google Scholar] [CrossRef]
- Ghatak, A.; Brar, M.J.; Agarwal, A.; Goel, N.; Rastogi, A.K.; Vaish, A.K.; Sircar, A.R.; Chandra, M. Oxy free radical system in heart failure and therapeutic role of vitamin E. Int. J. Cardiol. 1996, 57, 119–127. [Google Scholar] [CrossRef]
- Dhalla, A.K.; Hill, M.F.; Singal, P.K. Role of oxidative stress in transition of hypertrophy to heart failure. Am. J. Coll. Cardiol. 1996, 28, 506–514. [Google Scholar] [CrossRef]
- Duhamel, T.A.; Dhalla, N.S. New insight into the causes of heart failure. Drug Discov. Today Dis. Mech. 2017, 4, 175–184. [Google Scholar] [CrossRef]
- Dhalla, N.S.; Ademeova, A.; Kaur, M. Role of catecholamine oxidation in sudden cardiac death. Fund. Clin. Pharmacol. 2010, 24, 539–546. [Google Scholar] [CrossRef]
- Mialet-Perez, J.; Santin, Y.; Parini, A. Monoamine oxidase-A, serotonin and norepinephrine: Synergistic players in cardiac physiology and pathology. J. Neural Transm. 2018, 125, 1627–1634. [Google Scholar] [CrossRef]
- Li, B.; Tian, J.; Sun, Y.; Xu, T.-R.; Chi, R.-F.; Zhang, X.-L.; Hu, X.-L.; Zhang, Y.; Qin, F.-Z.; Zhang, W.-F. Activation of NADPH oxidase mediates increased endoplasmic reticulum stress and left ventricular remodeling after myocardial infarction in rabbits. Biochim. Biophys. Acta 2015, 1852, 805–815. [Google Scholar] [CrossRef]
- Nguyen Dinh Cat, A.; Montezano, A.C.; Burger, D.; Touyz, R.M. Angiotensin II, NADPH oxidase, and redox signaling in the vasculature. Antioxid. Redox. Signal 2013, 19, 1110–1120. [Google Scholar] [CrossRef] [PubMed]
- Ashrafian, H.; Frenneaux, M.P.; Opie, L.H. Metabolic mechanisms in heart failure. Circulation 2007, 116, 434–448. [Google Scholar] [CrossRef]
- Lopaschuk, G.D.; Ussher, J.R.; Folmes, C.D.; Jaswal, J.S.; Stanley, W.C. Myocardial fatty acid metabolism in health and disease. Physiol. Rev. 2010, 90, 207–258. [Google Scholar] [CrossRef]
- Wang, J.; Guo, X.; Dhalla, N.S. Modification of myosin protein and gene expression in failing hearts due to myocardial infarction by enalapril or losartan. Biochim. Biophys. Acta 2004, 1690, 177–184. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Shao, Q.; Ren, B.; Saini, H.K.; Netticadan, T.; Takeda, N.; Dhalla, N.S. Sarcoplasmic reticulum Ca2+ transport and gene expression in congestive heart failure are modified by imidapril treatment. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H1674–H1682. [Google Scholar] [CrossRef] [PubMed]
- Shao, Q.; Ren, B.; Elimban, V.; Tappia, P.S.; Takeda, N.; Dhalla, N.S. Modification of sarcolemmal Na+-K+-ATPase and Na+/Ca2+ exchanger expression in heart failure by blockade of renin-angiotensin system. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H2637–H2646. [Google Scholar] [CrossRef]
- Muller, A.L.; Dhalla, N.S. Role of various proteases in cardiac remodeling and progression of heart failure. Heart Fail. Rev. 2012, 17, 395–409. [Google Scholar] [CrossRef]
- Cantor, E.J.; Babick, A.P.; Vasanji, Z.; Dhalla, N.S.; Netticadan, T. A comparative serial echocardiographic analysis of cardiac structure and function in rats subjected to pressure or volume overload. J. Moll. Cell. Cardiol. 2005, 38, 777–786. [Google Scholar] [CrossRef]
- Carabello, B.A. Concentric versus eccentric remodeling. J. Card. Fail. 2002, 8, S258–S263. [Google Scholar] [CrossRef] [PubMed]
