
Long-Term Treatment of Cuban Policosanol Attenuates Abnormal Oxidative Stress and Inflammatory Response via Amyloid Plaques Reduction in 5xFAD Mice



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
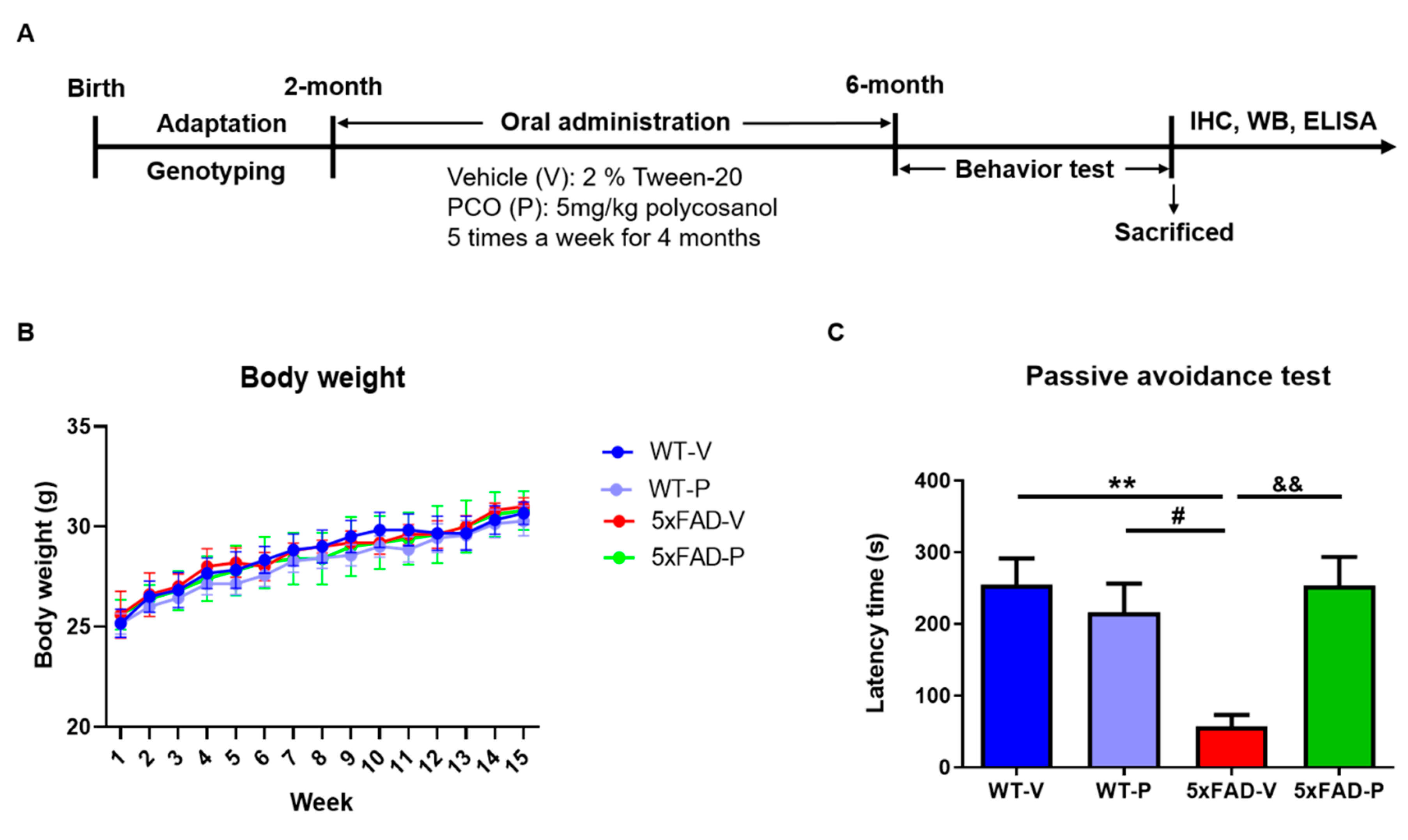
2.1. Animals
2.2. Policosanol Treatment
2.3. Passive Avoidance Test
2.4. Histological Preparation
