
Impaired Brain Mitochondrial Bioenergetics in the Ts65Dn Mouse Model of Down Syndrome Is Restored by Neonatal Treatment with the Polyphenol 7,8-Dihydroxyflavone





{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Subjects and Treatment
2.2. Brain Tissue Dissection and Mitochondria Isolation
2.3. Measurements of Mitochondrial ATP Production
2.4. Measurement of Mouse Brain ATP Levels
2.5. Measurement of Mitochondrial Respiratory Chain Complex (MRC) Activities
2.6. Detection of Reactive Oxygen Species (ROS) Levels
2.7. Western Blotting
2.8. Statistical Analysis
3. Results
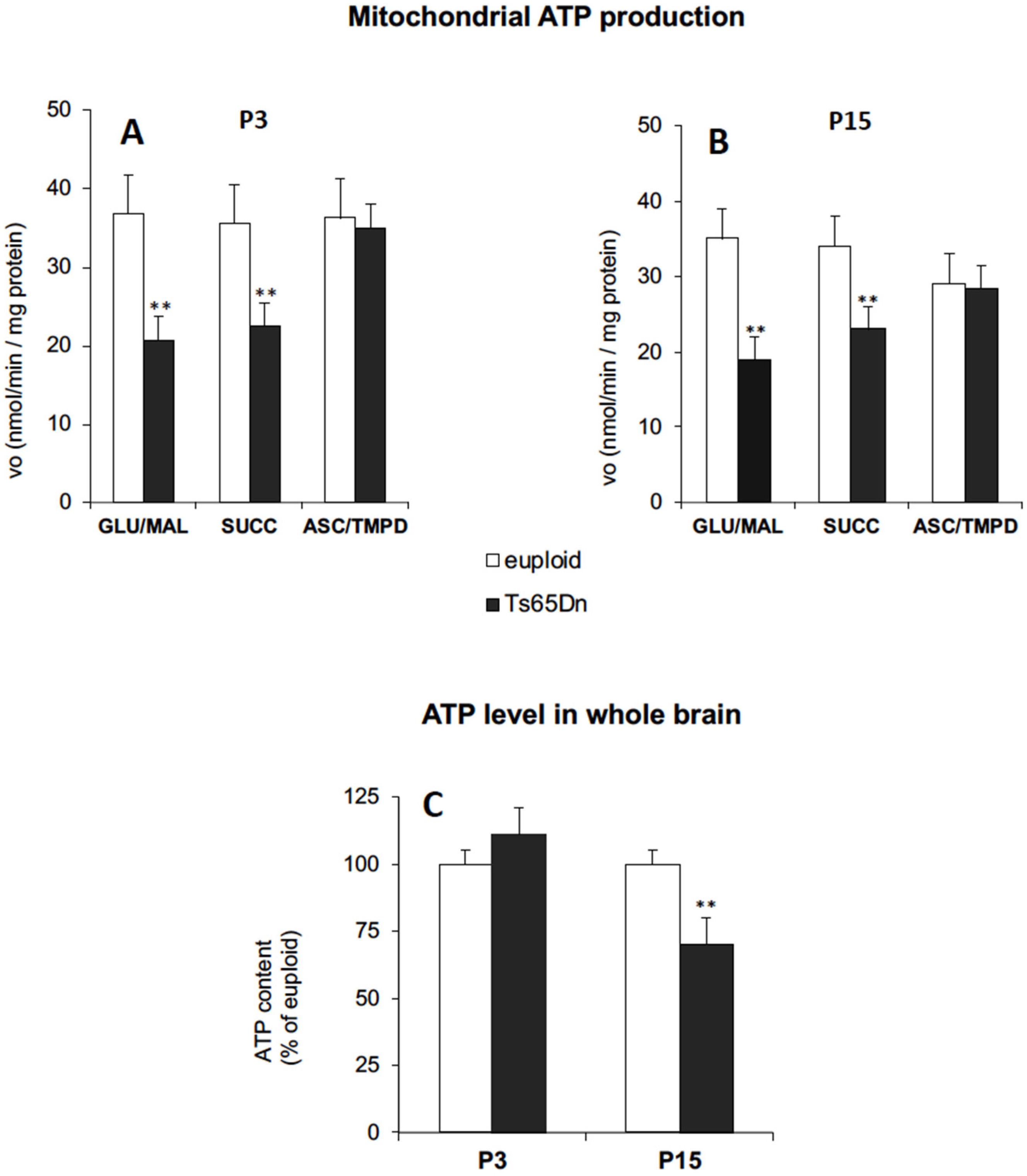
3.1. Brain Mitochondrial Bioenergetics in Ts65Dn Pups
3.2. Effect of Early Treatment with 7,8-DHF on Mitochondrial Bioenergetics
3.3. Effect of Early Treatment with 7,8-DHF on PGC-1α Levels
4. Discussion
4.1. Early Alterations in Mitochondrial Function in Ts65Dn Mice
4.2. Early Treatment with 7,8-DHF Restores Brain Mitochondrial Bioenergetics
4.3. Early Treatment with 7,8-DHF Restores PGC-1α Levels in Ts65Dn Mice
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kazemi, M.; Salehi, M.; Kheirollahi, M. Down syndrome: Current status, challenges and future perspectives. Int. J. Mol. Cell. Med. 2016, 5, 125–133. [Google Scholar] [PubMed]
- Rachidi, M.; Lopes, C. Mental retardation and human chromosome 21 gene overdosage: From functional genomics and molecular mechanisms towards preventionand treatment of the neuropathogenesis of Down syndrome. In Genomics, Proteomics, and the Nervous System; Clelland, J.D., Ed.; Springer: New York, NY, USA, 2011. [Google Scholar]
- Kitamura, T.; Saitoh, Y.; Takashima, N.; Murayama, A.; Niibori, Y.; Ageta, H.; Sekiguchi, M.; Sugiyama, H.; Inokuchi, K. Adult neurogenesis modulates the hippocampus-dependent period of associative fear memory. Cell 2009, 139, 814–827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spalding, K.L.; Bergmann, O.; Alkass, K.; Bernard, S.; Salehpour, M.; Huttner, H.B.; Boström, E.; Westerlund, I.; Vial, C.; Buchholz, B.A.; et al. Dynamics of hippocampal neurogenesis in adult humans. Cell 2013, 153, 1219–1227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stagni, F.; Giacomini, A.; Emili, M.; Guidi, S.; Bartesaghi, R. Neurogenesis impairment: An early developmental defect in Down syndrome. Free Radic. Biol. Med. 2018, 114, 15–32. [Google Scholar] [CrossRef] [PubMed]
- Bartesaghi, R.; Guidi, S.; Ciani, E. Is it possible to improve neurodevelopmental abnormalities in Down syndrome? Rev. Neurosci. 2011, 22, 419–455. [Google Scholar] [CrossRef]
- Rueda, N.; Florez, J.; Martinez-Cue, C. Mouse models of Down syndrome as a tool to unravel the causes of mental disabilities. Neural Plast. 2012, 2012, 584071. [Google Scholar] [CrossRef] [PubMed]