- Norton, G.R.; Woodiwiss, A.J.; Gaasch, W.H.; Mela, T.; Chung, E.S.; Aurigemma, G.P.; Meyer, T.E. Heart failure in pressure overload hypertrophy. The relative roles of ventricular remodeling and myocardial dysfunction. J. Am. Coll. Cardiol. 2002, 39, 664–671. [Google Scholar] [CrossRef]
- Wang, X.; Ren, B.; Liu, S.; Sentex, E.; Tappia, P.S.; Dhalla, N.S. Characterization of cardiac hypertrophy and heart failure due to volume overload in the rat. J. Appl. Physiol. 2003, 94, 752–763. [Google Scholar] [CrossRef] [PubMed]
- Dhalla, N.S.; Golfman, L.; Liu, X.; Sasaki, H.; Elimban, V.; Rapp, H. Subcellular remodeling and heart dysfunction in cardiac hypertrophy due to pressure overload. Ann. N. Y. Acad. Sci. 1999, 874, 100–110. [Google Scholar] [CrossRef]
- Frey, N.; Olson, E.N. Cardiac hypertrophy: The good, the bad, and the ugly. Annu. Rev. Physiol. 2003, 65, 45–79. [Google Scholar] [CrossRef]
- Ruzicka, M.; Leenen, F.H.H. Relevance of blockade of cardiac and circulatory angiotensin-converting enzyme for the prevention of volume overload-induced cardiac hypertrophy. Circulation 1995, 91, 16–19. [Google Scholar] [CrossRef]
- Ruzicka, M.; Keeley, F.W.; Leenen, F.H. The renin-angiotensin system and volume overload-induced changes in cardiac collagen and elastin. Circulation 1994, 90, 1989–1996. [Google Scholar] [CrossRef]
- Ganguly, P.K.; Lee, S.L.; Beamish, R.E.; Dhalla, N.S. Altered sympathetic system and adrenoceptors during the development of cardiac hypertrophy. Am. Heart J. 1989, 118, 520–525. [Google Scholar] [CrossRef]
- Lindpaintner, K.; Ganten, D. The cardiac renin-angiotensin system. An appraisal of present experimental and clinical evidence. Circ. Res. 1991, 68, 905–921. [Google Scholar] [CrossRef] [PubMed]
- Kromer, E.P.; Elsner, D.; Riegger, G.A. Role of neurohormonal systems for pressure induced left ventricular hypertrophy in experimental supravalvular aortic stenosis in rats. Am. J. Hypertens. 1991, 4, 521–524. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Sentex, E.; Golfman, L.; Takeda, S.; Osada, M.; Dhalla, N.S. Modification of cardiac subcellular remodeling due to pressure overload by captopril and losartan. Clin. Exp. Hypertens. 1999, 21, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Tarazi, R.C.; Sen, S.; Saragoca, M.; Khairallah, P. The multifactorial role of catecholamines in hypertensive cardiac hypertrophy. Eur. Heart J. 1982, 3 (Suppl. A), 103–110. [Google Scholar] [CrossRef]
- Kallfelt, B.J.; Hjalmarson, A.C.; Isaksson, O.G. In vitro effects of catecholamines on protein synthesis in perfused rat heart. J. Mol. Cell. Cardiol. 1976, 8, 787–802. [Google Scholar] [CrossRef]
- Takeo, S.; Elmoselhi, A.B.; Goel, R.; Sentex, E.; Wang, J.; Dhalla, N.S. Attenuation of changes in sarcoplasmic reticular and gene expression in cardiac hypertrophy by propranolol and verapamil. Mol. Cell. Biochem. 2000, 213, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Singal, T.; Dhalla, N.S.; Tappia, P.S. Phospholipase C may be involved in norepinephrine-induced cardiac hypertrophy. Biochem. Biophys. Res. Commun. 2004, 320, 1015–1019. [Google Scholar] [CrossRef] [PubMed]
- Ju, H.; Scammell-LaFleur, T.; Dixon, I.M.C. Altered mRNA abundance of calcium transport genes in cardiac myocytes in induced by angiotensin II. J. Mol. Cell. Cardiol. 1996, 28, 1119–1128. [Google Scholar] [CrossRef] [PubMed]
