
Parkinson’s Disease and the Metal–Microbiome–Gut–Brain Axis: A Systems Toxicology Approach

 , and
, and
Abstract
:1. Introduction
1.1. What Is Parkinson’s Disease?
1.2. Gut–Brain Axis: Braak’s Hypothesis
1.3. Neurotoxicity and Parkinson’s Disease
1.3.1. Manganese (Mn)
1.3.2. (Methyl) Mercury (MeHg; Hg)
1.3.3. Iron (Fe)
2. Microbiome–Gut–Brain Communication in Health and Disease
2.1. Neuroendocrine and Neuroactive Metabolites Crosstalk
2.2. Immunomodulation and Dysregulation
3. Heavy Metals and the Gut–Brain Axis
3.1. Metal Transport, Movement, and Molecular Mimicry
3.2. Oxidative Stress and Inflammation in the Gut and Brain
3.3. Microbes and Heavy Metals in Gut–Brain Bioremediation
4. Systems Toxicology
4.1. Towards Systems Toxicology
4.2. Whole-Body Models Integration with Heavy Metals Bioremediation
5. Conclusions: Fermented Functional Probiotics and Oxidative Stress
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tysnes, O.B.; Storstein, A. Epidemiology of Parkinson’s disease. J. Neural Transm. 2017, 124, 901–905. [Google Scholar] [CrossRef]
- Qamar, M.A.; Sauerbier, A.; Politis, M.; Carr, H.; Loehrer, P.; Chaudhuri, K.R. Presynaptic dopaminergic terminal imaging and non-motor symptoms assessment of Parkinson’s disease: Evidence for dopaminergic basis? NPJ Parkinsons Dis. 2017, 3, 5. [Google Scholar] [CrossRef]
- Pfeiffer, R.F. Non-motor symptoms in Parkinson’s disease. Park. Relat. Disord. 2015, 22, S119–S122. [Google Scholar] [CrossRef]
- Brudek, T. Inflammatory Bowel Diseases and Parkinson’s Disease. J. Parkinsons Dis. 2019, 9, S331–S344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulak, A.; Bonaz, B. Brain-gut-microbiota axis in Parkinson’s disease. World J. Gastroenterol. 2015, 21, 10609. [Google Scholar] [CrossRef] [PubMed]
- Bellou, V.; Belbasis, L.; Tzoulaki, I.; Evangelou, E.; Ioannidis, J.P.A. Environmental risk factors and Parkinson’s disease: An umbrella review of meta-analyses. Parkinsonism Relat. Disord. 2015, 23, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Ritz, B. The Search for Environmental Causes of Parkinson’s Disease: Moving Forward. J. Parkinson Dis. 2018, 8, S9–S17. [Google Scholar] [CrossRef] [Green Version]
- Kouli, A.; Torsney, K.M.; Kuan, W.-L. Parkinson’s Disease: Etiology, Neuropathology, and Pathogenesis. In Parkinson’s Disease: Pathogenesis and Clinical Aspects; Exon Publications: Brisbane, Australia, 2018; ISBN 9780994438164. [Google Scholar]
- Sampson, T.R.; Debelius, J.W.; Thron, T.; Janssen, S.; Shastri, G.G.; Ilhan, Z.E.; Challis, C.; Schretter, C.E.; Rocha, S.; Gradinaru, V.; et al. Gut Microbiota Regulate Motor Deficits and Neuroinflammation in a Model of Parkinson’s Disease. Cell 2016, 167, 1469–1480.e12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quigley, E.M.M. Microbiota-Brain-Gut Axis and Neurodegenerative Diseases. Curr. Neurol. Neurosci. Rep. 2017, 17, 94. [Google Scholar] [CrossRef]
- Calvani, R.; Picca, A.; Lo Monaco, M.R.; Landi, F.; Bernabei, R.; Marzetti, E. Of Microbes and Minds: A Narrative Review on the Second Brain Aging. Front. Med. 2018, 5, 53. [Google Scholar] [CrossRef] [PubMed]
- Cryan, J.F.; O’Riordan, K.J.; Cowan, C.S.M.; Sandhu, K.V.; Bastiaanssen, T.F.S.; Boehme, M.; Codagnone, M.G.; Cussotto, S.; Fulling, C.; Golubeva, A.V.; et al. The Microbiota-Gut-Brain Axis. Physiol. Rev. 2019, 99, 1877–2013. [Google Scholar] [CrossRef] [PubMed]
