
The Impact of Oxidative Stress of Environmental Origin on the Onset of Placental Diseases



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
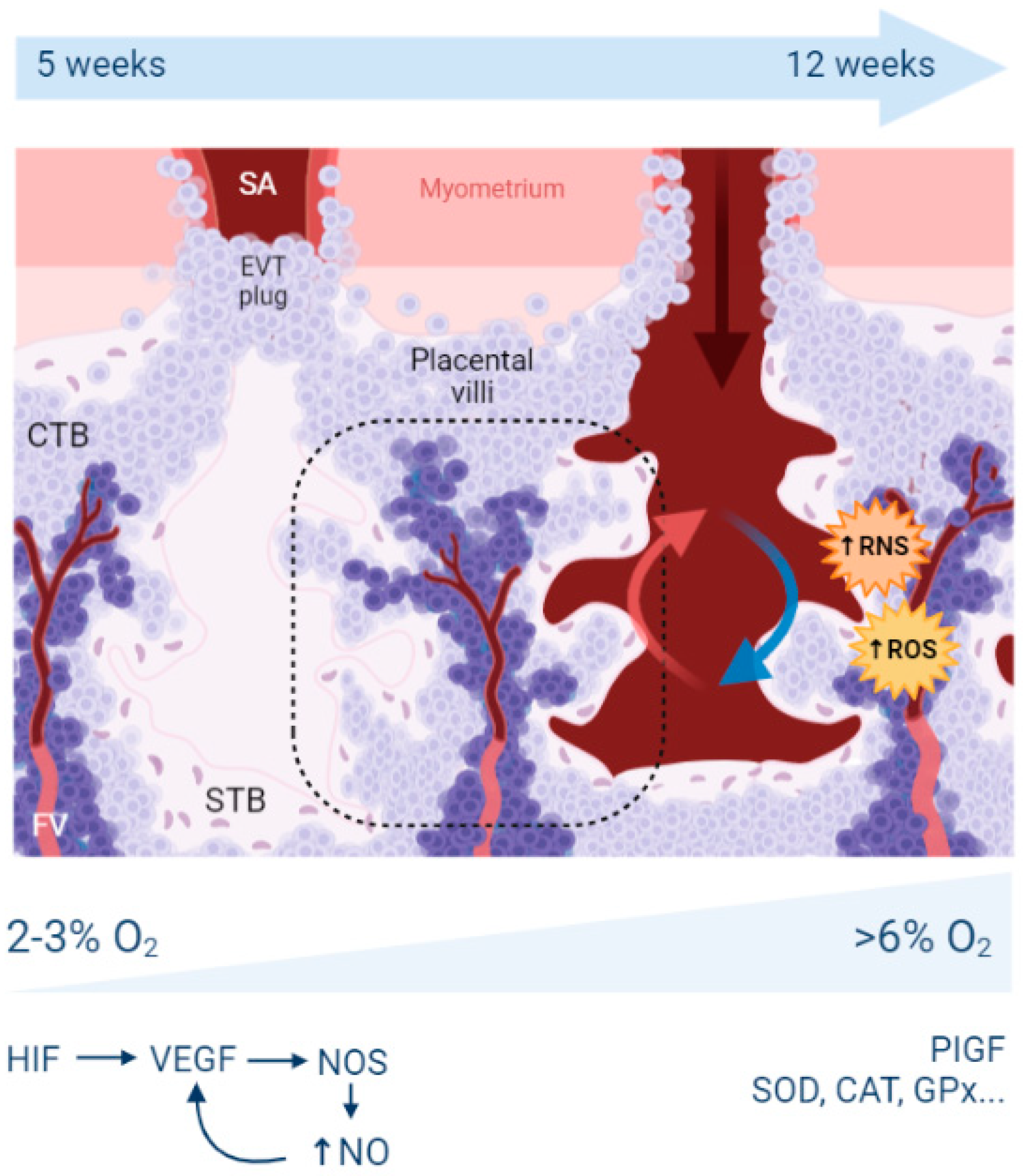
2. Reminders about Hypoxia, Oxidative Stress, and Placental Development
2.1. What Is the Origin of Oxidative Stress in the Placenta?
2.2. What Are the Antioxidant Mechanisms in the Placenta?
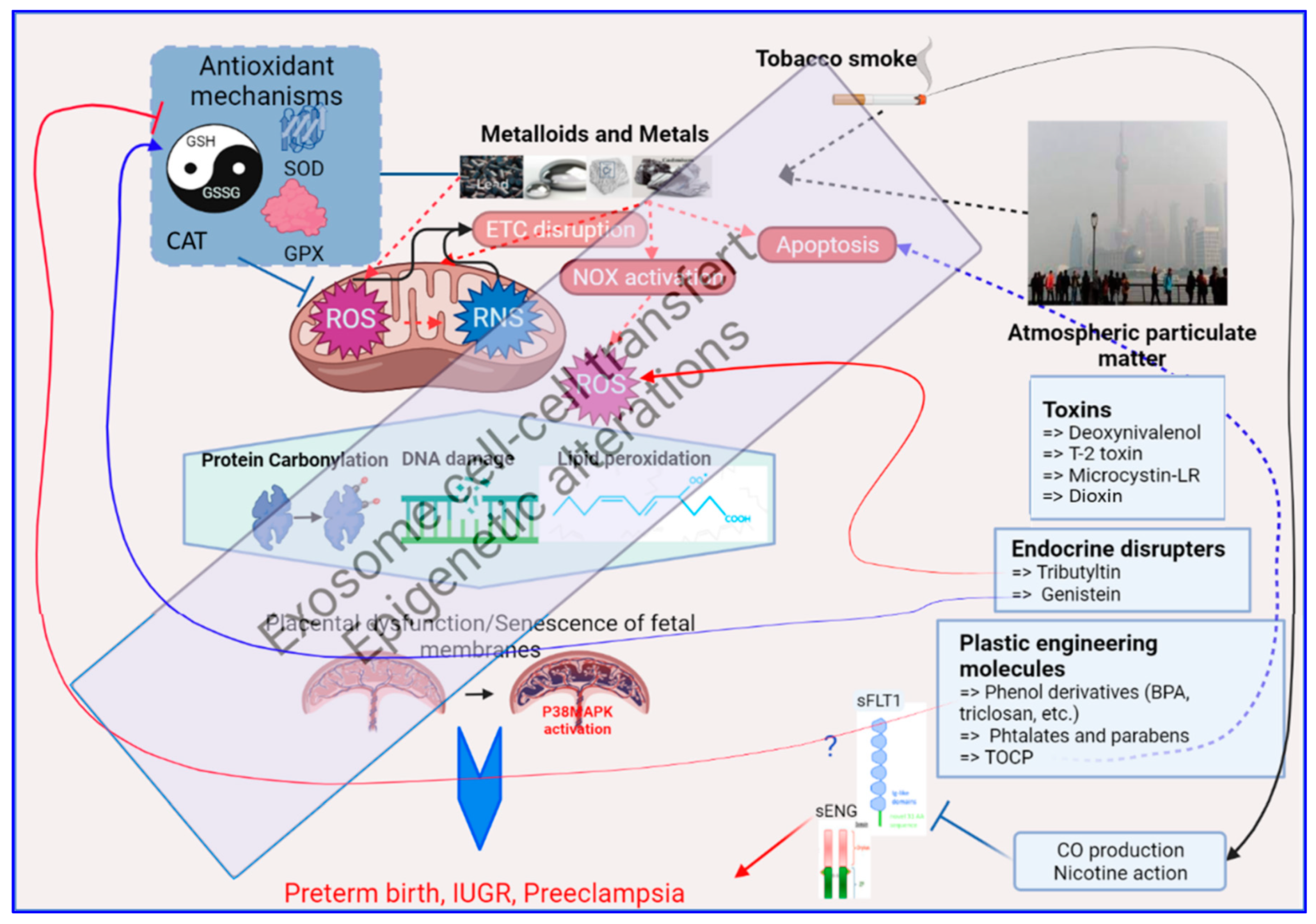
3. Mechanisms of Oxidative Stress Induction by Environmental Pollutants
3.1. Exposure to Environmental Metals
3.1.1. Cadmium
3.1.2. Mercury and Methyl-Mercury
3.1.3. Lead
3.1.4. Chromium
3.2. Exposure to Tobacco
3.3. Exposure to Airborne Particulate Matter
3.4. Exposure to Vexing Biomolecules and Riling Toxins
3.5. Exposure to Organometallic Molecules and Endocrine Disrupters
3.6. Plastic Modifiers: Phenols, Bisphenols and Parabens
4. Final Considerations and Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Zejnullahu, V.A.; Zejnullahu, V.A.; Kosumi, E. The role of oxidative stress in patients with recurrent pregnancy loss: A review. Reprod. Health 2021, 18, 207. [Google Scholar] [CrossRef]
- van Westering-Kroon, E.; Huizing, M.J.; Villamor-Martinez, E.; Villamor, E. Male Disadvantage in Oxidative Stress-Associated Complications of Prematurity: A Systematic Review, Meta-Analysis and Meta-Regression. Antioxidants 2021, 10, 1490. [Google Scholar] [CrossRef] [PubMed]
- Oke, S.L.; Hardy, D.B. The Role of Cellular Stress in Intrauterine Growth Restriction and Postnatal Dysmetabolism. Int. J. Mol. Sci. 2021, 22, 6986. [Google Scholar] [CrossRef] [PubMed]
- Ornoy, A.; Becker, M.; Weinstein-Fudim, L.; Ergaz, Z. Diabetes during Pregnancy: A Maternal Disease Complicating the Course of Pregnancy with Long-Term Deleterious Effects on the Offspring. A Clinical Review. Int. J. Mol. Sci. 2021, 22, 2956. [Google Scholar] [CrossRef]
- Han, C.; Huang, P.; Lyu, M.; Dong, J. Oxidative Stress and Preeclampsia-Associated Prothrombotic State. Antioxidants 2020, 9, 1139. [Google Scholar] [CrossRef]
- Brosens, J.J.; Pijnenborg, R.; Brosens, I.A. The myometrial junctional zone spiral arteries in normal and abnormal pregnancies: A review of the literature. Am. J. Obstet. Gynecol. 2002, 187, 1416–1423. [Google Scholar] [CrossRef] [PubMed]
- Fryer, B.H.; Simon, M.C. Hypoxia, HIF and the placenta. Cell Cycle 2006, 5, 495–498. [Google Scholar] [CrossRef]
- Krock, B.L.; Skuli, N.; Simon, M.C. Hypoxia-induced angiogenesis: Good and evil. Genes Cancer 2011, 2, 1117–1133. [Google Scholar] [CrossRef]
- D’Souza, V.; Rani, A.; Patil, V.; Pisal, H.; Randhir, K.; Mehendale, S.; Wagh, G.; Gupte, S.; Joshi, S. Increased oxidative stress from early pregnancy in women who develop preeclampsia. Clin. Exp. Hypertens. 2016, 38, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.K.; Georgiadis, A.; Copland, D.A.; Liyanage, S.; Luhmann, U.F.; Robbie, S.J.; Liu, J.; Wu, J.; Bainbridge, J.W.; Bates, D.O.; et al. IL-4 regulates specific Arg-1(+) macrophage sFlt-1-mediated inhibition of angiogenesis. Am. J. Pathol. 2015, 185, 2324–2335. [Google Scholar] [CrossRef] [PubMed]
- Jauniaux, E.; Watson, A.L.; Hempstock, J.; Bao, Y.P.; Skepper, J.N.; Burton, G.J. Onset of maternal arterial blood flow and placental oxidative stress. A possible factor in human early pregnancy failure. Am. J. Pathol. 2000, 157, 2111–2122. [Google Scholar] [CrossRef]
- Aouache, R.; Biquard, L.; Vaiman, D.; Miralles, F. Oxidative Stress in Preeclampsia and Placental Diseases. Int. J. Mol. Sci. 2018, 19, 1496. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Sferruzzi-Perri, A.N. Placental mitochondrial function in response to gestational exposures. Placenta 2021, 104, 124–137. [Google Scholar] [CrossRef] [PubMed]
- Fisher, J.J.; McKeating, D.R.; Cuffe, J.S.; Bianco-Miotto, T.; Holland, O.J.; Perkins, A.V. Proteomic Analysis of Placental Mitochondria Following Trophoblast Differentiation. Front. Physiol. 2019, 10, 1536. [Google Scholar] [CrossRef]
- Martinez, F.; Olvera-Sanchez, S.; Esparza-Perusquia, M.; Gomez-Chang, E.; Flores-Herrera, O. Multiple functions of syncytiotrophoblast mitochondria. Steroids 2015, 103, 11–22. [Google Scholar] [CrossRef]
- Redman, C.W.G.; Staff, A.C.; Roberts, J.M. Syncytiotrophoblast stress in preeclampsia: The convergence point for multiple pathways. Am. J. Obstet. Gynecol. 2020, S0002-9378(20), 31115–31117. [Google Scholar] [CrossRef]
- Ganguly, E.; Kirschenman, R.; Spaans, F.; Holody, C.D.; Phillips, T.E.J.; Case, C.P.; Cooke, C.M.; Murphy, M.P.; Lemieux, H.; Davidge, S.T. Nanoparticle-encapsulated antioxidant improves placental mitochondrial function in a sexually dimorphic manner in a rat model of prenatal hypoxia. FASEB J. 2021, 35, e21338. [Google Scholar] [CrossRef]
- Ferreira, R.C.; Fragoso, M.B.T.; Bueno, N.B.; Goulart, M.O.F.; de Oliveira, A.C.M. Oxidative stress markers in preeclamptic placentas: A systematic review with meta-analysis. Placenta 2020, 99, 89–100. [Google Scholar] [CrossRef]
- Li, M.; Cheng, W.; Zhang, L. Maternal selenium deficiency suppresses proliferation, induces autophagy dysfunction, apoptosis in the placenta of mice. Metallomics 2021, 13, mfab058. [Google Scholar] [CrossRef]
- Vanderlelie, J.; Gude, N.; Perkins, A.V. Antioxidant gene expression in preeclamptic placentae: A preliminary investigation. Placenta 2008, 29, 519–522. [Google Scholar] [CrossRef]