- Carabello, B.A.; Zile, M.R.; Tanaka, R.; Cooper, G. 4th. Left ventricular hypertrophy due to volume overload versus pressure overload. Am. J. Physiol. Heart Circ. Physiol. 1992, 263, H1137–H1144. [Google Scholar] [CrossRef]
- Litwin, S.E.; Katz, S.E.; Weinberg, E.O.; Lorell, B.H.; Aurigemma, G.P.; Douglas, P.S. Serial echocardiographic-Doppler assessment of left ventricular geometry and function in rats with pressure-overload hypertrophy. Chronic angiotensin-converting enzyme inhibition attenuates the transition to heart failure. Circulation 1995, 91, 2642–2654. [Google Scholar] [CrossRef]
- Mann, D.L.; Spann, J.F.; Cooper, G. Basic mechanisms and models in cardiac hypertrophy: Pathophysiological models. Mod. Concepts Cardiovasc. Dis. 1988, 57, 7–11. [Google Scholar]
- Modesti, P.A.; Vanni, S.; Bertolozzi, I.; Cecioni, I.; Polidori, G.; Paniccia, R.; Bandinelli, B.; Perna, A.; Liguori, P.; Boddi, M.; et al. Early sequence of cardiac adaptations and growth factor formation in pressure- and volume-overload hypertrophy. Am. J. Physiol. Heart Circ. Physiol. 2000, 279, H976–H985. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.F.; Brower, G.L.; Zhong, Q.; Murray, D.; Holland, M.; Janicki, J.S.; Zhong, J. Defective intracellular Ca2+ homeostasis contributes to myocytes dysfunction during ventricular remodeling induced by chronic volume overload in rats. Clin. Exp. Pharmacol. Physiol. 2008, 35, 827–835. [Google Scholar] [CrossRef] [PubMed]
- Spinale, F.G. Myocardial matrix remodeling and the matrix metalloproteinases: Influence on cardiac form and function. Physiol. Rev. 2007, 87, 1285–1342. [Google Scholar] [CrossRef] [PubMed]
- Dolgilevich, S.M.; Siri, F.M.; Atlas, S.A.; Eng, C. Changes in collagenase and collagen gene experession after induction of aortocaval fistula in rats. Am. J. Physiol. Heart Circ. Physiol. 2001, 281, H207–H214. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.M.; Tu, V.C. Apoptosis and heart failure: Mechanisms and therapeutic implications. Am. J. Cardiovasc. Drugs 2002, 2, 43–57. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Bing, O.H.; Long, X.; Robinson, K.G.; Lakatta, E.G. Increased cardiomyocyte apoptosis during the transition to heart failure in the spontaneously hypertensive rat. Am. J. Physiol. 1997, 272, H2213–H2319. [Google Scholar] [CrossRef]
- Dent, M.R.; Das, S.; Dhalla, N.S. Alterations in both death and survival signals for apoptosis in heart failure due to volume overload. J. Mol. Cell. Cardiol. 2007, 43, 726–732. [Google Scholar] [CrossRef]
- Vatner, D.E.; Asai, K.; Iwase, M.; Ishikawa, Y.; Shannon, R.P.; Homcy, C.J.; Vatner, S.F. β-Adrenergic receptors-G protein-adenylyl cyclase signal transduction in the failing heart. Am. J. Cardiol. 1999, 83, 80H–85H. [Google Scholar] [CrossRef]
- Vatner, D.E.; Vatner, S.F.; Fuji, A.M.; Homcy, C.J. Loss of high affinity cardiac β-Adrenergic receptors in dogs with heart failure. J. Clin. Investig. 1985, 76, 2259–2264. [Google Scholar] [CrossRef]
- Wang, X.; Sentex, E.; Chapman, D.; Dhalla, N.S. Alterations of adenylyl cyclase and G proteins in aortocaval shunt-induced heart failure. An. J. Physiol. Heart Circ. Physiol. 2004, 287, H118–H125. [Google Scholar] [CrossRef]
