
Protection against Amyloid-β Oligomer Neurotoxicity by Small Molecules with Antioxidative Properties: Potential for the Prevention of Alzheimer’s Disease Dementia


Abstract
:1. Introduction
2. AβOs and Oxidative Stress in AD
3. Small Molecules with Protective Activity against AβO Neurotoxicity
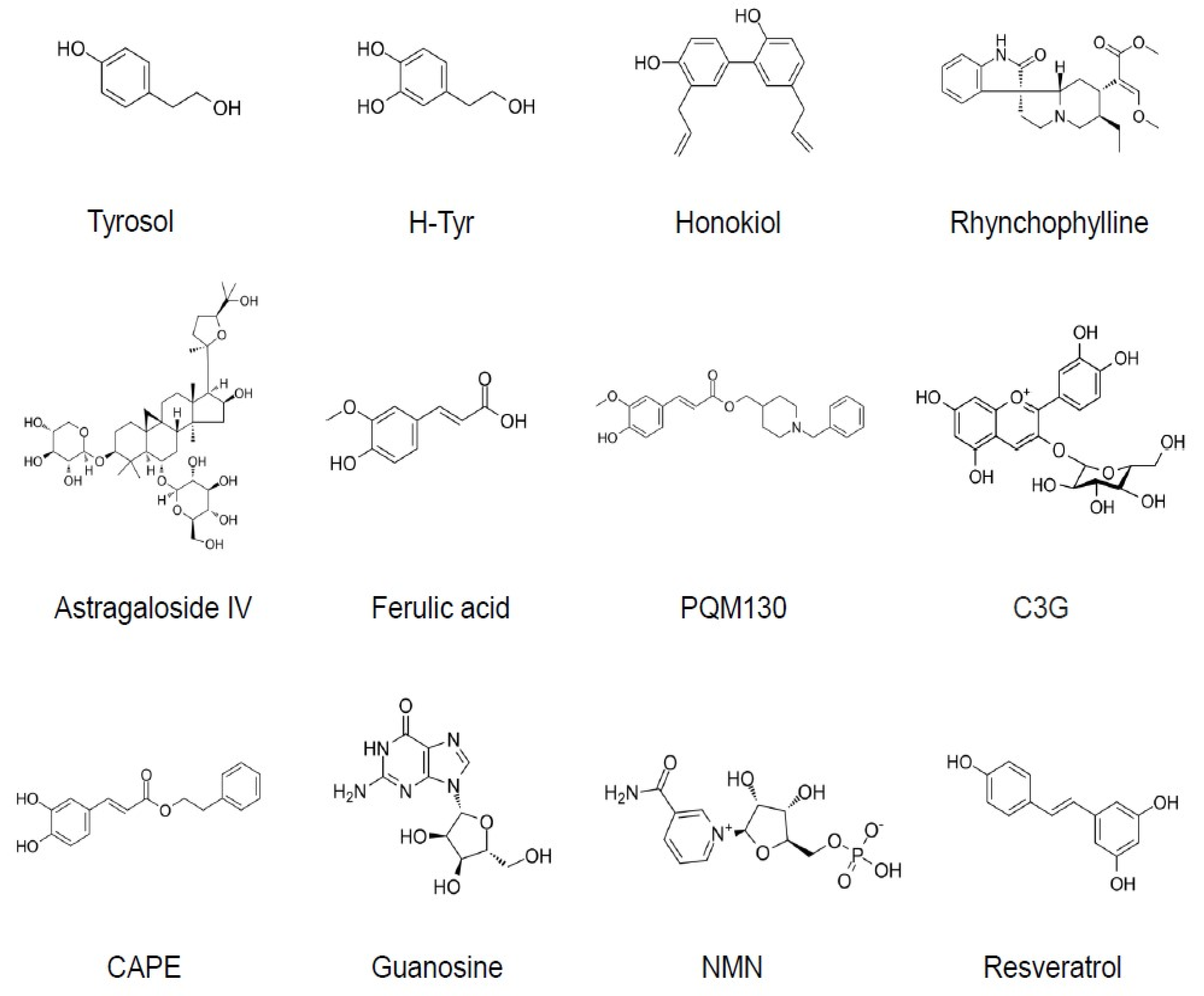
3.1. Tyrosol and Hydroxytyrosol
3.2. Honokiol
3.3. Rhynchophylline
3.4. Astragaloside IV
3.5. Ferulic Acid and Related Compounds
3.6. Anthocyanins
3.7. Caffeic Acid Phenyl Ester
3.8. Guanosine
3.9. Nicotinamide Mononucleotide (NMN) and Nicotinamide
3.10. Other Compounds
3.10.1. Resveratrol
3.10.2. Myricetin
3.10.3. 17 Oxo Sparteine and Lupanine
3.10.4. Esculetin
4. Antioxidative and Other Mechanisms Involved in Small-Molecule-Mediated Prevention of AβO Neurotoxicity
5. Issues in Translational Research and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Ballard, C.; Gauthier, S.; Corbett, A.; Brayne, C.; Aarsland, D.; Jones, E. Alzheimer’s disease. Lancet 2011, 377, 1019–1031. [Google Scholar] [CrossRef]
- Sperling, R.A.; Aisen, P.S.; Beckett, L.A.; Bennett, D.A.; Craft, S.; Fagan, A.M.; Iwatsubo, T.; Jack, C.R., Jr.; Kaye, J.; Montine, T.J.; et al. Toward defining the preclinical stages of Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dement. 2011, 7, 280–292. [Google Scholar] [CrossRef] [Green Version]
- Johnson, K.A.; Schultz, A.; Betensky, R.A.; Becker, J.A.; Sepulcre, J.; Rentz, D.; Mormino, E.; Chhatwal, J.; Amariglio, R.; Papp, K.; et al. Tau positron emission tomographic imaging in aging and early Alzheimer disease. Ann. Neurol. 2016, 79, 110–119. [Google Scholar] [CrossRef] [Green Version]
- Vanhaute, H.; Ceccarini, J.; Michiels, L.; Koole, M.; Sunaert, S.; Lemmens, R.; Triau, E.; Emsell, L.; Vandenbulcke, M.; Van Laere, K. In vivo synaptic density loss is related to tau deposition in amnestic mild cognitive impairment. Neurology 2020, 95, e545–e553. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef] [PubMed]
- De Strooper, B.; Vassar, R.; Golde, T. The secretases: Enzymes with therapeutic potential in Alzheimer disease. Nat. Rev. Neurol. 2010, 6, 99–107. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, S.T.; Klein, W.L. The Aβ oligomer hypothesis for synapse failure and memory loss in Alzheimer’s disease. Neurobiol. Learn. Mem. 2011, 96, 529–543. [Google Scholar] [CrossRef] [Green Version]
- Cline, E.N.; Bicca, M.A.; Viola, K.L.; Klein, W.L. The Amyloid-β Oligomer Hypothesis: Beginning of the Third Decade. J. Alzheimer’s Dis. 2018, 64, S567–S610. [Google Scholar] [CrossRef] [Green Version]
- Tanokashira, D.; Mamada, N.; Yamamoto, F.; Taniguchi, K.; Tamaoka, A.; Lakshmana, M.K.; Araki, W. The neurotoxicity of amyloid β-protein oligomers is reversible in a primary neuron model. Mol. Brain 2017, 10, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, T.W.; Lane, H.Y.; Lin, C.H. Novel Therapeutic Approachesfor Alzheimer’s Disease: An Updated Review. Int. J. Mol. Sci. 2021, 22, 8208. [Google Scholar] [CrossRef]
- Goure, W.F.; Krafft, G.A.; Jerecic, J.; Hefti, F. Targeting the proper amyloid-beta neuronal toxins: A path forward for Alzheimer’s disease immunotherapeutics. Alzheimer’s Res. Ther. 2014, 6, 42. [Google Scholar] [CrossRef] [Green Version]
- Tolar, M.; Abushakra, S.; Sabbagh, M. The path forward in Alzheimer’s disease therapeutics: Reevaluating the amyloid cascade hypothesis. Alzheimer’s Dement. 2020, 16, 1553–1560. [Google Scholar] [CrossRef]
