
A Combined Drug Treatment That Reduces Mitochondrial Iron and Reactive Oxygen Levels Recovers Insulin Secretion in NAF-1-Deficient Pancreatic Cells

 ,
, 

Abstract
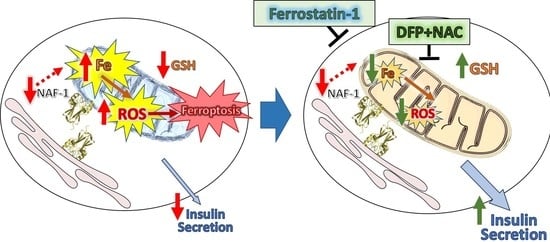
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Growth and NAF-1 Expression
2.2. Protein Blots
2.3. Lipid Peroxidation
2.4. Fluorescence Probes
2.5. Treatment of Cells with Ferrostatin-1, the Iron Chelator (DFP), and the Glutathione Precursor CGP (NAC)
2.6. Mitochondrial Bioenergetics, Oxygen Consumption Rate (OCR), Cellular Glycolysis, and Extracellular Acidification Rate (ECAR)
2.7. Insulin Secretion Measurement
2.8. Mitochondria and ER Structure
2.9. Statistics
3. Results
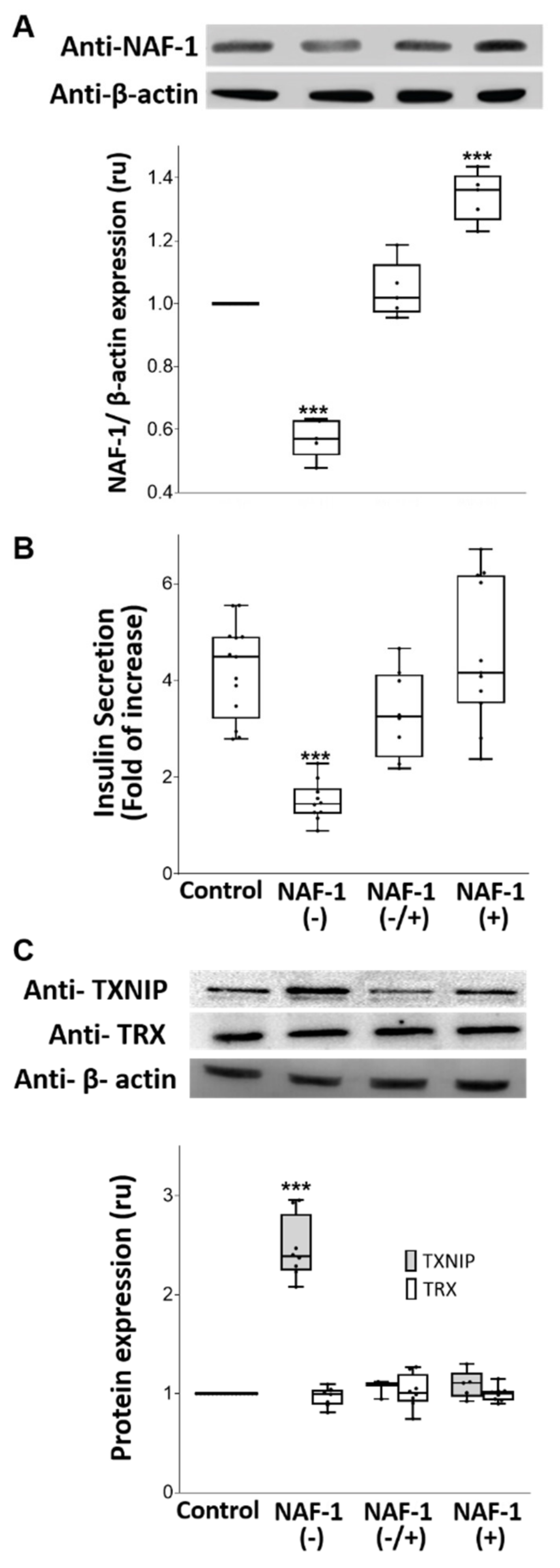
3.1. NAF-1-Suppressed Insulinoma Cells as a Model for Pancreatic β-Cells of WFS-T2
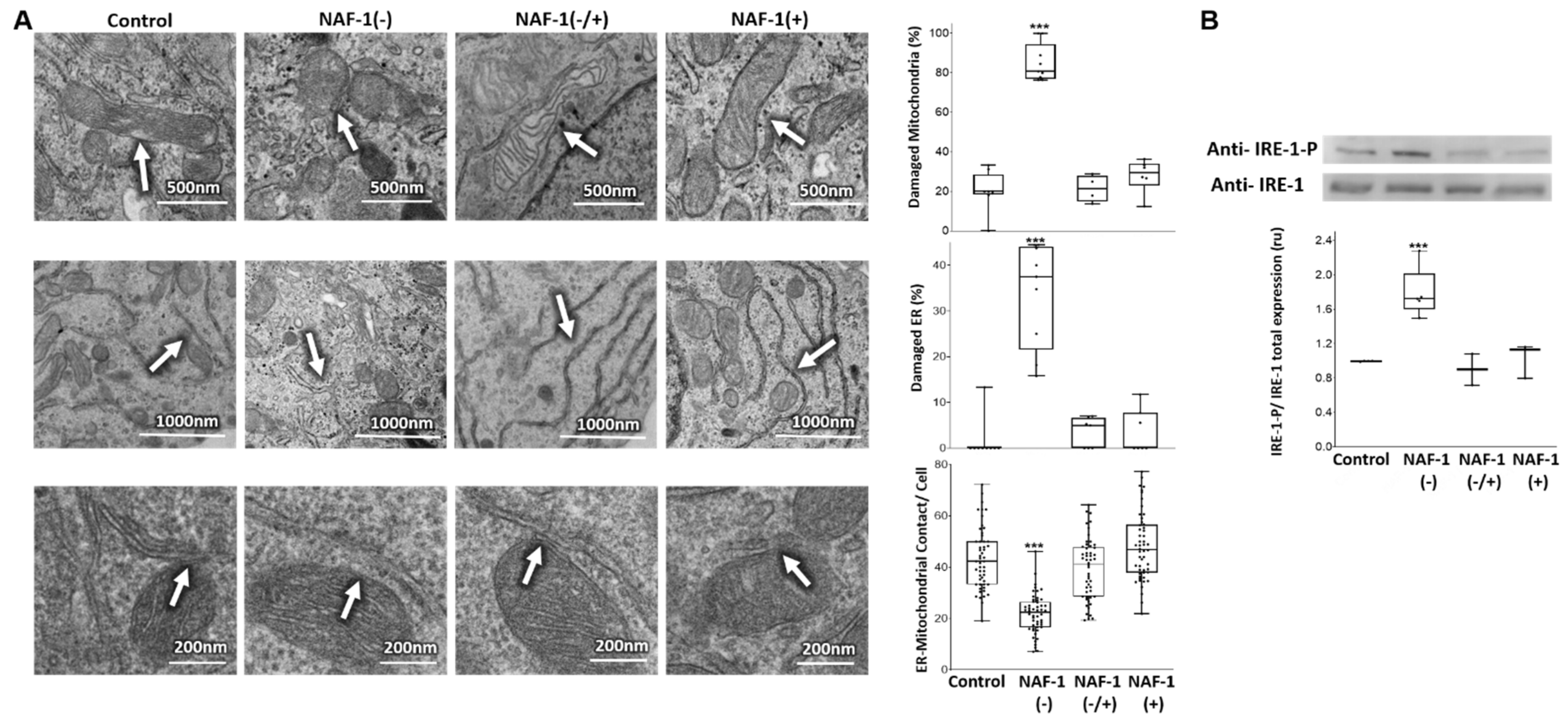
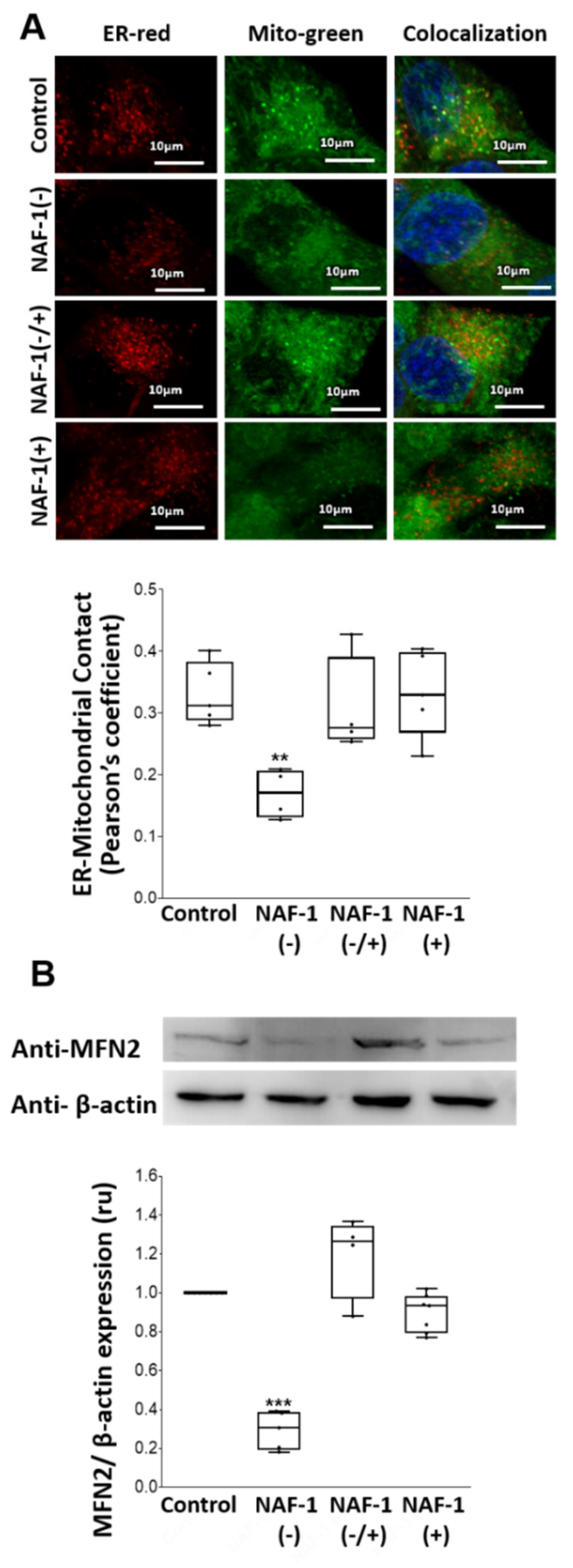
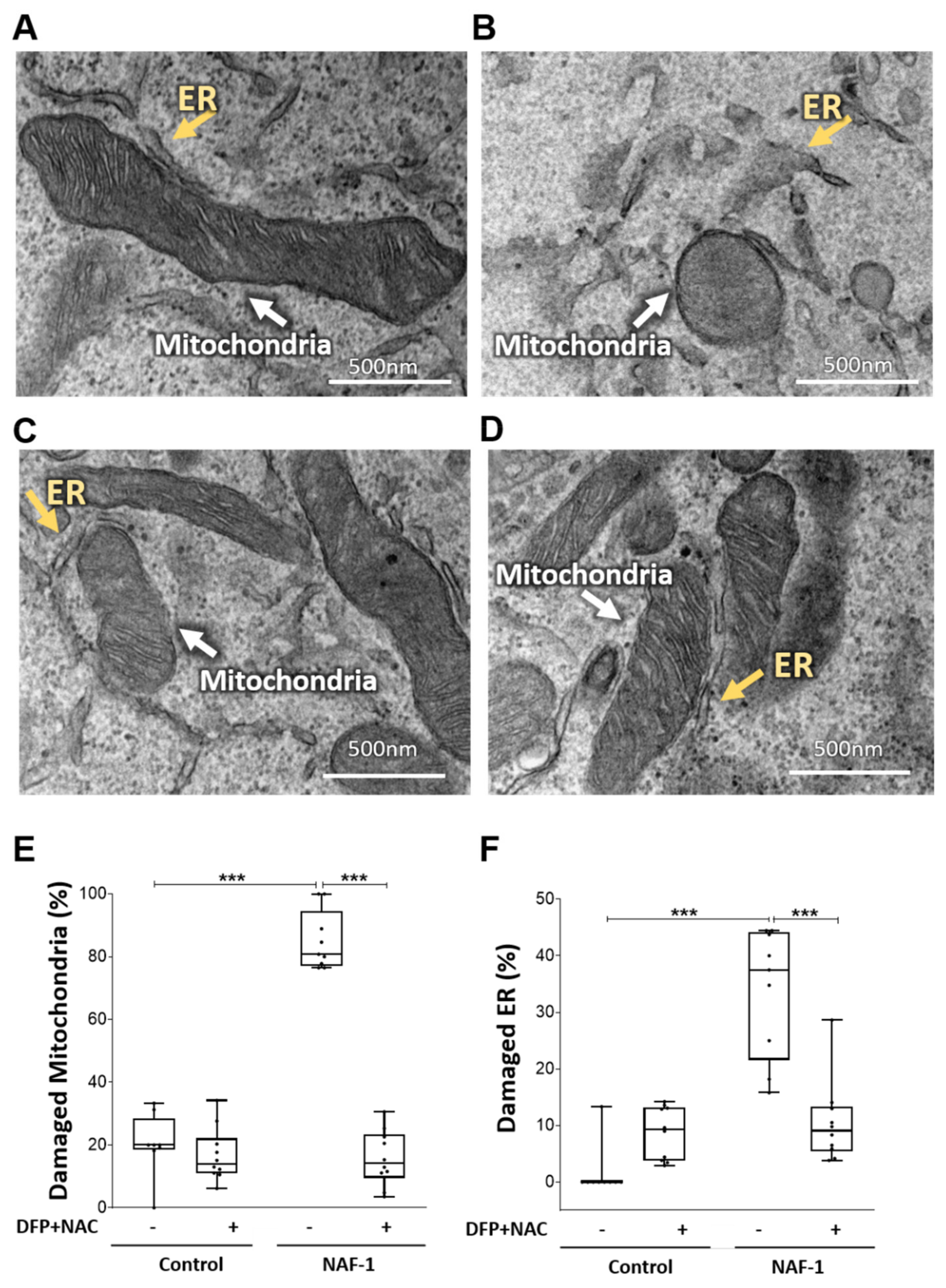
3.2. Mitochondrial, ER, and MAM Structural Changes in NAF-1(−) Cells
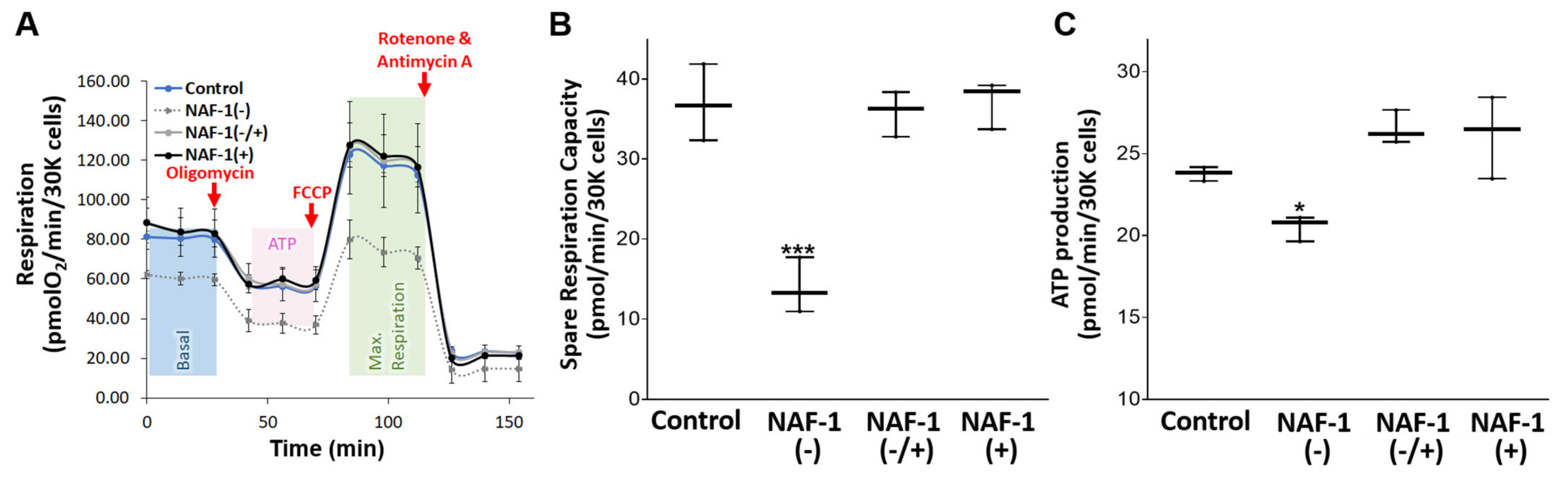
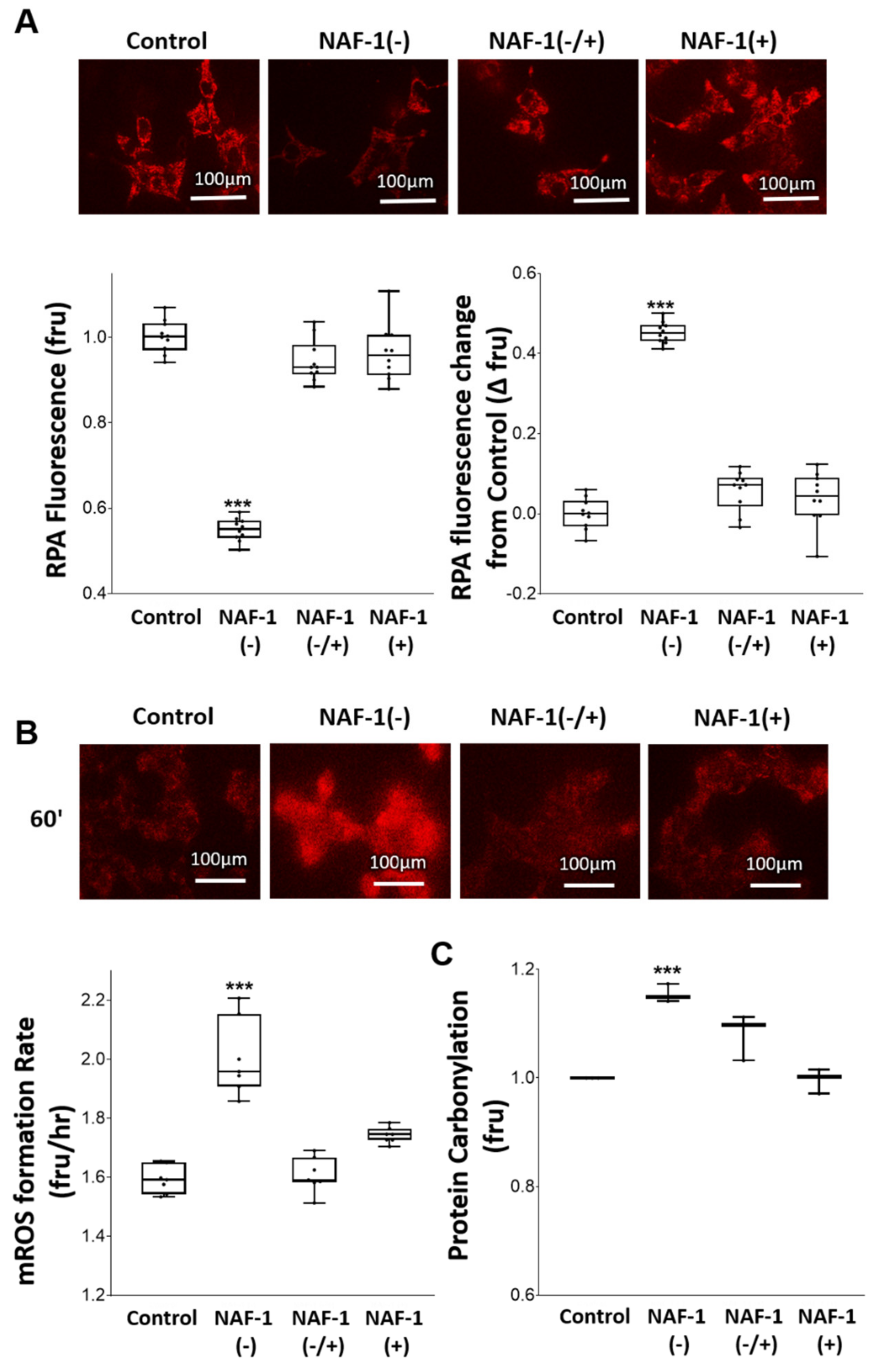
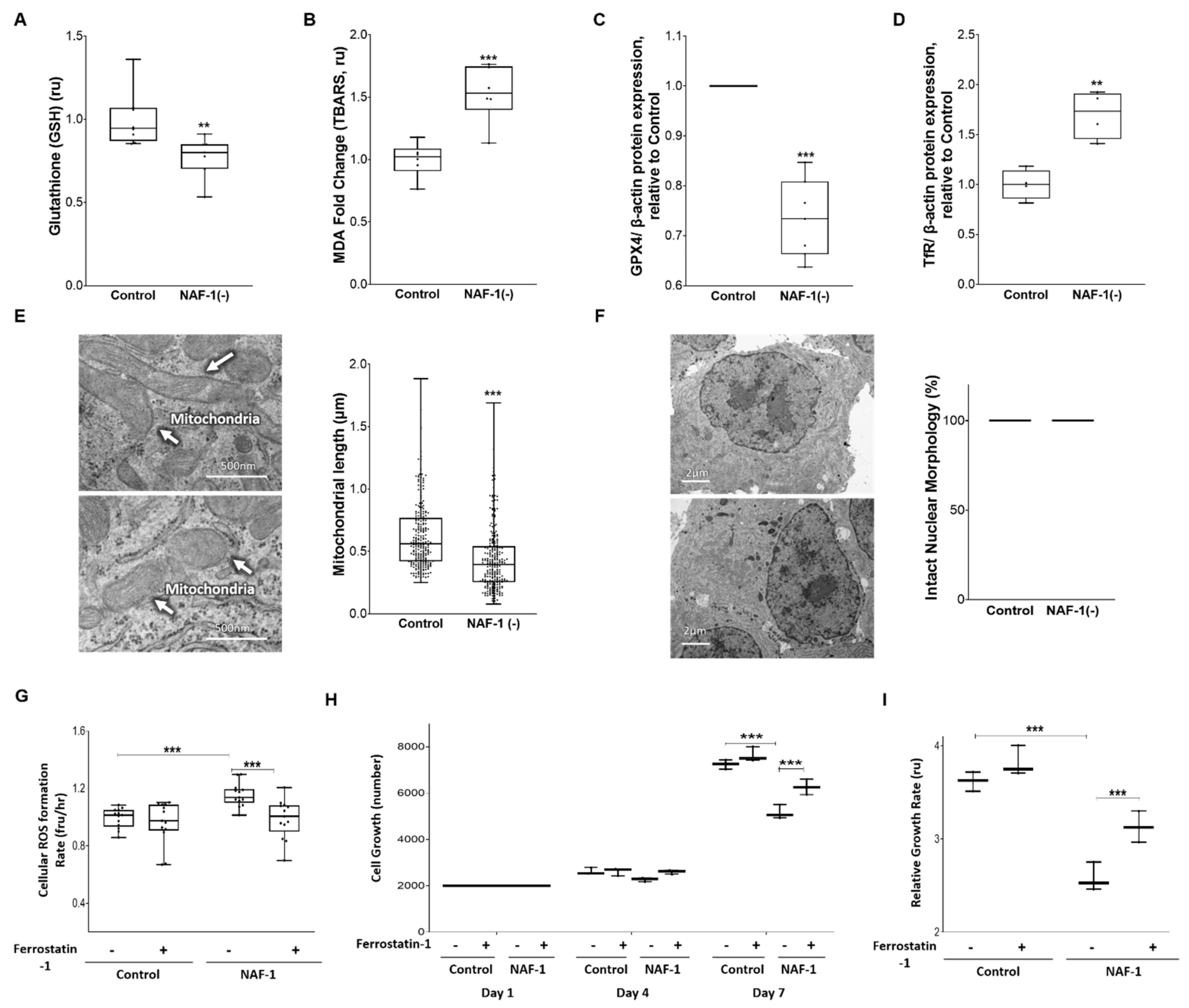
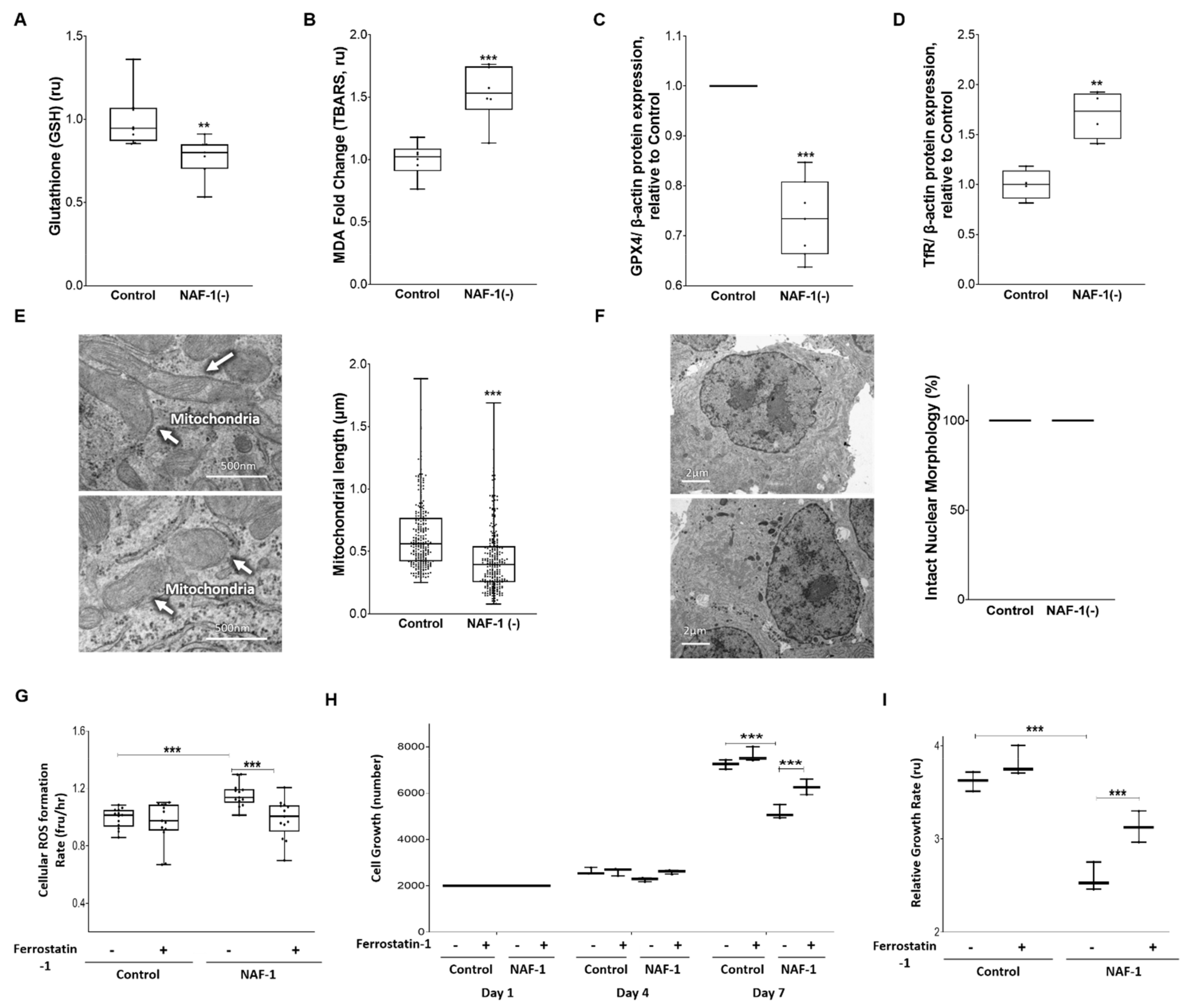
3.3. Biochemical and Physiological Changes in NAF-1(−) Cells
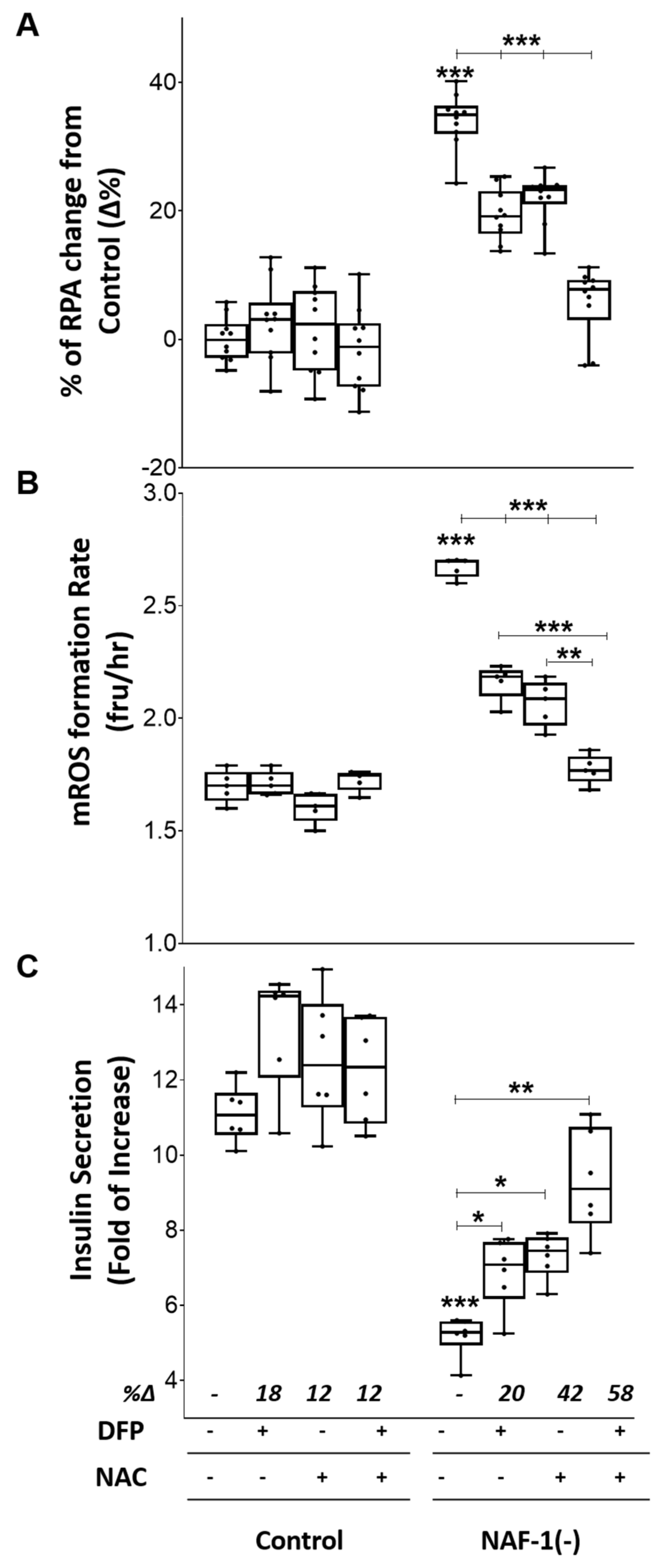
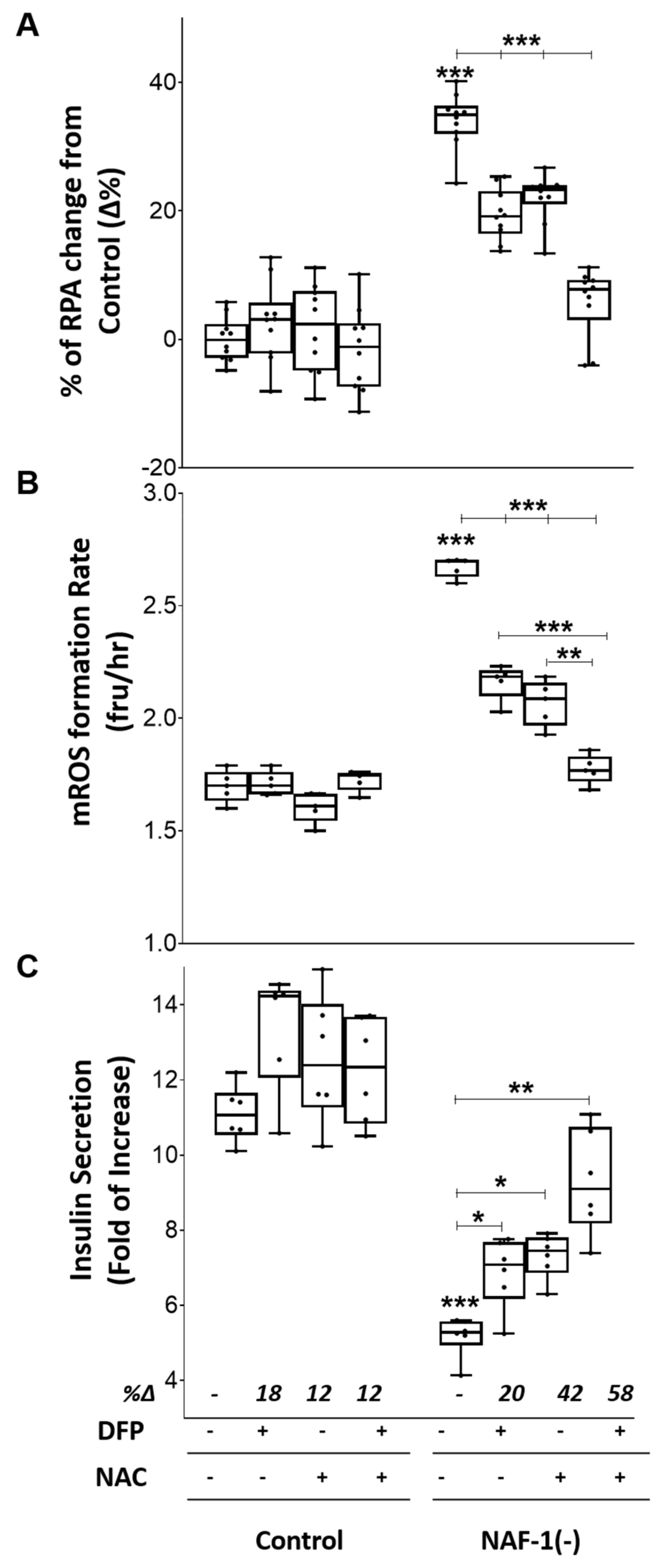
3.4. Pharmacological Amelioration of NAF-1 Depletion in INS-1E Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| CH | Coumarin hydrazine |
| DFP | Deferiprone |
| ECAR | Extracellular acidification rate |
| ER | Endoplasmic reticulum |
| GI | Gastro-intestinal |
| GPX4 | Glutathione peroxidase 4 |
| GSH | Glutathione |
| IP3R | Inositol 1,4,5-triphosphate receptor |
| IRE-1 | Serine/threonine-protein kinase/endoribonuclease IRE1 |
| KD | Knock down |
| MAM | Mitochondria-ER-associated membranes |
| MDA | Malondialdehyde |
| MFN2 | Mitofusin 2 |
| mLI | Mitochondrial labile iron |
| MMP | Mitochondrial membrane potential |
| mROS | Mitochondrial reactive oxygen species |
| NAC | N-acetyl cysteine |
| NAF-1 | Nutrient-deprivation autophagy factor-1 |
| OCR | Oxygen consumption rate |
| OMM | Outer mitochondrial membrane |
| ROS | Reactive oxygen species |
| RPA | Rhodamine B-[(1,10-phenanthrolin-5-yl) aminocarbonyl] benzyl ester |
| SERCA2 | Sarco/endoplasmic reticulum Ca2+-ATPase |
| TBARS | Thiobarbituric acid reactive substances |
| TEM | Transmission electron microscope |
| TfR | Transferrin receptor |
| TMRE | Tetramethylrhodamine ethyl ester |
| TRX | Thioredoxin |
| TXNIP | Thioredoxin-interacting protein |
| UPR | Unfolded protein response |
| WFS-T2 | Wolfram syndrome type 2 |
| WT | Wildtype |
References
- Amr, S.; Heisey, C.; Zhang, M.; Xia, X.-J.; Shows, K.H.; Ajlouni, K.; Pandya, A.; Satin, L.S.; El-Shanti, H.; Shiang, R. A Homozygous Mutation in a Novel Zinc-Finger Protein, ERIS, Is Responsible for Wolfram Syndrome 2. Am. J. Hum. Genet. 2007, 81, 673–683. [Google Scholar] [CrossRef] [Green Version]