- Suganya, K.; Koo, B.-S. Gut–Brain Axis: Role of Gut Microbiota on Neurological Disorders and How Probiotics/Prebiotics Beneficially Modulate Microbial and Immune Pathways to Improve Brain Functions. Int. J. Mol. Sci. 2020, 21, 7551. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Rüb, U.; Gai, W.P.; Del Tredici, K. Idiopathic Parkinson’s disease: Possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J. Neural Transm. 2003, 110, 517–536. [Google Scholar] [CrossRef] [PubMed]
- Hawkes, C.H.; Del Tredici, K.; Braak, H. Parkinson’s disease: A dual-hit hypothesis. Neuropathol. Appl. Neurobiol. 2007, 33, 599–614. [Google Scholar] [CrossRef] [PubMed]
- Ghaisas, S.; Maher, J.; Kanthasamy, A. Gut microbiome in health and disease: Linking the microbiome–gut–brain axis and environmental factors in the pathogenesis of systemic and neurodegenerative diseases. Pharmacol. Ther. 2015, 158, 52–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Visanji, N.P.; Brooks, P.L.; Hazrati, L.-N.; Lang, A.E. The prion hypothesis in Parkinson’s disease: Braak to the future. Acta Neuropathol. Commun. 2013, 1, 2. [Google Scholar] [CrossRef] [Green Version]
- Oertel, W.H. Recent advances in treating Parkinson’s disease. F1000Research 2017, 6, 260. [Google Scholar] [CrossRef] [Green Version]
- Chung, S.J.; Kim, J.; Lee, H.J.; Ryu, H.-S.; Kim, K.; Lee, J.H.; Jung, K.W.; Kim, M.J.; Kim, Y.J.; Yun, S.-C.; et al. Alpha-synuclein in gastric and colonic mucosa in Parkinson’s disease: Limited role as a biomarker. Mov. Disord. 2015, 31, 241–249. [Google Scholar] [CrossRef]
- Lebouvier, T.; Chaumette, T.; Paillusson, S.; Duyckaerts, C.; Bruley Des Varannes, S.; Neunlist, M.; Derkinderen, P. The second brain and Parkinson’s disease. Eur. J. Neurosci. 2009, 30, 735–741. [Google Scholar] [CrossRef]
- Lin, J.-C.; Lin, C.-S.; Hsu, C.-W.; Lin, C.-L.; Kao, C.-H. Association between Parkinson’s Disease and Inflammatory Bowel Disease: A nationwide Taiwanese retrospective cohort study. Inflamm. Bowel Dis. 2016, 22, 1049–1055. [Google Scholar] [CrossRef] [Green Version]
- Peter, I.; Dubinsky, M.; Bressman, S.; Park, A.; Lu, C.; Chen, N.; Wang, A. Anti–Tumor Necrosis Factor Therapy and Incidence of Parkinson Disease Among Patients With Inflammatory Bowel Disease. JAMA Neurol. 2018, 75, 939–946. [Google Scholar] [CrossRef]
- Villumsen, M.; Aznar, S.; Pakkenberg, B.; Jess, T.; Brudek, T. Inflammatory bowel disease increases the risk of Parkinson’s disease: A Danish nationwide cohort study 1977–2014. Gut 2018, 68, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Weimers, P.; Halfvarson, J.; Sachs, M.C.; Saunders-Pullman, R.; Ludvigsson, J.F.; Peter, I.; Burisch, J.; Olén, O. Inflammatory Bowel Disease and Parkinson’s Disease: A Nationwide Swedish Cohort Study. Inflamm. Bowel Dis. 2018, 25, 111–123. [Google Scholar] [CrossRef]
- Blaecher, C.; Smet, A.; Flahou, B.; Pasmans, F.; Ducatelle, R.; Taylor, D.; Weller, C.; Bjarnason, I.; Charlett, A.; Lawson, A.J.; et al. Significantly higher frequency ofHelicobacter suisin patients with idiopathic parkinsonism than in control patients. Aliment. Pharmacol. Ther. 2013, 38, 1347–1353. [Google Scholar] [CrossRef] [Green Version]
- Hashim, H.; Azmin, S.; Razlan, H.; Yahya, N.W.; Tan, H.J.; Manaf, M.R.A.; Ibrahim, N.M. Eradication of Helicobacter pylori Infection Improves Levodopa Action, Clinical Symptoms and Quality of Life in Patients with Parkinson’s Disease. PLoS ONE 2014, 9, e112330. [Google Scholar] [CrossRef] [Green Version]