- Fantini, J.; Chahinian, H.; Yahi, N. Progress toward Alzheimer’s disease treatment: Leveraging the Achilles’ heel of Abeta oligomers? Protein Sci. 2020, 29, 1748–1759. [Google Scholar] [CrossRef] [PubMed]
- Simunkova, M.; Alwasel, S.H.; Alhazza, I.M.; Jomova, K.; Kollar, V.; Rusko, M.; Valko, M. Management of oxidative stress and other pathologies in Alzheimer’s disease. Arch. Toxicol. 2019, 93, 2491–2513. [Google Scholar] [CrossRef] [Green Version]
- Tramutola, A.; Lanzillotta, C.; Perluigi, M.; Butterfield, D.A. Oxidative stress, protein modification and Alzheimer disease. Brain Res. Bull. 2017, 133, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Arslan, J.; Jamshed, H.; Qureshi, H. Early Detection and Prevention of Alzheimer’s Disease: Role of Oxidative Markers and Natural Antioxidants. Front. Aging Neurosci. 2020, 12, 231. [Google Scholar] [CrossRef]
- Di Domenico, F.; Tramutola, A.; Butterfield, D.A. Role of 4-hydroxy-2-nonenal (HNE) in the pathogenesis of alzheimer disease and other selected age-related neurodegenerative disorders. Free Radic. Biol. Med. 2017, 111, 253–261. [Google Scholar] [CrossRef]
- Cascella, R.; Cecchi, C. Calcium Dyshomeostasis in Alzheimer’s Disease Pathogenesis. Int. J. Mol. Sci. 2021, 22, 4914. [Google Scholar] [CrossRef] [PubMed]
- Calvo-Rodriguez, M.; Hou, S.S.; Snyder, A.C.; Kharitonova, E.K.; Russ, A.N.; Das, S.; Fan, Z.; Muzikansky, A.; Garcia-Alloza, M.; Serrano-Pozo, A.; et al. Increased mitochondrial calcium levels associated with neuronal death in a mouse model of Alzheimer’s disease. Nat. Commun. 2020, 11, 2146. [Google Scholar] [CrossRef]
- Oliver, D.M.A.; Reddy, P.H. Small molecules as therapeutic drugs for Alzheimer’s disease. Mol. Cell. Neurosci. 2019, 96, 47–62. [Google Scholar] [CrossRef]
- Chiang, H.M.; Chen, H.C.; Wu, C.S.; Wu, P.Y.; Wen, K.C. Rhodiola plants: Chemistry and biological activity. J. Food Drug Anal. 2015, 23, 359–369. [Google Scholar] [CrossRef] [Green Version]
- Osama, A.; Zhang, J.; Yao, J.; Yao, X.; Fang, J. Nrf2: A dark horse in Alzheimer’s disease treatment. Ageing Res. Rev. 2020, 64, 101206. [Google Scholar] [CrossRef]
- Ramsey, C.P.; Glass, C.A.; Montgomery, M.B.; Lindl, K.A.; Ritson, G.P.; Chia, L.A.; Hamilton, R.L.; Chu, C.T.; Jordan-Sciutto, K.L. Expression of Nrf2 in neurodegenerative diseases. J. Neuropathol. Exp. Neurol. 2007, 66, 75–85. [Google Scholar] [CrossRef]
- Tuck, K.L.; Hayball, P.J. Major phenolic compounds in olive oil: Metabolism and health effects. J. Nutr. Biochem. 2002, 13, 636–644. [Google Scholar] [CrossRef]
- Panossian, A.; Wikman, G.; Sarris, J. Rosenroot (Rhodiola rosea): Traditional use, chemical composition, pharmacology and clinical efficacy. Phytomedicine 2010, 17, 481–493. [Google Scholar] [CrossRef]
- Taniguchi, K.; Yamamoto, F.; Arai, T.; Yang, J.; Sakai, Y.; Itoh, M.; Mamada, N.; Sekiguchi, M.; Yamada, D.; Saitoh, A.; et al. Tyrosol reduces amyloid-β oligomer neurotoxicity and alleviates synaptic, oxidative, and cognitive disturbances in Alzheimer’s disease model mice. J. Alzheimer’s Dis. 2019, 70, 937–952. [Google Scholar] [CrossRef]
- Peng, Y.; Hou, C.; Yang, Z.; Li, C.; Jia, L.; Liu, J.; Tang, Y.; Shi, L.; Li, Y.; Long, J.; et al. Hydroxytyrosol mildly improve cognitive function independent of APP processing in APP/PS1 mice. Mol. Nutr. Food Res. 2016, 60, 2331–2342. [Google Scholar] [CrossRef]
- Arunsundar, M.; Shanmugarajan, T.S.; Ravichandran, V. 3,4-dihydroxyphenylethanol attenuates spatio-cognitive deficits in an Alzheimer’s disease mouse model: Modulation of the molecular signals in neuronal survival-apoptotic programs. Neurotox. Res. 2015, 27, 143–155. [Google Scholar] [CrossRef]
