
Effect of Chronic Stress Present in Fibroblasts Derived from Patients with a Sporadic Form of AD on Mitochondrial Function and Mitochondrial Turnover

Abstract
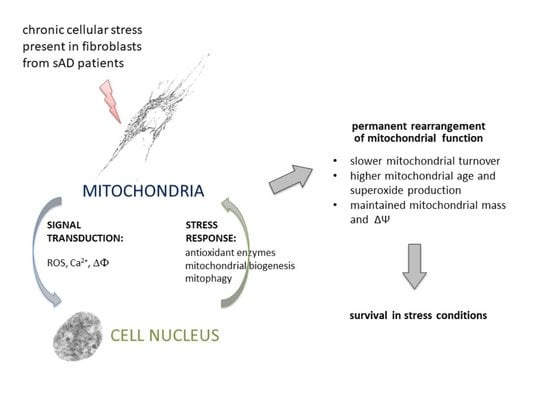
:
1. Introduction
2. Materials and Methods
2.1. Chemicals and Antibodies
2.2. Patients and Control Individuals
2.3. Primary Fibroblast Culture
2.4. Live Cell Studies
2.5. Whole-Cell Extracts and Immunoblotting
2.6. Visualization of Mitochondrial Age with MitoTimer Vector
- Green fluorescence: excitation/emission—490/500–540 nm, EM gain—600, exposure time—750 ms, laser power—4%;
- Red fluorescence: excitation/emission—550/580–640 nm, EM gain—700, exposure time—150 ms, laser power—4%.
2.7. Statistical Analysis
3. Results
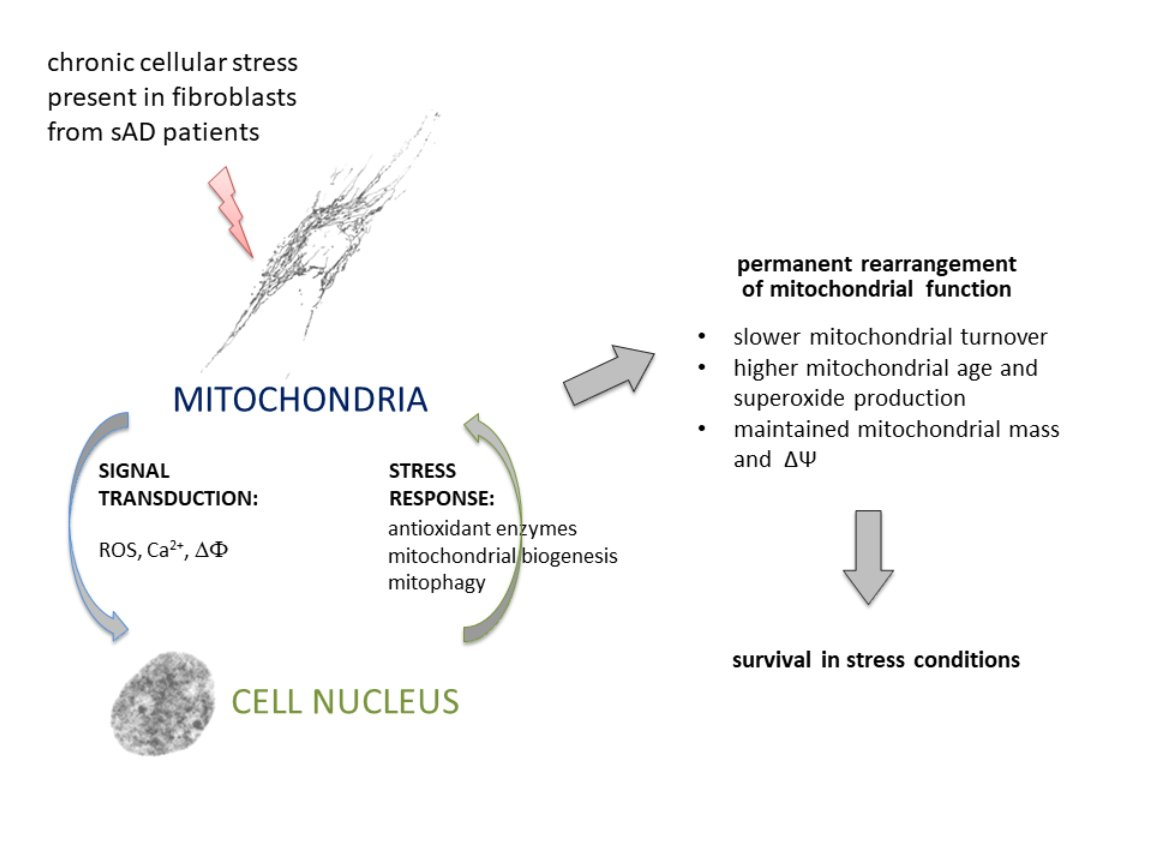
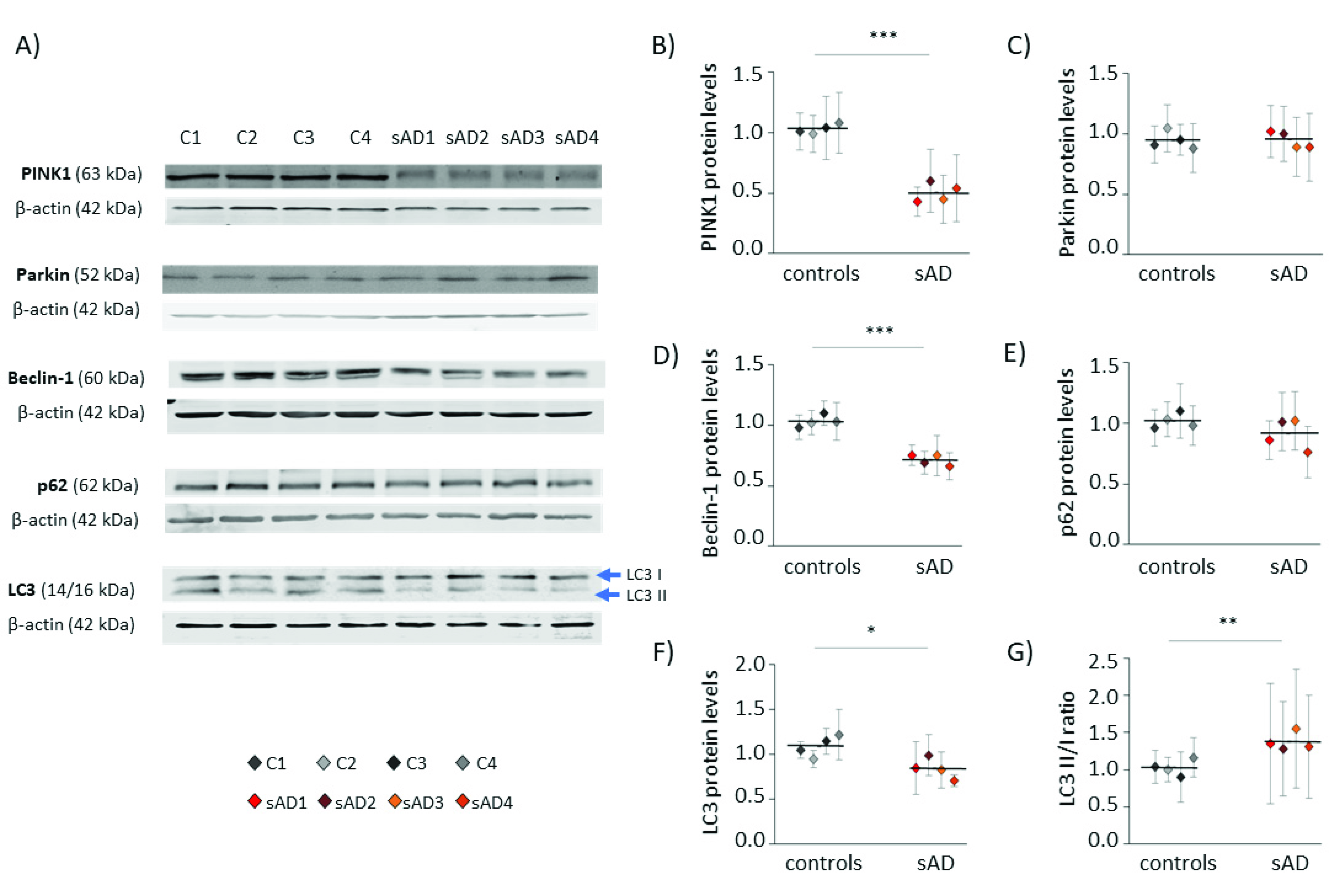
3.1. Fibroblasts from sAD Patients Show Signs of Increased Cellular Stress
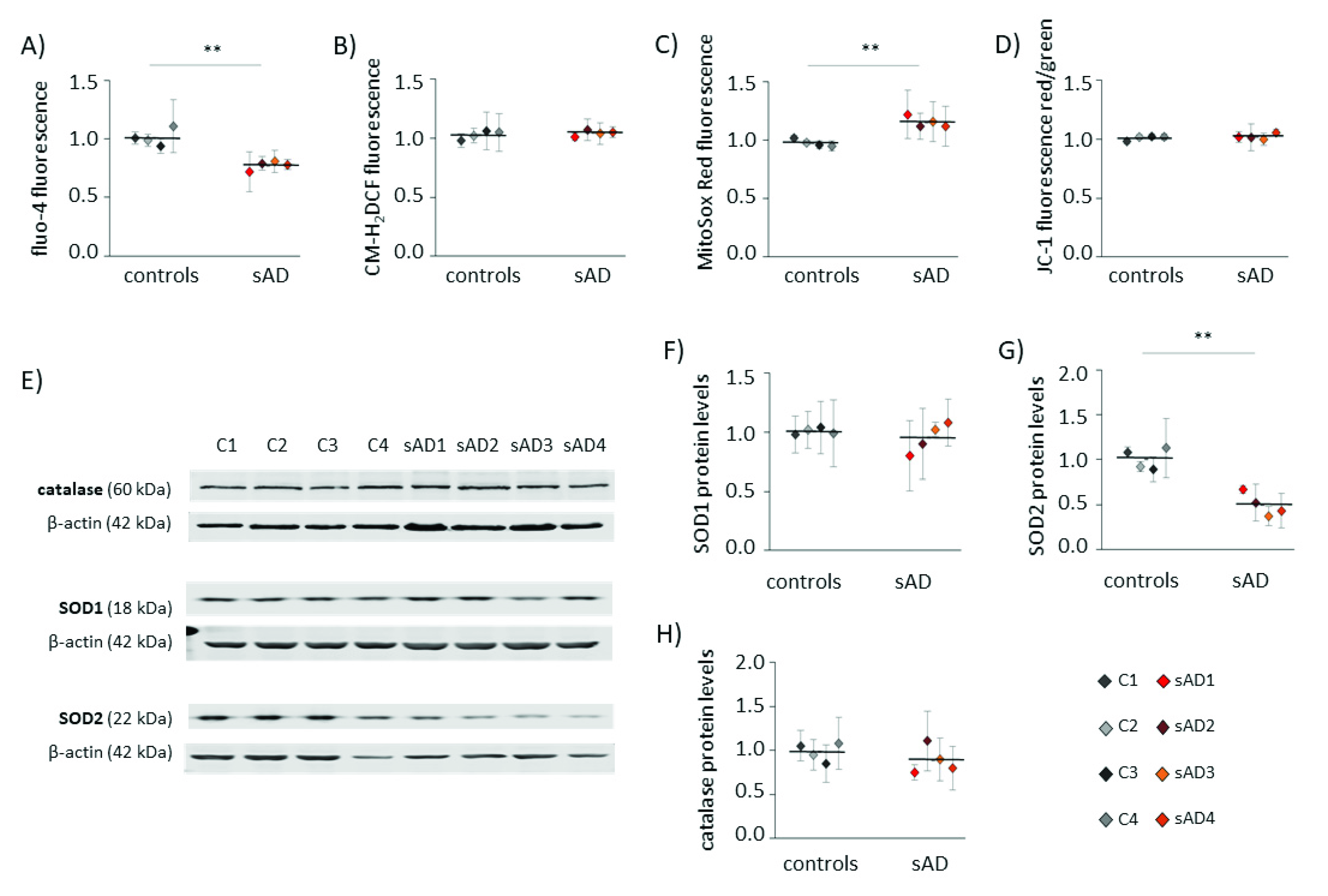
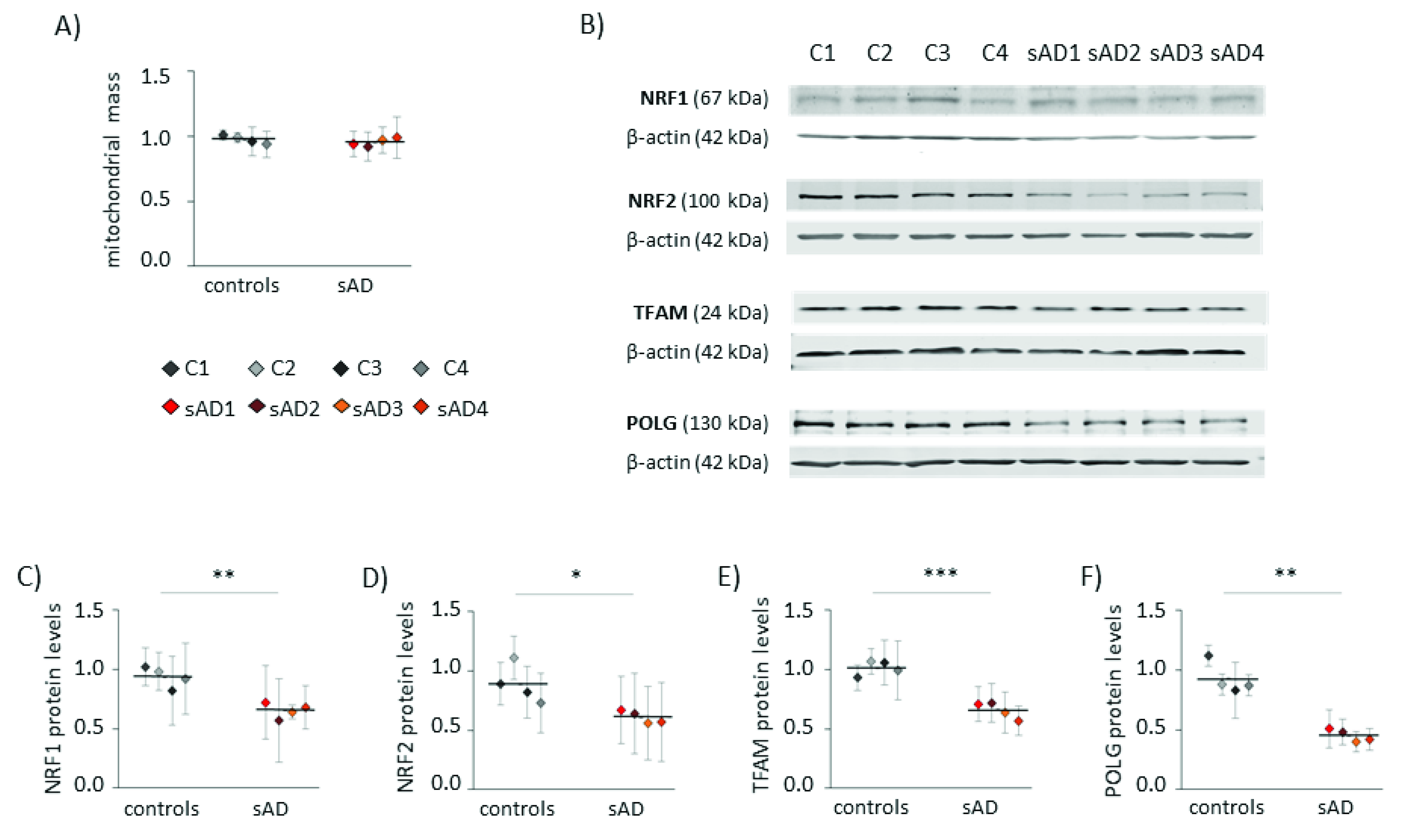
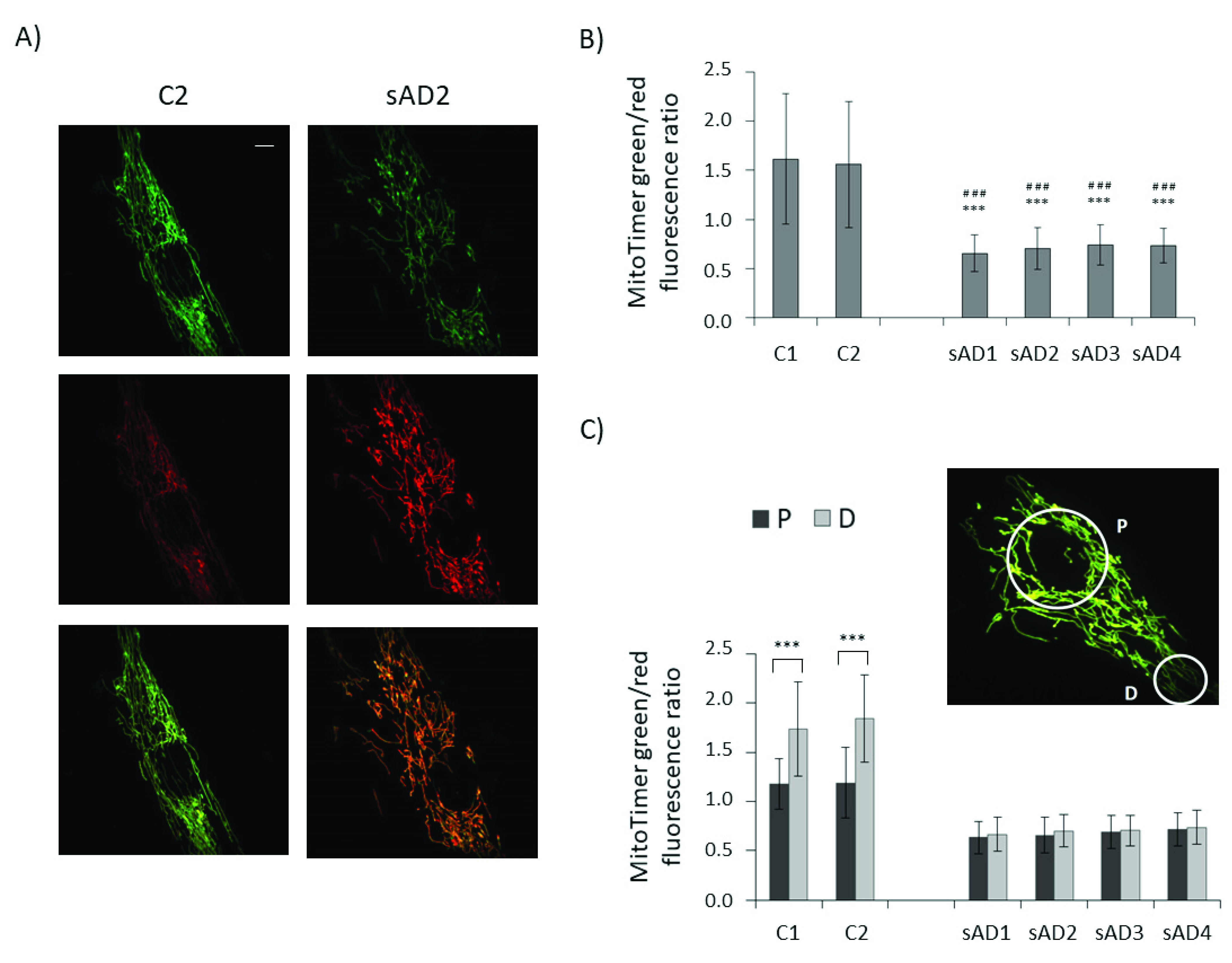
3.2. Mitochondrial Turnover in sAD Fibroblasts Is Slower Than That in Control Cells
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Area-Gomez, E.; Schon, E.A. Alzheimer Disease. Adv. Exp. Med. Biol. 2017, 997, 149–156. [Google Scholar] [CrossRef]
- Dos Santos Picanco, L.C.; Ozela, P.F.; de Fatima de Brito Brito, M.; Pinheiro, A.A.; Padilha, E.C.; Braga, F.S.; de Paula da Silva, C.H.T.; Dos Santos, C.B.R.; Rosa, J.M.C.; da Silva Hage-Melim, L.I. Alzheimer’s Disease: A Review from the Pathophysiology to Diagnosis, New Perspectives for Pharmacological Treatment. Curr. Med. Chem. 2018, 25, 3141–3159. [Google Scholar] [CrossRef] [PubMed]
- Gallardo, G.; Holtzman, D.M. Amyloid-β and Tau at the Crossroads of Alzheimer’s Disease. Adv. Exp. Med. Biol. 2019, 1184, 187–203. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.L.; Morstorf, T.; Zhong, K. Alzheimer’s Disease Drug-Development Pipeline: Few Candidates, Frequent Failures. Alzheimer’s Res. Ther. 2014, 6, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feldman, H.H.; Haas, M.; Gandy, S.; Schoepp, D.D.; Cross, A.J.; Mayeux, R.; Sperling, R.A.; Fillit, H.; van de Hoef, D.L.; Dougal, S.; et al. Alzheimer’s Disease Research and Development: A Call for a New Research Roadmap. Ann. N. Y. Acad. Sci. 2014, 1313, 1–16. [Google Scholar] [CrossRef] [PubMed]
- De Strooper, B. Lessons from a Failed γ-Secretase Alzheimer Trial. Cell 2014, 159, 721–726. [Google Scholar] [CrossRef] [Green Version]
- Swerdlow, R.H.; Khan, S.M. A “Mitochondrial Cascade Hypothesis” for Sporadic Alzheimer’s Disease. Med. Hypotheses 2004, 63, 8–20. [Google Scholar] [CrossRef]
- Perez Ortiz, J.M.; Swerdlow, R.H. Mitochondrial Dysfunction in Alzheimer’s Disease: Role in Pathogenesis and Novel Therapeutic Opportunities. Br. J. Pharmacol. 2019, 176, 3489–3507. [Google Scholar] [CrossRef]
- Galla, L.; Redolfi, N.; Pozzan, T.; Pizzo, P.; Greotti, E. Intracellular Calcium Dysregulation by the Alzheimer’s Disease-Linked Protein Presenilin 2. Int. J. Mol. Sci. 2020, 21, 770. [Google Scholar] [CrossRef] [Green Version]
- Xie, C.; Aman, Y.; Adriaanse, B.A.; Cader, M.Z.; Plun-Favreau, H.; Xiao, J.; Fang, E.F. Culprit or Bystander: Defective Mitophagy in Alzheimer’s Disease. Front. Cell Dev. Biol. 2020, 7, 391. [Google Scholar] [CrossRef]
- McGuire, P.J. Mitochondrial Dysfunction and the Aging Immune System. Biology 2019, 8, 26. [Google Scholar] [CrossRef] [Green Version]
- Sprenger, H.-G.; Langer, T. The Good and the Bad of Mitochondrial Breakups. Trends Cell Biol. 2019, 29, 888–900. [Google Scholar] [CrossRef]
- Zorov, D.B.; Krasnikov, B.F.; Kuzminova, A.E.; Vysokikh, M.Y.; Zorova, L.D. Mitochondria Revisited. Alternative Functions of Mitochondria. Biosci. Rep. 1997, 17, 507–520. [Google Scholar] [CrossRef] [Green Version]
- Meyer, J.N.; Leuthner, T.C.; Luz, A.L. Mitochondrial Fusion, Fission, and Mitochondrial Toxicity. Toxicology 2017, 391, 42–53. [Google Scholar] [CrossRef]