2.4.1. Tissue Preparation
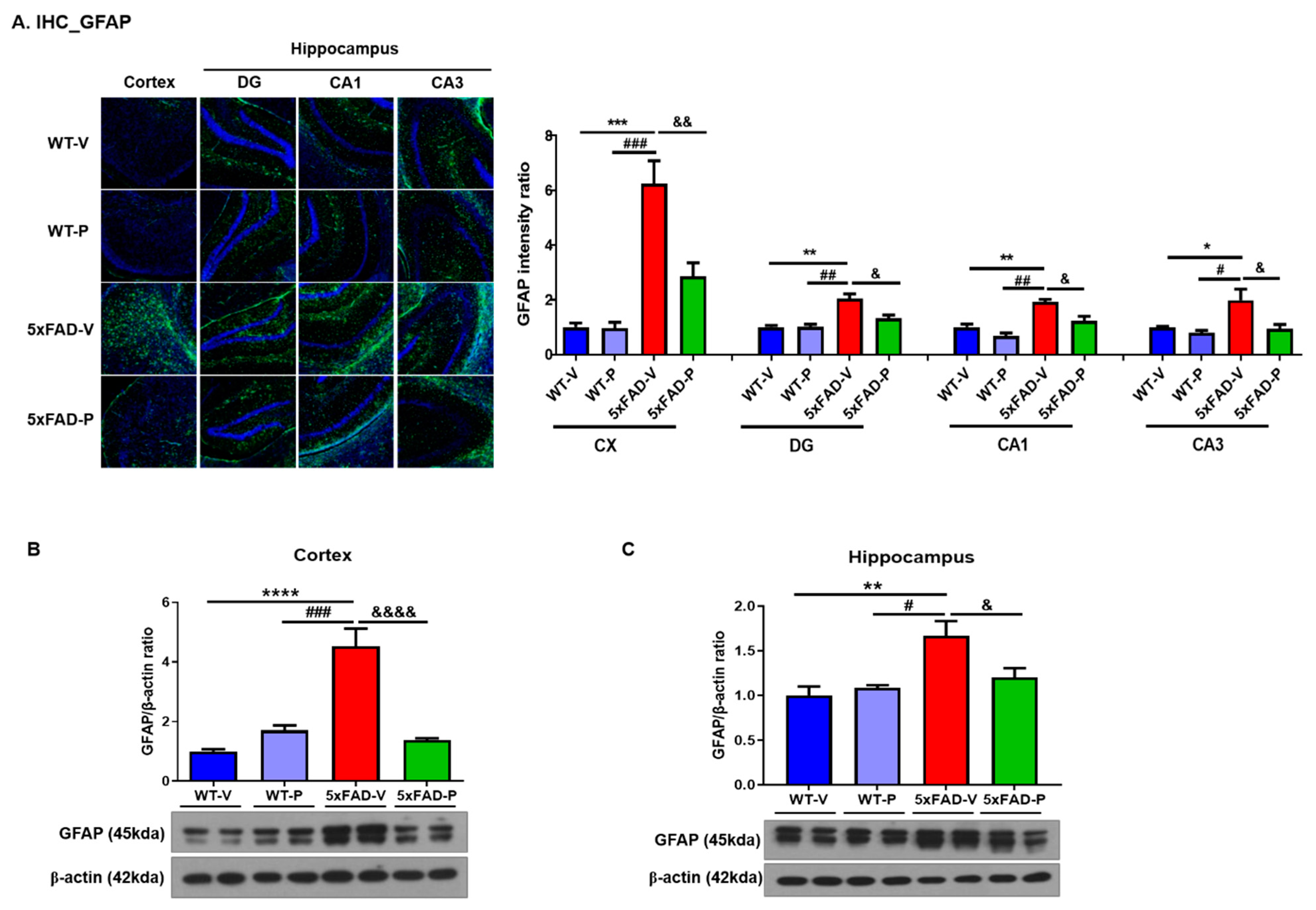
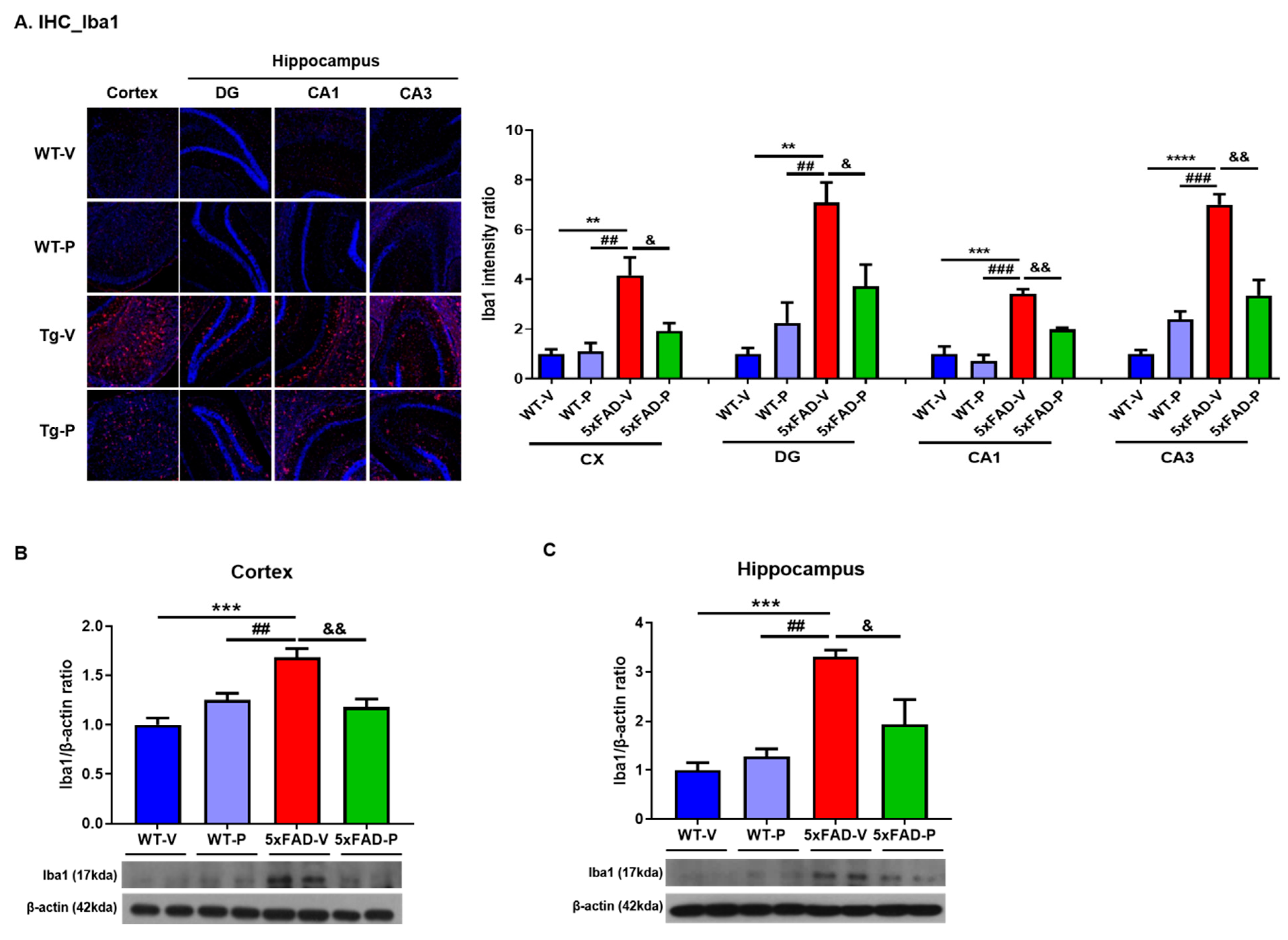
2.4.2. Immunohistochemistry and Quantification
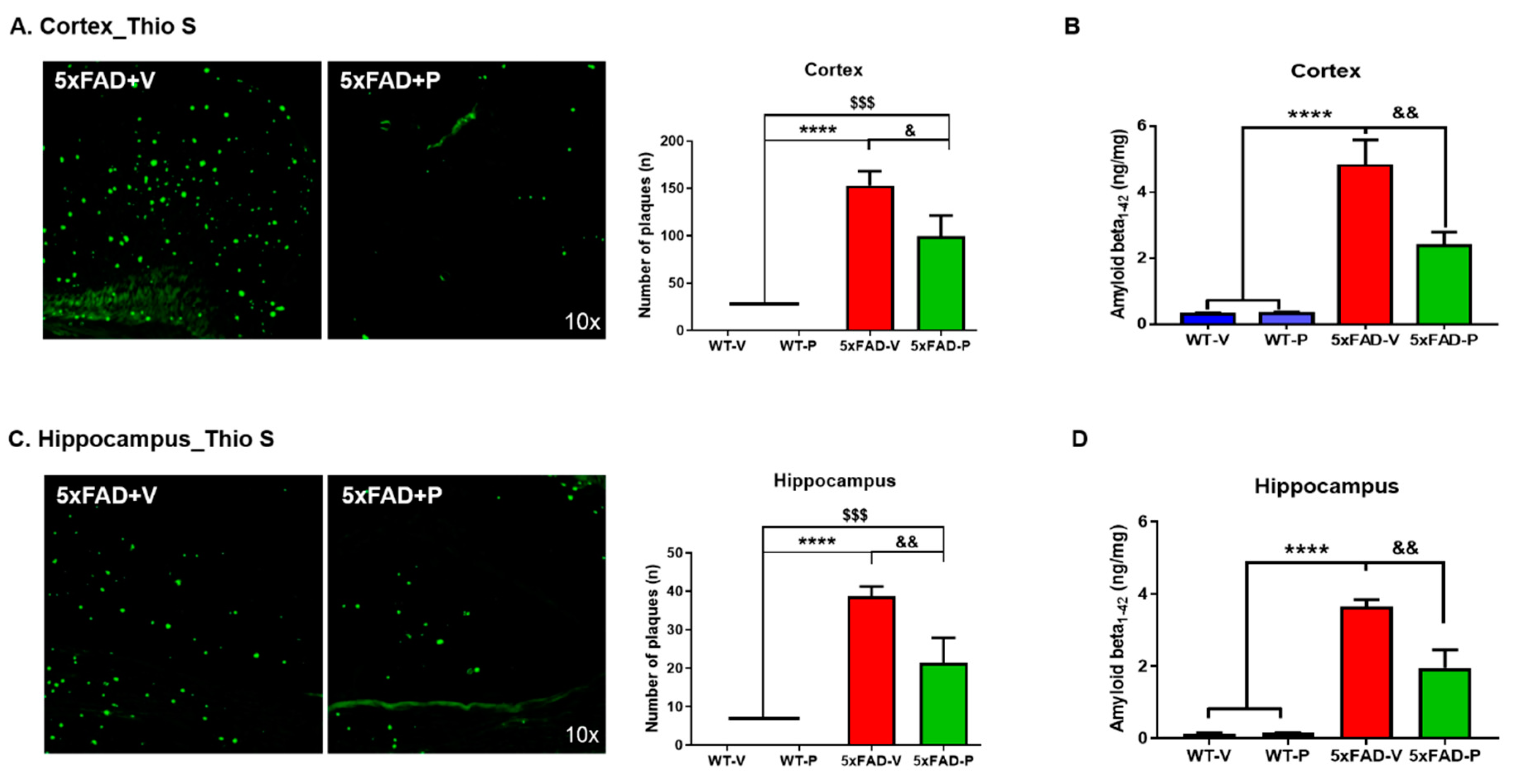
2.4.3. Amyloid Plaque Staining and Quantification
2.5. Western Blot
2.6. Enzyme-Linked Immunosorbent Assay
2.7. Statistical Analysis
3. Results
