
The Trypanosoma brucei-Derived Ketoacids, Indole Pyruvate and Hydroxyphenylpyruvate, Induce HO-1 Expression and Suppress Inflammatory Responses in Human Dendritic Cells

 , , ,
, , ,
Abstract
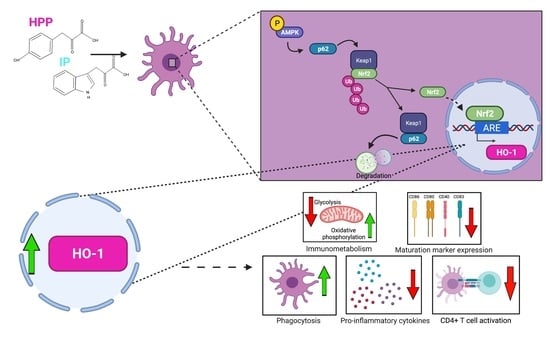
:
1. Introduction
2. Materials and Methods
2.1. Reagents and Chemicals
2.2. Human Blood Samples
2.3. Dendritic Cell Culture
2.4. Western Blot Experiments
2.5. Antioxidant Assay
2.6. DC Flow Cytometry Experiments
2.7. Quantitative Real-Time PCR
2.8. DC ELISA Experiments
2.9. DC-CD4+ T Cell Co-Cultures
2.10. Two-Photon Fluorescence Lifetime Imaging Microscopy (FLIM)
2.11. Metabolic Profiling Using Seahorse Analysis
2.12. IBD Patient PBMC Experiments
2.13. Assessment of Endotoxin Contamination
2.14. Statistical Analysis
3. Results
3.1. HPP and IP Upregulate HO-1 in Primary Human DC
3.2. HPP and IP Induce HO-1 through Nrf2 Activation
3.3. HPP and IP Reduce the Production of Pro-Inflammatory Cytokines in LPS-Stimulated Human DC
3.4. HPP Treatment Inhibits the Maturation of LPS-Stimulated Human DC, Resulting in Reduced Activation of CD4+ T Cells
3.5. HPP and IP Modulate Metabolic Reprogramming in LPS-Stimulated Human DC
3.6. HPP and IP Activate Autophagy-Related Proteins
3.7. HPP and IP Reduce Proliferation and Cytokine Expression in PBMC Isolated from IBD Patients
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Stijlemans, B.; Caljon, G.; Van Den Abbeele, J.; Van Ginderachter, J.A.; Magez, S.; De Trez, C. Immune Evasion Strategies of Trypanosoma brucei within the Mammalian Host: Progression to Pathogenicity. Front. Immunol. 2016, 7, 233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berger, B.J.; Dai, W.W.; Wang, H.; Stark, R.E.; Cerami, A. Aromatic amino acid transamination and methionine recycling in trypanosomatids. Proc. Natl. Acad. Sci. USA 1996, 93, 4126–4130. [Google Scholar] [CrossRef] [Green Version]
- Marciano, D.; Llorente, C.; Maugeri, D.A.; de la Fuente, C.; Opperdoes, F.; Cazzulo, J.J.; Nowicki, C. Biochemical characterization of stage-specific isoforms of aspartate aminotransferases from Trypanosoma cruzi and Trypanosoma brucei. Mol. Biochem. Parasitol. 2008, 161, 12–20. [Google Scholar] [CrossRef] [PubMed]
- McGettrick, A.F.; Corcoran, S.E.; Barry, P.J.G.; McFarland, J.; Crès, C.; Curtis, A.M.; Franklin, E.; Corr, S.C.; Mok, K.H.; Cummins, E.P.; et al. Trypanosoma brucei metabolite indolepyruvate decreases HIF-1α and glycolysis in macrophages as a mechanism of innate immune evasion. Proc. Natl. Acad. Sci. USA 2016, 113, E7778–E7787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, N.K.; Williams, D.G.; Fitzgerald, H.K.; Barry, P.J.; Cunningham, C.C.; Nolan, D.P.; Dunne, A. Trypanosoma brucei Secreted Aromatic Ketoacids Activate the Nrf2/HO-1 Pathway and Suppress Pro-inflammatory Responses in Primary Murine Glia and Macrophages. Front. Immunol. 2019, 10, 2137. [Google Scholar] [CrossRef]
- Aoki, R.; Aoki-Yoshida, A.; Suzuki, C.; Takayama, Y. Protective Effect of Indole-3-Pyruvate against Ultraviolet B-Induced Damage to Cultured HaCaT Keratinocytes and the Skin of Hairless Mice. PLoS ONE 2014, 9, e96804. [Google Scholar] [CrossRef]
- Aoki, R.; Aoki-Yoshida, A.; Suzuki, C.; Takayama, Y. Indole-3-Pyruvic Acid, an Aryl Hydrocarbon Receptor Activator, Suppresses Experimental Colitis in Mice. J. Immunol. 2018, 201, 3683–3693. [Google Scholar] [CrossRef] [Green Version]