- Remigante, A.; Morabito, R.; Marino, A. Band 3 protein function and oxidative stress in erythrocytes. J. Cell. Physiol. 2021, 236, 6225–6234. [Google Scholar] [CrossRef] [PubMed]
- Morabito, R.; Remigante, A.; Marino, A. Protective Role of Magnesium against Oxidative Stress on SO4(=) Uptake through Band 3 Protein in Human Erythrocytes. Cell. Physiol. Biochem. 2019, 52, 1292–1308. [Google Scholar] [PubMed]
- Yuan, J.; Yu, Y.; Zhu, T.; Lin, X.; Jing, X.; Zhang, J. Oral Magnesium Supplementation for the Prevention of Preeclampsia: A Meta-analysis or Randomized Controlled Trials. Biol. Trace Elem. Res. 2021. [Google Scholar] [CrossRef] [PubMed]
- Varshavsky, J.; Smith, A.; Wang, A.; Hom, E.; Izano, M.; Huang, H.; Padula, A.; Woodruff, T.J. Heightened susceptibility: A review of how pregnancy and chemical exposures influence maternal health. Reprod. Toxicol. 2020, 92, 14–56. [Google Scholar] [CrossRef]
- Robinson, D.L. Health damage from current air pollution levels. Aust. N. Z. J. Public Health 2015, 39, 208–209. [Google Scholar] [CrossRef]
- Al-Gubory, K.H.; Krawiec, A.; Grange, S.; Faure, P.; Garrel, C. Abortion-prone mating influences placental antioxidant status and adversely affects placental and foetal development. Free Radic. Res. 2014, 48, 1505–1513. [Google Scholar] [CrossRef]
- Wigle, D.T.; Arbuckle, T.E.; Turner, M.C.; Berube, A.; Yang, Q.; Liu, S.; Krewski, D. Epidemiologic evidence of relationships between reproductive and child health outcomes and environmental chemical contaminants. J. Toxicol. Environ. Health B Crit. Rev. 2008, 11, 373–517. [Google Scholar] [CrossRef]
- Saenen, N.D.; Martens, D.S.; Neven, K.Y.; Alfano, R.; Bove, H.; Janssen, B.G.; Roels, H.A.; Plusquin, M.; Vrijens, K.; Nawrot, T.S. Air pollution-induced placental alterations: An interplay of oxidative stress, epigenetics, and the aging phenotype? Clin. Epigenet. 2019, 11, 124. [Google Scholar] [CrossRef]
- Ruder, E.H.; Hartman, T.J.; Blumberg, J.; Goldman, M.B. Oxidative stress and antioxidants: Exposure and impact on female fertility. Hum. Reprod. Update 2008, 14, 345–357. [Google Scholar] [CrossRef]
- Singh, L.; Anand, M.; Singh, S.; Taneja, A. Environmental toxic metals in placenta and their effects on preterm delivery-current opinion. Drug Chem. Toxicol. 2020, 43, 531–538. [Google Scholar] [CrossRef]
- Xu, R.; Meng, X.; Pang, Y.; An, H.; Wang, B.; Zhang, L.; Ye, R.; Ren, A.; Li, Z.; Gong, J. Associations of maternal exposure to 41 metals/metalloids during early pregnancy with the risk of spontaneous preterm birth: Does oxidative stress or DNA methylation play a crucial role? Environ. Int. 2021, 158, 106966. [Google Scholar] [CrossRef] [PubMed]
- Pilger, A.; Rudiger, H.W. 8-Hydroxy-2′-deoxyguanosine as a marker of oxidative DNA damage related to occupational and environmental exposures. Int. Arch. Occup. Environ. Health 2006, 80, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Vangronsveld, J.; Van Assche, F.; Clijsters, H. Reclamation of a bare industrial area contaminated by non-ferrous metals: In situ metal immobilization and revegetation. Environ. Pollut. 1995, 87, 51–59. [Google Scholar] [CrossRef]
- Cuypers, A.; Plusquin, M.; Remans, T.; Jozefczak, M.; Keunen, E.; Gielen, H.; Opdenakker, K.; Nair, A.R.; Munters, E.; Artois, T.J.; et al. Cadmium stress: An oxidative challenge. Biometals 2010, 23, 927–940. [Google Scholar] [CrossRef]
- Rani, A.; Kumar, A.; Lal, A.; Pant, M. Cellular mechanisms of cadmium-induced toxicity: A review. Int. J. Environ. Health Res. 2014, 24, 378–399. [Google Scholar] [CrossRef]
- Jomova, K.; Valko, M. Importance of iron chelation in free radical-induced oxidative stress and human disease. Curr. Pharm. Des. 2011, 17, 3460–3473. [Google Scholar] [CrossRef] [PubMed]
- Branca, J.J.V.; Fiorillo, C.; Carrino, D.; Paternostro, F.; Taddei, N.; Gulisano, M.; Pacini, A.; Becatti, M. Cadmium-Induced Oxidative Stress: Focus on the Central Nervous System. Antioxidants 2020, 9, 492. [Google Scholar] [CrossRef]
- Liao, Y.; Zheng, H.; Wu, L.; He, L.; Wang, Y.; Ou, Y.; Yang, H.; Peng, S.; Chen, F.; Wang, X.; et al. Cadmium cytotoxicity and possible mechanisms in human trophoblast HTR-8/SV-neo cells. Environ. Toxicol. 2021, 36, 1111–1124. [Google Scholar] [CrossRef]
- Everson, T.M.; Kappil, M.; Hao, K.; Jackson, B.P.; Punshon, T.; Karagas, M.R.; Chen, J.; Marsit, C.J. Maternal exposure to selenium and cadmium, fetal growth, and placental expression of steroidogenic and apoptotic genes. Environ. Res. 2017, 158, 233–244. [Google Scholar] [CrossRef]
- Adebambo, O.A.; Ray, P.D.; Shea, D.; Fry, R.C. Toxicological responses of environmental mixtures: Environmental metal mixtures display synergistic induction of metal-responsive and oxidative stress genes in placental cells. Toxicol. Appl. Pharmacol. 2015, 289, 534–541. [Google Scholar] [CrossRef]
- Pan, J.; Plant, J.A.; Voulvoulis, N.; Oates, C.J.; Ihlenfeld, C. Cadmium levels in Europe: Implications for human health. Environ. Geochem. Health 2010, 32, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Talhout, R.; Schulz, T.; Florek, E.; van Benthem, J.; Wester, P.; Opperhuizen, A. Hazardous compounds in tobacco smoke. Int. J. Environ. Res. Public Health 2011, 8, 613–628. [Google Scholar] [CrossRef]
- Ganguly, K.; Levanen, B.; Palmberg, L.; Akesson, A.; Linden, A. Cadmium in tobacco smokers: A neglected link to lung disease? Eur. Respir. Rev. 2018, 27, 170122. [Google Scholar] [CrossRef] [PubMed]