- Gimeno, A.; García-Giménez, J.L.; Audí, L.; Toran, N.; Andaluz, P.; Dasí, F.; Viña, J.; Pallardó, F.V. Decreased cell proliferation and higher oxidative stress in fibroblasts from Down syndrome fetuses. Preliminary study. Biochim. Biophys. Acta-Mol. Basis Dis. 2014, 1842, 116–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hibaoui, Y.; Grad, I.; Letourneau, A.; Sailani, M.R.; Dahoun, S.; Santoni, F.A.; Gimelli, S.; Guipponi, M.; Pelte, M.F.; Béna, F.; et al. Modelling and rescuing neurodevelopmental defect of Down syndrome using induced pluripotent stem cells from monozygotic twins discordant for trisomy 21. EMBO Mol. Med. 2014, 6, 259–277. [Google Scholar] [CrossRef] [PubMed]
- Kann, O.; Kovács, R. Mitochondria and neuronal activity. Am. J. Physiol. Cell. Physiol. 2007, 292, C641–C657. [Google Scholar] [CrossRef]
- Xavier, J.M.; Rodrigues, C.M.P.; Solá, S. Mitochondria: Major regulators of neural development. Neurosci. Rev. J. Bring Neurobiol. Neurol. Psychiatry 2016, 22, 346–358. [Google Scholar] [CrossRef]
- Valenti, D.; de Bari, L.; De Filippis, B.; Henrion-Caude, A.; Vacca, R.A. Mitochondrial dysfunction as a central actor in intellectual disability-related diseases: An overview of Down syndrome, autism, Fragile X and Rett syndrome. Neurosci. Biobehav. Rev. 2014, 46, 202–217. [Google Scholar] [CrossRef] [PubMed]
- Valenti, D.; de Bari, L.; Vigli, D.; Bellomo, F.; Lacivita, E.; Leopoldo, M.; Laviola, G.; Vacca, R.A.; De Filippis, B. Stimulation of the brain serotonin receptor 7 rescues mitochondrial dysfunction in female mice from two models of Rett syndrome. Neuropharmacology 2017, 121, 79–88. [Google Scholar] [CrossRef]
- D’Antoni, S.; de Bari, L.; Valenti, D.; Borro, M.; Bonaccorso, C.M.; Simmaco, M.; Vacca, R.V.; Catania, M.V. Aberrant mitochondrial bioenergetics in the cerebral cortex of the Fmr1 knockout mouse model of fragile X syndrome. Biol. Chem. 2020, 401, 497–503. [Google Scholar] [CrossRef]
- Valenti, D.; Braidy, N.; De Rasmo, D.; Signorile, A.; Rossi, L.; Atanasov, A.G.; Volpicella, M.; Henrion-Caude, A.; Nabavi, S.M.; Vacca, R.A. Mitochondria as pharmacological targets in Down syndrome. Free Radic. Biol. Med. 2018, 114, 69–83. [Google Scholar] [CrossRef]
- Vacca, R.A.; Bawari, S.; Valenti, D.; Tewari, D.; Nabavi, S.F.; Shirooie, S.; Sah, A.N.; Volpicella, M.; Braidy, N.; Nabavi, S.M. Down syndrome: Neurobiological alterations and therapeutic targets. Neurosci. Biobehav. Rev. 2019, 98, 234–255. [Google Scholar] [CrossRef]
- Bayona-Bafaluy, M.P.; Garrido-Pérez, N.; Meade, P.; Iglesias, E.; Jiménez-Salvador, I.; Montoya, J.; Martínez-Cué, C.; Ruiz-Pesini, E. Down syndrome is an oxidative phosphorylation disorder. Redox Biol. 2021, 41, 101871. [Google Scholar] [CrossRef]