- Qin, C.; Hu, S.; Zhang, S.; Zhao, D.; Wang, Y.; Li, H.; Peng, Y.; Shi, L.; Xu, X.; Wang, C.; et al. Hydroxytyrosol Acetate Improves the Cognitive Function of APP/PS1 Transgenic Mice in ERbeta-dependent Manner. Mol. Nutr. Food Res. 2021, 65, e2000797. [Google Scholar] [CrossRef] [PubMed]
- St-Laurent-Thibault, C.; Arseneault, M.; Longpré, F.; Ramassamy, C. Tyrosol and hydroxytyrosol, two main components of olive oil, protect N2a cells against amyloid-β-induced toxicity. Involvement of the NF-κB signaling. Curr. Alzheimer Res. 2011, 8, 543–551. [Google Scholar] [CrossRef]
- Dewapriya, P.; Himaya, S.W.; Li, Y.X.; Kim, S.K. Tyrosol exerts a protective effect against dopaminergic neuronal cell death in in vitro model of Parkinson’s disease. Food Chem. 2013, 141, 1147–1157. [Google Scholar] [CrossRef]
- Garcia-Moreno, J.C.; Porta de la Riva, M.; Martínez-Lara, E.; Siles, E.; Cañuelo, A. Tyrosol, a simple phenol from EVOO, targets multiple pathogenic mechanisms of neurodegeneration in a C. elegans model of Parkinson’s disease. Neurobiol. Aging 2019, 82, 60–68. [Google Scholar] [CrossRef]
- Peng, S.; Zhang, B.; Yao, J.; Duan, D.; Fang, J. Dual protection of hydroxytyrosol, an olive oil polyphenol, against oxidative damage in PC12 cells. Food Funct. 2015, 6, 2091–2100. [Google Scholar] [CrossRef]
- Wang, W.C.; Xia, Y.M.; Yang, B.; Su, X.N.; Chen, J.K.; Li, W.; Jiang, T. Protective Effects of Tyrosol against LPS-Induced Acute Lung Injury via Inhibiting NF-kappaB and AP-1 Activation and Activating the HO-1/Nrf2 Pathways. Biol. Pharm. Bull. 2017, 40, 583–593. [Google Scholar] [CrossRef] [Green Version]
- Muriana, F.J.G.; Montserrat-de la Paz, S.; Lucas, R.; Bermudez, B.; Jaramillo, S.; Morales, J.C.; Abia, R.; Lopez, S. Tyrosol and its metabolites as antioxidative and anti-inflammatory molecules in human endothelial cells. Food Funct. 2017, 8, 2905–2914. [Google Scholar] [CrossRef] [Green Version]
- Hoi, C.P.; Ho, Y.P.; Baum, L.; Chow, A.H. Neuroprotective effect of honokiol and magnolol, compounds from Magnolia officinalis, on beta-amyloid-induced toxicity in PC12 cells. Phytother. Res. 2010, 24, 1538–1542. [Google Scholar] [CrossRef]
- Xian, Y.F.; Ip, S.P.; Mao, Q.Q.; Lin, Z.X. Neuroprotective effects of honokiol against beta-amyloid-induced neurotoxicity via GSK-3β and β-catenin signaling pathway in PC12 cells. Neurochem. Int. 2016, 97, 8–14. [Google Scholar] [CrossRef]
- Wang, M.; Li, Y.; Ni, C.; Song, G. Honokiol Attenuates Oligomeric Amyloid beta1-42-Induced Alzheimer’s Disease in Mice Through Attenuating Mitochondrial Apoptosis and Inhibiting the Nuclear Factor Kappa-B Signaling Pathway. Cell Physiol. Biochem. 2017, 43, 69–81. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Jia, J.; Wang, W.; Hou, T.; Tian, Y.; Wu, Q.; Xu, L.; Wei, Y.; Wang, X. Honokiol Alleviates Cognitive Deficits of Alzheimer’s Disease (PS1V97L) Transgenic Mice by Activating Mitochondrial SIRT3. J. Alzheimer’s Dis. 2018, 64, 291–302. [Google Scholar] [CrossRef]
- Wang, D.; Dong, X.; Wang, C. Honokiol Ameliorates Amyloidosis and Neuroinflammation and Improves Cognitive Impairment in Alzheimer’s Disease Transgenic Mice. J. Pharmacol. Exp. Ther. 2018, 366, 470–478. [Google Scholar] [CrossRef]
- Geetha, R.G.; Ramachandran, S. Recent Advances in the Anti-Inflammatory Activity of Plant-Derived Alkaloid Rhynchophylline in Neurological and Cardiovascular Diseases. Pharmaceutics 2021, 13, 1170. [Google Scholar] [CrossRef]