3.1. Long-Term Administration of PCO Recovers the Memory Deficit in 5xFAD Mice
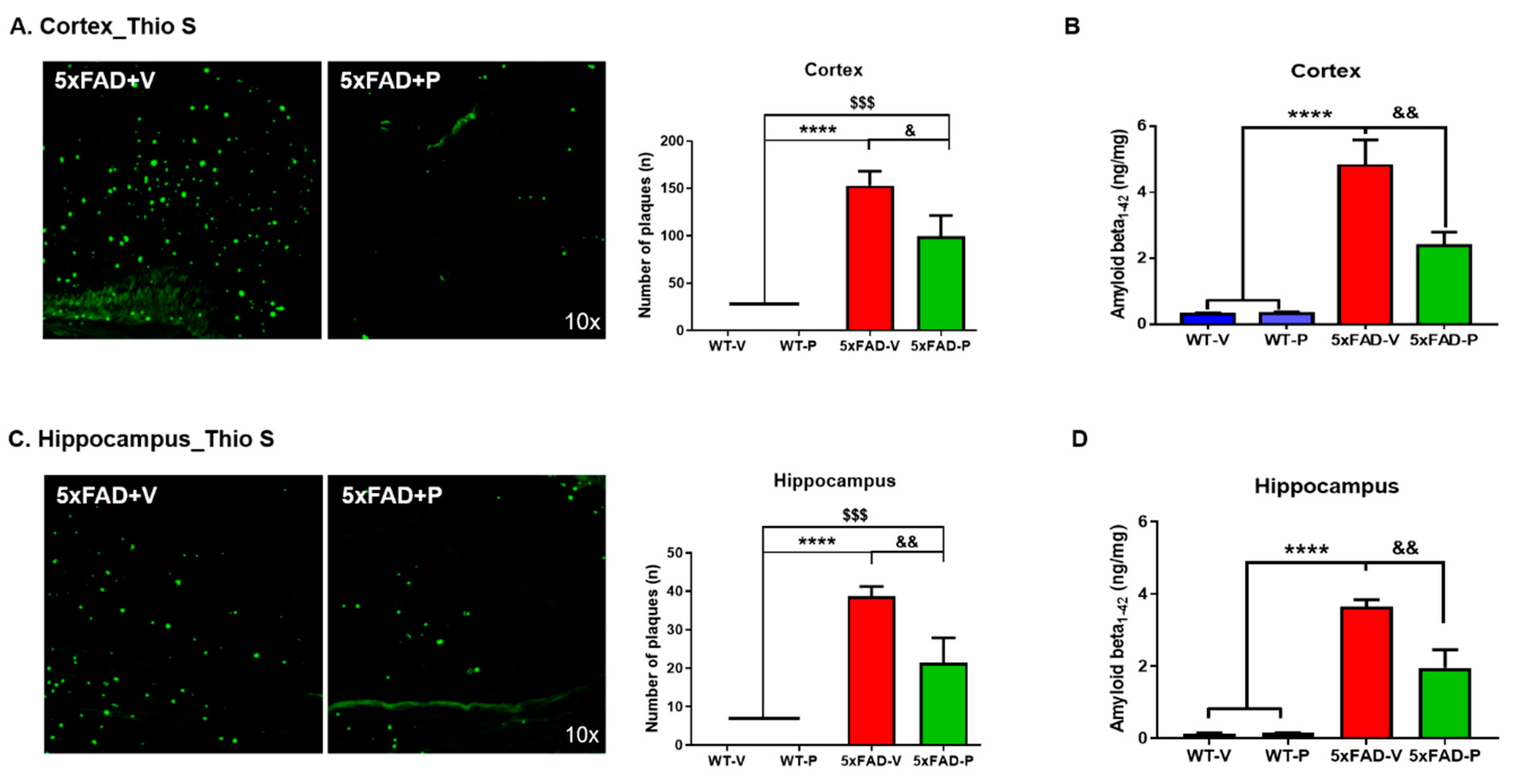
3.2. PCO Treatment Decreases the Formation of Amyloid Plaques in the Cortex and Hippocampus of 5xFAD Mice
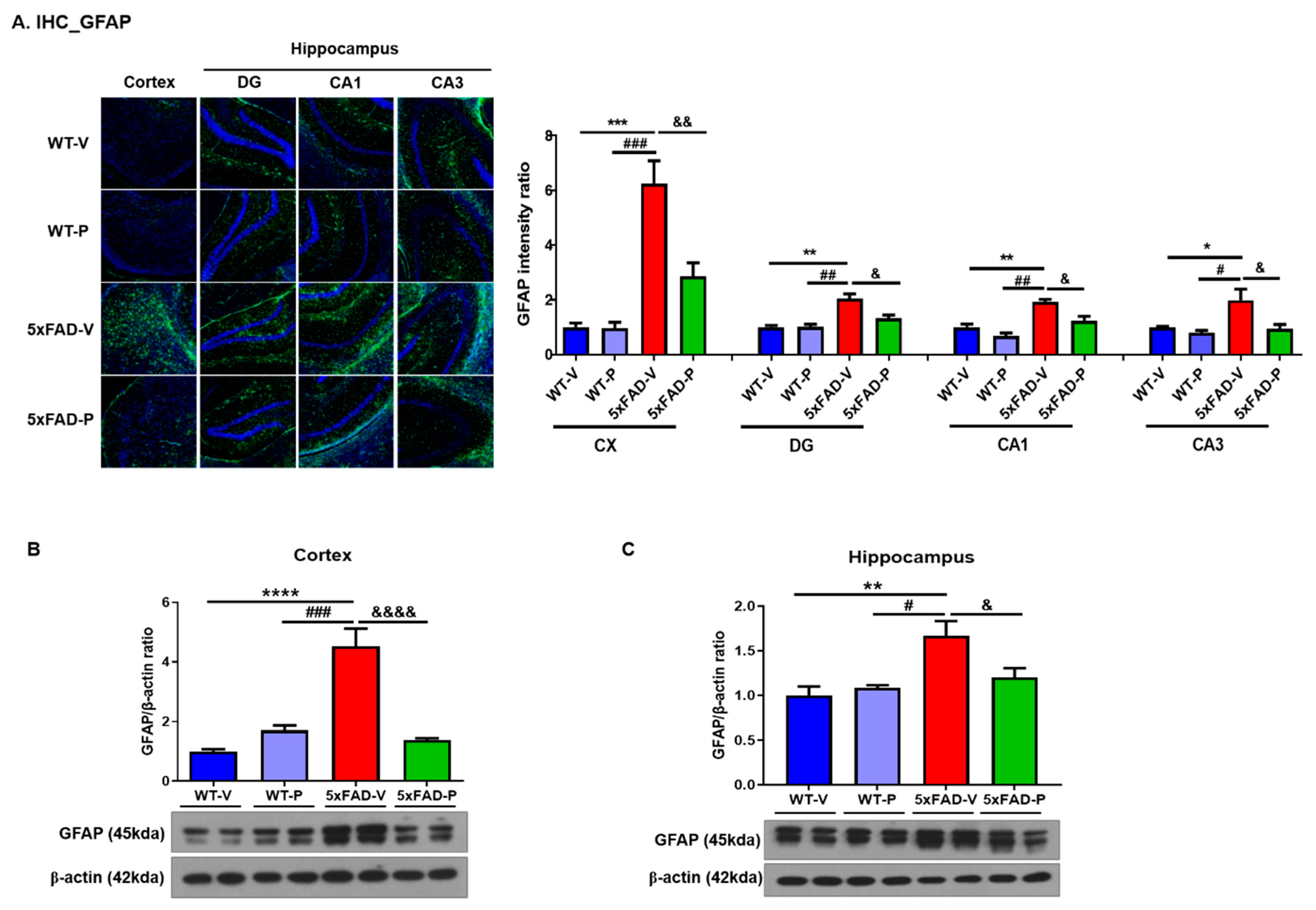
3.3. Long-Term Administration of PCO Attenuated Gliosis in the Brain of 5xFAD Mice
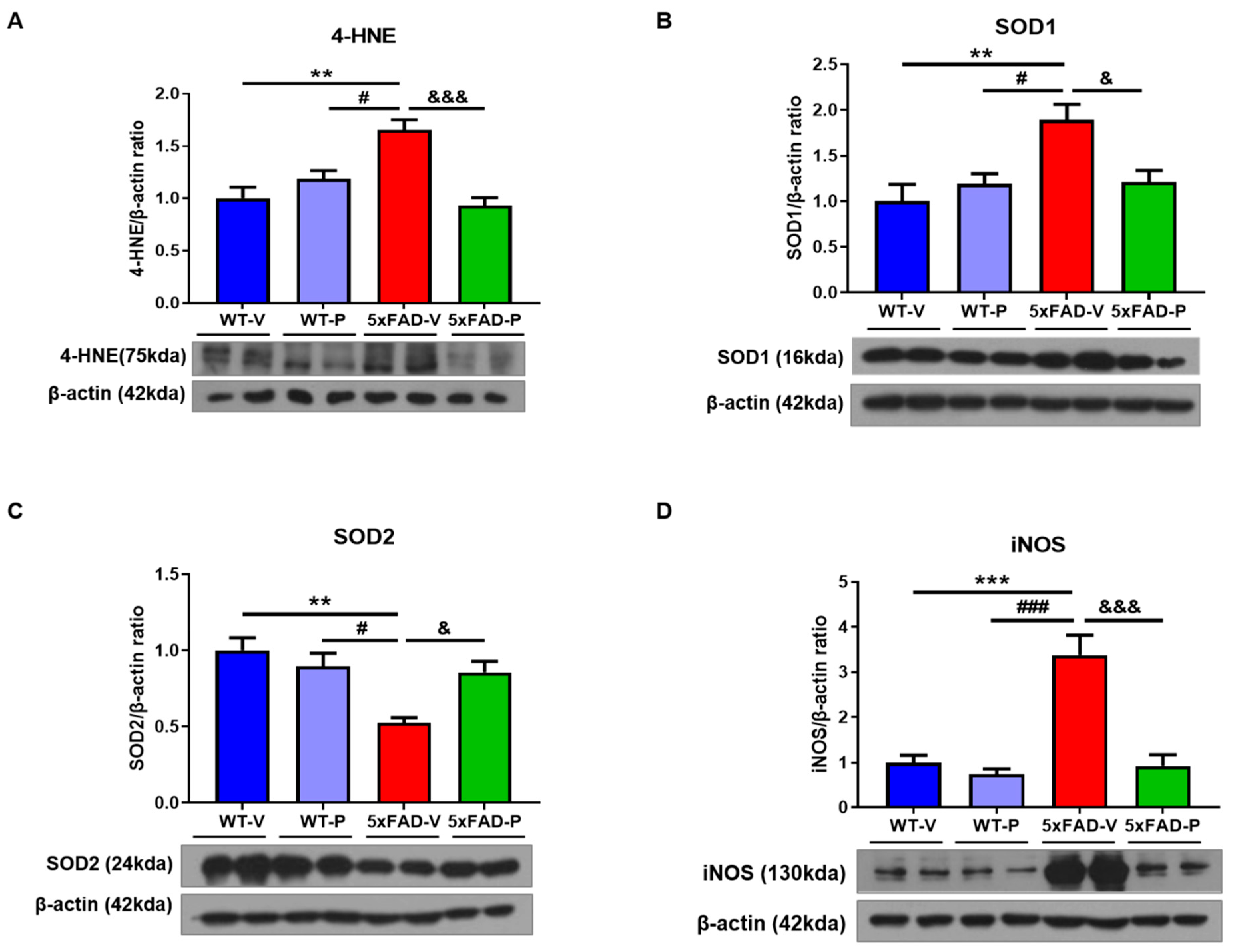