- Wang, X.; Sentex, E.; Saini, H.K.; Chapman, D.; Dhalla, N.S. Upregulation of β-Adrenergic receptors in heart failure due to volume overload. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H151–H159. [Google Scholar] [CrossRef]
- Prasad, K.; Gupta, J.B.; Kalra, J.; Lee, P.; Mantha, S.V.; Bharadwaj, B. Oxidative stress as a mechanism of cardiac failure in chronic volume overload in canine model. J. Mol. Cell. Cardiol. 1996, 28, 375–385. [Google Scholar] [CrossRef]
- Keith, M.; Geranmayegan, A.; Sole, M.J.; Kurian, R.; Robinson, A.; Omran, A.S.; Jeejeebhoy, K.N. Increased oxidative stress in patients with congestive heart failure. J. Am. Cell. Cardiol. 1998, 31, 1352–1356. [Google Scholar] [CrossRef]
- Singal, P.K.; Khaper, N.; Palace, V.; Kumar, D. The role of oxidative stress in the genesis of heart disease. Cardiovasc. Res. 1998, 3, 426–432. [Google Scholar] [CrossRef]
- Prasad, K.; Gupta, J.B.; Kalra, J.; Bharadwaj, B. Oxygen free radicals in volume overload heart failure. Mol. Cell. Biochem. 1992, 111, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Singal, P.K.; Khaper, N.; Farahmand, F.; Bello-Klein, A. Oxidative stress in congestive heart failure. Curr. Cardiol. Rep. 2000, 2, 206–211. [Google Scholar] [CrossRef] [PubMed]
- Takimoto, E.; Kass, D.A. Role of oxidative stress in cardiac hypertrophy and remodeling. Hypertension 2007, 49, 241–248. [Google Scholar] [CrossRef]
- Seddon, M.; Looi, Y.H.; Shah, A.M. Oxidative stress and redox signaling in cardiac hypertrophy and heart failure. Heart 2007, 93, 903–907. [Google Scholar] [CrossRef]
- Tsutsui, H.; Kinugawa, S.; Matsushima, S. Oxidative stress and heart failure. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H2181–H2190. [Google Scholar] [CrossRef]
- Maulik, S.K.; Kumar, S. Oxidative stress and cardiac hypertrophy: A review. Toxicol. Mech. Methods 2012, 22, 359–366. [Google Scholar] [CrossRef]
- Tsutsui, H. Mitochondrial oxidative stress and heart failure. Intern. Med. 2006, 45, 809–813. [Google Scholar] [CrossRef] [PubMed]
- Ramachandra, C.J.A.; Cong, S.; Chan, X. Oxidative stress in cardiac hypertrophy: From molecular mechanisms to novel therapeutic targets. Free Radic. Biol. Med. 2021, 166, 297–312. [Google Scholar] [CrossRef]
- Tsutsui, H.; Kinugawa, S.; Matsushima, S. Oxidative stress and mitochondrial DNA damage in heart failure. Circ. J. 2008, 72 (Suppl. A), A31–A37. [Google Scholar] [CrossRef] [PubMed]
- Antognelli, C.; Trapani, E.; Delle Monache, S.; Perrelli, A.; Daga, M.; Pizzimenti, S.; Barrera, G.; Cassoni, P.; Angelucci, A.; Trabalzini, L.; et al. KRIT1 loss-of-function induces a chronic Nrf2-mediated adaptive homeostasis that sensitizes cells to oxidative stress: Implication for cerebral cavernous malformation disease. Free Radic. Biol. Med. 2018, 115, 202–218. [Google Scholar] [CrossRef] [PubMed]
- Ziolo, M.T.; Houser, S.R. Abnormal Ca2+ cycling in failing ventricular myocytes: Role of NOS1-mediated Nitroso-Redox balance. Antioxid. Redox Signal. 2014, 21, 2044–2059. [Google Scholar] [CrossRef]
- Dhalla, N.S.; Das, P.K.; Sharma, G.P. Subcellular basis of cardiac contractile failure. J. Mol. Cell. Cardiol. 1978, 10, 363–385. [Google Scholar] [CrossRef]
- Dhalla, N.S.; Shao, Q.; Panagia, V. Remodeling of cardiac membranes during the development of congestive heart failure. Heart Fail. Rev. 1998, 2, 261–272. [Google Scholar] [CrossRef]