- Hunt, R.J.; Bateman, J.M. Mitochondrial Retrograde Signaling in the Nervous System. FEBS Lett. 2018, 592, 663–678. [Google Scholar] [CrossRef] [Green Version]
- da Cunha, F.M.; Torelli, N.Q.; Kowaltowski, A.J. Mitochondrial Retrograde Signaling: Triggers, Pathways, and Outcomes. Oxidative Med. Cell. Longev. 2015, 2015, 482582. [Google Scholar] [CrossRef] [Green Version]
- Strobbe, D.; Sharma, S.; Campanella, M. Links between Mitochondrial Retrograde Response and Mitophagy in Pathogenic Cell Signalling. Cell. Mol. Life Sci. 2021, 78, 3767–3775. [Google Scholar] [CrossRef]
- Hauptmann, S.; Scherping, I.; Dröse, S.; Brandt, U.; Schulz, K.L.; Jendrach, M.; Leuner, K.; Eckert, A.; Müller, W.E. Mitochondrial Dysfunction: An Early Event in Alzheimer Pathology Accumulates with Age in AD Transgenic Mice. Neurobiol. Aging 2009, 30, 1574–1586. [Google Scholar] [CrossRef]
- Stockburger, C.; Gold, V.A.M.; Pallas, T.; Kolesova, N.; Miano, D.; Leuner, K.; Müller, W.E. A Cell Model for the Initial Phase of Sporadic Alzheimer’s Disease. J. Alzheimer’s Dis. 2014, 42, 395–411. [Google Scholar] [CrossRef]
- Baker, A.C.; Ko, L.-W.; Blass, J.P. Systemic Manifestations of Alzheimer’s Disease. AGE 1988, 11, 60–65. [Google Scholar] [CrossRef]
- Cecchi, C.; Fiorillo, C.; Sorbi, S.; Latorraca, S.; Nacmias, B.; Bagnoli, S.; Nassi, P.; Liguri, G. Oxidative Stress and Reduced Antioxidant Defenses in Peripheral Cells from Familial Alzheimer’s Patients. Free Radic. Biol. Med. 2002, 33, 1372–1379. [Google Scholar] [CrossRef]
- Khan, T.K.; Alkon, D.L. Peripheral Biomarkers of Alzheimer’s Disease. J. Alzheimer’s Dis. 2015, 44, 729–744. [Google Scholar] [CrossRef] [Green Version]
- Trushina, E. Alzheimer’s Disease Mechanisms in Peripheral Cells: Promises and Challenges. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2019, 5, 652–660. [Google Scholar] [CrossRef]