- Heintz-Buschart, A.; Pandey, U.; Wicke, T.; Sixel-Döring, F.; Janzen, A.; Sittig-Wiegand, E.; Trenkwalder, C.; Oertel, W.H.; Mollenhauer, B.; Wilmes, P. The nasal and gut microbiome in Parkinson’s disease and idiopathic rapid eye movement sleep behavior disorder. Mov. Disord. 2017, 33, 88–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vandeputte, D.; Falony, G.; Vieira-Silva, S.; Tito, R.Y.; Joossens, M.; Raes, J. Stool consistency is strongly associated with gut microbiota richness and composition, enterotypes and bacterial growth rates. Gut 2016, 65, 57–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gobert, A.P.; Sagrestani, G.; Delmas, E.; Wilson, K.T.; Verriere, T.G.; Dapoigny, M.; Del’Homme, C.; Bernalier-Donadille, A. The human intestinal microbiota of constipated-predominant irritable bowel syndrome patients exhibits anti-inflammatory properties. Sci. Rep. 2016, 6, 39399. [Google Scholar] [CrossRef]
- Reunanen, J.; Kainulainen, V.; Huuskonen, L.; Ottman, N.; Belzer, C.; Huhtinen, H.; de Vos, W.M.; Satokari, R. Akkermansia muciniphila Adheres to Enterocytes and Strengthens the Integrity of the Epithelial Cell Layer. Appl. Environ. Microbiol. 2015, 81, 3655–3662. [Google Scholar] [CrossRef] [Green Version]
- Ottman, N.; Reunanen, J.; Meijerink, M.; Pietilä, T.E.; Kainulainen, V.; Klievink, J.; Huuskonen, L.; Aalvink, S.; Skurnik, M.; Boeren, S.; et al. Pili-like proteins of Akkermansia muciniphila modulate host immune responses and gut barrier function. PLoS ONE 2017, 12, e0173004. [Google Scholar] [CrossRef] [PubMed]
- Rolli-Derkinderen, M.; Leclair-Visonneau, L.; Bourreille, A.; Coron, E.; Neunlist, M.; Derkinderen, P. Is Parkinson’s disease a chronic low-grade inflammatory bowel disease? J. Neurol. 2020, 267, 2207–2213. [Google Scholar] [CrossRef]
- Feng, P.; Ye, Z.; Kakade, A.; Virk, A.K.; Li, X.; Liu, P. A Review on Gut Remediation of Selected Environmental Contaminants: Possible Roles of Probiotics and Gut Microbiota. Nutrients 2019, 11, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mrvčić, J.; Stanzer, D.; Šolić, E.; Stehlik-Tomas, V. Interaction of lactic acid bacteria with metal ions: Opportunities for improving food safety and quality. World J. Microbiol. Biotechnol. 2012, 28, 2771–2782. [Google Scholar] [CrossRef]
- Claus, S.P.; Guillou, H.; Ellero-Simatos, S. The gut microbiota: A major player in the toxicity of environmental pollutants? NPJ Biofilms Microbiomes 2016, 2, 16003. [Google Scholar] [CrossRef]
- Daisley, B.A.; Monachese, M.; Trinder, M.; Bisanz, J.E.; Chmiel, J.A.; Burton, J.P.; Reid, G. Immobilization of cadmium and lead by Lactobacillus rhamnosus GR-1 mitigates apical-to-basolateral heavy metal translocation in a Caco-2 model of the intestinal epithelium. Gut Microbes 2019, 10, 321–333. [Google Scholar] [CrossRef] [Green Version]
- Monroy-Torres, R.; Antonio Hernández-Luna, M.; Sofía Ramírez-Gómez, X.; López-Briones, S. Role of the Microbiome as the First Metal Detoxification Mechanism. In Prebiotics and Probiotics—Potential Benefits in Nutrition and Health; IntechOpen: London, UK, 2020; Available online: https://www.intechopen.com/chapters/69664 (accessed on 3 November 2021). [CrossRef] [Green Version]
- Breton, J.; Daniel, C.; Vignal, C.; Body-Malapel, M.; Garat, A.; Plé, C.; Foligne, B. Does oral exposure to cadmium and lead mediate susceptibility to colitis? The dark-and-bright sides of heavy metals in gut ecology. Sci. Rep. 2016, 6, 19200. [Google Scholar] [CrossRef] [Green Version]
- Andreini, C.; Bertini, I.; Cavallaro, G.; Holliday, G.L.; Thornton, J.M. Metal ions in biological catalysis: From enzyme databases to general principles. JBIC J. Biol. Inorg. Chem. 2008, 13, 1205–1218. [Google Scholar] [CrossRef] [PubMed]