- Dhalla, N.S.; Rangi, S.; Babick, A.P.; Zieroth, S.; Elimban, V. Cardiac remodeling and subcellular defects in heart failure due to myocardial infarction and aging. Heart Fail. Rev. 2012, 17, 671–681. [Google Scholar] [CrossRef]
- Dhingra, S.; Sharma, A.K.; Singla, D.K.; Singal, P.K. P38 and ERK ½ MAPKs mediate the interplay of TNF-alpha and IL-10 in regulating oxidative stress and cardiac myocyte apoptosis. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H3524–H3531. [Google Scholar] [CrossRef]
- Milinkovic, I.; Polovina, M.; Simeunovic, D.S.; Ašanin, M.; Seferović, P.M. Oxidative stress and inflammation in heart failure: The best is yet to come. Eur. J. Prev. Cardiol. 2020, 27, 490–493. [Google Scholar] [CrossRef]
- Neri, M.; Fineschi, V.; Di Paolo, M.; Pomara, C.; Riezzo, I.; Turillazzi, E.; Cerretani, D. Cardiac oxidative stress and inflammatory cytokines response after myocardial infarction. Curr. Vasc. Pharmacol. 2015, 13, 26–36. [Google Scholar] [CrossRef]
- Ayoub, K.F.; Pothineni, N.V.K.; Rutland, J.; Ding, Z.; Mehta, J.L. Immunity, inflammation, and oxidative stress in heart failure: Emerging molecular targets. Cardiovasc. Drugs Ther. 2017, 31, 593–608. [Google Scholar] [CrossRef]
- van der Pol, A.; van Gilst, W.H.; Voors, A.A.; van der Meer, P. Treating oxidative stress in heart failure: Past, present and future. Eur. J. Heart Fail. 2019, 21, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Bartekova, M.; Adameova, A.; Gorbe, A.; Ferenczyová, K.; Pecháňová, O.; Lazou, A.; Dhalla, N.S.; Ferdinandy, P.; Giricz, Z. Natural and synthetic antioxidants targeting cardiac oxidative stress and redox signaling in cardiometabolic diseases. Free Radic. Biol. Med. 2021, 169, 446–477. [Google Scholar] [CrossRef] [PubMed]
- Dhalla, N.S.; Saini, H.K.; Tappia, P.S.; Sethi, R.; Mengi, S.A.; Gupta, S.K. Potential role and mechanism of subcellular remodeling in cardiac function due to ischemic heart disease. J. Cardiovasc. Med. 2007, 8, 238–250. [Google Scholar] [CrossRef] [PubMed]
- Dhalla, N.S.; Shah, A.K.; Tappia, P.S. Role of oxidative stress in metabolic and subcellular abnormalities in diabetic cardiomyopathy. Int. J. Mol. Sci. 2020, 21, 2413. [Google Scholar] [CrossRef]
- Ducas, A.; Bartekova, M.; Dhalla, N.S. Ischemia-reperfusion injury of the heart: Moving forward with our knowledge. J. Heart Health 2015, 1. [Google Scholar] [CrossRef]
- Dhalla, N.S.; Golfman, L.; Takeda, S.; Takeda, N.; Nagano, M. Evidence for the role of oxidative stress in acute ischemic heart disease: A brief review. Can. J. Cardiol. 1999, 15, 587–593. [Google Scholar]
- Dhalla, N.S.; Panagia, V.; Singal, P.K.; Makino, N.; Dixon, I.M.C.; Eyolfson, D.A. Alterations in heart membrane calcium transport during the development of ischemia-reperfusion injury. J. Moll. Cell. Cardiol. 1988, 20 (Suppl. 2), 3–13. [Google Scholar] [CrossRef]
- Burton, K.P.; McCord, J.M.; Ghai, G. Myocardial alterations due to free-radical generation. Am. J. Physiol. 1984, 246, H776–H783. [Google Scholar] [CrossRef]
- Gupta, M.; Singal, P.K. Oxygen radical injury in the presence of desferal, a specific iron-chelating agent. Biochem. Pharmacol. 1987, 36, 3774–3777. [Google Scholar] [CrossRef]
- Hammond, B.; Hess, M.L. The oxygen free radical system: Potential mediator of myocardial injury. J. Am. Coll. Cardiol. 1985, 6, 215–220. [Google Scholar] [CrossRef]