- Mozzillo, E.; Delvecchio, M.; Carella, M.; Grandone, E.; Palumbo, P.; Salina, A.; Aloi, C.; Buono, P.; Izzo, A.; D’Annunzio, G.; et al. A novel CISD2 intragenic deletion, optic neuropathy and platelet aggregation defect in Wolfram syndrome type 2. BMC Med. Genet. 2014, 15, 88. [Google Scholar] [CrossRef] [Green Version]
- Rondinelli, M.; Novara, F.; Calcaterra, V.; Zuffardi, O.; Genovese, S. Wolfram syndrome 2: A novel CISD2 mutation identified in Italian siblings. Acta Diabetol. 2015, 52, 175–178. [Google Scholar] [CrossRef]
- Rouzier, C.; Moore, D.; Delorme, C.; Lacas-Gervais, S.; Ait-El-Mkadem, S.; Fragaki, K.; Burté, F.; Serre, V.; Bannwarth, S.; Chaussenot, A.; et al. A novel CISD2 mutation associated with a classical Wolfram syndrome phenotype alters Ca2+ homeostasis and ER-mitochondria interactions. Hum. Mol. Genet. 2017, 26, 1599–1611. [Google Scholar] [CrossRef] [PubMed]
- Akturk, H.K.; Msc, S.Y. Previously unreported abnormalities in Wolfram Syndrome Type 2. Pediatr. Endocrinol. Diabetes Metab. 2017, 23, 107–110. [Google Scholar] [CrossRef] [PubMed]
- Cattaneo, M.; La Sala, L.; Rondinelli, M.; Errichiello, E.; Zuffardi, O.; Puca, A.A.; Genovese, S.; Ceriello, A. A donor splice site mutation in CISD2 generates multiple truncated, non-functional isoforms in Wolfram syndrome type 2 patients. BMC Med. Genet. 2017, 18, 147. [Google Scholar] [CrossRef]
- Pourreza, M.R.; Sobhani, M.; Rahimi, A.; Aramideh, M.; Kajbafzadeh, A.-M.; Noori-Daloii, M.R.; Tabatabaiefar, M.A. Homozygosity mapping and direct sequencing identify a novel pathogenic variant in the CISD2 gene in an Iranian Wolfram syndrome family. Acta Diabetol. 2020, 57, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Ajlouni, K.; Jarrah, N.; El-Khateeb, M.; El-Zaheri, M.; El-Shanti, H.; Lidral, A. Wolfram syndrome: Identification of a phenotypic and genotypic variant from Jordan. Am. J. Med. Genet. 2002, 115, 61–65. [Google Scholar] [CrossRef]
- Rigoli, L.; Di Bella, C. Wolfram syndrome 1 and Wolfram syndrome 2. Curr. Opin. Pediatr. 2012, 24, 512–517. [Google Scholar] [CrossRef] [PubMed]
- Urano, F. Wolfram Syndrome: Diagnosis, Management, and Treatment. Curr. Diabetes Rep. 2016, 16, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danielpur, L.; Sohn, Y.-S.; Karmi, O.; Fogel, C.; Zinger, A.; Abu-Libdeh, A.; Israeli, T.; Riahi, Y.; Pappo, O.; Birk, R.; et al. GLP-1-RA Corrects Mitochondrial Labile Iron Accumulation and Improves β-Cell Function in Type 2 Wolfram Syndrome. J. Clin. Endocrinol. Metab. 2016, jc20162240. [Google Scholar] [CrossRef] [PubMed]
- Tamir, S.; Paddock, M.L.; Darash-Yahana-Baram, M.; Holt, S.H.; Sohn, Y.S.; Agranat, L.; Michaeli, D.; Stofleth, J.; Lipper, C.H.; Morcos, F.; et al. Structure–function analysis of NEET proteins uncovers their role as key regulators of iron and ROS homeostasis in health and disease. Biochim. Biophys. Acta 2015, 1853, 1294–1315. [Google Scholar] [CrossRef] [Green Version]