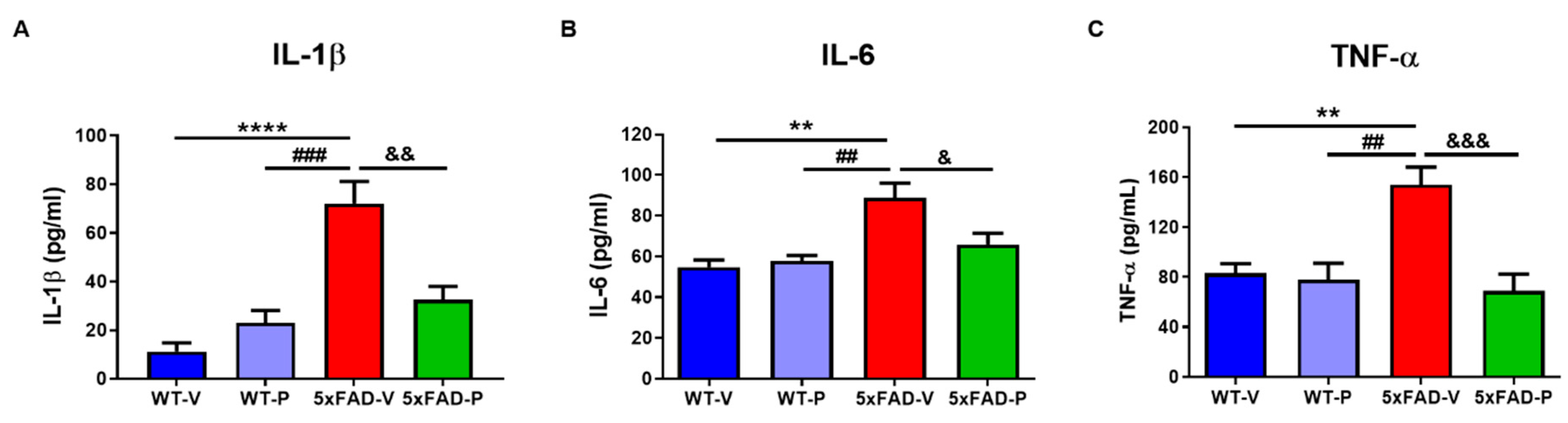
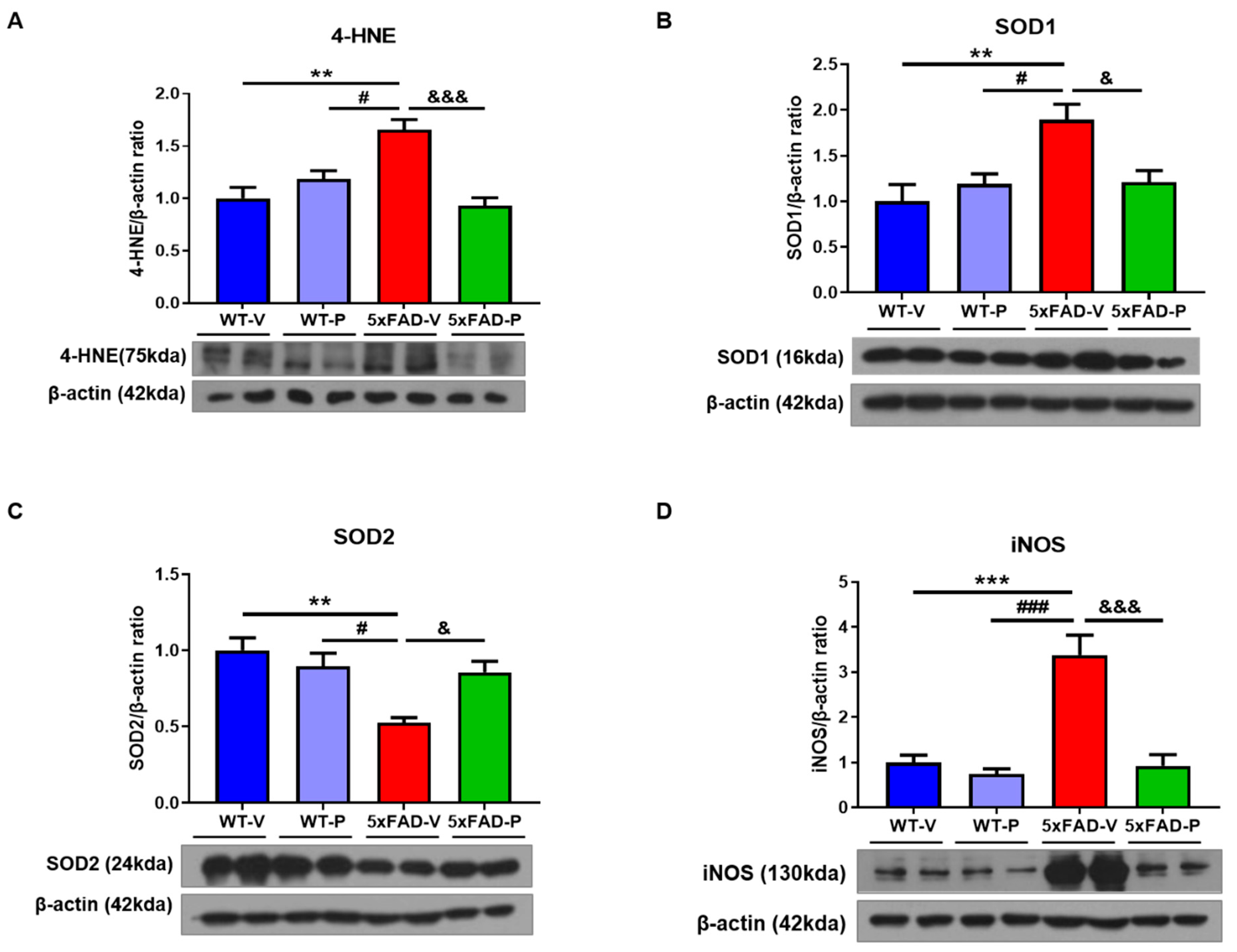
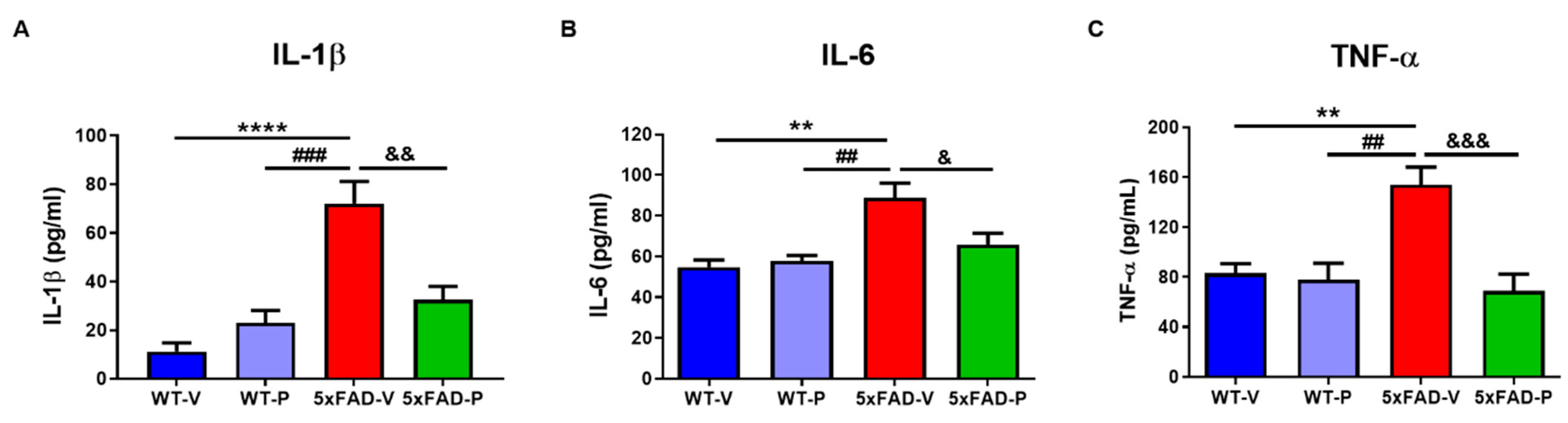
3.4. PCO Has the Antioxidant and Anti-Inflammatory Effect in the Brain of 5xFAD
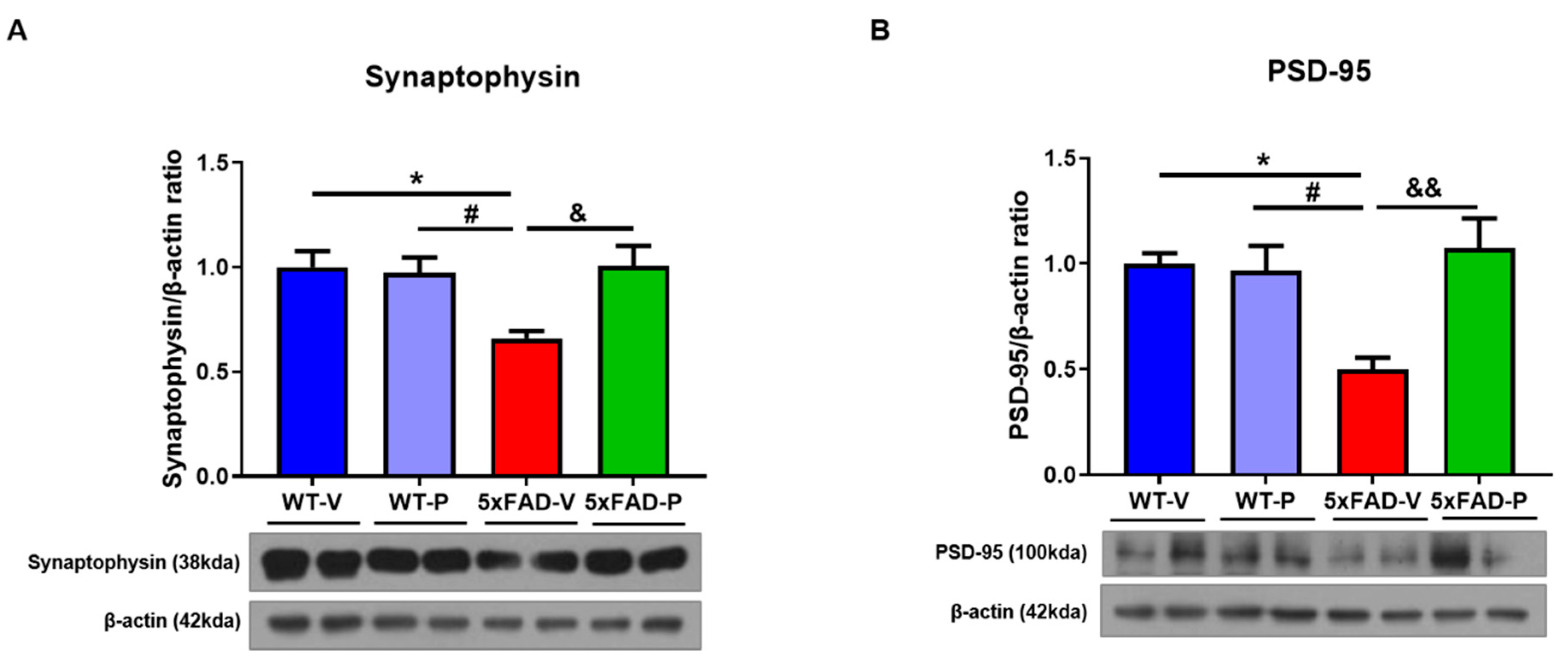
3.5. PCO Treatment Attenuates Synaptic loss in 5xFAD Mice
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- MartinPrince, A.C.-H. Martin Knapp, Maëlenn Guerchet, Maria Karagiannidou. World Alzheimer Report 2016: Impoving healthcare for people living with dementia: Coverage, quality and costs now and in the future. Report © 2016 Alzheimer’s Disease International. Available online: http://www.alz.co.uk/ (accessed on 18 August 2021).