- Gasparini, L.; Racchi, M.; Binetti, G.; Trabucchi, M.; Solerte, S.B.; Alkon, D.; Etcheberrigaray, R.; Gibson, G.; Blass, J.; Paoletti, R.; et al. Peripheral Markers in Testing Pathophysiological Hypotheses and Diagnosing Alzheimer’s Disease. FASEB J. 1998, 12, 17–34. [Google Scholar] [CrossRef] [Green Version]
- Pickles, S.; Vigié, P.; Youle, R.J. Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr. Biol. 2018, 28, R170–R185. [Google Scholar] [CrossRef] [Green Version]
- Dodson, M.; Darley-Usmar, V.; Zhang, J. Cellular Metabolic and Autophagic Pathways: Traffic Control by Redox Signaling. Free Radic. Biol. Med. 2013, 63, 207–221. [Google Scholar] [CrossRef] [Green Version]
- Okatsu, K.; Saisho, K.; Shimanuki, M.; Nakada, K.; Shitara, H.; Sou, Y.; Kimura, M.; Sato, S.; Hattori, N.; Komatsu, M.; et al. P62/SQSTM1 Cooperates with Parkin for Perinuclear Clustering of Depolarized Mitochondria. Genes Cells 2010, 15, 887–900. [Google Scholar] [CrossRef] [Green Version]
- Hernandez, G.; Thornton, C.; Stotland, A.; Lui, D.; Sin, J.; Ramil, J.; Magee, N.; Andres, A.; Quarato, G.; Carreira, R.S.; et al. MitoTimer: A Novel Tool for Monitoring Mitochondrial Turnover. Autophagy 2013, 9, 1852–1861. [Google Scholar] [CrossRef] [Green Version]
- Ferree, A.W.; Trudeau, K.; Zik, E.; Benador, I.Y.; Twig, G.; Gottlieb, R.A.; Shirihai, O.S. MitoTimer Probe Reveals the Impact of Autophagy, Fusion, and Motility on Subcellular Distribution of Young and Old Mitochondrial Protein and on Relative Mitochondrial Protein Age. Autophagy 2013, 9, 1887–1896. [Google Scholar] [CrossRef] [Green Version]
- Trudeau, K.M.; Gottlieb, R.A.; Shirihai, O.S. Measurement of Mitochondrial Turnover and Life Cycle Using MitoTimer. Methods Enzymol. 2014, 547, 21–38. [Google Scholar] [CrossRef]
- Walczak, J.; Dębska-Vielhaber, G.; Vielhaber, S.; Szymański, J.; Charzyńska, A.; Duszyński, J.; Szczepanowska, J. Distinction of Sporadic and Familial Forms of ALS Based on Mitochondrial Characteristics. FASEB J. 2019, 33, 4388–4403. [Google Scholar] [CrossRef]