- Karmi, O.; Marjault, H.-B.; Pesce, L.; Carloni, P.; Onuchic, J.N.; Jennings, P.A.; Mittler, R.; Nechushtai, R. The unique fold and lability of the [2Fe-2S] clusters of NEET proteins mediate their key functions in health and disease. JBIC J. Biol. Inorg. Chem. 2018, 23, 599–612. [Google Scholar] [CrossRef] [Green Version]
- Mittler, R.; Darash-Yahana, M.; Sohn, Y.S.; Bai, F.; Song, L.; Cabantchik, I.Z.; Jennings, P.A.; Onuchic, J.N.; Nechushtai, R. NEET Proteins: A New Link Between Iron Metabolism, Reactive Oxygen Species, and Cancer. Antioxid. Redox Signal 2019, 30, 1083–1095. [Google Scholar] [CrossRef]
- Nechushtai, R.; Karmi, O.; Zuo, K.; Marjault, H.-B.; Darash-Yahana, M.; Sohn, Y.-S.; King, S.D.; Zandalinas, S.I.; Carloni, P.; Mittler, R. The balancing act of NEET proteins: Iron, ROS, calcium and metabolism. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2020, 1867, 118805. [Google Scholar] [CrossRef]
- Sohn, Y.-S.; Tamir, S.; Song, L.; Michaeli, D.; Matouk, I.; Conlan, A.R.; Harir, Y.; Holt, S.H.; Shulaev, V.; Paddock, M.L.; et al. NAF-1 and mitoNEET are central to human breast cancer proliferation by maintaining mitochondrial homeostasis and promoting tumor growth. Proc. Natl. Acad. Sci. USA 2013, 110, 14676–14681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Xia, M.; Wang, J.; Zhang, W.; Zhang, Y.; He, M. CISD2 expression is a novel marker correlating with pelvic lymph node metastasis and prognosis in patients with early-stage cervical cancer. Med. Oncol. 2014, 31, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Darash-Yahana, M.; Pozniak, Y.; Lu, M.; Sohn, Y.-S.; Karmi, O.; Tamir, S.; Bai, F.; Song, L.; Jennings, P.A.; Pikarsky, E.; et al. Breast cancer tumorigenicity is dependent on high expression levels of NAF-1 and the lability of its Fe-S clusters. Proc. Natl. Acad. Sci. USA 2016, 113, 10890–10895. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Hong, S.; Wang, Y.; He, Z.; Liang, S.; Chen, H.; He, S.; Wu, S.; Song, L.; Chen, Y. A novel prognostic score model incorporating CDGSH iron sulfur domain2 (CISD2) predicts risk of disease progression in laryngeal squamous cell carcinoma. Oncotarget 2016, 7, 22720–22732. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Ouyang, F.; Liu, X.; Wu, S.; Wu, H.-M.; Xu, Y.; Wang, B.; Zhu, J.; Xu, X.; Zhang, L. Overexpressed CISD2 has prognostic value in human gastric cancer and promotes gastric cancer cell proliferation and tumorigenesis via AKT signaling pathway. Oncotarget 2016, 7, 3791–3805. [Google Scholar] [CrossRef] [Green Version]