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect Med. 2011, 1, a006189. [Google Scholar] [CrossRef] [PubMed]
- Haque, R.U.; Levey, A.I. Alzheimer’s disease: A clinical perspective and future nonhuman primate research opportunities. Proc. Natl. Acad. Sci. USA 2019, 116, 26224–26229. [Google Scholar] [CrossRef] [Green Version]
- Campora, M.; Francesconi, V.; Schenone, S.; Tasso, B.; Tonelli, M. Journey on Naphthoquinone and Anthraquinone Derivatives: New Insights in Alzheimer’s Disease. Pharmaceuticals 2021, 14, 33. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.F.; Xu, T.H.; Yan, Y.; Zhou, Y.R.; Jiang, Y.; Melcher, K.; Xu, H.E. Amyloid beta: Structure, biology and structure-based therapeutic development. Acta Pharmacol. Sin. 2017, 38, 1205–1235. [Google Scholar] [CrossRef] [PubMed]
- Bloom, G.S. Amyloid-beta and tau: The trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol. 2014, 71, 505–508. [Google Scholar] [CrossRef] [Green Version]
- Selkoe, D.J. Alzheimer’s disease: Genes, proteins, and therapy. Physiol. Rev. 2001, 81, 741–766. [Google Scholar] [CrossRef]
- Iwatsubo, T.; Odaka, A.; Suzuki, N.; Mizusawa, H.; Nukina, N.; Ihara, Y. Visualization of A beta 42(43) and A beta 40 in senile plaques with end-specific A beta monoclonals: Evidence that an initially deposited species is A beta 42(43). Neuron 1994, 13, 45–53. [Google Scholar] [CrossRef]
- Guo, T.; Zhang, D.; Zeng, Y.; Huang, T.Y.; Xu, H.; Zhao, Y. Molecular and cellular mechanisms underlying the pathogenesis of Alzheimer’s disease. Mol. Neurodegener. 2020, 15, 40. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Kinney, J.W.; Bemiller, S.M.; Murtishaw, A.S.; Leisgang, A.M.; Salazar, A.M.; Lamb, B.T. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimers Dement. 2018, 4, 575–590. [Google Scholar] [CrossRef]
- Olabarria, M.; Noristani, H.N.; Verkhratsky, A.; Rodriguez, J.J. Concomitant astroglial atrophy and astrogliosis in a triple transgenic animal model of Alzheimer’s disease. Glia 2010, 58, 831–838. [Google Scholar] [CrossRef] [PubMed]
- Stalder, M.; Phinney, A.; Probst, A.; Sommer, B.; Staufenbiel, M.; Jucker, M. Association of microglia with amyloid plaques in brains of APP23 transgenic mice. Am. J. Pathol. 1999, 154, 1673–1684. [Google Scholar] [CrossRef] [Green Version]
- Swardfager, W.; Lanctot, K.; Rothenburg, L.; Wong, A.; Cappell, J.; Herrmann, N. A meta-analysis of cytokines in Alzheimer’s disease. Biol. Psychiatry 2010, 68, 930–941. [Google Scholar] [CrossRef] [PubMed]
- Frost, G.R.; Li, Y.M. The role of astrocytes in amyloid production and Alzheimer’s disease. Open Biol. 2017, 7. [Google Scholar] [CrossRef] [Green Version]
- Parajuli, B.; Sonobe, Y.; Horiuchi, H.; Takeuchi, H.; Mizuno, T.; Suzumura, A. Oligomeric amyloid beta induces IL-1beta processing via production of ROS: Implication in Alzheimer’s disease. Cell Death Dis. 2013, 4, e975. [Google Scholar] [CrossRef] [Green Version]
- Markesbery, W.R. The role of oxidative stress in Alzheimer disease. Arch. Neurol. 1999, 56, 1449–1452. [Google Scholar] [CrossRef] [Green Version]
- Persson, T.; Popescu, B.O.; Cedazo-Minguez, A. Oxidative stress in Alzheimer’s disease: Why did antioxidant therapy fail? Oxid. Med. Cell Longev. 2014, 2014, 427318. [Google Scholar] [CrossRef] [Green Version]
- Chauhan, V.; Chauhan, A. Oxidative stress in Alzheimer’s disease. Pathophysiology 2006, 13, 195–208. [Google Scholar] [CrossRef]
- Wang, W.; Zhao, F.; Ma, X.; Perry, G.; Zhu, X. Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: Recent advances. Mol. Neurodegener. 2020, 15, 30. [Google Scholar] [CrossRef]
- Sayre, L.M.; Zelasko, D.A.; Harris, P.L.; Perry, G.; Salomon, R.G.; Smith, M.A. 4-Hydroxynonenal-derived advanced lipid peroxidation end products are increased in Alzheimer’s disease. J. Neurochem. 1997, 68, 2092–2097. [Google Scholar] [CrossRef]
- Mark, R.J.; Lovell, M.A.; Markesbery, W.R.; Uchida, K.; Mattson, M.P. A role for 4-hydroxynonenal, an aldehydic product of lipid peroxidation, in disruption of ion homeostasis and neuronal death induced by amyloid beta-peptide. J. Neurochem. 1997, 68, 255–264. [Google Scholar] [CrossRef]
- Zemlan, F.P.; Thienhaus, O.J.; Bosmann, H.B. Superoxide dismutase activity in Alzheimer’s disease: Possible mechanism for paired helical filament formation. Brain Res. 1989, 476, 160–162. [Google Scholar] [CrossRef]
- Murakami, K.; Murata, N.; Noda, Y.; Tahara, S.; Kaneko, T.; Kinoshita, N.; Hatsuta, H.; Murayama, S.; Barnham, K.J.; Irie, K.; et al. SOD1 (copper/zinc superoxide dismutase) deficiency drives amyloid beta protein oligomerization and memory loss in mouse model of Alzheimer disease. J. Biol. Chem. 2011, 286, 44557–44568. [Google Scholar] [CrossRef] [Green Version]
- Gong, J.; Qin, X.; Yuan, F.; Hu, M.; Chen, G.; Fang, K.; Wang, D.; Jiang, S.; Li, J.; Zhao, Y.; et al. Efficacy and safety of sugarcane policosanol on dyslipidemia: A meta-analysis of randomized controlled trials. Mol. Nutr. Food Res. 2018, 62. [Google Scholar] [CrossRef] [PubMed]
- Janikula, M. Policosanol: A new treatment for cardiovascular disease? Altern. Med. Rev. 2002, 7, 203–217. [Google Scholar]
- Park, H.J.; Yadav, D.; Jeong, D.J.; Kim, S.J.; Bae, M.A.; Kim, J.R.; Cho, K.H. Short-Term Consumption of Cuban Policosanol Lowers Aortic and Peripheral Blood Pressure and Ameliorates Serum Lipid Parameters in Healthy Korean Participants: Randomized, Double-Blinded, and Placebo-Controlled Study. Int. J. Environ. Res. Public Health 2019, 16, 809. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.J.; Yadav, D.; Park, H.J.; Kim, J.R.; Cho, K.H. Long-Term Consumption of Cuban Policosanol Lowers Central and Brachial Blood Pressure and Improves Lipid Profile With Enhancement of Lipoprotein Properties in Healthy Korean Participants. Front. Physiol. 2018, 9, 412. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.Y.; Yoo, J.A.; Lim, S.M.; Cho, K.H. Anti-Aging and Tissue Regeneration Ability of Policosanol Along with Lipid-Lowering Effect in Hyperlipidemic Zebrafish via Enhancement of High-Density Lipoprotein Functionality. Rejuvenation Res. 2016, 19, 149–158. [Google Scholar] [CrossRef] [Green Version]
- Kang, S.; Kim, J.; Chang, K.A. Spatial memory deficiency early in 6xTg Alzheimer’s disease mouse model. Sci. Rep. 2021, 11, 1334. [Google Scholar] [CrossRef]
- Crouzin, N.; Baranger, K.; Cavalier, M.; Marchalant, Y.; Cohen-Solal, C.; Roman, F.S.; Khrestchatisky, M.; Rivera, S.; Feron, F.; Vignes, M. Area-specific alterations of synaptic plasticity in the 5XFAD mouse model of Alzheimer’s disease: Dissociation between somatosensory cortex and hippocampus. PLoS ONE 2013, 8, e74667. [Google Scholar] [CrossRef]