- Geng, H.X.; Wang, L. Cadmium: Toxic effects on placental and embryonic development. Environ. Toxicol. Pharmacol. 2019, 67, 102–107. [Google Scholar] [CrossRef]
- Kippler, M.; Tofail, F.; Gardner, R.; Rahman, A.; Hamadani, J.D.; Bottai, M.; Vahter, M. Maternal cadmium exposure during pregnancy and size at birth: A prospective cohort study. Environ. Health Perspect. 2012, 120, 284–289. [Google Scholar] [CrossRef] [PubMed]
- Nishijo, M.; Nakagawa, H.; Honda, R.; Tanebe, K.; Saito, S.; Teranishi, H.; Tawara, K. Effects of maternal exposure to cadmium on pregnancy outcome and breast milk. Occup. Environ. Med. 2002, 59, 394–396; discussion 397. [Google Scholar] [CrossRef] [PubMed]
- Samuel, J.B.; Stanley, J.A.; Princess, R.A.; Shanthi, P.; Sebastian, M.S. Gestational cadmium exposure-induced ovotoxicity delays puberty through oxidative stress and impaired steroid hormone levels. J. Med. Toxicol. 2011, 7, 195–204. [Google Scholar] [CrossRef]
- Dong, F.; Xiao, P.; Li, X.; Chang, P.; Zhang, W.; Wang, L. Cadmium triggers oxidative stress and mitochondrial injury mediated apoptosis in human extravillous trophoblast HTR-8/SV-neo cells. Reprod. Toxicol. 2021, 101, 18–27. [Google Scholar] [CrossRef]
- Llanos, M.N.; Ronco, A.M. Fetal growth restriction is related to placental levels of cadmium, lead and arsenic but not with antioxidant activities. Reprod. Toxicol. 2009, 27, 88–92. [Google Scholar] [CrossRef]
- Khanam, R.; Kumar, I.; Oladapo-Shittu, O.; Twose, C.; Islam, A.A.; Biswal, S.S.; Raqib, R.; Baqui, A.H. Prenatal Environmental Metal Exposure and Preterm Birth: A Scoping Review. Int. J. Environ. Res. Public Health 2021, 18, 573. [Google Scholar] [CrossRef]
- Xiong, Y.W.; Xu, X.F.; Zhu, H.L.; Cao, X.L.; Yi, S.J.; Shi, X.T.; Zhu, K.H.; Nan, Y.; Zhao, L.L.; Zhang, C.; et al. Environmental exposure to cadmium impairs fetal growth and placental angiogenesis via GCN-2-mediated mitochondrial stress. J. Hazard. Mater. 2021, 401, 123438. [Google Scholar] [CrossRef]
- Morabito, R.; Remigante, A.; Arcuri, B.; Giammanco, M.; La Spada, G.; Marino, A. Effect of cadmium on anion exchange capability through Band 3 protein in human erythrocytes. J. Biol. Res. 2018, 91, 1–7. [Google Scholar] [CrossRef]
- Qu, M.; Nan, X.; Gao, Z.; Guo, B.; Liu, B.; Chen, Z. Protective effects of lycopene against methylmercury-induced neurotoxicity in cultured rat cerebellar granule neurons. Brain Res. 2013, 1540, 92–102. [Google Scholar] [CrossRef]
- Franco, J.L.; Posser, T.; Dunkley, P.R.; Dickson, P.W.; Mattos, J.J.; Martins, R.; Bainy, A.C.; Marques, M.R.; Dafre, A.L.; Farina, M. Methylmercury neurotoxicity is associated with inhibition of the antioxidant enzyme glutathione peroxidase. Free Radic. Biol. Med. 2009, 47, 449–457. [Google Scholar] [CrossRef] [PubMed]
- Branco, V.; Godinho-Santos, A.; Goncalves, J.; Lu, J.; Holmgren, A.; Carvalho, C. Mitochondrial thioredoxin reductase inhibition, selenium status, and Nrf-2 activation are determinant factors modulating the toxicity of mercury compounds. Free Radic. Biol. Med. 2014, 73, 95–105. [Google Scholar] [CrossRef]
- Choi, B.H.; Lapham, L.W.; Amin-Zaki, L.; Saleem, T. Abnormal neuronal migration, deranged cerebral cortical organization, and diffuse white matter astrocytosis of human fetal brain: A major effect of methylmercury poisoning in utero. J. Neuropathol. Exp. Neurol. 1978, 37, 719–733. [Google Scholar] [CrossRef] [PubMed]
- Straka, E.; Ellinger, I.; Balthasar, C.; Scheinast, M.; Schatz, J.; Szattler, T.; Bleichert, S.; Saleh, L.; Knofler, M.; Zeisler, H.; et al. Mercury toxicokinetics of the healthy human term placenta involve amino acid transporters and ABC transporters. Toxicology 2016, 340, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Bridges, C.C.; Zalups, R.K. Mechanisms involved in the transport of mercuric ions in target tissues. Arch. Toxicol. 2017, 91, 63–81. [Google Scholar] [CrossRef] [PubMed]
- Granitzer, S.; Widhalm, R.; Forsthuber, M.; Ellinger, I.; Desoye, G.; Hengstschlager, M.; Zeisler, H.; Salzer, H.; Gundacker, C. Amino Acid Transporter LAT1 (SLC7A5) Mediates MeHg-Induced Oxidative Stress Defense in the Human Placental Cell Line HTR-8/SV-neo. Int. J. Mol. Sci. 2021, 22, 1707. [Google Scholar] [CrossRef] [PubMed]
- Tucker, E.K.; Nowak, R.A. Methylmercury alters proliferation, migration, and antioxidant capacity in human HTR8/SV-neo trophoblast cells. Reprod. Toxicol. 2018, 78, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Ferlemi, A.V.; Avgoustatos, D.; Kokkosis, A.G.; Protonotarios, V.; Constantinou, C.; Margarity, M. Lead-induced effects on learning/memory and fear/anxiety are correlated with disturbances in specific cholinesterase isoform activity and redox imbalance in adult brain. Physiol. Behav. 2014, 131, 115–122. [Google Scholar] [CrossRef]
- Pal, A.; Roy, D.; Adhikary, S.; Roy, A.; Dasgupta, M.; Mandal, A.K. A prospective study for the prediction of preeclampsia with urinary calcium level. J. Obstet. Gynaecol. India 2012, 62, 312–316. [Google Scholar] [CrossRef]
- Shafiq-ur-Rehman. Effect of lead on lipid peroxidation, phospholipids composition, and methylation in erythrocyte of human. Biol. Trace Elem. Res. 2013, 154, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, Z.L.; Liu, B.; Cai, J. Pregnancy outcome of overweight and obese Chinese women with gestational diabetes. J. Obstet. Gynaecol. 2014, 34, 662–665. [Google Scholar] [CrossRef] [PubMed]