- Li, S.-M.; Chen, C.-H.; Chen, Y.-W.; Yen, Y.-C.; Fang, W.-T.; Tsai, F.-Y.; Chang, J.-L.; Shen, Y.-Y.; Huang, S.-F.; Chuu, C.-P.; et al. Upregulation of CISD2 augments ROS homeostasis and contributes to tumorigenesis and poor prognosis of lung adenocarcinoma. Sci. Rep. 2017, 7, 11893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Jiang, Y.; Huang, J.; Chen, H.; Liao, Y.; Yang, Z. CISD2 enhances the chemosensitivity of gastric cancer through the enhancement of 5-FU-induced apoptosis and the inhibition of autophagy by AKT/mTOR pathway. Cancer Med. 2017, 6, 2331–2346. [Google Scholar] [CrossRef] [Green Version]
- Sun, A.-G.; Meng, F.-G.; Wang, M.-G. CISD2 promotes the proliferation of glioma cells via suppressing beclin-1-mediated autophagy and is targeted by microRNA-449a. Mol. Med. Rep. 2017, 16, 7939–7948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, E.H.; Shin, D.; Lee, J.; Jung, A.R.; Roh, J.-L. CISD2 inhibition overcomes resistance to sulfasalazine-induced ferroptotic cell death in head and neck cancer. Cancer Lett. 2018, 432, 180–190. [Google Scholar] [CrossRef]
- Yuan, H.; Li, X.; Zhang, X.; Kang, R.; Tang, D. CISD1 inhibits ferroptosis by protection against mitochondrial lipid peroxidation. Biochem. Biophys. Res. Commun. 2016, 478, 838–844. [Google Scholar] [CrossRef] [PubMed]
- Furihata, T.; Takada, S.; Kakutani, N.; Maekawa, S.; Tsuda, M.; Matsumoto, J.; Mizushima, W.; Fukushima, A.; Yokota, T.; Enzan, N.; et al. Cardiac-specific loss of mitoNEET expression is linked with age-related heart failure. Commun. Biol. 2021, 4, 1–11. [Google Scholar] [CrossRef]
- Pandolfo, M.; Hausmann, L. Deferiprone for the treatment of Friedreich’s ataxia. J. Neurochem. 2013, 126, 142–146. [Google Scholar] [CrossRef]
- Agrawal, S.; Fox, J.; Thyagarajan, B.; Fox, J.H. Brain mitochondrial iron accumulates in Huntington’s disease, mediates mitochondrial dysfunction, and can be removed pharmacologically. Free. Radic. Biol. Med. 2018, 120, 317–329. [Google Scholar] [CrossRef]
- Wongjaikam, S.; Kumfu, S.; Khamseekaew, J.; Chattipakorn, S.C.; Chattipakorn, N. Restoring the impaired cardiac calcium homeostasis and cardiac function in iron overload rats by the combined deferiprone and N-acetyl cysteine. Sci. Rep. 2017, 7, srep44460. [Google Scholar] [CrossRef] [Green Version]
- Mohammadi, H.; Sayad, A.; Mohammadi, M.; Niknahad, H.; Heidari, R. N-acetyl cysteine treatment pre-serves mitochondrial indices of functionality in the brain of hyperammonemic mice. Clin. Exp. Hepatol. 2020, 6, 106. [Google Scholar] [CrossRef]
- Ommati, M.M.; Amjadinia, A.; Mousavi, K.; Azarpira, N.; Jamshidzadeh, A.; Heidari, R. N-acetyl cysteine treatment mitigates biomarkers of oxidative stress in different tissues of bile duct ligated rats. Stress 2021, 24, 213–228. [Google Scholar] [CrossRef]