- Valenti, D.; Tullo, A.; Caratozzolo, M.F.; Merafina, R.S.; Scartezzini, P.; Marra, E.; Vacca, R.A. Impairment of F1Fo-ATPase, adenine nucleotide translocator and adenylate kinase causes mitochondrial energy deficit in human skin fibroblasts with chromosome 21 trisomy. Biochem. J. 2010, 431, 299–310. [Google Scholar] [CrossRef]
- Valenti, D.; Manente, G.A.; Moro, L.; Marra, E.; Vacca, R.A. Deficit of complex I activity in human skin fibroblasts with chromosome 21 trisomy and overproduction of reactive oxygen species by mitochondria: Involvement of the cAMP/PKA signaling pathway. Biochem. J. 2011, 435, 679–688. [Google Scholar] [CrossRef] [Green Version]
- Piccoli, C.; Izzo, A.; Scrima, R.; Bonfiglio, F.; Manco, R.; Negri, R.; Quarato, G.; Cela, O.; Ripoli, M.; Prisco, M.; et al. Chronic pro-oxidative state and mitochondrial dysfunctions are more pronounced in fibroblasts from Down syndrome foeti with congenital heart defects. Hum. Mol. Genet. 2013, 22, 1218–1232. [Google Scholar] [CrossRef] [Green Version]
- Izzo, A.; Nitti, M.; Mollo, N.; Paladino, S.; Procaccini, C.; Faicchia, D.; Calì, G.; Genesio, R.; Bonfigli, F.; Cicatiello, R.; et al. Metformin restores the mitochondrial network and reverses mitochondrial dysfunction in Down syndrome cells. Hum. Mol. Genet. 2017, 26, 1056–1069. [Google Scholar] [CrossRef] [Green Version]
- Valenti, D.; de Bari, L.; de Rasmo, D.; Signorile, A.; Henrion-Caud, A.; Contestabile, A.; Vacca, R.A. The polyphenols resveratrol and epigallocatechin-3-gallate restore the severe impairment of mitochondria in hippocampal progenitor cells from a Down syndrome mouse model. Biochim. Biophys. Acta 2016, 1862, 093–1104. [Google Scholar] [CrossRef]
- Valenti, D.; Rossi, L.; Marzulli, D.; Bellomo, F.; De Rasmo, D.; Signorile, A.; Vacca, R.A. Inhibition of Drp1-mediated mitochondrial fission improves mitochondrial dynamics and bioenergetics stimulating neurogenesis in hippocampal progenitor cells from a Down syndrome mouse model. Biochim. Biophys. Acta 2017, 1863, 3117–3127. [Google Scholar] [CrossRef]
- Scala, I.; Valenti, D.; Scotto D’Aniello, V.; Marino, M.; Riccio, M.P.; Bravaccio, C.; Vacca, R.A.; Strisciuglio, P. Epigallocatechin-3-Gallate Plus Omega-3 Restores the Mitochondrial Complex I and FoF1-ATP Synthase Activities in PBMCs of Young Children with Down Syndrome: A Pilot Study of Safety and Efficacy. Antioxidants 2021, 10, 469. [Google Scholar] [CrossRef]
- Spencer, J.P. Flavonoids: Modulators of brain function? Br. J. Nutr. 2008, 99 (Suppl. 1), ES60–ES77. [Google Scholar] [CrossRef] [Green Version]
- Williams, R.J.; Spencer, J.P. Flavonoids, cognition, and dementia: Actions, mechanisms and potential therapeutic utility for Alzheimer disease. Free Radic. Biol. Med. 2012, 52, 35–45. [Google Scholar] [CrossRef]