- Diskin, C.; Corcoran, S.E.; Tyrrell, V.J.; McGettrick, A.F.; Zaslona, Z.; O’Donnell, V.B.; Nolan, D.P.; O’Neill, L.A.J. The Trypanosome-Derived Metabolite Indole-3-Pyruvate Inhibits Prostaglandin Production in Macrophages by Targeting COX2. J. Immunol. 2021, 207, 2551–2560. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Komoda, Y.; Nakajima, H. Biliverdin-IXα reductase and biliverdin-IXβ reductase from human liver. Purification and characterization. J. Biol. Chem. 1994, 269, 24343–24348. [Google Scholar] [CrossRef]
- Singleton, J.W.; Laster, L. Biliverdin reductase of guinea pig liver. J. Biol. Chem. 1965, 240, 4780–4789. [Google Scholar] [CrossRef]
- Radhakrishnan, N.; Yadav, S.P.; Sachdeva, A.; Pruthi, P.K.; Sawhney, S.; Piplani, T.; Wada, T.; Yachie, A. Human heme oxygenase-1 deficiency presenting with hemolysis, nephritis, and asplenia. J. Pediatr. Hematol. Oncol. 2011, 33, 74–78. [Google Scholar] [CrossRef] [PubMed]
- Kapturczak, M.H.; Wasserfall, C.; Brusko, T.; Campbell-Thompson, M.; Ellis, T.M.; Atkinson, M.A.; Agarwal, A. Heme oxygenase-1 modulates early inflammatory responses: Evidence from the heme oxygenase-1-deficient mouse. Am. J. Pathol. 2004, 165, 1045–1053. [Google Scholar] [CrossRef]
- Poss, K.D.; Tonegawa, S. Reduced stress defense in heme oxygenase 1-deficient cells. Proc. Natl. Acad. Sci. USA 1997, 94, 10925–10930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, N.K.; Fitzgerald, H.K.; Dunne, A. Regulation of inflammation by the antioxidant haem oxygenase 1. Nat. Rev. Immunol. 2021, 21, 411–425. [Google Scholar] [CrossRef] [PubMed]
- Neto, N.; Dmitriev, R.I.; Monaghan, M.G. Seeing Is Believing: Noninvasive Microscopic Imaging Modalities for Tissue Engineering and Regenerative Medicine. In Cell Engineering and Regeneration; Springer: Cham, Switzerland, 2020; pp. 599–638. [Google Scholar]
- Floudas, A.; Neto, N.; Marzaioli, V.; Murray, K.; Moran, B.; Monaghan, M.G.; Low, C.; Mullan, R.H.; Rao, N.; Krishna, V.; et al. Pathogenic, glycolytic PD-1+ B cells accumulate in the hypoxic RA joint. JCI Insight 2020, 5, e139032. [Google Scholar] [CrossRef]
- El Sawalhy, A.; Seed, J.R.; Hall, J.E.; El Attar, H. Increased excretion of aromatic amino acid catabolites in animals infected with Trypanosoma brucei evansi. J. Parasitol. 1998, 84, 469–473. [Google Scholar] [CrossRef]
- Campbell, N.K.; Fitzgerald, H.K.; Malara, A.; Hambly, R.; Sweeney, C.M.; Kirby, B.; Fletcher, J.M.; Dunne, A. Naturally derived Heme-Oxygenase 1 inducers attenuate inflammatory responses in human dendritic cells and T cells: Relevance for psoriasis treatment. Sci. Rep. 2018, 8, 10287. [Google Scholar] [CrossRef]
- Chauveau, C.; Rémy, S.; Royer, P.J.; Hill, M.; Tanguy-Royer, S.S.; Hubert, F.-X.F.X.; Tesson, L.; Brion, R.R.; Beriou, G.G.; Gregoire, M.; et al. Heme oxygenase-1 expression inhibits dendritic cell maturation and proinflammatory function but conserves IL-10 expression. Blood 2005, 106, 1694–1702. [Google Scholar] [CrossRef] [Green Version]
- Hull, T.D.; Agarwal, A.; George, J.F. The mononuclear phagocyte system in homeostasis and disease: A role for heme oxygenase-1. Antioxid. Redox Signal. 2014, 20, 1770–1788. [Google Scholar] [CrossRef] [Green Version]
- O’Neill, L.A.J.; Kishton, R.J.; Rathmell, J. A guide to immunometabolism for immunologists. Nat. Rev. Immunol. 2016, 16, 553–565. [Google Scholar] [CrossRef] [Green Version]