- Anthonymuthu, T.S.; Tyurina, Y.Y.; Sun, W.-Y.; Mikulska-Ruminska, K.; Shrivastava, I.H.; Tyurin, V.A.; Cinemre, F.B.; Dar, H.H.; VanDemark, A.P.; Holman, T.R.; et al. Resolving the paradox of ferroptotic cell death: Ferrostatin-1 binds to 15LOX/PEBP1 complex, suppresses generation of peroxidized ETE-PE, and protects against ferroptosis. Redox Biol. 2021, 38, 101744. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Feng, Y.; Li, H.; Chen, X.; Wang, G.; Xu, S.; Li, Y.; Zhao, L. Ferrostatin-1 alleviates lipopolysaccharide-induced acute lung injury via inhibiting ferroptosis. Cell. Mol. Biol. Lett. 2020, 25, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holt, S.H.; Darash-Yahana, M.; Sohn, Y.S.; Song, L.; Karmi, O.; Tamir, S.; Michaeli, D.; Luo, Y.; Paddock, M.L.; Jennings, P.A.; et al. Activation of apoptosis in NAF-1-deficient human epithelial breast cancer cells. J. Cell Sci. 2016, 129, 155–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukherjee, K.; Chio, T.I.; Sackett, D.L.; Bane, S.L. Detection of oxidative stress-induced carbonylation in live mammalian cells. Free. Radic. Biol. Med. 2015, 84, 11–21. [Google Scholar] [CrossRef] [Green Version]
- Bharath, S.; Hsu, M.; Kaur, D.; Rajagopalan, S.; Andersen, J.K. Glutathione, iron and Parkinson’s disease. Biochem. Pharmacol. 2002, 64, 1037–1048. [Google Scholar] [CrossRef]
- Lu, S.C. Regulation of glutathione synthesis. Mol. Asp. Med. 2009, 30, 42–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schieber, M.; Chandel, N.S. ROS Function in Redox Signaling and Oxidative Stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [Green Version]
- Divakaruni, A.S.; Paradyse, A.; Ferrick, D.A.; Murphy, A.N.; Jastroch, M. Analysis and Interpretation of Microplate-Based Oxygen Consumption and pH Data. In Methods in Enzymology; Elsevier BV: Amsterdam, The Netherlands, 2014; Volume 547, pp. 309–335. [Google Scholar] [CrossRef]
- Xu, W.; Li, T.; Gao, L.; Zheng, J.; Yan, J.; Zhang, J.; Shao, A. Apelin-13/APJ system attenuates early brain injury via suppression of endoplasmic reticulum stress-associated TXNIP/NLRP3 inflammasome activation and oxidative stress in a AMPK-dependent manner after subarachnoid hemorrhage in rats. J. Neuroinflamm. 2019, 16, 1–14. [Google Scholar] [CrossRef]
- Yoshihara, E.; Masaki, S.; Matsuo, Y.; Chen, Z.; Tian, H.; Yodoi, J. Thioredoxin/Txnip: Redoxisome, as a Redox Switch for the Pathogenesis of Diseases. Front. Immunol. 2013, 4, 514. [Google Scholar] [CrossRef] [Green Version]
- Zhou, R.; Tardivel, A.; Thorens, B.; Choi, I.; Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 2010, 11, 136–140. [Google Scholar] [CrossRef]
- Schrepfer, E.; Scorrano, L. Mitofusins, from Mitochondria to Metabolism. Mol. Cell 2016, 61, 683–694. [Google Scholar] [CrossRef] [Green Version]
- De Brito, O.M.; Scorrano, L. Mitofusin-2 regulates mitochondrial and endoplasmic reticulum morphology and tethering: The role of Ras. Mitochondrion 2009, 9, 222–226. [Google Scholar] [CrossRef] [PubMed]