- Genoud, S.; Roberts, B.R.; Gunn, A.P.; Halliday, G.M.; Lewis, S.J.G.; Ball, H.J.; Hare, D.J.; Double, K.L. Subcellular compartmentalisation of copper, iron, manganese, and zinc in the Parkinson’s disease brain. Metallomics 2017, 9, 1447–1455. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharyya, A.; Chattopadhyay, R.; Mitra, S.; Crowe, S.E. Oxidative Stress: An Essential Factor in the Pathogenesis of Gastrointestinal Mucosal Diseases. Physiol. Rev. 2014, 94, 329–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Möller, H.E.; Bossoni, L.; Connor, J.R.; Crichton, R.R.; Does, M.D.; Ward, R.J.; Zecca, L.; Zucca, F.A.; Ronen, I. Iron, Myelin, and the Brain: Neuroimaging Meets Neurobiology. Trends Neurosci. 2019, 42, 384–401. [Google Scholar] [CrossRef]
- Barnham, K.J.; Bush, A.I. Metals in Alzheimer’s and Parkinson’s Diseases. Curr. Opin. Chem. Biol. 2008, 12, 222–228. [Google Scholar] [CrossRef]
- Hegde, M.L.; Shanmugavelu, P.; Vengamma, B.; Rao, T.S.S.; Menon, R.B.; Rao, R.V.; Rao, K.S.J. Serum trace element levels and the complexity of inter-element relations in patients with Parkinson’s disease. J. Trace Elem. Med. Biol. 2004, 18, 163–171. [Google Scholar] [CrossRef]
- Abramov, A.Y.; Potapova, E.V.; Dremin, V.V.; Dunaev, A.V. Interaction of Oxidative Stress and Misfolded Proteins in the Mechanism of Neurodegeneration. Life 2020, 10, 101. [Google Scholar] [CrossRef]
- Carmona, A.; Roudeau, S.; Ortega, R. Molecular Mechanisms of Environmental Metal Neurotoxicity: A Focus on the Interactions of Metals with Synapse Structure and Function. Toxics 2021, 9, 198. [Google Scholar] [CrossRef] [PubMed]
- Harischandra, D.S.; Ghaisas, S.; Zenitsky, G.; Jin, H.; Kanthasamy, A.; Anantharam, V.; Kanthasamy, A.G. Manganese-Induced Neurotoxicity: New Insights into the Triad of Protein Misfolding, Mitochondrial Impairment, and Neuroinflammation. Front. Neurosci. 2019, 13, 654. [Google Scholar] [CrossRef] [Green Version]
- Fitsanakis, V.A.; Piccola, G.; Dos Santos, A.P.M.; Aschner, J.L.; Aschner, M. Putative proteins involved in manganese transport across the blood-brain barrier. Hum. Exp. Toxicol. 2007, 26, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Bornhorst, J.; Aschner, M. Manganese metabolism in humans. Front. Biosci. 2018, 23, 1655–1679. [Google Scholar] [CrossRef] [Green Version]
- Bouabid, S.; Delaville, C.; De Deurwaerdère, P.; Lakhdar-Ghazal, N.; Benazzouz, A. Manganese-Induced Atypical Parkinsonism Is Associated with Altered Basal Ganglia Activity and Changes in Tissue Levels of Monoamines in the Rat. PLoS ONE 2014, 9, e98952. [Google Scholar] [CrossRef] [Green Version]
- O’Neal, S.L.; Zheng, W. Manganese Toxicity upon Overexposure: A Decade in Review. Curr. Environ. Health Rep. 2015, 2, 315–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunter, T.E.; Gerstner, B.; Gunter, K.K.; Malecki, J.; Gelein, R.; Valentine, W.M.; Aschner, M.; Yule, D.I. Manganese transport via the transferrin mechanism. Neurotoxicology 2013, 34, 118–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Finley, E.J.; Chakraborty, S.; Fretham, S.J.B.; Aschner, M. Cellular transport and homeostasis of essential and nonessential metals. Metallomics 2012, 4, 593–605. [Google Scholar] [CrossRef] [Green Version]
- Ingrassia, R.; Garavaglia, B.; Memo, M. DMT1 Expression and Iron Levels at the Crossroads between Aging and Neurodegeneration. Front. Neurosci. 2019, 13, 575. [Google Scholar] [CrossRef] [Green Version]
- Tai, Y.K.; Chew, K.C.M.; Tan, B.W.Q.; Lim, K.-L.; Soong, T.W. Iron mitigates DMT1-mediated manganese cytotoxicity via the ASK1-JNK signaling axis: Implications of iron supplementation for manganese toxicity. Sci. Rep. 2016, 6, 21113. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Sun, L.; Cai, T.; Zhang, Y.; Lv, S.; Wang, Y.; Ye, L. α-Synuclein overexpression during manganese-induced apoptosis in SH-SY5Y neuroblastoma cells. Brain Res. Bull. 2010, 81, 428–433. [Google Scholar] [CrossRef] [PubMed]