- Hess, M.L.; Manson, N.H.; Okabe, E. Involvement of free radicals in pathophysiology of ischemic heart disease. Can. J. Physiol. Pharmacol. 1982, 60, 1382–1389. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Akera, T.O. O2 free radicals: Cause of ischemia-reperfusion injury to cardiac Na+-K+ ATPase. Am. J. Physiol. 1987, 256, H368–H374. [Google Scholar] [CrossRef]
- Zweier, J.L.; Flaherty, J.T.; Weisfeldt, M.L. Direct measurement of free radical generation following reperfusion of ischemic myocardium. Proc. Nat. Acad. Sci. USA 1987, 84, 1404–1407. [Google Scholar] [CrossRef] [PubMed]
- Ostadal, P.; Elmoselhi, A.B.; Zdobnicka, I.; Lukas, A.; Elimban, V.; Dhalla, N.S. Role of oxidative stress in ischemia-reperfusion-induced changes in, Na+,K+-ATPase isoform expression in rat heart. Antioxid. Redox. Signal. 2007, 6, 914–923. [Google Scholar] [CrossRef] [PubMed]
- Temsah, R.M.; Netticadan, T.; Chapman, D.; Takeda, S.; Mochizuki, S.; Dhalla, N.S. Alterations in sarcoplasmic reticulum function and gene expression in ischemic-reperfused rat heart. Am. J. Physiol. 1999, 277, H584–H594. [Google Scholar] [CrossRef] [PubMed]
- Makazan, Z.; Saini, H.K.; Dhalla, N.S. Role of oxidative stress in alterations of mitochondrial function in ischemic-reperfused hearts. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H1986–H1994. [Google Scholar] [CrossRef] [PubMed]
- Madikka, S.; Elimban, V.; Chapman, D.; Dhalla, N.S. Role of oxidative stress in ischemia-reperfusion-induced alterations in myofibrillar ATPase activities and gene expression in the heart. Can. J. Physiol. Pharmacol. 2009, 87, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Ostadal, P.; Elmoselhi, A.B.; Zdbonicka, I.; Lukas, A.; Chapman, D.; Dhalla, N.S. Ischemia-reperfusion alters gene expression of Na+-K+ ATPase isoforms in rat heart. Biochem. Biophys. Res. Commun. 2003, 306, 457–462. [Google Scholar] [CrossRef]
- Dixon, I.M.C.; Kaneko, M.; Hata, T.; Panagia, V.; Dhalla, N.S. Alterations in cardiac membrane Ca2+ transport during oxidative stress. Mol. Cell. Biochem. 1990, 99, 125–133. [Google Scholar] [CrossRef]
- Persad, S.; Takeda, S.; Panagia, V.; Dhalla, N.S. β-adrenoceptor-linked signal transduction in ischemic-reperfused heart and scavenging of oxyradicals. J. Moll. Cell. Cardiol. 1997, 29, 545–558. [Google Scholar] [CrossRef]
- Netticadan, T.; Temsah, R.; Osada, M.; Dhalla, N.S. Status of Ca2+/calmodulin protein kinase phosphorylation of cardiac SR proteins in ischemia-reperfusion. Am. J. Physiol. 1999, 277, C384–C391. [Google Scholar] [CrossRef]
- Bartekova, M.; Barancik, M.; Ferenczyova, K.; Dhalla, N.S. Beneficial effects of N-acetylcysteine and N-mercaptopropionylglycine in ischemia reperfusion injury in the heart. Curr. Med. Chem. 2018, 25, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Elmoselhi, A.B.; Lukas, A.; Ostadal, P.; Dhalla, N.S. Preconditioning attenuates ischemia-reperfusion-induced remodeling of Na+-K+-ATPase in hearts. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H1055–H1063. [Google Scholar] [CrossRef]
- Osada, M.; Netticadan, T.; Kawabata, K.; Tamura, K.; Dhalla, N.S. Ischemic preconditioning prevents I/R-induced alterations in SR calcium-calmodulin protein kinase II. Am. J. Physiol. Heart Circ. Physiol. 2000, 278, H1791–H1798. [Google Scholar] [CrossRef] [PubMed]