- Naon, D.; Zaninello, M.; Giacomello, M.; Varanita, T.; Grespi, F.; Lakshminaranayan, S.; Serafini, A.; Semenzato, M.; Herkenne, S.; Hernández-Alvarez, M.I.; et al. Critical reappraisal confirms that Mitofusin 2 is an endoplasmic reticulum–mitochondria tether. Proc. Natl. Acad. Sci. USA 2016, 113, 11249–11254. [Google Scholar] [CrossRef] [Green Version]
- Abdalkader, M.; Lampinen, R.; Kanninen, K.M.; Malm, T.; Liddell, J.R. Targeting Nrf2 to Suppress Ferroptosis and Mitochondrial Dysfunction in Neurodegeneration. Front. Neurosci. 2018, 12, 466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Codenotti, S.; Poli, M.; Asperti, M.; Zizioli, D.; Marampon, F.; Fanzani, A. Cell growth potential drives ferroptosis susceptibility in rhabdomyosarcoma and myoblast cell lines. J. Cancer Res. Clin. Oncol. 2018, 144, 1717–1730. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wang, Y.; Wu, J.; Yang, J.; Li, M.; Chen, Q. The Potential Value of Targeting Ferroptosis in Early Brain Injury After Acute CNS Disease. Front. Mol. Neurosci. 2020, 13, 110. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Pratte, J.; Giardina, C. Ferroptosis and its potential as a therapeutic target. Biochem. Pharmacol. 2021, 186, 114486. [Google Scholar] [CrossRef]
- Li, Z.-J.; Dai, H.-Q.; Huang, X.-W.; Feng, J.; Deng, J.-H.; Wang, Z.-X.; Yang, X.-M.; Liu, Y.-J.; Wu, Y.; Chen, P.-H.; et al. Artesunate synergizes with sorafenib to induce ferroptosis in hepatocellular carcinoma. Acta Pharmacol. Sin. 2021, 42, 301–310. [Google Scholar] [CrossRef]
- Wang, C.-H.; Chen, Y.-F.; Wu, C.-Y.; Wu, P.-C.; Huang, Y.-L.; Kao, C.-H.; Lin, C.-H.; Kao, L.-S.; Tsai, T.-F.; Wei, Y.-H. Cisd2 modulates the differentiation and functioning of adipocytes by regulating intracellular Ca2+ homeostasis. Hum. Mol. Genet. 2014, 23, 4770–4785. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Stimpson, S.; Fernandez-Bueno, G.A.; Mathews, C.E. Mitochondrial Reactive Oxygen Species and Type 1 Diabetes. Antioxid. Redox Signal. 2018, 29, 1361–1372. [Google Scholar] [CrossRef] [PubMed]
- Brozzi, F.; Eizirik, D.L. ER stress and the decline and fall of pancreatic beta cells in type 1 diabetes. Upsala J. Med. Sci. 2016, 121, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Lerner, A.G.; Upton, J.-P.; Praveen, P.; Ghosh, R.; Nakagawa, Y.; Igbaria, A.; Shen, S.; Nguyen, V.; Backes, B.J.; Heiman, M.; et al. IRE1α Induces Thioredoxin-Interacting Protein to Activate the NLRP3 Inflammasome and Promote Programmed Cell Death under Irremediable ER Stress. Cell Metab. 2012, 16, 250–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rutter, G.; Pinton, P. Mitochondria-associated endoplasmic reticulum membranes in insulin signaling. Diabetes 2014, 63, 3163–3165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, N.C.; Nguyen, M.; Germain, M.; Shore, G.C. Antagonism of Beclin 1-dependent autophagy by BCL-2 at the endoplasmic reticulum requires NAF-1. EMBO J. 2009, 29, 606–618. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.-Q.; Chen, Y.-F.; Chen, J.