- Drummond, E.; Wisniewski, T. Alzheimer’s Disease: Experimental Models and Reality. Acta Neuropathol. 2017, 133, 155–175. [Google Scholar] [CrossRef]
- Israel, M.A.; Yuan, S.H.; Bardy, C.; Reyna, S.M.; Mu, Y.; Herrera, C.; Hefferan, M.P.; Van Gorp, S.; Nazor, K.L.; Boscolo, F.S.; et al. Probing Sporadic and Familial Alzheimer’s Disease Using Induced Pluripotent Stem Cells. Nature 2012, 482, 216–220. [Google Scholar] [CrossRef]
- Bialopiotrowicz, E.; Kuzniewska, B.; Kachamakova-Trojanowska, N.; Barcikowska, M.; Kuznicki, J.; Wojda, U. Cell Cycle Regulation Distinguishes Lymphocytes from Sporadic and Familial Alzheimer’s Disease Patients. Neurobiol. Aging 2011, 32, 2319.e13–2319.e26. [Google Scholar] [CrossRef]
- Piaceri, I.; Nacmias, B.; Sorbi, S. Genetics of Familial and Sporadic Alzheimer’s Disease. Front. Biosci. 2013, 5, 167–177. [Google Scholar] [CrossRef] [Green Version]
- LaFerla, F.M.; Green, K.N. Animal Models of Alzheimer Disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006320. [Google Scholar] [CrossRef] [Green Version]
- Martín-Maestro, P.; Gargini, R.; García, E.; Perry, G.; Avila, J.; García-Escudero, V. Slower Dynamics and Aged Mitochondria in Sporadic Alzheimer’s Disease. Oxidative Med. Cell. Longev. 2017, 2017, 9302761. [Google Scholar] [CrossRef] [Green Version]
- Ramamoorthy, M.; Sykora, P.; Scheibye-Knudsen, M.; Dunn, C.; Kasmer, C.; Zhang, Y.; Becker, K.G.; Croteau, D.L.; Bohr, V.A. Sporadic Alzheimer’s Disease Fibroblasts Display an Oxidative Stress Phenotype. Free Radic. Biol. Med. 2012, 53, 1371–1380. [Google Scholar] [CrossRef] [Green Version]
- Palotás, A.; Kálmán, J.; Laskay, G.; Juhász, A.; Janka, Z.; Penke, B. Change of fibroblast calcium levels caused by beta-amyloid peptide in Alzheimer disease. Ideggyogy Szle 2002, 55, 164–167. [Google Scholar]
- Peterson, C.; Ratan, R.R.; Shelanski, M.L.; Goldman, J.E. Altered Response of Fibroblasts from Aged and Alzheimer Donors to Drugs That Elevate Cytosolic Free Calcium. Neurobiol. Aging 1988, 9, 261–266. [Google Scholar] [CrossRef]