- Yang, Y.; Ji, W.G.; Zhu, Z.R.; Wu, Y.L.; Zhang, Z.Y.; Qu, S.C. Rhynchophylline suppresses soluble Abeta(1-42)-induced impairment of spatial cognition function via inhibiting excessive activation of extrasynaptic NR2B-containing NMDA receptors. Neuropharmacology 2018, 135, 100–112. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Chen, L.; Xu, J.; Liu, W.; Feng, F.; Qu, W. Neuroprotective Effects of Rhynchophylline Against Abeta(1-42)-Induced Oxidative Stress, Neurodegeneration, and Memory Impairment Via Nrf2-ARE Activation. Neurochem. Res. 2021, 46, 2439–2450. [Google Scholar] [CrossRef]
- Wang, X.; Wang, Y.; Hu, J.P.; Yu, S.; Li, B.K.; Cui, Y.; Ren, L.; Zhang, L.D. Astragaloside IV, a Natural PPARgamma Agonist, Reduces Abeta Production in Alzheimer’s Disease Through Inhibition of BACE1. Mol. Neurobiol. 2017, 54, 2939–2949. [Google Scholar] [CrossRef]
- Gu, D.M.; Lu, P.H.; Zhang, K.; Wang, X.; Sun, M.; Chen, G.Q.; Wang, Q. EGFR mediates astragaloside IV-induced Nrf2 activation to protect cortical neurons against in vitro ischemia/reperfusion damages. Biochem. Biophys. Res. Commun. 2015, 457, 391–397. [Google Scholar] [CrossRef]
- Wang, X.; Xu, W.; Chen, H.; Li, W.; Li, W.; Zhu, G. Astragaloside IV prevents Abeta(1-42) oligomers-induced memory impairment and hippocampal cell apoptosis by promoting PPARgamma/BDNF signaling pathway. Brain Res. 2020, 1747, 147041. [Google Scholar] [CrossRef]
- Thapliyal, S.; Singh, T.; Handu, S.; Bisht, M.; Kumari, P.; Arya, P.; Srivastava, P.; Gandham, R. A Review on Potential Footprints of Ferulic Acid for Treatment of Neurological Disorders. Neurochem. Res. 2021, 46, 1043–1057. [Google Scholar] [CrossRef] [PubMed]
- Sultana, R.; Ravagna, A.; Mohmmad-Abdul, H.; Calabrese, V.; Butterfield, D.A. Ferulic acid ethyl ester protects neurons against amyloid beta- peptide(1-42)-induced oxidative stress and neurotoxicity: Relationship to antioxidant activity. J. Neurochem. 2005, 92, 749–758. [Google Scholar] [CrossRef]
- Picone, P.; Bondi, M.L.; Montana, G.; Bruno, A.; Pitarresi, G.; Giammona, G.; Di Carlo, M. Ferulic acid inhibits oxidative stress and cell death induced by Ab oligomers: Improved delivery by solid lipid nanoparticles. Free. Radic. Res. 2009, 43, 1133–1145. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, J.; Rong, H.; Zhang, X.; Dong, M. Ferulic Acid Ameliorates MPP(+)/MPTP-Induced Oxidative Stress via ERK1/2-Dependent Nrf2 Activation: Translational Implication for Parkinson Disease Treatment. Mol. Neurobiol. 2020, 57, 2981–2995. [Google Scholar] [CrossRef]
- Mori, T.; Koyama, N.; Guillot-Sestier, M.V.; Tan, J.; Town, T. Ferulic acid is a nutraceutical beta-secretase modulator that improves behavioral impairment and alzheimer-like pathology in transgenic mice. PLoS ONE 2013, 8, e55774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, J.J.; Jung, J.S.; Kim, T.K.; Hasan, A.; Hong, C.W.; Nam, J.S.; Song, D.K. Protective effects of ferulic acid in amyloid precursor protein plus presenilin-1 transgenic mouse model of Alzheimer disease. Biol. Pharm. Bull. 2013, 36, 140–143. [Google Scholar] [CrossRef] [Green Version]
- Morroni, F.; Sita, G.; Graziosi, A.; Ravegnini, G.; Molteni, R.; Paladini, M.S.; Dias, K.S.T.; Dos Santos, A.F.; Viegas CJr Camps, I.; Pruccoli, L.; et al. PQM130, a Novel Feruloyl-Donepezil Hybrid Compound, Effectively Ameliorates the Cognitive Impairments and Pathology in a Mouse Model of Alzheimer’s Disease. Front. Pharmacol. 2019, 10, 658. [Google Scholar] [CrossRef]