- Atanasov, A.G.; Zotchev, S.B.; Verena, M.N.; Dirsch, V.M. The International Natural Product Sciences Taskforce & Supuran CT. Natural products in drug discovery: Advances and opportunities. Nat. Rev. Drug Discov. 2021, 20, 200–216. [Google Scholar] [PubMed]
- Vacca, R.A.; Valenti, D.; Caccamese, S.; Daglia, M.; Braidy, N.; Nabavi, S.M. Plant polyphenols as natural drugs for the management of Down syndrome and related disorders. Neurosci. Biobehav. Rev. 2016, 71, 865–877. [Google Scholar] [CrossRef]
- Emili, M.; Guidi, S.; Uguagliati, B.; Giacomini, A.; Bartesaghi, R.; Stagni, F. Treatment with the flavonoid 7,8-Dihydroxyflavone: A promising strategy for a constellation of body and brain disorders. Crit. Rev. Food Sci. Nutr. 2020, 11, 1–38. [Google Scholar] [CrossRef]
- Guedj, F.; Siegel, A.E.; Pennings, J.L.A.; Alsebaa, F.; Massingham, L.J.; Tantravahi, U.; Bianchi, D.W. Apigenin as a Candidate Prenatal Treatment for Trisomy 21: Effects in Human Amniocytes and the Ts1Cje Mouse Model. Am. J. Hum. Genet. 2020, 107, 911–931. [Google Scholar] [CrossRef]
- Stagni, F.; Giacomini, A.; Guidi, S.; Ciani, E.; Bartesaghi, R. Timing of therapies for Down syndrome: The sooner, the better. Front. Behav. Neurosci. 2015, 9, 265. [Google Scholar] [CrossRef] [Green Version]
- Stagni, F.; Giacomini, A.; Emili, M.; Trazzi, S.; Guidi, S.; Sassi, M.; Ciani, E.; Rimondini, R.; Bartesaghi, R. Short- and long-term effects of neonatal pharmacotherapy with epigallocatechin-3-gallate on hippocampal development in the Ts65Dn mouse model of Down syndrome. Neuroscience 2016, 333, 277–301. [Google Scholar] [CrossRef] [PubMed]
- Stagni, F.; Giacomini, A.; Guidi, S.; Emili, M.; Uguagliati, B.; Salvalai, M.E.; Bortolotto, V.; Grilli, M.; Rimondini, R.; Bartesaghi, R. A flavonoid agonist of the TrkB receptor for BDNF improves hippocampal neurogenesis and hippocampus-dependent memory in the Ts65Dn mouse model of DS. Exp. Neurol. 2017, 298 Pt A, 79–96. [Google Scholar] [CrossRef]
- Stagni, F.; Giacomini, A.; Emili, M.; Guidi, S.; Ciani, E.; Bartesaghi, R. Epigallocatechin gallate: A useful therapy for cognitive disability in Down syndrome? Neurogenesis 2017, 4, e1270383. [Google Scholar] [CrossRef] [PubMed]
- Arrázola, M.S.; Andraini, T.; Szelechowski, M.; Mouledous, L.; Arnauné-Pelloquin, L.; Davezac, N.; Belenguer, P.; Rampon, C.; Miquel, M.C. Mitochondria in Developmental and Adult Neurogenesis. Neurotox. Res. 2019, 36, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Valenti, D.; de Bari, L.; De Filippis, B.; Ricceri, L.; Vacca, R.A. Preservation of mitochondrial functional integrity in mitochondria isolated from small cryopreserved mouse brain areas. Anal. Biochem. 