- Baranowska-Bosiacka, I.; Gutowska, I.; Marchlewicz, M.; Marchetti, C.; Kurzawski, M.; Dziedziejko, V.; Kolasa, A.; Olszewska, M.; Rybicka, M.; Safranow, K.; et al. Disrupted pro- and antioxidative balance as a mechanism of neurotoxicity induced by perinatal exposure to lead. Brain Res. 2012, 1435, 56–71. [Google Scholar] [CrossRef] [PubMed]
- Gottipolu, R.R.; Davuljigari, C.B. Perinatal exposure to lead: Reduction in alterations of brain mitochondrial antioxidant system with calcium supplement. Biol. Trace Elem. Res. 2014, 162, 270–277. [Google Scholar] [CrossRef]
- Zhong, J.; Cayir, A.; Trevisi, L.; Sanchez-Guerra, M.; Lin, X.; Peng, C.; Bind, M.A.; Prada, D.; Laue, H.; Brennan, K.J.; et al. Traffic-Related Air Pollution, Blood Pressure, and Adaptive Response of Mitochondrial Abundance. Circulation 2016, 133, 378–387. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Guerra, M.; Peng, C.; Trevisi, L.; Cardenas, A.; Wilson, A.; Osorio-Yanez, C.; Niedzwiecki, M.M.; Zhong, J.; Svensson, K.; Acevedo, M.T.; et al. Altered cord blood mitochondrial DNA content and pregnancy lead exposure in the PROGRESS cohort. Environ. Int. 2019, 125, 437–444. [Google Scholar] [CrossRef] [PubMed]
- Barceloux, D.G. Chromium. J. Toxicol. Clin. Toxicol. 1999, 37, 173–194. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, H.; Xiang, X.H.; Liu, F.Y. Outline of occupational chromium poisoning in China. Bull. Environ. Contam. Toxicol. 2013, 90, 742–749. [Google Scholar] [CrossRef] [PubMed]
- Hemminki, K.; Kyyronen, P.; Niemi, M.L.; Koskinen, K.; Sallmen, M.; Vainio, H. Spontaneous abortions in an industrialized community in Finland. Am. J. Public Health 1983, 73, 32–37. [Google Scholar] [CrossRef]
- Quansah, R.; Jaakkola, J.J. Paternal and maternal exposure to welding fumes and metal dusts or fumes and adverse pregnancy outcomes. Int. Arch. Occup. Environ. Health 2009, 82, 529–537. [Google Scholar] [CrossRef] [PubMed]
- Sivakumar, K.K.; Stanley, J.A.; Arosh, J.A.; Pepling, M.E.; Burghardt, R.C.; Banu, S.K. Prenatal exposure to chromium induces early reproductive senescence by increasing germ cell apoptosis and advancing germ cell cyst breakdown in the F1 offspring. Dev. Biol. 2014, 388, 22–34. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Liu, K.J.; Shi, X. In vivo reduction of chromium (VI) and its related free radical generation. Mol. Cell. Biochem. 2001, 222, 41–47. [Google Scholar] [CrossRef]
- Banu, S.K.; Stanley, J.A.; Sivakumar, K.K.; Arosh, J.A.; Taylor, R.J.; Burghardt, R.C. Chromium VI—Induced developmental toxicity of placenta is mediated through spatiotemporal dysregulation of cell survival and apoptotic proteins. Reprod. Toxicol. 2017, 68, 171–190. [Google Scholar] [CrossRef]
- Gould, G.S.; Havard, A.; Lim, L.L.; The Psanz Smoking In Pregnancy Expert Group; Kumar, R. Exposure to Tobacco, Environmental Tobacco Smoke and Nicotine in Pregnancy: A Pragmatic Overview of Reviews of Maternal and Child Outcomes, Effectiveness of Interventions and Barriers and Facilitators to Quitting. Int. J. Environ. Res. Public Health 2020, 17, 2034. [Google Scholar] [CrossRef]
- Rousseaux, S.; Seyve, E.; Chuffart, F.; Bourova-Flin, E.; Benmerad, M.; Charles, M.A.; Forhan, A.; Heude, B.; Siroux, V.; Slama, R.; et al. Immediate and durable effects of maternal tobacco consumption alter placental DNA methylation in enhancer and imprinted gene-containing regions. BMC Med. 2020, 18, 306. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.A.; Blythe, N.M. Cardiac Fibroblast p38 MAPK: A Critical Regulator of Myocardial Remodeling. J. Cardiovasc. Dev. Dis. 2019, 6, 27. [Google Scholar] [CrossRef]
- Sinzato, Y.K.; Bevilacqua, E.M.; Volpato, G.T.; Hernandez-Pando, R.E.; Rudge, M.V.C.; Damasceno, D.C. Maternal Oxidative Stress, Placental Morphometry, and Fetal Growth in Diabetic Rats Exposed to Cigarette Smoke. Reprod. Sci. 2019, 26, 1287–1293. [Google Scholar] [CrossRef]
- Menon, R.; Fortunato, S.J.; Yu, J.; Milne, G.L.; Sanchez, S.; Drobek, C.O.; Lappas, M.; Taylor, R.N. Cigarette smoke induces oxidative stress and apoptosis in normal term fetal membranes. Placenta 2011, 32, 317–322. [Google Scholar] [CrossRef]
- Sheller, S.; Papaconstantinou, J.; Urrabaz-Garza, R.; Richardson, L.; Saade, G.; Salomon, C.; Menon, R. Amnion-Epithelial-Cell-Derived Exosomes Demonstrate Physiologic State of Cell under Oxidative Stress. PLoS ONE 2016, 11, e0157614. [Google Scholar]
- Hadley, E.E.; Sheller-Miller, S.; Saade, G.; Salomon, C.; Mesiano, S.; Taylor, R.N.; Taylor, B.D.; Menon, R. Amnion epithelial cell-derived exosomes induce inflammatory changes in uterine cells. Am. J. Obstet. Gynecol. 2018, 219, 478.e1–478.e21. [Google Scholar] [CrossRef]
- Shinjo, A.; Ventura, W.; Koide, K.; Hori, K.; Yotsumoto, J.; Matsuoka, R.; Ichizuka, K.; Sekizawa, A. Maternal smoking and placental expression of a panel of genes related to angiogenesis and oxidative stress in early pregnancy. Fetal Diagn. Ther. 2014, 35, 289–295. [Google Scholar] [CrossRef]
- Dittrich, R.; Schibel, A.; Hoffmann, I.; Mueller, A.; Beckmann, M.W.; Cupisti, S. Influence of maternal smoking during pregnancy on oxidant status in amniotic fluid. In Vivo 2012, 26, 813–818. [Google Scholar]