- Gouni-Berthold, I.; Berthold, H.K. Policosanol: Clinical pharmacology and therapeutic significance of a new lipid-lowering agent. Am. Heart J. 2002, 143, 356–365. [Google Scholar] [CrossRef]
- Cho, K.H.; Kim, S.J.; Yadav, D.; Kim, J.Y.; Kim, J.R. Consumption of Cuban Policosanol Improves Blood Pressure and Lipid Profile via Enhancement of HDL Functionality in Healthy Women Subjects: Randomized, Double-Blinded, and Placebo-Controlled Study. Oxid. Med. Cell Longev. 2018, 2018, 4809525. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.Y.; Kim, S.M.; Kim, S.J.; Lee, E.Y.; Kim, J.R.; Cho, K.H. Consumption of policosanol enhances HDL functionality via CETP inhibition and reduces blood pressure and visceral fat in young and middle-aged subjects. Int. J. Mol. Med. 2017, 39, 889–899. [Google Scholar] [CrossRef] [Green Version]
- Molina, V.; Ravelo, Y.; Noa, M.; Mas, R.; Perez, Y.; Oyarzabal, A.; Mendoza, N.; Valle, M.; Jimenez, S.; Sanchez, J. Therapeutic Effects of Policosanol and Atorvastatin against Global Brain Ischaemia-Reperfusion Injury in Gerbils. Indian J. Pharm Sci. 2013, 75, 635–641. [Google Scholar]
- Alzheimer’s, A. 2016 Alzheimer’s disease facts and figures. Alzheimers Dement. 2016, 12, 459–509. [Google Scholar] [CrossRef]
- Marsillach, J.; Adorni, M.P.; Zimetti, F.; Papotti, B.; Zuliani, G.; Cervellati, C. HDL Proteome and Alzheimer’s Disease: Evidence of a Link. Antioxidants 2020, 9, 1224. [Google Scholar] [CrossRef] [PubMed]
- Wingo, T.S.; Cutler, D.J.; Wingo, A.P.; Le, N.A.; Rabinovici, G.D.; Miller, B.L.; Lah, J.J.; Levey, A.I. Association of Early-Onset Alzheimer Disease With Elevated Low-Density Lipoprotein Cholesterol Levels and Rare Genetic Coding Variants of APOB. JAMA Neurol. 2019, 76, 809–817. [Google Scholar] [CrossRef]
- Zhou, Z.; Liang, Y.; Zhang, X.; Xu, J.; Lin, J.; Zhang, R.; Kang, K.; Liu, C.; Zhao, C.; Zhao, M. Low-Density Lipoprotein Cholesterol and Alzheimer’s Disease: A Systematic Review and Meta-Analysis. Front. Aging Neurosci. 2020, 12, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reitz, C.; Tang, M.X.; Schupf, N.; Manly, J.J.; Mayeux, R.; Luchsinger, J.A. Association of higher levels of high-density lipoprotein cholesterol in elderly individuals and lower risk of late-onset Alzheimer disease. Arch. Neurol. 2010, 67, 1491–1497. [Google Scholar] [CrossRef] [Green Version]
- Lewis, T.L.; Cao, D.; Lu, H.; Mans, R.A.; Su, Y.R.; Jungbauer, L.; Linton, M.F.; Fazio, S.; LaDu, M.J.; Li, L. Overexpression of human apolipoprotein A-I preserves cognitive function and attenuates neuroinflammation and cerebral amyloid angiopathy in a mouse model of Alzheimer disease. J. Biol. Chem. 2010, 285, 36958–36968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dal Magro, R.; Simonelli, S.; Cox, A.; Formicola, B.; Corti, R.; Cassina, V.; Nardo, L.; Mantegazza, F.; Salerno, D.; Grasso, G.; et al. The Extent of Human Apolipoprotein A-I Lipidation Strongly Affects the beta-Amyloid Efflux Across the Blood-Brain Barrier in vitro. Front. Neurosci. 2019, 13, 419. [Google Scholar] [CrossRef]
- Paterno, R.; Ruocco, A.; Postiglione, A.; Hubsch, A.; Andresen, I.; Lang, M.G. Reconstituted high-density lipoprotein exhibits neuroprotection in two rat models of stroke. Cerebrovasc. Dis. 2004, 17, 204–211. [Google Scholar] [CrossRef] [PubMed]
- Button, E.B.; Boyce, G.K.; Wilkinson, A.; Stukas, S.; Hayat, A.; Fan, J.; Wadsworth, B.J.; Robert, J.; Martens, K.M.; Wellington, C.L. ApoA-I deficiency increases cortical amyloid deposition, cerebral amyloid angiopathy, cortical and hippocampal astrogliosis, and amyloid-associated astrocyte reactivity in APP/PS1 mice. Alzheimers Res. Ther. 2019, 11, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Button, E.B.; Robert, J.; Caffrey, T.M.; Fan, J.; Zhao, W.; Wellington, C.L. HDL from an Alzheimer’s disease perspective. Curr. Opin. Lipidol. 2019, 30, 224–234. [Google Scholar] [CrossRef] [PubMed]
- Zimetti, F.; Adorni, M.P.; Marsillach, J.; Marchi, C.; Trentini, A.; Valacchi, G.; Cervellati, C. Connection between the Altered HDL Antioxidant and Anti-Inflammatory Properties and the Risk to Develop Alzheimer’s Disease: A Narrative Review. Oxid. Med. Cell Longev. 2021, 2021, 6695796. [Google Scholar] [CrossRef]
- Sasaguri, H.; Nilsson, P.; Hashimoto, S.; Nagata, K.; Saito, T.; De Strooper, B.; Hardy, J.; Vassar, R.; Winblad, B.; Saido, T.C. APP mouse models for Alzheimer’s disease preclinical studies. EMBO J. 2017, 36, 2473–2487. [Google Scholar] [CrossRef]
- Ries, M.; Sastre, M. Mechanisms of Abeta Clearance and Degradation by Glial Cells. Front. Aging Neurosci. 2016, 8, 160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minter, M.R.; Taylor, J.M.; Crack, P.J. The contribution of neuroinflammation to amyloid toxicity in Alzheimer’s disease. J. Neurochem. 2016, 136, 457–474. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Akama, K.T.; Krafft, G.A.; Chromy, B.A.; Van Eldik, L.J. Amyloid-beta peptide activates cultured astrocytes: Morphological alterations, cytokine induction and nitric oxide release. Brain Res. 1998, 785, 195–206. [Google Scholar] [CrossRef]
- Fuhrmann, M.; Bittner, T.; Jung, C.K.; Burgold, S.; Page, R.M.; Mitteregger, G.; Haass, C.; LaFerla, F.M.; Kretzschmar, H.; Herms, J. Microglial Cx3cr1 knockout prevents neuron loss in a mouse model of Alzheimer’s disease. Nat. Neurosci. 2010, 13, 411–413. [Google Scholar] [CrossRef] [Green Version]
- Mandrekar-Colucci, S.; Landreth, G.E. Microglia and inflammation in Alzheimer’s disease. CNS Neurol. Disord. Drug Targets 2010, 9, 156–167. [Google Scholar] [CrossRef] [PubMed]
- Villa, V.; Thellung, S.; Bajetto, A.; Gatta, E.; Robello, M.; Novelli, F.; Tasso, B.; Tonelli, M.; Florio, T. Novel celecoxib analogues inhibit glial production of prostaglandin E2, nitric oxide, and oxygen radicals reverting the neuroinflammatory responses induced by misfolded prion protein fragment 90-231 or lipopolysaccharide. Pharmacol. Res. 2016, 113, 500–514. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.J.; Zhang, X.; Chen, W.W. Role of oxidative stress in Alzheimer’s disease. Biomed. Rep. 2016, 4, 519–522. [Google Scholar] [CrossRef] [Green Version]
- Butterfield, D.A.; Boyd-Kimball, D. Oxidative Stress, Amyloid-beta Peptide, and Altered Key Molecular Pathways in the Pathogenesis and Progression of Alzheimer’s Disease. J. Alzheimers Dis. 2018, 62, 1345–1367. [Google Scholar] [CrossRef] [Green Version]