2014, 444, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Trazzi, S.; Mitrugno, V.M.; Valli, E.; Fuchs, C.; Rizzi, S.; Guidi, S.; Perini, G.; Bartesaghi, R.; Ciani, E. APP-dependent up-regulation of Ptch1 underlies proliferation impairment of neural precursors in Down syndrome. Hum. Mol. Genet. 2011, 20, 1560–1573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanzillotta, C.; Tramutola, A.; Di Giacomo, G.; Marini, F.; Butterfield, D.A.; Di Domenico, F.; Perluigi, M.; Barone, E. Insulin resistance, oxidative stress and mitochondrial defects in Ts65dn mice brain: A harmful synergistic path in Down syndrome. Free Radic. Biol. Med. 2021, 165, 152–170. [Google Scholar] [CrossRef]
- Beckervordersandforth, R. Mitochondrial metabolism-mediated regulation of adult neurogenesis. Brain Plast. 2017, 3, 73–87. [Google Scholar] [CrossRef] [Green Version]
- Aylward, E.H.; Habbak, R.; Warren, A.C.; Pulsifer, M.B.; Barta, P.E.; Jerram, M.; Pearlson, G.D. Cerebellar volume in adults with Down syndrome. Arch. Neurol. 1997, 54, 209–212. [Google Scholar] [CrossRef]
- Guidi, S.; Ciani, E.; Bonasoni, P.; Santini, D.; Bartesaghi, R. Widespread proliferation impairment and hypocellularity in the cerebellum of fetuses with Down syndrome. Brain Pathol. 2011, 21, 361–373. [Google Scholar] [CrossRef]
- Lambert, A.J.; Brand, M.D. Reactive oxygen species production by mitochondria. Methods Mol. Biol. 2009, 554, 165–181. [Google Scholar]
- Hroudová, J.; Fišar, Z. Control mechanisms in mitochondrial oxidative phosphorylation. Neural Regen. Res. 2013, 8, 363–375. [Google Scholar]
- Austin, S.; St-Pierre, J. PGC1α and mitochondrial metabolism—Emerging concepts and relevance in ageing and neurodegenerative disorders. J. Cell. Sci. 2012, 125 Pt 21, 4963–4971. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Marcos, P.J.; Auwerx, J. Regulation of PGC-1α, a nodal regulator of mitochondrial biogenesis. Am. J. Clin. Nutr. 2011, 93, 884S–890S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izzo, A.; Mollo, N.; Nitti, M.; Paladino, S.; Cali, G.; Genesio, R.; Bonfiglio, F.; Cicatiello, R.; Barbato, M.; Sarnataro, V.; et al. Mitochondrial dysfunction in Down syndrome: Molecular mechanisms and therapeutic targets. Mol. Med. 2018, 24, 2. [Google Scholar] [CrossRef] [Green Version]