- Polettini, J.; Richardson, L.S.; Menon, R. Oxidative stress induces senescence and sterile inflammation in murine amniotic cavity. Placenta 2018, 63, 26–31. [Google Scholar] [CrossRef]
- Jin, J.; Richardson, L.; Sheller-Miller, S.; Zhong, N.; Menon, R. Oxidative stress induces p38MAPK-dependent senescence in the feto-maternal interface cells. Placenta 2018, 67, 15–23. [Google Scholar] [CrossRef]
- Ayad, M.T.; Taylor, B.D.; Menon, R. Regulation of p38 mitogen-activated kinase-mediated fetal membrane senescence by statins. Am. J. Reprod. Immunol. 2018, 80, e12999. [Google Scholar] [CrossRef]
- Wu, D.; Yuan, Y.; Lin, Z.; Lai, T.; Chen, M.; Li, W.; Lv, Q.; Yuan, B.; Li, D.; Wu, B. Cigarette smoke extract induces placental growth factor release from human bronchial epithelial cells via ROS/MAPK (ERK-1/2)/Egr-1 axis. Int. J. Chron. Obstruct. Pulmon. Dis. 2016, 11, 3031–3042. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado, A.; Nebreda, A.R. Mechanisms and functions of p38 MAPK signalling. Biochem J. 2010, 429, 403–417. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Janmohamed, K.; Jiang, J.; Ainooson, J.; Billings, A.; Chen, G.Q.; Chumo, F.; Cueto, L.; Niaura, R.; Zhang, A. Tobacco cessation in low- to middle-income countries: A scoping review of randomized controlled trials. Addict. Behav. 2021, 112, 106612. [Google Scholar] [CrossRef] [PubMed]
- Aycicek, A.; Ipek, A. Maternal active or passive smoking causes oxidative stress in cord blood. Eur. J. Pediatr. 2008, 167, 81–85. [Google Scholar] [CrossRef]
- Richardson, L.S.; Radnaa, E.; Urrabaz-Garza, R.; Lavu, N.; Menon, R. Stretch, scratch, and stress: Suppressors and supporters of senescence in human fetal membranes. Placenta 2020, 99, 27–34. [Google Scholar] [CrossRef]
- Sbrana, E.; Suter, M.A.; Abramovici, A.R.; Hawkins, H.K.; Moss, J.E.; Patterson, L.; Shope, C.; Aagaard-Tillery, K. Maternal tobacco use is associated with increased markers of oxidative stress in the placenta. Am. J. Obstet. Gynecol. 2011, 205, 246.e1–246.e7. [Google Scholar] [CrossRef]
- Shimada, T. Xenobiotic-metabolizing enzymes involved in activation and detoxification of carcinogenic polycyclic aromatic hydrocarbons. Drug Metab. Pharmacokinet. 2006, 21, 257–276. [Google Scholar] [CrossRef]
- Board, P.G.; Menon, D. Glutathione transferases, regulators of cellular metabolism and physiology. Biochim. Biophys. Acta 2013, 1830, 3267–3288. [Google Scholar] [CrossRef]
- Florek, E.; Ignatowicz, E.; Piekoszewski, W.; Wachowiak, A.; Wrzosek, J.; Moczko, J.; Czekaj, P.; Slusarska, E. Tobacco smoke effects the activity of superoxide dismutase, glutathione peroxidase and total antioxidant status in pregnant and non-pregnant animals. Przegl. Lek. 2004, 61, 1104–1108. [Google Scholar] [PubMed]
- Wang, X.; Lee, N.L.; Burstyn, I. Maternal smoking and gestational hypertension: Heterogeneous effect by timing of the exposure. Pregnancy Hypertens. 2019, 15, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Kondracki, A.J.; Hofferth, S.L. A gestational vulnerability window for smoking exposure and the increased risk of preterm birth: How timing and intensity of maternal smoking matter. Reprod. Health 2019, 16, 43. [Google Scholar] [CrossRef]
- Machaalani, R.; Ghazavi, E.; Hinton, T.; Makris, A.; Hennessy, A. Immunohistochemical expression of the nicotinic acetylcholine receptor (nAChR) subunits in the human placenta, and effects of cigarette smoking and preeclampsia. Placenta 2018, 71, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Laule, C.F.; Wing, C.R.; Odean, E.J.; Wilcox, J.A.; Gilbert, J.S.; Regal, J.F. Effect of nicotine on placental ischemia-induced complement activation and hypertension in the rat. J. Immunotoxicol. 2017, 14, 235–240. [Google Scholar] [CrossRef]
- Zhao, L.; Ma, R.; Zhang, L.; Yuan, X.; Wu, J.; He, L.; Liu, G.; Du, R. Inhibition of HIF-1a-mediated TLR4 activation decreases apoptosis and promotes angiogenesis of placental microvascular endothelial cells during severe pre-eclampsia pathogenesis. Placenta 2019, 83, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A. Molecular mechanisms and therapeutic implications of the carbon monoxide/hmox1 and the hydrogen sulfide/CSE pathways in the prevention of pre-eclampsia and fetal growth restriction. Pregnancy Hypertens. 2014, 4, 243–244. [Google Scholar] [CrossRef] [PubMed]
- Bainbridge, S.A.; Smith, G.N. HO in pregnancy. Free Radic. Biol. Med. 2005, 38, 979–988. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Lei, F.; Yan, X.; Zhang, S.; Wang, W.; Zheng, Y. Protective Effects of Hydrogen Sulfide against Cigarette Smoke Exposure-Induced Placental Oxidative Damage by Alleviating Redox Imbalance via Nrf2 Pathway in Rats. Cell. Physiol. Biochem. 2018, 48, 1815–1828. [Google Scholar] [CrossRef]
- Feng, S.; Gao, D.; Liao, F.; Zhou, F.; Wang, X. The health effects of ambient PM2.5 and potential mechanisms. Ecotoxicol. Environ. Saf. 2016, 128, 67–74. [Google Scholar] [CrossRef]
- Bove, H.; Bongaerts, E.; Slenders, E.; Bijnens, E.M.; Saenen, N.D.; Gyselaers, W.; Van Eyken, P.; Plusquin, M.; Roeffaers, M.B.J.; Ameloot, M.; et al. Ambient black carbon particles reach the fetal side of human placenta. Nat. Commun. 2019, 10, 3866. [Google Scholar] [CrossRef]
- Risom, L.; Moller, P.; Loft, S. Oxidative stress-induced DNA damage by particulate air pollution. Mutat. Res. 2005, 592, 119–137. [Google Scholar] [CrossRef]