-R.; Jou, Y.-S.; Wu, P.-C.; Kao, C.-H.; Wang, C.-H.; Huang, Y.-L.; Chen, C.-F.; Huang, T.-S.; et al. CISD2 Haploinsufficiency Disrupts Calcium Homeostasis, Causes Nonalcoholic Fatty Liver Disease, and Promotes Hepatocellular Carcinoma. Cell Rep. 2017, 21, 2198–2211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Z.-Q.; Huang, Y.-L.; Tsai, T.-F. Cisd2 haploinsufficiency: A driving force for hepatocellular carcinoma. Mol. Cell. Oncol. 2018, 5, e1441627. [Google Scholar] [CrossRef] [Green Version]
- Rutter, G.A.; Tsuboi, T.; Ravier, M. Ca2+ microdomains and the control of insulin secretion. Cell Calcium 2006, 40, 539–551. [Google Scholar] [CrossRef] [PubMed]
- Wiley, S.E.; Andreyev, A.Y.; Divakaruni, A.S.; Karisch, R.; Perkins, G.; Wall, E.A.; van der Geer, P.; Chen, Y.F.; Tsai, T.F.; Simon, M.I.; et al. Wolfram Syndrome protein, Miner1, regulates sulphydryl redox status, the unfolded protein response, and Ca2+ homeostasis. EMBO Mol. Med. 2013, 5, 904–918. [Google Scholar] [CrossRef]
- Shen, Z.-Q.; Huang, Y.-L.; Teng, Y.-C.; Wang, T.-W.; Kao, C.-H.; Yeh, C.-H.; Tsai, T.-F. CISD2 maintains cellular homeostasis. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2021, 1868, 118954. [Google Scholar] [CrossRef]
- Delprat, B.; Maurice, T.; Delettre, C. Wolfram syndrome: MAMs’ connection? Cell Death Dis. 2018, 9, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Zheng, Y.; Wang, C.; Liu, Y. Glutathione depletion induces ferroptosis, autophagy, and premature cell senescence in retinal pigment epithelial cells. Cell Death Dis. 2018, 9, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Cao, F.; Yin, H.-l.; Huang, Z.-j.; Lin, Z.-t.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, present and future. Cell Death Disease 2020, 11, 1–13. [Google Scholar] [CrossRef]
- Berndt, C.; Lillig, C.H. Glutathione, Glutaredoxins, and Iron. Antioxid. Redox Signal. 2017, 27, 1235–1251. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.; et al. Regulation of Ferroptotic Cancer Cell Death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Gutierrez, E.; Kovacevic, Z.; Saletta, F.; Obeidy, P.; Rahmanto, Y.S.; Richardson, D. Iron Chelators for the Treatment of Cancer. Curr. Med. Chem. 2012, 19, 2689–2702. [Google Scholar] [CrossRef] [PubMed]
- Fiorillo, M.; Tóth, F.; Brindisi, M.; Sotgia, F.; Lisanti, M.P. Deferiprone (DFP) Targets Cancer Stem cell (CSC) Propagation by Inhibiting Mitochondrial Metabolism and Inducing ROS Production. Cells 2020, 9, 1529. [Google Scholar] [CrossRef]
- Buss, J.L.; Greene, B.T.; Turner, J.; Torti, F.M.; Torti, S.V. Iron chelators in cancer chemotherapy. Curr. Top. Med. Chem. 2004, 4, 1623–1635. [Google Scholar] [CrossRef]
- Merkofer, M.; Kissner, R.; Hider, R.C.; Brunk, U.T.; Koppenol, W.H. Fenton Chemistry and Iron Chelation under Physiologically Relevant Conditions: Electrochemistry and Kinetics. Chem. Res. Toxicol. 2006, 19, 1263–1269. [Google Scholar] [CrossRef]