- Bambrick, L.L.; Fiskun, G. Mitochondrial dysfunction in mouse trisomy 16 brain. Brain Res. 2008, 1188, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Reeves, R.H.; Irving, N.G.; Moran, T.H.; Wohn, A.; Kitt, C.; Sisodia, S.S.; Schmidt, C.; Bronson, R.T.; Davisson, M.T. A mouse model for Down syndrome exhibits learning and behaviour deficits. Nat Genet. 1995, 11, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Valenti, D.; De Rasmo, D.; Signorile, A.; Rossi, L.; de Bari, L.; Scala, I.; Granese, B.; Papa, S.; Vacca, R.A. Epigallocatechin-3-gallate prevents oxidative phosphorylation deficit and promotes mitochondrial biogenesis in human cells from subjects with Down’s syndrome. Biochim. Biophys. Acta 2013, 1832, 542–552. [Google Scholar] [CrossRef] [Green Version]
- Coskun, P.E.; Busciglio, J. Oxidative stress and mitochondrial dysfunction in Down’s syndrome: Relevance to aging and dementia. Curr. Gerontol. Geriatr. Res. 2012, 2012, 383170. [Google Scholar] [CrossRef]
- Schuchmann, S.; Heinemann, U. Increased mitochondrial superoxide generation in neurons from trisomy 16 mice: A model of Down’s syndrome. Free Radic. Biol. Med. 2000, 28, 235–250. [Google Scholar] [CrossRef]
- Capone, G.; Kim, P.; Jovanovich, S.; Payne, L.; Freund, L.; Welch, K.; Miller, E.; Trush, M. Evidence for increased mitochondrial superoxide production in Down syndrome. Life Sci. 2002, 70, 2885–2895. [Google Scholar] [CrossRef]
- Wood, J.; Tse, M.C.L.; Yang, X.; Brobst, D.; Liu, Z.; Pang, B.P.S.; Chan, W.S.; Zaw, A.M.; Chow, B.K.C.; Ye, K.; et al. BDNF mimetic alleviates body weight gain in obese mice by enhancing mitochondrial biogenesis in skeletal muscle. Metabolism 2018, 87, 113–122. [Google Scholar] [CrossRef]
- Zhao, J.; Du, J.; Pan, Y.; Chen, T.; Zhao, L.; Zhu, Y.; Chen, Y.; Zheng, Y.; Liu, Y.; Sun, L.; et al. Activation of cardiac TrkB receptor by its small molecular agonist 7,8-dihydroxyflavone inhibits doxorubicin-induced cardiotoxicity via enhancing mitochondrial oxidative phosphorylation. Free Radic. Biol. Med. 2016, 130, 557–567. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, S.; Qun Shao, Q.; Li, P.; Sun, Y.; Luo, L.; Yan, X.; Fan, Z.; Hu, J.; Zhaod, J.; et al. Brain-derived neurotrophic factor mimetic, 7,8-dihydroxyflavone, protects against myocardial ischemia by rebalancing optic atrophy 1 processing. Free Radic. Biol. Med. 2019, 145, 187–197. [Google Scholar] [CrossRef]
- Agrawal, R.; Noble, E.; Tyagi, E.; Zhuang, Y.; Ying, Z.; Gomez-Pinilla, F. Flavonoid derivative 7,8-DHF attenuates TBI pathology via TrkB activation. Biochim. Biophys. Acta 2015, 1852, 862–872. [Google Scholar] [CrossRef] [Green Version]
- Bryant, J.; Andhavarapu, S.; Bever, C.; Guda, P.; Katuri, A.; Gupta, U.; Arvas, M.; Asemu, G.; Heredia, A.; Gerzanich, V.; et al. 7,8-Dihydroxyflavone improves neuropathological changes in the brain of Tg26 mice, a model for HIV-associated neurocognitive disorder. Sci. Rep. 2021, 11, 18519. [Google Scholar] [CrossRef]