- Saenen, N.D.; Vrijens, K.; Janssen, B.G.; Madhloum, N.; Peusens, M.; Gyselaers, W.; Vanpoucke, C.; Lefebvre, W.; Roels, H.A.; Nawrot, T.S. Placental Nitrosative Stress and Exposure to Ambient Air Pollution during Gestation: A Population Study. Am. J. Epidemiol. 2016, 184, 442–449. [Google Scholar] [CrossRef] [PubMed]
- Bandookwala, M.; Sengupta, P. 3-Nitrotyrosine: A versatile oxidative stress biomarker for major neurodegenerative diseases. Int. J. Neurosci. 2020, 130, 1047–1062. [Google Scholar] [CrossRef] [PubMed]
- Abraham, E.; Rousseaux, S.; Agier, L.; Giorgis-Allemand, L.; Tost, J.; Galineau, J.; Hulin, A.; Siroux, V.; Vaiman, D.; Charles, M.A.; et al. Pregnancy exposure to atmospheric pollution and meteorological conditions and placental DNA methylation. Environ. Int. 2018, 118, 334–347. [Google Scholar] [CrossRef]
- Cabalin, C.; Villalobos-Labra, R.; Toledo, F.; Sobrevia, L. Involvement of A2B adenosine receptors as anti-inflammatory in gestational diabesity. Mol. Asp. Med. 2019, 66, 31–39. [Google Scholar] [CrossRef]
- Wojcik, M.; Zieleniak, A.; Mac-Marcjanek, K.; Wozniak, L.A.; Cypryk, K. The elevated gene expression level of the A(2B) adenosine receptor is associated with hyperglycemia in women with gestational diabetes mellitus. Diabetes Metab. Res. Rev. 2014, 30, 42–53. [Google Scholar] [CrossRef]
- Squadrito, G.L.; Cueto, R.; Dellinger, B.; Pryor, W.A. Quinoid redox cycling as a mechanism for sustained free radical generation by inhaled airborne particulate matter. Free Radic. Biol. Med. 2001, 31, 1132–1138. [Google Scholar] [CrossRef]
- Grevendonk, L.; Janssen, B.G.; Vanpoucke, C.; Lefebvre, W.; Hoxha, M.; Bollati, V.; Nawrot, T.S. Mitochondrial oxidative DNA damage and exposure to particulate air pollution in mother-newborn pairs. Environ. Health 2016, 15, 10. [Google Scholar] [CrossRef]
- Janssen, B.G.; Byun, H.M.; Gyselaers, W.; Lefebvre, W.; Baccarelli, A.A.; Nawrot, T.S. Placental mitochondrial methylation and exposure to airborne particulate matter in the early life environment: An ENVIRONAGE birth cohort study. Epigenetics 2015, 10, 536–544. [Google Scholar] [CrossRef]
- Rotter, B.A.; Prelusky, D.B.; Pestka, J.J. Toxicology of deoxynivalenol (vomitoxin). J. Toxicol. Environ. Health 1996, 48, 1–34. [Google Scholar] [CrossRef]
- Sobrova, P.; Adam, V.; Vasatkova, A.; Beklova, M.; Zeman, L.; Kizek, R. Deoxynivalenol and its toxicity. Interdiscip. Toxicol. 2010, 3, 94–99. [Google Scholar] [CrossRef]
- Nielsen, J.K.; Vikstrom, A.C.; Turner, P.; Knudsen, L.E. Deoxynivalenol transport across the human placental barrier. Food Chem. Toxicol. 2011, 49, 2046–2052. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Chen, L.; Peng, Z.; Wang, D.; Song, Y.; Wang, H.; Yao, P.; Yan, H.; Nussler, A.K.; Liu, L.; et al. Embryotoxicity Caused by DON-Induced Oxidative Stress Mediated by Nrf2/HO-1 Pathway. Toxins 2017, 9, 188. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Wei, Z.Y.; Xu, Z.H.; Pan, J.Q.; Chen, J.H. Oxidative Damage and Nrf2 Translocation Induced by Toxicities of Deoxynivalenol on the Placental and Embryo on Gestation Day 12.5 d and 18.5 d. Toxins 2018, 10, 370. [Google Scholar] [CrossRef]
- Yu, M.; Chen, L.; Peng, Z.; Nussler, A.K.; Wu, Q.; Liu, L.; Yang, W. Mechanism of deoxynivalenol effects on the reproductive system and fetus malformation: Current status and future challenges. Toxicol. In Vitro 2017, 41, 150–158. [Google Scholar] [CrossRef]
- Ryter, S.W.; Tyrrell, R.M. The heme synthesis and degradation pathways: Role in oxidant sensitivity. Heme oxygenase has both pro- and antioxidant properties. Free Radic. Biol. Med. 2000, 28, 289–309. [Google Scholar] [CrossRef]
- Yang, X.; Liu, P.; Cui, Y.; Xiao, B.; Liu, M.; Song, M.; Huang, W.; Li, Y. Review of the Reproductive Toxicity of T-2 Toxin. J. Agric. Food Chem. 2020, 68, 727–734. [Google Scholar] [CrossRef]
- Rousseaux, C.G.; Nicholson, S.; Schiefer, H.B. Fatal placental hemorrhage in pregnant CD-1 mice following one oral dose of T-2 toxin. Can. J. Comp. Med. 1985, 49, 95–98. [Google Scholar] [PubMed]
- Sehata, S.; Kiyosawa, N.; Sakuma, K.; Ito, K.; Yamoto, T.; Teranishi, M.; Uetsuka, K.; Nakayama, H.; Doi, K. Gene expression profiles in pregnant rats treated with T-2 toxin. Exp. Toxicol. Pathol. 2004, 55, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Du, X.; Liu, H.; Losiewic, M.D.; Chen, X.; Ma, Y.; Wang, R.; Tian, Z.; Shi, L.; Guo, H.; et al. The latest advances in the reproductive toxicity of microcystin-LR. Environ. Res. 2021, 192, 110254. [Google Scholar] [CrossRef]
- Harada, K.; Oshikata, M.; Uchida, H.; Suzuki, M.; Kondo, F.; Sato, K.; Ueno, Y.; Yu, S.Z.; Chen, G.; Chen, G.C. Detection and identification of microcystins in the drinking water of Haimen City, China. Nat. Toxins 1996, 4, 277–283. [Google Scholar] [CrossRef]
- Zhao, S.; Zhong, S.; Wang, F.; Wang, H.; Xu, D.; Li, G. Microcystin-LR exposure decreased the fetal weight of mice by disturbance of placental development and ROS-mediated endoplasmic reticulum stress in the placenta. Environ. Pollut. 2020, 256, 113362. [Google Scholar] [CrossRef]
- Wang, Y.; Hu, C.; Fang, T.; Jin, Y.; Wu, R. Perspective on prenatal polychlorinated biphenyl exposure and the development of the progeny nervous system (Review). Int. J. Mol. Med. 2021, 48, 150. [Google Scholar] [CrossRef]