- Osada, M.; Netticadan, T.; Tamura, K.; Dhalla, N.S. Modification of ischemia-reperfusion-induced changes in cardiac sarcoplasmic reticulum by preconditioning. Am. J. Physiol. 1998, 274, H2025–H2034. [Google Scholar] [CrossRef]
- Temsah, R.M.; Kawabata, K.; Chapman, D.; Dhalla, N.S. Preconditioning prevents alterations in cardiac SR gene expression due to ischemia-reperfusion. Am. J. Physiol. Heart Circ. Physiol. 2002, 282, H1461–H1466. [Google Scholar] [CrossRef]
- Saini, H.K.; Machackova, J.; Dhalla, N.S. Role of reactive oxygen species in ischemic preconditioning of subcellular organelles in the heart. Antioxid. Redox Signal. 2004, 6, 393–404. [Google Scholar] [CrossRef]
- Muller, A.L.; Dhalla, N.S. Mechanisms of the beneficial actions of ischemic preconditioning on subcellular remodeling in ischemic-reperfused heart. Curr. Cardiol. Rev. 2010, 6, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.B.; Hryshko, L.; Freed, D.; Dhalla, N.S. Activation of proteolytic enzymes and depression of the sarcolemmal Na+/K+-ATPase in ischemia-reperfused heart may be mediated through oxidative stress. Can. J. Physiol. Pharmacol. 2012, 90, 249–260. [Google Scholar] [CrossRef] [PubMed]
- Muller, A.L.; Freed, D.; Dhalla, N.S. Activation of proteases and changes in Na+-K+-ATPase subunits in hearts subjected to ischemia-reperfusion. J. Appl. Physiol. 2013, 114, 351–360. [Google Scholar] [CrossRef][Green Version]
- Singh, R.B.; Dhalla, N.S. Ischemia-reperfusion-induced changes in sarcolemmal Na+/K+-ATPase are due to the activation of calpain in the heart. Can. J. Physiol. Pharmacol. 2010, 88, 388–397. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.B.; Chohan, P.K.; Dhalla, N.S.; Netticadan, T. The sarcoplasmic reticulum proteins are targets for calpain action in the ischemic-reperfused heart. J. Moll. Cell. Cardiol. 2004, 37, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Dhalla, N.S.; Elmoselhi, A.B.; Hata, T.; Makino, N. Status of myocardial antioxidants in ischemia-reperfusion injury. Cardiovsac. Res. 2000, 47, 446–456. [Google Scholar] [CrossRef]
- Matsubara, T.; Dhalla, N.S. Relationship between mechanical dysfunction and depression of sarcolemmal Ca2+-pump activity in hearts perfused with oxygen free radicals. Mol. Cell. Biochem. 1996, 160–161, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, T.; Dhalla, N.S. Effect of oxygen free radicals on cardiac contractile activity and sarcolemmal Na+-Ca2+ exchange. J. Cardiovasc. Pharmacol. Therapeut. 1996, 1, 211–218. [Google Scholar] [CrossRef]
- Persad, S.; Panagia, V.; Dhalla, N.S. Role of H2O2 in changing β-adrenoceptor and adenylyl cyclase in ischemia-reperfused hearts. Mol. Cell. Biochem. 1998, 186, 99–106. [Google Scholar] [CrossRef]
- Persad, S.; Takeda, S.; Dhalla, N.S. Alterations in β-adrenoceptor mechanisms in hearts perfused with xanthine plus xanthine oxidase. J. Cardiovasc. Pharmacol. Therapeut. 1997, 2, 115–124. [Google Scholar] [CrossRef]
- Kaneko, M.; Beamish, R.E.; Dhalla, N.S. Depression of heart sarcolemmal Ca2+-pump activity by oxygen free radicals. Am. J. Physiol. 1989, 256, H368–H374. [Google Scholar] [CrossRef]
- Kaneko, M.; Elimban, V.; Dhalla, N.S. Mechanism for depression of heart sarcolemmal Ca2+ pump by oxygen free radicals. Am. J. Physiol. 1989, 257, H804–H811. [Google Scholar] [CrossRef]