- Raefsky, S.M.; Mattson, M.P. Adaptive responses of neuronal mitochondria to bioenergetic challenges: Roles in neuroplasticity and disease resistance. Free Radic. Biol. Med. 2017, 102, 203–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stagni, F.; Uguagliati, B.; Emili, M.; Giacomini, A.; Bartesaghi, R.; Guidi, S. The flavonoid 7,8-DHF fosters prenatal brain proliferation potency in a mouse model of Down syndrome. Sci. Rep. 2021, 11, 6300. [Google Scholar] [CrossRef]
- Bimonte-Nelson, H.A.; Hunter, C.L.; Nelson, M.E.; Granholm, A.C. Frontal cortex BDNF levels correlate with working memory in an animal model of Down syndrome. Behav. Brain Res. 2003, 139, 47–57. [Google Scholar] [CrossRef]
- Giacomini, A.; Stagni, F.; Emili, M.; Uguagliati, B.; Rimondini, R.; Bartesaghi, R.; Guidi, S. Timing of Treatment with the Flavonoid 7,8-DHF Critically Impacts on Its Effects on Learning and Memory in the Ts65Dn Mouse. Antioxidants 2019, 8, 163. [Google Scholar] [CrossRef] [Green Version]
- Cheng, A.; Wan, R.; Yang, J.L.; Kamimura, N.; Son, T.G.; Ouyang, X.; Luo, Y.; Okun, E.; Mattson, M.P. Involvement of PGC-1α in the formation and maintenance of neuronal dendritic spines. Nat. Commun. 2012, 3, 1250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahuja, P.; Ng, C.F.; Pang, B.P.S.; Chan, W.S.; Tse, M.C.L.; Bi, X.; Kwan, H.R.; Brobst, D.; Herlea-Pana, O.; Yang, X.; et al. Muscle-generated BDNF (brain derived neurotrophic factor) maintains mitochondrial quality control in female mice. Autophagy 2021, 25, 1–18. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Valenti, D.; Stagni, F.; Emili, M.; Guidi, S.; Bartesaghi, R.; Vacca, R.A. Impaired Brain Mitochondrial Bioenergetics in the Ts65Dn Mouse Model of Down Syndrome Is Restored by Neonatal Treatment with the Polyphenol 7,8-Dihydroxyflavone. Antioxidants 2022, 11, 62. https://doi.org/10.3390/antiox11010062
Valenti D, Stagni F, Emili M, Guidi S, Bartesaghi R, Vacca RA. Impaired Brain Mitochondrial Bioenergetics in the Ts65Dn Mouse Model of Down Syndrome Is Restored by Neonatal Treatment with the Polyphenol 7,8-Dihydroxyflavone. Antioxidants. 2022; 11(1):62. https://doi.org/10.3390/antiox11010062
Chicago/Turabian StyleValenti, Daniela, Fiorenza Stagni, Marco Emili, Sandra Guidi, Renata Bartesaghi, and Rosa Anna Vacca. 2022. "Impaired Brain Mitochondrial Bioenergetics in the Ts65Dn Mouse Model of Down Syndrome Is Restored by Neonatal Treatment with the Polyphenol 7,8-Dihydroxyflavone" Antioxidants 11, no. 1: 62. https://doi.org/10.3390/antiox11010062
APA StyleValenti, D., Stagni, F., Emili, M., Guidi, S., Bartesaghi, R., & Vacca, R. A. (2022). Impaired Brain Mitochondrial Bioenergetics in the Ts65Dn Mouse Model of Down Syndrome Is Restored by Neonatal Treatment with the Polyphenol 7,8-Dihydroxyflavone. Antioxidants, 11(1), 62. https://doi.org/10.3390/antiox11010062