- Iqbal, K.; Dhakal, P.; Pierce, S.H.; Soares, M.J. Catechol-O-methyltransferase and Pregnancy Outcome: An Appraisal in Rat. Reprod. Sci. 2021, 28, 462–469. [Google Scholar] [CrossRef]
- Liu, H.; Jiang, W.; Ye, Y.; Yang, B.; Shen, X.; Lu, S.; Zhu, J.; Liu, M.; Yang, C.; Kuang, H. Maternal exposure to tributyltin during early gestation increases adverse pregnancy outcomes by impairing placental development. Environ. Toxicol. 2021, 36, 1303–1315. [Google Scholar] [CrossRef]
- Podratz, P.L.; Merlo, E.; de Araujo, J.F.P.; Ayub, J.G.M.; Pereira, A.F.Z.; Freitas-Lima, L.C.; da Costa, M.B.; Miranda-Alves, L.; Cassa, S.G.S.; Carneiro, M.; et al. Disruption of fertility, placenta, pregnancy outcome, and multigenerational inheritance of hepatic steatosis by organotin exposure from contaminated seafood in rats. Sci. Total Environ. 2020, 723, 138000. [Google Scholar] [CrossRef]
- Awobajo, F.O.; Morakinyo, A.O.; Samuel, T.A.; Oyelowo, O.T.; Ogunsola, A.O.; Onyekwele, P.U.; Okedina, M.E.; Ogunbanwo, O.O. Dynamics of inflammatory reaction and oxidative stress across maternal serum, placenta and amniotic fluid in laboratory rats and the role played by genistein aglycone. J. Basic Clin. Physiol. Pharmacol. 2018, 30, 37–45. [Google Scholar] [CrossRef]
- Song, S.; He, Y.; Zhang, T.; Zhu, H.; Huang, X.; Bai, X.; Zhang, B.; Kannan, K. Profiles of parabens and their metabolites in paired maternal-fetal serum, urine and amniotic fluid and their implications for placental transfer. Ecotoxicol. Environ. Saf. 2020, 191, 110235. [Google Scholar] [CrossRef]
- Valle-Sistac, J.; Molins-Delgado, D.; Diaz, M.; Ibanez, L.; Barcelo, D.; Silvia Diaz-Cruz, M. Determination of parabens and benzophenone-type UV filters in human placenta. First description of the existence of benzyl paraben and benzophenone-4. Environ. Int. 2016, 88, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Vela-Soria, F.; Gallardo-Torres, M.E.; Ballesteros, O.; Diaz, C.; Perez, J.; Navalon, A.; Fernandez, M.F.; Olea, N. Assessment of parabens and ultraviolet filters in human placenta tissue by ultrasound-assisted extraction and ultra-high performance liquid chromatography-tandem mass spectrometry. J. Chromatogr. A 2017, 1487, 153–161. [Google Scholar] [CrossRef]
- Rochester, J.R. Bisphenol A and human health: A review of the literature. Reprod. Toxicol. 2013, 42, 132–155. [Google Scholar] [CrossRef]
- Shin, B.; Kwon, J.A.; Park, E.K.; Kang, S.; Kim, S.; Park, E.; Kim, B. Prenatal Exposure to Parabens Affects Birth Outcomes through Maternal Glutathione S-Transferase (GST) Polymorphisms: From the Mothers and Kids Environmental Health (MAKE) Study. Int. J. Environ. Res. Public Health 2021, 18, 3012. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, K.K.; Cantonwine, D.E.; Rivera-Gonzalez, L.O.; Loch-Caruso, R.; Mukherjee, B.; Anzalota Del Toro, L.V.; Jimenez-Velez, B.; Calafat, A.M.; Ye, X.; Alshawabkeh, A.N.; et al. Urinary phthalate metabolite associations with biomarkers of inflammation and oxidative stress across pregnancy in Puerto Rico. Environ. Sci. Technol. 2014, 48, 7018–7025. [Google Scholar] [CrossRef]
- Strakovsky, R.S.; Schantz, S.L. Impacts of bisphenol A (BPA) and phthalate exposures on epigenetic outcomes in the human placenta. Environ. Epigenet. 2018, 4, dvy022. [Google Scholar] [CrossRef] [PubMed]
- Philippat, C.; Heude, B.; Botton, J.; Alfaidy, N.; Calafat, A.M.; Slama, R.; Group, E.M.-C.C.S. Prenatal Exposure to Select Phthalates and Phenols and Associations with Fetal and Placental Weight among Male Births in the EDEN Cohort (France). Environ. Health Perspect. 2019, 127, 17002. [Google Scholar] [CrossRef]
- Zong, T.; Lai, L.; Hu, J.; Guo, M.; Li, M.; Zhang, L.; Zhong, C.; Yang, B.; Wu, L.; Zhang, D.; et al. Maternal exposure to di-(2-ethylhexyl) phthalate disrupts placental growth and development in pregnant mice. J. Hazard. Mater. 2015, 297, 25–33. [Google Scholar] [CrossRef] [PubMed]
- LaRocca, J.; Binder, A.M.; McElrath, T.F.; Michels, K.B. The impact of first trimester phthalate and phenol exposure on IGF2/H19 genomic imprinting and birth outcomes. Environ. Res. 2014, 133, 396–406. [Google Scholar] [CrossRef] [PubMed]
- Jedynak, P.; Tost, J.; Calafat, A.M.; Bourova-Flin, E.; Busato, F.; Forhan, A.; Heude, B.; Jakobi, M.; Rousseaux, S.; Schwartz, J.; et al. Pregnancy exposure to synthetic phenols and placental DNA methylation—An epigenome-wide association study in male infants from the EDEN cohort. Environ. Pollut. 2021, 290, 118024. [Google Scholar] [CrossRef]
- Li, Q.; Yao, B.; Endler, A.; Chen, L.; Shibasaki, F.; Cheng, H. Int6/eIF3e Silencing Promotes Placenta Angiogenesis in a Rat Model of Pre-eclampsia. Sci. Rep. 2018, 8, 8944. [Google Scholar] [CrossRef] [PubMed]
- Yavasoglu, N.U.; Koksal, C.; Dagdeviren, M.; Aktug, H.; Yavasoglu, A. Induction of oxidative stress and histological changes in liver by subacute doses of butyl cyclohexyl phthalate. Environ. Toxicol. 2014, 29, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Shah, K.H.; Verma, R.J. Butyl p-hydroxybenzoic acid induces oxidative stress in mice liver—An in vivo study. Acta Pol. Pharm. 2011, 68, 875–879. [Google Scholar]
- Yang, B.; Wang, X.; Ma, Y.; Yan, L.; Ren, Y.; Yu, D.; Qiao, B.; Shen, X.; Liu, H.; Zhang, D.; et al. Tri-ortho-cresyl phosphate (TOCP)-induced reproductive toxicity involved in placental apoptosis, autophagy and oxidative stress in pregnant mice. Environ. Toxicol. 2020, 35, 97–107. [Google Scholar] [CrossRef]