- Dias, K.S.; de Paula, C.T.; Dos Santos, T.; Souza, I.N.; Boni, M.S.; Guimarães, M.J.; da Silva, F.M.; Castro, N.G.; Neves, G.A.; Veloso, C.C.; et al. Design, synthesis and evaluation of novel feruloyl-donepezil hybrids as potential multitarget drugs for the treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2017, 130, 440–457. [Google Scholar] [CrossRef]
- Winter, A.N.; Bickford, P.C. Anthocyanins and Their Metabolites as Therapeutic Agents for Neurodegenerative Disease. Antioxidants 2019, 8, 333. [Google Scholar] [CrossRef] [Green Version]
- Badshah, H.; Kim, T.H.; Kim, M.O. Protective effects of anthocyanins against amyloid beta-induced neurotoxicity in vivo and in vitro. Neurochem. Int. 2015, 80, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Ali, T.; Kim, T.; Rehman, S.U.; Khan, M.S.; Amin, F.U.; Khan, M.; Ikram, M.; Kim, M.O. Natural Dietary Supplementation of Anthocyanins via PI3K/Akt/Nrf2/HO-1 Pathways Mitigate Oxidative Stress, Neurodegeneration, and Memory Impairment in a Mouse Model of Alzheimer’s Disease. Mol. Neurobiol. 2018, 55, 6076–6093. [Google Scholar] [CrossRef]
- Song, N.; Zhang, L.; Chen, W.; Zhu, H.; Deng, W.; Han, Y.; Guo, J.; Qin, C. Cyanidin 3-O-beta-glucopyranoside activates peroxisome proliferator-activated receptor-gamma and alleviates cognitive impairment in the APP(swe)/PS1(deltaE9) mouse model. Biochim. Biophys. Acta 2016, 1862, 1786–1800. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.; Li, B.; Li, D.; Wang, Y.; Lin, Y.; Meng, X.; Sun, X.; Liu, N. Cyanidin-3-O-glucoside attenuates amyloid-beta (1–40)-induced oxidative stress and apoptosis in SH-SY5Y cells through a Nrf2 mechanism. J. Funct. Foods 2017, 38, 474–485. [Google Scholar] [CrossRef]
- Kulkarni, N.P.; Vaidya, B.; Narula, A.S.; Sharma, S.S. Neuroprotective Potential of Caffeic Acid Phenethyl Ester (CAPE) in CNS Disorders: Mechanistic and Therapeutic Insights. Curr. Neuropharmacol. 2021, 19, 1401–1415. [Google Scholar] [CrossRef] [PubMed]
- Morroni, F.; Sita, G.; Graziosi, A.; Turrini, E.; Fimognari, C.; Tarozzi, A.; Hrelia, P. Neuroprotective Effect of Caffeic Acid Phenethyl Ester in A Mouse Model of Alzheimer’s Disease Involves Nrf2/HO-1 Pathway. Aging Dis. 2018, 9, 605–622. [Google Scholar] [CrossRef] [Green Version]
- Tsai, C.F.; Kuo, Y.H.; Yeh, W.L.; Wu, C.Y.; Lin, H.Y.; Lai, S.W.; Liu, Y.S.; Wu, L.H.; Lu, J.K.; Lu, D.Y. Regulatory effects of caffeic acid phenethyl ester on neuroinflammation in microglial cells. Int. J. Mol. Sci. 2015, 16, 5572–5589. [Google Scholar] [CrossRef] [Green Version]
- Lanznaster, D.; Dal-Cim, T.; Piermartiri, T.C.; Tasca, C.I. Guanosine: A Neuromodulator with Therapeutic Potential in Brain Disorders. Aging Dis. 2016, 7, 657–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarozzi, A.; Merlicco, A.; Morroni, F.; Bolondi, C.; Di Iorio, P.; Ciccarelli, R.; Romano, S.; Giuliani, P.; Hrelia, P. Guanosine protects human neuroblastoma cells from oxidative stress and toxicity induced by Amyloid-beta peptide oligomers. J. Biol. Regul. Homeost. Agents 2010, 24, 297–306. [Google Scholar]
- da Silva, J.S.; Nonose, Y.; Rohden, F.; Lukasewicz Ferreira, P.C.; Fontella, F.U.; Rocha, A.; Brochier, A.W.; Apel, R.V.; de Lima, T.M.; Seminotti, B.; et al. Guanosine Neuroprotection of Presynaptic Mitochondrial Calcium Homeostasis in a Mouse Study with Amyloid-beta Oligomers. Mol. Neurobiol. 2020, 57, 4790–4809. [Google Scholar] [CrossRef]