- Krawczyk, C.M.; Holowka, T.; Sun, J.; Blagih, J.; Amiel, E.; DeBerardinis, R.J.; Cross, J.R.; Jung, E.; Thompson, C.B.; Jones, R.G.; et al. Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood 2010, 115, 4742–4749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komatsu, M.; Kurokawa, H.; Waguri, S.; Taguchi, K.; Kobayashi, A.; Ichimura, Y.; Sou, Y.S.; Ueno, I.; Sakamoto, A.; Tong, K.I.; et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 2010, 12, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Katsuragi, Y.; Ichimura, Y.; Komatsu, M. Regulation of the Keap1–Nrf2 pathway by p62/SQSTM1. Curr. Opin. Toxicol. 2016, 1, 54–61. [Google Scholar] [CrossRef] [Green Version]
- Jiang, T.; Harder, B.; Rojo De La Vega, M.; Wong, P.K.; Chapman, E.; Zhang, D.D. p62 links autophagy and Nrf2 signaling. Free Radic. Biol. Med. 2015, 88, 199. [Google Scholar] [CrossRef] [Green Version]
- Dauchy, F.A.; Contin-Bordes, C.; Nzoumbou-Boko, R.; Bonhivers, M.; Landrein, N.; Robinson, D.R.; Rambert, J.; Courtois, P.; Daulouède, S.; Vincendeau, P. Trypanosoma brucei gambiense excreted/secreted factors impair lipopolysaccharide-induced maturation and activation of human monocyte-derived dendritic cells. Parasite Immunol. 2019, 41, e12632. [Google Scholar] [CrossRef] [PubMed]
- Garzón, E.; Holzmuller, P.; Bras-Gonçalves, R.; Vincendeau, P.; Cuny, G.; Lemesre, J.L.; Geiger, A. The Trypanosoma brucei gambiense Secretome Impairs Lipopolysaccharide-Induced Maturation, Cytokine Production, and Allostimulatory Capacity of Dendritic Cells. Infect. Immun. 2013, 81, 3300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreau, A.; Hill, M.; Thébault, P.; Deschamps, J.Y.; Chiffoleau, E.; Chauveau, C.; Moullier, P.; Anegon, I.; Alliot-Licht, B.; Cuturi, M.C. Tolerogenic dendritic cells actively inhibit T cells through heme oxygenase-1 in rodents and in nonhuman primates. FASEB J. 2009, 23, 3070–3077. [Google Scholar] [CrossRef] [PubMed]
- Andreou, N.P.; Legaki, E.; Gazouli, M. Inflammatory bowel disease pathobiology: The role of the interferon signature. Ann. Gastroenterol. 2020, 33, 125. [Google Scholar] [CrossRef] [PubMed]
- Campbell, N.K.; Fitzgerald, H.K.; Fletcher, J.M.; Dunne, A. Plant-derived polyphenols modulate human dendritic cell metabolism and immune function via AMPK-dependent induction of heme oxygenase-1. Front. Immunol. 2019, 10, 345. [Google Scholar] [CrossRef]
- Sheikh, S.Z.; Hegazi, R.A.; Kobayashi, T.; Onyiah, J.C.; Russo, S.M.; Matsuoka, K.; Sepulveda, A.R.; Li, F.; Otterbein, L.E.; Plevy, S.E. An Anti-Inflammatory Role for Carbon Monoxide and Heme Oxygenase-1 in Chronic Th2-Mediated Murine Colitis. J. Immunol. 2011, 186, 5506–5513. [Google Scholar] [CrossRef]
- Hegazi, R.A.F.; Rao, K.N.; Mayle, A.; Sepulveda, A.R.; Otterbein, L.E.; Plevy, S.E. Carbon monoxide ameliorates chronic murine colitis through a heme oxygenase 1–dependent pathway. J. Exp. Med. 2005, 202, 1703–1713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Zhang, Y.; Zhong, W.; Di, C.; Lin, X.; Xia, Z. Heme oxygenase-1 ameliorates dextran sulfate sodiuminduced acute murine colitis by regulating Th17/Treg cell balance. J. Biol. Chem. 2014, 289, 26847–26858. [Google Scholar] [CrossRef] [Green Version]