- Perez-Albaladejo, E.; Fernandes, D.; Lacorte, S.; Porte, C. Comparative toxicity, oxidative stress and endocrine disruption potential of plasticizers in JEG-3 human placental cells. Toxicol. In Vitro 2017, 38, 41–48. [Google Scholar] [CrossRef]
- Ponniah, M.; Billett, E.E.; De Girolamo, L.A. Bisphenol A increases BeWo trophoblast survival in stress-induced paradigms through regulation of oxidative stress and apoptosis. Chem. Res. Toxicol. 2015, 28, 1693–1703. [Google Scholar] [CrossRef]
- Ferguson, K.K.; McElrath, T.F.; Ko, Y.A.; Mukherjee, B.; Meeker, J.D. Variability in urinary phthalate metabolite levels across pregnancy and sensitive windows of exposure for the risk of preterm birth. Environ. Int. 2014, 70, 118–124. [Google Scholar] [CrossRef] [PubMed]
- Zen, M.; Padmanabhan, S.; Zhang, K.; Kirby, A.; Cheung, N.W.; Lee, V.W.; Alahakoon, T.I. Urinary and Serum Angiogenic Markers in Women with Preexisting Diabetes during Pregnancy and Their Role in Preeclampsia Prediction. Diabetes Care 2020, 43, 67–73. [Google Scholar] [CrossRef]
- Doridot, L.; Passet, B.; Mehats, C.; Rigourd, V.; Barbaux, S.; Ducat, A.; Mondon, F.; Vilotte, M.; Castille, J.; Breuiller-Fouche, M.; et al. Preeclampsia-like symptoms induced in mice by fetoplacental expression of STOX1 are reversed by aspirin treatment. Hypertension 2013, 61, 662–668. [Google Scholar] [CrossRef]
- Doridot, L.; Chatre, L.; Ducat, A.; Vilotte, J.L.; Lombes, A.; Mehats, C.; Barbaux, S.; Calicchio, R.; Ricchetti, M.; Vaiman, D. Nitroso-redox balance and mitochondrial homeostasis are regulated by STOX1, a pre-eclampsia-associated gene. Antioxid. Redox. Signal. 2014, 21, 819–834. [Google Scholar] [CrossRef]
- Taysi, S.; Tascan, A.S.; Ugur, M.G.; Demir, M. Radicals, Oxidative/Nitrosative Stress and Preeclampsia. Mini Rev. Med. Chem. 2019, 19, 178–193. [Google Scholar] [CrossRef]
- Saif, J.; Ahmad, S.; Rezai, H.; Litvinova, K.; Sparatore, A.; Alzahrani, F.A.; Wang, K.; Ahmed, A. Hydrogen sulfide releasing molecule MZe786 inhibits soluble Flt-1 and prevents preeclampsia in a refined RUPP mouse model. Redox. Biol. 2021, 38, 101814. [Google Scholar] [CrossRef] [PubMed]
- Thieme, R.; Schramel, P.; Klose, B.J.; Waidl, E. Trace elements in the human placenta (author’s transl). Geburtshilfe Frauenheilkd 1975, 35, 349–353. [Google Scholar]
- Herlin, M.; Broberg, K.; Igra, A.M.; Li, H.; Harari, F.; Vahter, M. Exploring telomere length in mother-newborn pairs in relation to exposure to multiple toxic metals and potential modifying effects by nutritional factors. BMC Med. 2019, 17, 77. [Google Scholar] [CrossRef] [PubMed]
- Harari, F.; Langeen, M.; Casimiro, E.; Bottai, M.; Palm, B.; Nordqvist, H.; Vahter, M. Environmental exposure to lithium during pregnancy and fetal size: A longitudinal study in the Argentinean Andes. Environ. Int. 2015, 77, 48–54. [Google Scholar] [CrossRef]
- Rao, H.; Bai, Y.; Zhang, F.; Li, Q.; Zhuang, B.; Luo, X.; Qi, H. The role of SATB1 in HTR8/SV-neo cells and pathological mechanism of preeclampsia. J. Matern. Fetal Neonatal Med. 2019, 32, 2069–2078. [Google Scholar] [CrossRef]
- Issah, I.; Arko-Mensah, J.; Agyekum, T.P.; Dwomoh, D.; Fobil, J.N. Electronic waste exposure and DNA damage: A systematic review and meta-analysis. Rev. Environ. Health 2021. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Huo, X.; Zhang, Q.; Fan, X.; Du, L.; Xu, X.; Qiu, S.; Zhang, Y.; Wang, Y.; Gu, J. Short placental telomere was associated with cadmium pollution in an electronic waste recycling town in China. PLoS ONE 2013, 8, e60815. [Google Scholar] [CrossRef] [PubMed]
- von Zglinicki, T. Oxidative stress shortens telomeres. Trends Biochem. Sci. 2002, 27, 339–344. [Google Scholar] [CrossRef]
- Cai, K.; Song, Q.; Yuan, W.; Ruan, J.; Duan, H.; Li, Y.; Li, J. Human exposure to PBDEs in e-waste areas: A review. Environ. Pollut. 2020, 267, 115634. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Zhang, G.; Wang, J.; Zhang, W.; Liu, L.; Lin, K. Polybrominated diphenyl ethers in air and fallouts from an e-waste polluted region in southeast China: Insight into levels, compositional profiles, and seasonal variation. Environ. Sci. Pollut. Res. Int. 2015, 22, 19676–19686. [Google Scholar] [CrossRef]
- Song, Q.; Li, J. A systematic review of the human body burden of e-waste exposure in China. Environ. Int. 2014, 68, 82–93. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruano, C.S.M.; Miralles, F.; Méhats, C.; Vaiman, D. The Impact of Oxidative Stress of Environmental Origin on the Onset of Placental Diseases. Antioxidants 2022, 11, 106. https://doi.org/10.3390/antiox11010106
Ruano CSM, Miralles F, Méhats C, Vaiman D. The Impact of Oxidative Stress of Environmental Origin on the Onset of Placental Diseases. Antioxidants. 2022; 11(1):106. https://doi.org/10.3390/antiox11010106
Chicago/Turabian StyleRuano, Camino San Martin, Francisco Miralles, Céline Méhats, and Daniel Vaiman. 2022. "The Impact of Oxidative Stress of Environmental Origin on the Onset of Placental Diseases" Antioxidants 11, no. 1: 106. https://doi.org/10.3390/antiox11010106
APA StyleRuano, C. S. M., Miralles, F., Méhats, C., & Vaiman, D. (2022). The Impact of Oxidative Stress of Environmental Origin on the Onset of Placental Diseases. Antioxidants, 11(1), 106. https://doi.org/10.3390/antiox11010106