- Prabhakaran, K.; Chapman, G.D.; Gunasekar, P.G. α-Synuclein overexpression enhances manganese-induced neurotoxicity through the NF-κB-mediated pathway. Toxicol. Mech. Methods 2011, 21, 435–443. [Google Scholar] [CrossRef] [PubMed]
- Moons, R.; Konijnenberg, A.; Mensch, C.; Van Elzen, R.; Johannessen, C.; Maudsley, S.; Lambeir, A.-M.; Sobott, F. Metal ions shape α-synuclein. Sci. Rep. 2020, 10, 16293. [Google Scholar] [CrossRef] [PubMed]
- Cai, T.; Yao, T.; Zheng, G.; Chen, Y.; Du, K.; Cao, Y.; Shen, X.; Chen, J.; Luo, W. Manganese induces the overexpression of α-synuclein in PC12 cells via ERK activation. Brain Res. 2010, 1359, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Harischandra, D.S.; Jin, H.; Anantharam, V.; Kanthasamy, A.; Kanthasamy, A.G. α-Synuclein Protects Against Manganese Neurotoxic Insult During the Early Stages of Exposure in a Dopaminergic Cell Model of Parkinson’s Disease. Toxicol. Sci. 2014, 143, 454–468. [Google Scholar] [CrossRef] [Green Version]
- Guilarte, T.R.; Burton, N.C.; McGlothan, J.L.; Verina, T.; Zhou, Y.; Alexander, M.; Pham, L.; Griswold, M.; Wong, D.F.; Syversen, T.; et al. Impairment of nigrostriatal dopamine neurotransmission by manganese is mediated by pre-synaptic mechanism(s): Implications to manganese-induced parkinsonism. J. Neurochem. 2008, 107, 1236–1247. [Google Scholar] [CrossRef] [Green Version]
- Guilarte, T.R.; Chen, M.-K.; McGlothan, J.L.; Verina, T.; Wong, D.F.; Zhou, Y.; Alexander, M.; Rohde, C.A.; Syversen, T.; DeCamp, E. Nigrostriatal dopamine system dysfunction and subtle motor deficits in manganese-exposed non-human primates. Exp. Neurol. 2006, 202, 381–390. [Google Scholar] [CrossRef]
- Ratner, M.H.; Fitzgerald, E. Understanding of the role of manganese in parkinsonism and Parkinson disease. Neurology 2016, 88, 338–339. [Google Scholar] [CrossRef]
- Mallet, N.; Delgado, L.; Chazalon, M.; Miguelez, C.; Baufreton, J. Cellular and Synaptic Dysfunctions in Parkinson’s Disease: Stepping out of the Striatum. Cells 2019, 8, 1005. [Google Scholar] [CrossRef] [Green Version]
- Aschner, M.; Erikson, K.M.; Hernández, E.H.; Tjalkens, R. Manganese and its role in Parkinson’s disease: From transport to neuropathology. Neuromol. Med. 2009, 11, 252–266. [Google Scholar] [CrossRef] [PubMed]
- Roth, J.A.; Li, Z.; Sridhar, S.; Khoshbouei, H. The effect of manganese on dopamine toxicity and dopamine transporter (DAT) in control and DAT transfected HEK cells. Neurotoxicology 2013, 35, 121–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torrey, E.F.; Barci, B.M.; Webster, M.J.; Bartko, J.J.; Meador-Woodruff, J.H.; Knable, M.B. Neurochemical markers for schizophrenia, bipolar disorder, and major depression in postmortem brains. Biol. Psychiatry 2005, 57, 252–260. [Google Scholar] [CrossRef]
- Fitsanakis, V.A.; Au, C.; Erikson, K.M.; Aschner, M. The effects of manganese on glutamate, dopamine and γ-aminobutyric acid regulation. Neurochem. Int. 2006, 48, 426–433. [Google Scholar] [CrossRef]
- Gandhi, D.; Sivanesan, S.; Kannan, K. Manganese-Induced Neurotoxicity and Alterations in Gene Expression in Human Neuroblastoma SH-SY5Y Cells. Biol. Trace Elem. Res. 2017, 183, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Chen, L.; Chen, T.; Zhang, Y.; Ma, J.; Ji, H.; Jia, J.; Huang, Y.; Guo, C.; Xiao, Z.; et al. Expression Profiles of mRNAs in Manganese-Induced Acute and Chronic Neurotoxicity by Bioinformatics Analysis of Gene Microarray. SSRN Electron. J. 2020. [Google Scholar] [CrossRef]