- Qin, Y.; Zhu, W.; Zhan, C.; Zhao, L.; Wang, J.; Tian, Q.; Wang, W. Investigation on positive correlation of increased brain iron deposition with cognitive impairment in Alzheimer disease by using quantitative MR R2’ mapping. J. Huazhong Univ. Sci. Technolog. Med. Sci. 2011, 31, 578. [Google Scholar] [CrossRef]
- Nizzari, M.; Thellung, S.; Corsaro, A.; Villa, V.; Pagano, A.; Porcile, C.; Russo, C.; Florio, T. Neurodegeneration in Alzheimer disease: Role of amyloid precursor protein and presenilin 1 intracellular signaling. J. Toxicol. 2012, 2012, 187297. [Google Scholar] [CrossRef] [PubMed]
- Sultana, R.; Perluigi, M.; Butterfield, D.A. Lipid peroxidation triggers neurodegeneration: A redox proteomics view into the Alzheimer disease brain. Free Radic Biol. Med. 2013, 62, 157–169. [Google Scholar] [CrossRef] [Green Version]
- Montine, T.J.; Neely, M.D.; Quinn, J.F.; Beal, M.F.; Markesbery, W.R.; Roberts, L.J.; Morrow, J.D. Lipid peroxidation in aging brain and Alzheimer’s disease. Free Radic. Biol. Med. 2002, 33, 620–626. [Google Scholar] [CrossRef]
- Pratico, D.; Uryu, K.; Leight, S.; Trojanoswki, J.Q.; Lee, V.M. Increased lipid peroxidation precedes amyloid plaque formation in an animal model of Alzheimer amyloidosis. J. Neurosci. 2001, 21, 4183–4187. [Google Scholar] [CrossRef] [Green Version]
- Butterfield, D.A.; Lauderback, C.M. Lipid peroxidation and protein oxidation in Alzheimer’s disease brain: Potential causes and consequences involving amyloid beta-peptide-associated free radical oxidative stress. Free Radic Biol. Med. 2002, 32, 1050–1060. [Google Scholar] [CrossRef]
- Mecocci, P.; MacGarvey, U.; Beal, M.F. Oxidative damage to mitochondrial DNA is increased in Alzheimer’s disease. Ann. Neurol. 1994, 36, 747–751. [Google Scholar] [CrossRef]
- Aksenov, M.Y.; Aksenova, M.V.; Butterfield, D.A.; Geddes, J.W.; Markesbery, W.R. Protein oxidation in the brain in Alzheimer’s disease. Neuroscience 2001, 103, 373–383. [Google Scholar] [CrossRef]
- Markesbery, W.R.; Lovell, M.A. Four-hydroxynonenal, a product of lipid peroxidation, is increased in the brain in Alzheimer’s disease. Neurobiol. Aging 1998, 19, 33–36. [Google Scholar] [CrossRef]
- McGrath, L.T.; McGleenon, B.M.; Brennan, S.; McColl, D.; Mc, I.S.; Passmore, A.P. Increased oxidative stress in Alzheimer’s disease as assessed with 4-hydroxynonenal but not malondialdehyde. QJM 2001, 94, 485–490. [Google Scholar] [CrossRef] [PubMed]
- Lovell, M.A.; Xie, C.; Markesbery, W.R. Acrolein is increased in Alzheimer’s disease brain and is toxic to primary hippocampal cultures. Neurobiol. Aging 2001, 22, 187–194. [Google Scholar] [CrossRef]
- Shoeb, M.; Ansari, N.H.; Srivastava, S.K.; Ramana, K.V. 4-Hydroxynonenal in the pathogenesis and progression of human diseases. Curr. Med. Chem. 2014, 21, 230–237. [Google Scholar] [CrossRef]
- Marcus, D.L.; Thomas, C.; Rodriguez, C.; Simberkoff, K.; Tsai, J.S.; Strafaci, J.A.; Freedman, M.L. Increased peroxidation and reduced antioxidant enzyme activity in Alzheimer’s disease. Exp. Neurol. 1998, 150, 40–44. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhao, B. Oxidative stress and the pathogenesis of Alzheimer’s disease. Oxid Med. Cell Longev. 2013, 2013, 316523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, J.; Rees, H.D.; Weintraub, S.T.; Levey, A.I.; Chin, L.S.; Li, L. Oxidative modifications and aggregation of Cu,Zn-superoxide dismutase associated with Alzheimer and Parkinson diseases. J. Biol. Chem. 2005, 280, 11648–11655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grinan-Ferre, C.; Sarroca, S.; Ivanova, A.; Puigoriol-Illamola, D.; Aguado, F.; Camins, A.; Sanfeliu, C.; Pallas, M. Epigenetic mechanisms underlying cognitive impairment and Alzheimer disease hallmarks in 5XFAD mice. Aging 2016, 8, 664–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esposito, L.; Raber, J.; Kekonius, L.; Yan, F.; Yu, G.Q.; Bien-Ly, N.; Puolivali, J.; Scearce-Levie, K.; Masliah, E.; Mucke, L. Reduction in mitochondrial superoxide dismutase modulates Alzheimer’s disease-like pathology and accelerates the onset of behavioral changes in human amyloid precursor protein transgenic mice. J. Neurosci. 2006, 26, 5167–5179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.P.; Pancholi, N.; Esposito, L.; Previll, L.A.; Wang, X.; Zhu, X.; Smith, M.A.; Lee, H.G. Early induction of oxidative stress in mouse model of Alzheimer disease with reduced mitochondrial superoxide dismutase activity. PLoS ONE 2012, 7, e28033. [Google Scholar] [CrossRef] [PubMed]
- Tonnies, E.; Trushina, E. Oxidative Stress, Synaptic Dysfunction, and Alzheimer’s Disease. J. Alzheimers Dis. 2017, 57, 1105–1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferretti, M.T.; Iulita, M.F.; Cavedo, E.; Chiesa, P.A.; Schumacher Dimech, A.; Santuccione Chadha, A.; Baracchi, F.; Girouard, H.; Misoch, S.; Giacobini, E.; et al. Sex differences in Alzheimer disease—the gateway to precision medicine. Nat. Rev. Neurol. 2018, 14, 457–469. [Google Scholar] [CrossRef]
- Svensson, T.; Sawada, N.; Mimura, M.; Nozaki, S.; Shikimoto, R.; Tsugane, S. The association between midlife serum high-density lipoprotein and mild cognitive impairment and dementia after 19 years of follow-up. Transl. Psychiatry 2019, 9, 26. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.-H.; Lim, D.-K.; Suh, Y.-H.; Chang, K.-A. Long-Term Treatment of Cuban Policosanol Attenuates Abnormal Oxidative Stress and Inflammatory Response via Amyloid Plaques Reduction in 5xFAD Mice. Antioxidants 2021, 10, 1321. https://doi.org/10.3390/antiox10081321
Kim J-H, Lim D-K, Suh Y-H, Chang K-A. Long-Term Treatment of Cuban Policosanol Attenuates Abnormal Oxidative Stress and Inflammatory Response via Amyloid Plaques Reduction in 5xFAD Mice. Antioxidants. 2021; 10(8):1321. https://doi.org/10.3390/antiox10081321
Chicago/Turabian StyleKim, Jin-Ho, Dong-Kyun Lim, Yoo-Hun Suh, and Keun-A Chang. 2021. "Long-Term Treatment of Cuban Policosanol Attenuates Abnormal Oxidative Stress and Inflammatory Response via Amyloid Plaques Reduction in 5xFAD Mice" Antioxidants 10, no. 8: 1321. https://doi.org/10.3390/antiox10081321
APA StyleKim, J.-H., Lim, D.-K., Suh, Y.-H., & Chang, K.-A. (2021). Long-Term Treatment of Cuban Policosanol Attenuates Abnormal Oxidative Stress and Inflammatory Response via Amyloid Plaques Reduction in 5xFAD Mice. Antioxidants, 10(8), 1321. https://doi.org/10.3390/antiox10081321