- Hata, T.; Kaneko, M.; Beamish, R.E.; Dhalla, N.S. Influence of oxygen free radicals on heart sarcolemmal Na+-Ca2+ exchange. Coron. Artery Dis. 1991, 2, 397–407. [Google Scholar] [CrossRef]
- Shao, Q.; Matsubara, T.; Bhatt, S.K.; Dhalla, N.S. Inhibition of cardiac sarcolemma Na+-K+ ATPase by oxyradical generating systems. Mol. Cell. Biochem. 1995, 147, 139–144. [Google Scholar] [CrossRef]
- Kramer, J.H.; Mak, I.T.; Weglicki, W.B. Differential sensitivity of canine cardiac sarcolemmal and microsomal enzymes to inhibition by free radical-induced lipid peroxidation. Circ. Res. 1984, 55, 120–124. [Google Scholar] [CrossRef]
- Okabe, E.; Hess, M.L.; Oyama, M.; Ito, H. Characterization of free radical-mediated damage of canine cardiac sarcoplasmic reticulum. Arch. Biochem. Biophys. 1983, 225, 164–177. [Google Scholar] [CrossRef]
- Rowe, G.T.; Manson, N.H.; Caplan, M.; Hess, M.L. Hydrogen peroxide and hydroxyl radical mediation of activated leukocyte depression of cardiac sarcoplasmic reticulum. Circ. Res. 1983, 53, 584–591. [Google Scholar] [CrossRef] [PubMed]
- Scherer, N.M.; Deamer, D.W. Oxidative stress impairs the function of sarcoplasmic reticulum by oxidation of sulfhydryl groups in the Ca2+-ATPase. Arch. Biochem. Biophys. 1986, 246, 589–610. [Google Scholar] [CrossRef]
- Kaneko, M.; Lee, S.L.; Wolf, C.M.; Dhalla, N.S. Reduction of calcium channel antagonist binding sites by oxygen free radicals in rat heart. J. Moll. Cell. Cardiol. 1989, 21, 935–943. [Google Scholar] [CrossRef]
- Kaneko, M.; Singal, P.K.; Dhalla, N.S. Alterations in heart sarcolemmal Ca2+-ATPase and Ca2+-binding activities due to oxygen free radicals. Basic Res. Cardiol. 1990, 85, 45–54. [Google Scholar] [CrossRef]
- Kaneko, M.; Chapman, D.C.; Ganguly, P.K.; Beamish, R.E.; Dhalla, N.S. Modification of cardiac adrenergic receptors by oxygen free radicals. Am. J. Physiol. 1991, 260, H821–H826. [Google Scholar] [CrossRef]
- Persad, S.; Rupp, H.; Jindal, R.; Arneja, J.; Dhalla, N.S. Modification of cardiac β-adrenoceptor mechanisms by H2O2. Am. J. Physiol. 1998, 274, H416–H423. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Kaneko, M.; Chapman, D.C.; Dhalla, N.S. Alterations in cardiac contractile proteins due to oxygen free radicals. Biochim. Biophys. Acta 1991, 1074, 95–100. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shah, A.K.; Bhullar, S.K.; Elimban, V.; Dhalla, N.S. Oxidative Stress as A Mechanism for Functional Alterations in Cardiac Hypertrophy and Heart Failure. Antioxidants 2021, 10, 931. https://doi.org/10.3390/antiox10060931
Shah AK, Bhullar SK, Elimban V, Dhalla NS. Oxidative Stress as A Mechanism for Functional Alterations in Cardiac Hypertrophy and Heart Failure. Antioxidants. 2021; 10(6):931. https://doi.org/10.3390/antiox10060931
Chicago/Turabian StyleShah, Anureet K., Sukhwinder K. Bhullar, Vijayan Elimban, and Naranjan S. Dhalla. 2021. "Oxidative Stress as A Mechanism for Functional Alterations in Cardiac Hypertrophy and Heart Failure" Antioxidants 10, no. 6: 931. https://doi.org/10.3390/antiox10060931
APA StyleShah, A. K., Bhullar, S. K., Elimban, V., & Dhalla, N. S. (2021). Oxidative Stress as A Mechanism for Functional Alterations in Cardiac Hypertrophy and Heart Failure. Antioxidants, 10(6), 931. https://doi.org/10.3390/antiox10060931