- Wang, X.; He, H.J.; Xiong, X.; Zhou, S.; Wang, W.W.; Feng, L.; Han, R.; Xie, C.L. NAD+ in Alzheimer’s Disease: Molecular Mechanisms and Systematic Therapeutic Evidence Obtained in vivo. Front. Cell Dev. Biol. 2021, 9, 668491. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Hu, X.; Yang, Y.; Takata, T.; Sakurai, T. Nicotinamide mononucleotide protects against beta-amyloid oligomer-induced cognitive impairment and neuronal death. Brain Res. 2016, 1643, 1–9. [Google Scholar] [CrossRef]
- Yao, Z.; Yang, W.; Gao, Z.; Jia, P. Nicotinamide mononucleotide inhibits JNK activation to reverse Alzheimer disease. Neurosci. Lett. 2017, 647, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Rehman, I.U.; Ahmad, R.; Khan, I.; Lee, H.J.; Park, J.; Ullah, R.; Choi, M.J.; Kang, H.Y.; Kim, M.O. Nicotinamide Ameliorates Amyloid Beta-Induced Oxidative Stress-Mediated Neuroinflammation and Neurodegeneration in Adult Mouse Brain. Biomedicines 2021, 9, 408. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Pitta, M.; Jiang, H.; Lee, J.H.; Zhang, G.; Chen, X.; Kawamoto, E.M.; Mattson, M.P. Nicotinamide forestalls pathology and cognitive decline in Alzheimer mice: Evidence for improved neuronal bioenergetics and autophagy procession. Neurobiol. Aging 2013, 34, 1564–1580. [Google Scholar] [CrossRef] [Green Version]
- Jęśko, H.; Wencel, P.; Strosznajder, R.P.; Strosznajder, J.B. Sirtuins and Their Roles in Brain Aging and Neurodegenerative Disorders. Neurochem. Res. 2017, 42, 876–890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Y.; Wang, X.P.; Yang, S.G.; Wang, Y.J.; Zhang, X.; Du, X.T.; Sun, X.X.; Zhao, M.; Huang, L.; Liu, R.T. Resveratrol inhibits beta-amyloid oligomeric cytotoxicity but does not prevent oligomer formation. Neurotoxicology 2009, 30, 986–995. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.T.; Cao, K.; Tan, L.C.; Wang, X.L.; Qi, X.L.; Xiao, Y.; Guan, Z.Z. Stimulation of SIRT1 Attenuates the Level of Oxidative Stress in the Brains of APP/PS1 Double Transgenic Mice and in Primary Neurons Exposed to Oligomers of the Amyloid-beta Peptide. J. Alzheimer’s Dis. 2018, 63, 283–301. [Google Scholar] [CrossRef] [PubMed]
- Donmez, G.; Wang, D.; Cohen, D.E.; Gaurente, L. SIRT1 suppresses beta-amyloid production by activating the alpha secretase gene ADAM10. Cell 2010, 142, 320–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semwal, D.K.; Semwal, R.B.; Combrinck, S.; Viljoen, A. Myricetin: A Dietary Molecule with Diverse Biological Activities. Nutrients 2016, 8, 90. [Google Scholar] [CrossRef] [Green Version]
- Ono, K.; Li, L.; Takamura, Y.; Yoshiike, Y.; Zhu, L.; Han, F.; Mao, X.; Ikeda, T.; Takasaki, J.; Nishijo, H.; et al. Phenolic compounds prevent amyloid beta-protein oligomerization and synaptic dysfunction by site-specific binding. J. Biol. Chem. 2012, 287, 14631–14643. [Google Scholar] [CrossRef] [Green Version]
- Kimura, A.M.; Tsuji, M.; Yasumoto, T.; Mori, Y.; Oguchi, T.; Tsuji, Y.; Umino, M.; Umino, A.; Nishikawa, T.; Nakamura, S.; et al. Myricetin prevents high molecular weight Abeta(1-42) oligomer-induced neurotoxicity through antioxidant effects in cell membranes and mitochondria. Free Radic. Biol. Med. 2021, 171, 232–244. [Google Scholar] [CrossRef]
- Gavilan, J.; Mennickent, D.; Ramirez-Molina, O.; Triviño, S.; Perez, C.; Silva-Grecchi, T.; Godoy, P.A.; Becerra, J.; Aguayo, L.G.; Moraga-Cid, G.; et al. 17 Oxo Sparteine and Lupanine, Obtained from Cytisus scoparius, Exert a Neuroprotection against Soluble Oligomers of Amyloid-beta Toxicity by Nicotinic Acetylcholine Receptors. J. Alzheimer’s Dis. 2019, 67, 343–356. [Google Scholar] [CrossRef]
- Kadakol, A.; Sharma, N.; Kulkarni, Y.A.; Gaikwad, A.B. Esculetin: A phytochemical endeavor fortifying effect against non-communicable diseases. Biomed. Pharmacother. 2016, 84, 1442–1448. [Google Scholar] [CrossRef]