- Naderi, J.; Lopez, C.; Pandey, S. Chronically Increased Oxidative Stress in Fibroblasts from Alzheimer’s Disease Patients Causes Early Senescence and Renders Resistance to Apoptosis by Oxidative Stress. Mech. Ageing Dev. 2006, 127, 25–35. [Google Scholar] [CrossRef]
- Wang, X.; Su, B.; Fujioka, H.; Zhu, X. Dynamin-like Protein 1 Reduction Underlies Mitochondrial Morphology and Distribution Abnormalities in Fibroblasts from Sporadic Alzheimer’s Disease Patients. Am. J. Pathol. 2008, 173, 470–482. [Google Scholar] [CrossRef] [Green Version]
- Birnbaum, J.H.; Wanner, D.; Gietl, A.F.; Saake, A.; Kündig, T.M.; Hock, C.; Nitsch, R.M.; Tackenberg, C. Oxidative Stress and Altered Mitochondrial Protein Expression in the Absence of Amyloid-β and Tau Pathology in IPSC-Derived Neurons from Sporadic Alzheimer’s Disease Patients. Stem Cell Res. 2018, 27, 121–130. [Google Scholar] [CrossRef]
- Zelko, I.N.; Mariani, T.J.; Folz, R.J. Superoxide Dismutase Multigene Family: A Comparison of the CuZn-SOD (SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3) Gene Structures, Evolution, and Expression. Free Radic. Biol. Med. 2002, 33, 337–349. [Google Scholar] [CrossRef]
- Kitada, M.; Xu, J.; Ogura, Y.; Monno, I.; Koya, D. Manganese Superoxide Dismutase Dysfunction and the Pathogenesis of Kidney Disease. Front. Physiol. 2020, 11, 755. [Google Scholar] [CrossRef]
- Flynn, J.M.; Melov, S. SOD2 in Mitochondrial Dysfunction and Neurodegeneration. Free Radic. Biol. Med. 2013, 62, 4–12. [Google Scholar] [CrossRef] [Green Version]
- Schieber, M.; Chandel, N.S. ROS Function in Redox Signaling and Oxidative Stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [Green Version]
- English, J.; Son, J.M.; Cardamone, M.D.; Lee, C.; Perissi, V. Decoding the Rosetta Stone of Mitonuclear Communication. Pharmacol. Res. 2020, 161, 105161. [Google Scholar] [CrossRef]