- Paul, G.; Bataille, F.; Obermeier, F.; Bock, J.; Klebl, F.; Strauch, U.; Lochbaum, D.; Rümmele, P.; Farkas, S.; Schölmerich, J.; et al. Analysis of intestinal haem-oxygenase-1 (HO-1) in clinical and experimental colitis. Clin. Exp. Immunol. 2005, 140, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Longhi, M.S.; Vuerich, M.; Kalbasi, A.; Kenison, J.E.; Yeste, A.; Csizmadia, E.; Vaughn, B.; Feldbrugge, L.; Mitsuhashi, S.; Wegiel, B.; et al. Bilirubin suppresses Th17 immunity in colitis by upregulating CD39. JCI Insight 2017, 2, e92791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mowat, C.; Cole, A.; Windsor, A.; Ahmad, T.; Arnott, I.; Driscoll, R.; Mitton, S.; Orchard, T.; Rutter, M.; Younge, L.; et al. Guidelines for the management of inflammatory bowel disease in adults. Gut 2011, 60, 571–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Klionsky, D.J. Mammalian autophagy: Core molecular machinery and signaling regulation. Curr. Opin. Cell Biol. 2010, 22, 124–131. [Google Scholar] [CrossRef] [Green Version]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Y.; He, D.; Yao, Z.; Klionsky, D.J. The machinery of macroautophagy. Cell Res. 2014, 24, 24–41. [Google Scholar] [CrossRef] [Green Version]
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.A.; Outzen, H.; Øvervatn, A.; Bjørkøy, G.; Johansen, T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 2007, 282, 24131–24145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghislat, G.; Lawrence, T. Autophagy in dendritic cells. Cell. Mol. Immunol. 2018, 15, 944–952. [Google Scholar] [CrossRef] [PubMed]
- Carroll, K.C.; Viollet, B.; Suttles, J. AMPKα1 deficiency amplifies proinflammatory myeloid APC activity and CD40 signaling. J. Leukoc. Biol. 2013, 94, 1113–1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer | Reverse Primer |
|---|---|---|
| NQO1 | 5′ TGAAGAAGAAAGGATGGGAG 3′ | 5′ TTTACCTGTGATGTCCTTTC 3′ |
| GSR | 5′ GACCTATTCAACGAGCTTTAC 3′ | 5′ CAACCACCTTTTCTTCCTTG 3′ |
| β-actin | 5′ GGACTTCGAGCAAGAGATGG 3′ | 5′ AGCACTGTGTTGGCGTACAG 3′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fitzgerald, H.K.; O’Rourke, S.A.; Desmond, E.; Neto, N.G.B.; Monaghan, M.G.; Tosetto, M.; Doherty, J.; Ryan, E.J.; Doherty, G.A.; Nolan, D.P.; et al. The Trypanosoma brucei-Derived Ketoacids, Indole Pyruvate and Hydroxyphenylpyruvate, Induce HO-1 Expression and Suppress Inflammatory Responses in Human Dendritic Cells. Antioxidants 2022, 11, 164. https://doi.org/10.3390/antiox11010164
Fitzgerald HK, O’Rourke SA, Desmond E, Neto NGB, Monaghan MG, Tosetto M, Doherty J, Ryan EJ, Doherty GA, Nolan DP, et al. The Trypanosoma brucei-Derived Ketoacids, Indole Pyruvate and Hydroxyphenylpyruvate, Induce HO-1 Expression and Suppress Inflammatory Responses in Human Dendritic Cells. Antioxidants. 2022; 11(1):164. https://doi.org/10.3390/antiox11010164
Chicago/Turabian StyleFitzgerald, Hannah K., Sinead A. O’Rourke, Eva Desmond, Nuno G. B. Neto, Michael G. Monaghan, Miriam Tosetto, Jayne Doherty, Elizabeth J. Ryan, Glen A. Doherty, Derek P. Nolan, and et al. 2022. "The Trypanosoma brucei-Derived Ketoacids, Indole Pyruvate and Hydroxyphenylpyruvate, Induce HO-1 Expression and Suppress Inflammatory Responses in Human Dendritic Cells" Antioxidants 11, no. 1: 164. https://doi.org/10.3390/antiox11010164
APA StyleFitzgerald, H. K., O’Rourke, S. A., Desmond, E., Neto, N. G. B., Monaghan, M. G., Tosetto, M., Doherty, J., Ryan, E. J., Doherty, G. A., Nolan, D. P., Fletcher, J. M., & Dunne, A. (2022). The Trypanosoma brucei-Derived Ketoacids, Indole Pyruvate and Hydroxyphenylpyruvate, Induce HO-1 Expression and Suppress Inflammatory Responses in Human Dendritic Cells. Antioxidants, 11(1), 164. https://doi.org/10.3390/antiox11010164