- Pruccoli, L.; Morroni, F.; Sita, G.; Hrelia, P.; Tarozzi, A. Esculetin as a Bifunctional Antioxidant Prevents and Counteracts the Oxidative Stress and Neuronal Death Induced by Amyloid Protein in SH-SY5Y Cells. Antioxidants 2020, 9, 551. [Google Scholar] [CrossRef]
- Ali, M.Y.; Jannat, S.; Jung, H.A.; Choi, R.J.; Roy, A.; Choi, J.S. Anti-Alzheimer’s disease potential of coumarins from Angelica decursiva and Artemisia capillaris and structure-activity analysis. Asian Pac. J. Trop. Med. 2016, 9, 103–111. [Google Scholar] [CrossRef] [Green Version]
- Leopoldini, M.; Russo, N.; Toscano, M. The molecular basis of working mechanism of natural polyphenolic antioxidants. Food Chem. 2011, 125, 288–306. [Google Scholar] [CrossRef]
- Zhou, Y.; Xie, N.; Li, L.; Zou, Y.; Zhang, X.; Dong, M. Puerarin alleviates cognitive impairment and oxidative stress in APP/PS1 transgenic mice. Int. J. Neuropsychopharmacol. 2014, 17, 635–644. [Google Scholar] [CrossRef] [Green Version]
- Kanninen, K.; Malm, T.M.; Jyrkkänen, H.K.; Goldsteins, G.; Keksa-Goldsteine, V.; Tanila, H.; Yamamoto, M.; Ylä-Herttuala, S.; Levonen, A.L.; Koistinaho, J. Nuclear factor erythroid 2-related factor 2 protects against beta amyloid. Mol. Cell. Neurosci. 2008, 39, 302–313. [Google Scholar] [CrossRef] [PubMed]
- Yarza, R.; Vela, S.; Solas, M.; Ramirez, M.J. c-Jun N-terminal Kinase (JNK) Signaling as a Therapeutic Target for Alzheimer’s Disease. Front. Pharmacol. 2016, 6, 321. [Google Scholar] [CrossRef] [Green Version]
- Seo, E.J.; Fischer, N.; Efferth, T. Phytochemicals as inhibitors of NF-kappaB for treatment of Alzheimer’s disease. Pharmacol. Res. 2018, 129, 262–273. [Google Scholar] [CrossRef]
- Liao, Y.; Qi, X.L.; Cao, Y.; Yu, W.F.; Ravid, R.; Winblad, B.; Pei, J.J.; Guan, Z.Z. Elevations in the Levels of NF-kappaB and Inflammatory Chemotactic Factors in the Brains with Alzheimer’s Disease—One Mechanism May Involve alpha3 Nicotinic Acetylcholine Receptor. Curr. Alzheimer Res. 2016, 13, 1290–1301. [Google Scholar] [CrossRef] [PubMed]
- Wardyn, J.D.; Ponsford, A.H.; Sanderson, C.M. Dissecting molecular cross-talk between Nrf2 and NF-κB response pathways. Biochem. Soc. Trans. 2015, 43, 621–626. [Google Scholar] [CrossRef] [Green Version]
- Salvatori, I.; Valle, C.; Ferri, A.; Carrì, M.T. SIRT3 and mitochondrial metabolism in neurodegenerative diseases. Neurochem. Int. 2017, 109, 184–192. [Google Scholar] [CrossRef]
- Nicolakakis, N.; Hamel, E. The Nuclear Receptor PPARgamma as a Therapeutic Target for Cerebrovascular and Brain Dysfunction in Alzheimer’s Disease. Front. Aging Neurosci. 2010, 2, 21. [Google Scholar] [CrossRef] [Green Version]
- Fuenzalida, K.; Quintanilla, R.; Ramos, P.; Piderit, D.; Fuentealba, R.A.; Martinez, G.; Inestrosa, N.C.; Bronfman, M. Peroxisome proliferator-activated receptor gamma up-regulates the Bcl-2 anti-apoptotic protein in neurons and induces mitochondrial stabilization and protection against oxidative stress and apoptosis. J. Biol. Chem. 2007, 282, 37006–37015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inestrosa, N.C.; Godoy, J.A.; Quintanilla, R.A.; Koenig, C.S.; Bronfman, M. Peroxisome proliferator-activated receptor gamma is expressed in hippocampal neurons and its activation prevents beta-amyloid neurodegeneration: Role of Wnt signaling. Exp. Cell Res. 2005, 304, 91–104. [Google Scholar] [CrossRef]