- Sebastián, D.; Palacín, M.; Zorzano, A. Mitochondrial Dynamics: Coupling Mitochondrial Fitness with Healthy Aging. Trends Mol. Med. 2017, 23, 201–215. [Google Scholar] [CrossRef]
- Qin, W.; Haroutunian, V.; Katsel, P.; Cardozo, C.P.; Ho, L.; Buxbaum, J.D.; Pasinetti, G.M. PGC-1alpha Expression Decreases in the Alzheimer Disease Brain as a Function of Dementia. Arch. Neurol. 2009, 66, 352–361. [Google Scholar] [CrossRef]
- Sheng, B.; Wang, X.; Su, B.; Lee, H.; Casadesus, G.; Perry, G.; Zhu, X. Impaired Mitochondrial Biogenesis Contributes to Mitochondrial Dysfunction in Alzheimer’s Disease. J. Neurochem. 2012, 120, 419–429. [Google Scholar] [CrossRef] [PubMed]
- Fang, E.F.; Hou, Y.; Palikaras, K.; Adriaanse, B.A.; Kerr, J.S.; Yang, B.; Lautrup, S.; Hasan-Olive, M.M.; Caponio, D.; Dan, X.; et al. Mitophagy Inhibits Amyloid-β and Tau Pathology and Reverses Cognitive Deficits in Models of Alzheimer’s Disease. Nat. Neurosci. 2019, 22, 401–412. [Google Scholar] [CrossRef] [PubMed]
- Li, P.A.; Hou, X.; Hao, S. Mitochondrial biogenesis in neurodegeneration. J. Neurosci. Res. 2017, 95, 2025–2029. [Google Scholar] [CrossRef]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional Regulation by Nrf2. Antioxid. Redox Signal. 2018, 29, 1727–1745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drabik, K.; Piecyk, K.; Wolny, A.; Szulc-Dąbrowska, L.; Dębska-Vielhaber, G.; Vielhaber, S.; Duszyński, J.; Malińska, D.; Szczepanowska, J. Adaptation of Mitochondrial Network Dynamics and Velocity of Mitochondrial Movement to Chronic Stress Present in Fibroblasts Derived from Patients with Sporadic Form of Alzheimer’s Disease. FASEB J. 2021, 35, e21586. [Google Scholar] [CrossRef] [PubMed]
- Venditti, P.; Costagliola, I.R.; Di Meo, S. H2O2 Production and Response to Stress Conditions by Mitochondrial Fractions from Rat Liver. J. Bioenerg. Biomembr. 2002, 34, 115–125. [Google Scholar] [CrossRef]