- Lin, Y.; Vogt, R.; Larssen, T. Environmental mercury in China: A review. Environ. Toxicol. Chem. 2012, 31, 2431–2444. [Google Scholar] [CrossRef] [Green Version]
- Fung, Y.K.; Meade, A.G.; Rack, E.P.; Blotcky, A.J. Brain Mercury in Neurodegenerative Disorders. J. Toxicol. Clin. Toxicol. 1997, 35, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Finkelman, R.B.; Orem, W.; Castranova, V.; Tatu, C.A.; Belkin, H.E.; Zheng, B.; Lerch, H.E.; Maharaj, S.V.; Bates, A.L. Health impacts of coal and coal use: Possible solutions. Int. J. Coal Geol. 2002, 50, 425–443. [Google Scholar] [CrossRef]
- Cariccio, V.L.; Samà, A.; Bramanti, P.; Mazzon, E. Mercury Involvement in Neuronal Damage and in Neurodegenerative Diseases. Biol. Trace Elem. Res. 2018, 187, 341–356. [Google Scholar] [CrossRef]
- Arvidson, B. Accumulation of mercury in brainstem nuclei of mice after retrograde axonal transport. Acta Neurol. Scand. 2009, 82, 234–237. [Google Scholar] [CrossRef]
- Arvidson, B. A review of axonal transport of metals. Toxicology 1994, 88, 1–14. [Google Scholar] [CrossRef]
- Sarafian, T.A.; Bredesen, D.E.; Verity, M.A. Cellular resistance to methylmercury. Neurotoxicology 1996, 17, 27–36. [Google Scholar]
- Pendergrass, J.C.; Haley, B.E. Inhibition of brain tubulin-guanosine 5′-triphosphate interactions by mercury: Similarity to observations in Alzheimer’s diseased brain. Met. Ions Boil. Syst. 1997, 34, 461–478. [Google Scholar]
- Pendergrass, J.C.; Haley, B.E.; Vimy, M.J.; Winfield, S.A.; Lorscheider, F.L. Mercury vapor inhalation inhibits binding of GTP to tubulin in rat brain: Similarity to a molecular lesion in Alzheimer diseased brain. Neurotoxicology 1997, 18, 315–324. [Google Scholar] [PubMed]
- Breydo, L.; Wu, J.W.; Uversky, V.N. α-Synuclein misfolding and Parkinson’s disease. Biochim. Biophys. Acta Mol. Basis Dis. 2012, 1822, 261–285. [Google Scholar] [CrossRef] [Green Version]
- Wallin, C.; Friedemann, M.; Sholts, S.B.; Noormägi, A.; Svantesson, T.; Jarvet, J.; Roos, P.M.; Palumaa, P.; Gräslund, A.; Wärmländer, S.K.T.S. Mercury and Alzheimer’s Disease: Hg(II) Ions Display Specific Binding to the Amyloid-β Peptide and Hinder Its Fibrillization. Biomolecules 2020, 10, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceccatelli, S.; Daré, E.; Moors, M. Methylmercury-induced neurotoxicity and apoptosis. Chem. Biol. Interact. 2010, 188, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Sian-Hülsmann, J.; Mandel, S.; Youdim, M.B.H.; Riederer, P. The relevance of iron in the pathogenesis of Parkinson’s disease. J. Neurochem. 2010, 118, 939–957. [Google Scholar] [CrossRef]
- Muñoz, P.; Humeres, A. Iron deficiency on neuronal function. BioMetals 2012, 25, 825–835. [Google Scholar] [CrossRef] [PubMed]
- Savica, R.; Grossardt, B.R.; Carlin, J.M.; Icen, M.; Bower, J.H.; Ahlskog, J.E.; Maraganore, D.M.; Steensma, D.P.; Rocca, W.A. Anemia or low hemoglobin levels preceding Parkinson disease: A case-control study. Neurology 2009, 73, 1381–1387. [Google Scholar] [CrossRef] [Green Version]
- Dev, S.; Babitt, J.L. Overview of iron metabolism in health and disease. Hemodial. Int. 2017, 21, S6–S20. [Google Scholar] [CrossRef]
- Hare, D.J.; Arora, M.; Jenkins, N.L.; Finkelstein, D.I.; Doble, P.A.; Bush, A.I. Is early-life iron exposure critical in neurodegeneration? Nat. Rev. Neurol. 2015, 11, 536–544. [Google Scholar] [CrossRef] [PubMed]