- Kuo, K.; Mrkobrada, M. A Systematic Review and Meta-Analysis of Deferiprone Monotherapy and in Combination with Deferoxamine for Reduction of Iron Overload in Chronically Transfused Patients with β-Thalassemia. Hemoglobin 2014, 38, 409–421. [Google Scholar] [CrossRef]
- Fernández-Real, J.M.; López-Bermejo, A.; Ricart, W. Cross-Talk Between Iron Metabolism and Diabetes. Diabetes 2002, 51, 2348–2354. [Google Scholar] [CrossRef] [Green Version]
- Stroh, M.; Swerdlow, R.H.; Zhu, H. Common defects of mitochondria and iron in neurodegeneration and diabetes (MIND): A paradigm worth exploring. Biochem. Pharmacol. 2014, 88, 573–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swaminathan, S.; Fonseca, V.A.; Alam, M.G.; Shah, S.V. The Role of Iron in Diabetes and Its Complications. Diabetes Care 2007, 30, 1926–1933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wachnowsky, C.; Fidai, I.; Cowan, J.A. Iron–sulfur cluster biosynthesis and trafficking–impact on human disease conditions. Metallomics 2018, 10, 9–29. [Google Scholar] [CrossRef] [PubMed]
- Petronek, M.S.; Spitz, D.R.; Buettner, G.R.; Allen, B.G. Linking Cancer Metabolic Dysfunction and Genetic Instability through the Lens of Iron Metabolism. Cancers 2019, 11, 1077. [Google Scholar] [CrossRef] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karmi, O.; Sohn, Y.-S.; Marjault, H.-B.; Israeli, T.; Leibowitz, G.; Ioannidis, K.; Nahmias, Y.; Mittler, R.; Cabantchik, I.Z.; Nechushtai, R. A Combined Drug Treatment That Reduces Mitochondrial Iron and Reactive Oxygen Levels Recovers Insulin Secretion in NAF-1-Deficient Pancreatic Cells. Antioxidants 2021, 10, 1160. https://doi.org/10.3390/antiox10081160
Karmi O, Sohn Y-S, Marjault H-B, Israeli T, Leibowitz G, Ioannidis K, Nahmias Y, Mittler R, Cabantchik IZ, Nechushtai R. A Combined Drug Treatment That Reduces Mitochondrial Iron and Reactive Oxygen Levels Recovers Insulin Secretion in NAF-1-Deficient Pancreatic Cells. Antioxidants. 2021; 10(8):1160. https://doi.org/10.3390/antiox10081160
Chicago/Turabian StyleKarmi, Ola, Yang-Sung Sohn, Henri-Baptiste Marjault, Tal Israeli, Gil Leibowitz, Konstantinos Ioannidis, Yaakov Nahmias, Ron Mittler, Ioav Z. Cabantchik, and Rachel Nechushtai. 2021. "A Combined Drug Treatment That Reduces Mitochondrial Iron and Reactive Oxygen Levels Recovers Insulin Secretion in NAF-1-Deficient Pancreatic Cells" Antioxidants 10, no. 8: 1160. https://doi.org/10.3390/antiox10081160
APA StyleKarmi, O., Sohn, Y. -S., Marjault, H. -B., Israeli, T., Leibowitz, G., Ioannidis, K., Nahmias, Y., Mittler, R., Cabantchik, I. Z., & Nechushtai, R. (2021). A Combined Drug Treatment That Reduces Mitochondrial Iron and Reactive Oxygen Levels Recovers Insulin Secretion in NAF-1-Deficient Pancreatic Cells. Antioxidants, 10(8), 1160. https://doi.org/10.3390/antiox10081160