- Schofield, J.H.; Schafer, Z.T. Mitochondrial Reactive Oxygen Species and Mitophagy: A Complex and Nuanced Relationship. Antioxid. Redox Signal. 2021, 34, 517–530. [Google Scholar] [CrossRef] [PubMed]
- Tran, M.; Reddy, P.H. Defective Autophagy and Mitophagy in Aging and Alzheimer’s Disease. Front. Neurosci. 2020, 14, 612757. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Symbol | Sex 1 | Age |
|---|---|---|
| sAD patients | ||
| sAD1 | M | 74 |
| sAD2 | F | 74 |
| sAD3 | M | 83 |
| sAD4 | F | 71 |
| Controls | ||
| C1 | F | 68 |
| C2 | F | 71 |
| C3 | F | 63 |
| C4 | F | 67 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Drabik, K.; Malińska, D.; Piecyk, K.; Dębska-Vielhaber, G.; Vielhaber, S.; Duszyński, J.; Szczepanowska, J. Effect of Chronic Stress Present in Fibroblasts Derived from Patients with a Sporadic Form of AD on Mitochondrial Function and Mitochondrial Turnover. Antioxidants 2021, 10, 938. https://doi.org/10.3390/antiox10060938
Drabik K, Malińska D, Piecyk K, Dębska-Vielhaber G, Vielhaber S, Duszyński J, Szczepanowska J. Effect of Chronic Stress Present in Fibroblasts Derived from Patients with a Sporadic Form of AD on Mitochondrial Function and Mitochondrial Turnover. Antioxidants. 2021; 10(6):938. https://doi.org/10.3390/antiox10060938
Chicago/Turabian StyleDrabik, Karolina, Dominika Malińska, Karolina Piecyk, Grażyna Dębska-Vielhaber, Stefan Vielhaber, Jerzy Duszyński, and Joanna Szczepanowska. 2021. "Effect of Chronic Stress Present in Fibroblasts Derived from Patients with a Sporadic Form of AD on Mitochondrial Function and Mitochondrial Turnover" Antioxidants 10, no. 6: 938. https://doi.org/10.3390/antiox10060938
APA StyleDrabik, K., Malińska, D., Piecyk, K., Dębska-Vielhaber, G., Vielhaber, S., Duszyński, J., & Szczepanowska, J. (2021). Effect of Chronic Stress Present in Fibroblasts Derived from Patients with a Sporadic Form of AD on Mitochondrial Function and Mitochondrial Turnover. Antioxidants, 10(6), 938. https://doi.org/10.3390/antiox10060938