- Hare, D.J.; Cardoso, B.R.; Raven, E.P.; Double, K.L.; Finkelstein, D.I.; Szymlek-Gay, E.A.; Biggs, B.-A. Excessive early-life dietary exposure: A potential source of elevated brain iron and a risk factor for Parkinson’s disease. NPJ Parkinson Dis. 2017, 3, 1. [Google Scholar] [CrossRef] [PubMed]
- Costa-Mallen, P.; Gatenby, C.; Friend, S.; Maravilla, K.R.; Hu, S.-C.; Cain, K.C.; Agarwal, P.; Anzai, Y. Brain iron concentrations in regions of interest and relation with serum iron levels in Parkinson disease. J. Neurol. Sci. 2017, 378, 38–44. [Google Scholar] [CrossRef]
- Logroscino, G.; Marder, K.; Graziano, J.; Freyer, G.; Slavkovich, V.; LoIacono, N.; Cote, L.; Mayeux, R. Altered systemic iron metabolism in Parkinson’s disease. Neurology 1997, 49, 714–717. [Google Scholar] [CrossRef]
- Fasano, M.; Bergamasco, B.; Lopiano, L. Modifications of the iron-neuromelanin system in Parkinson’s disease. J. Neurochem. 2006, 96, 909–916. [Google Scholar] [CrossRef] [PubMed]
- Gerlach, M.; Double, K.L.; Youdim, M.B.H.; Riederer, P. Potential sources of increased iron in the substantia nigra of parkinsonian patients. Parkinson Dis. Relat. Disord. 2006, 70, 133–142. [Google Scholar] [CrossRef]
- Lotfipour, A.K.; Wharton, S.; Schwarz, S.T.; Gontu, V.; Schäfer, A.; Peters, A.M.; Bowtell, R.W.; Auer, D.P.; Gowland, P.A.; Bajaj, N.P.S. High resolution magnetic susceptibility mapping of the substantia nigra in Parkinson’s disease. J. Magn. Reson. Imaging 2012, 35, 48–55. [Google Scholar] [CrossRef]
- Levi, S.; Finazzi, D. Neurodegeneration with brain iron accumulation: Update on pathogenic mechanisms. Front. Pharmacol. 2014, 5, 99. [Google Scholar] [CrossRef] [Green Version]
- Xuan, M.; Guan, X.; Gu, Q.; Shen, Z.; Yu, X.; Qiu, T.; Luo, X.; Song, R.; Jiaerken, Y.; Xu, X.; et al. Different iron deposition patterns in early- and middle-late-onset Parkinson’s disease. Parkinsonism Relat. Disord. 2017, 44, 23–27. [Google Scholar] [CrossRef]
- Dexter, D.T.; Wells, F.R.; Lee, A.J.; Agid, F.; Agid, Y.; Jenner, P.; Marsden, C.D. Increased Nigral Iron Content and Alterations in Other Metal Ions Occurring in Brain in Parkinson’s Disease. J. Neurochem. 1989, 52, 1830–1836. [Google Scholar] [CrossRef]
- Ulla, M.; Bonny, J.M.; Ouchchane, L.; Rieu, I.; Claise, B.; Durif, F. Is R2* a New MRI Biomarker for the Progression of Parkinson’s Disease? A Longitudinal Follow-Up. PLoS ONE 2013, 8, e57904. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Du, T.; Song, N.; He, Q.; Shen, Y.; Jiang, H.; Xie, J. Decreased iron levels in the temporal cortex in postmortem human brains with Parkinson disease. Neurology 2013, 80, 492–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vancamelbeke, M.; Vermeire, S. The intestinal barrier: A fundamental role in health and disease. Expert Rev. Gastroenterol. Hepatol. 2017, 11, 821–834. [Google Scholar] [CrossRef]
- Turner, J.R. Intestinal mucosal barrier function in health and disease. Nat. Rev. Immunol. 2009, 9, 799–809. [Google Scholar] [CrossRef] [PubMed]
- Takiishi, T.; Fenero, C.I.M.; Câmara, N.O.S. Intestinal barrier and gut microbiota: Shaping our immune responses throughout life. Tissue Barriers 2017, 5, e1373208. [Google Scholar] [CrossRef] [PubMed]
- Pott, J.; Hornef, M. Innate immune signalling at the intestinal epithelium in homeostasis and disease. EMBO Rep. 2012, 13, 684–698. [Google Scholar] [CrossRef] [Green Version]