- Sastre, M.; Dewachter, I.; Rossner, S.; Bogdanovic, N.; Rosen, E.; Borghgraef, P.; Evert, B.O.; Dumitrescu-Ozimek, L.; Thal, D.R.; Landreth, G.; et al. Nonsteroidal anti-inflammatory drugs repress beta-secretase gene promoter activity by the activation of PPARgamma. Proc. Natl. Acad. Sci. USA 2006, 103, 443–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mota, S.I.; Ferreira, I.L.; Rego, A.C. Dysfunctional synapse in Alzheimer’s disease—A focus on NMDA receptors. Neuropharmacology 2014, 76, 16–26. [Google Scholar] [CrossRef]
- Webers, A.; Heneka, M.T.; Gleeson, P.A. The role of innate immune responses and neuroinflammation in amyloid accumulation and progression of Alzheimer’s disease. Immunol. Cell Biol. 2020, 98, 28–41. [Google Scholar] [CrossRef]
- Pardridge, W.M. Treatment of Alzheimer’s Disease and Blood-Brain Barrier Drug Delivery. Pharmaceuticals 2020, 13, 394. [Google Scholar] [CrossRef] [PubMed]
- Bucci, M.; Chiotis, K.; Nordberg, A.; Alzheimer’s Disease Neuroimaging Initiative. Alzheimer’s disease profiled by fluid and imaging markers: Tau PET best predicts cognitive decline. Mol. Psychiatry 2021. [Google Scholar] [CrossRef]
- Turner, R.S.; Thomas, R.G.; Craft, S.; van Dyck, C.H.; Mintzer, J.; Reynolds, B.A.; Brewer, J.B.; Rissman, R.A.; Raman, R.; Aisen, P.S. A randomized, double-blind, placebo-controlled trial of resveratrol for Alzheimer disease. Alzheimer’s Disease Cooperative Study. Neurology 2015, 85, 1383–1391. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Compound | MW | Experimental Models | Route | Ameliorative Effects | Refs | ||||
|---|---|---|---|---|---|---|---|---|---|
| In Vitro | In Vivo | OS | Apoptosis | Cognitive Impairment | Aβ Accumulation | ||||
| Tyrosol | 138 | Primary neurons | 5XFAD | oral | + | + | + | − | [26] |
| H-Tyr | 154 | APP/PS1 | oral | + | + | + | − | [27,29] | |
| AβO + ia injection | [28] | ||||||||
| Honokiol | 266 | Primary neurons | AβO injection PS1V97L APP/PS1 | ip | + | + | + | + | [38,39,40] |
| Rhynchophylline | 384 | AβO injection | ip | + | + | + | n.d. | [42,43] | |
| Astragaloside IV | 785 | HT22 | AβO injection | ip | n.d. | + | + | + | [46] |
| APP/PS1 | [44] | ||||||||
| Ferulic acid | 194 | LAN5 | APP/PS1 | oral | + | n.d. | + | + | [49,51,52] |
| PQM130 | 381 | SH-SY5Y | AβO injection | ip | + | + | + | n.d. | [53,54] |
| Anthocyanins | 450< | HT22 | APP/PS1 | ip | + | + | + | n.d. | [57] |
| CAPE | 284 | AβO injection | ip | + | + | + | n.d. | [61] | |
| Guanosine | 283 | SH-SY5Y | AβO injection | oral | + | + | + | n.d. | [64,65] |
| NMN | 334 | Slice cultures | AβO injection | ip | + | n.d. | + | + | [67] |
| APP/PS1 | subcutaneous | [68] | |||||||
| Resveratrol | 228 | SH-SY5Y | [72] | ||||||
| Primary neurons | APP/PS1 | oral | + | n.d. | n.d. | + | [73] | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Araki, W.; Kametani, F. Protection against Amyloid-β Oligomer Neurotoxicity by Small Molecules with Antioxidative Properties: Potential for the Prevention of Alzheimer’s Disease Dementia. Antioxidants 2022, 11, 132. https://doi.org/10.3390/antiox11010132
Araki W, Kametani F. Protection against Amyloid-β Oligomer Neurotoxicity by Small Molecules with Antioxidative Properties: Potential for the Prevention of Alzheimer’s Disease Dementia. Antioxidants. 2022; 11(1):132. https://doi.org/10.3390/antiox11010132
Chicago/Turabian StyleAraki, Wataru, and Fuyuki Kametani. 2022. "Protection against Amyloid-β Oligomer Neurotoxicity by Small Molecules with Antioxidative Properties: Potential for the Prevention of Alzheimer’s Disease Dementia" Antioxidants 11, no. 1: 132. https://doi.org/10.3390/antiox11010132