- Hiippala, K.; Jouhten, H.; Ronkainen, A.; Hartikainen, A.; Kainulainen, V.; Jalanka, J.; Satokari, R. The Potential of Gut Commensals in Reinforcing Intestinal Barrier Function and Alleviating Inflammation. Nutrients 2018, 10, 988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romero, E.S.; Cotoner, C.A.; Camacho, C.P.; Bedmar, M.C.; Vicario, M. The intestinal barrier function and its involvement in digestive disease. Rev. Esp. Enferm. Dig. 2015, 108, 686–696. [Google Scholar] [CrossRef] [Green Version]
- Breton, J.Ô.; Daniel, C.; Dewulf, J.; Pothion, S.; Froux, N.; Sauty, M.; Thomas, P.; Pot, B.; Foligne, B. Gut microbiota limits heavy metals burden caused by chronic oral exposure. Toxicol. Lett. 2013, 222, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Breton, J.; Le Clère, K.; Daniel, C.; Sauty, M.; Nakab, L.; Chassat, T.; Dewulf, J.; Penet, S.; Carnoy, C.; Thomas, P.; et al. Chronic ingestion of cadmium and lead alters the bioavailability of essential and heavy metals, gene expression pathways and genotoxicity in mouse intestine. Arch. Toxicol. 2013, 87, 1787–1795. [Google Scholar] [CrossRef]
- Li, X.; Brejnrod, A.; Ernst, M.; Rykær, M.; Herschend, J.; Olsen, N.M.C.; Dorrestein, P.C.; Rensing, C.; Sørensen, S.J. Heavy metal exposure causes changes in the metabolic health-associated gut microbiome and metabolites. Environ. Int. 2019, 126, 454–467. [Google Scholar] [CrossRef] [PubMed]
- Tinkov, A.A.; Gritsenko, V.A.; Skalnaya, M.G.; Cherkasov, S.V.; Aaseth, J.; Skalny, A.V. Gut as a target for cadmium toxicity. Environ. Pollut. 2018, 235, 429–434. [Google Scholar] [CrossRef]
- Martin, C.R.; Osadchiy, V.; Kalani, A.; Mayer, E.A. The Brain-Gut-Microbiome Axis. Cell. Mol. Gastroenterol. Hepatol. 2018, 6, 133–148. [Google Scholar] [CrossRef] [Green Version]
- Carabotti, M.; Scirocco, A.; Maselli, M.A.; Severi, C. The gut-brain axis: Interactions between enteric microbiota, central and enteric nervous systems. Ann. Gastroenterol. 2015, 28, 203–209. [Google Scholar]
- Coryell, M.; Roggenbeck, B.A.; Walk, S.T. The Human Gut Microbiome’s Influence on Arsenic Toxicity. Curr. Pharmacol. Rep. 2019, 5, 491–504. [Google Scholar] [CrossRef] [Green Version]
- Assefa, S.; Köhler, G. Intestinal microbiome and metal toxicity. Curr. Opin. Toxicol. 2019, 19, 21–27. [Google Scholar] [CrossRef]
- Breit, S.; Kupferberg, A.; Rogler, G.; Hasler, G. Vagus Nerve as Modulator of the Brain–Gut Axis in Psychiatric and Inflammatory Disorders. Front. Psychiatry 2018, 9, 44. [Google Scholar] [CrossRef] [Green Version]
- Galland, L. The Gut Microbiome and the Brain. J. Med. Food 2014, 17, 1261–1272. [Google Scholar] [CrossRef] [PubMed]
- Olmo, B.M.G.; Butler, M.J.; Barrientos, R.M. Evolution of the Human Diet and Its Impact on Gut Microbiota, Immune Responses, and Brain Health. Nutrients 2021, 13, 196. [Google Scholar] [CrossRef]
- Tsavkelova, E.A.; Botvinko, I.V.; Kudrin, V.S.; Oleskin, A.V. Detection of neurotransmitter amines in microorganisms with the use of high-performance liquid chromatography. Dokl. Biochem. 2000, 372, 115–117. [Google Scholar] [PubMed]
- Özoğul, F. Production of biogenic amines by Morganella morganii, Klebsiella pneumoniae and Hafnia alvei using a rapid HPLC method. Eur. Food Res. Technol. 2004, 219, 465–469. [Google Scholar] [CrossRef]
- Shishov, V.A.; Kirovskaya, T.A.; Kudrin, V.S.; Oleskin, A.V. Amine neuromediators, their precursors, and oxidation products in the culture of Escherichia coli K-12. Appl. Biochem. Microbiol. 2009, 45, 494–497. [Google Scholar] [CrossRef]
- Strandwitz, P. Neurotransmitter modulation by the gut microbiota. Brain Res. 2018, 1693, 128–133. [Google Scholar] [CrossRef]
- Bonaz, B.; Sinniger, V.; Hoffmann, D.; Clarençon, D.; Mathieu, N.; Dantzer, C.; Vercueil, L.; Picq, C.; Trocmé, C.; Faure, P.; et al. Chronic vagus nerve stimulation in Crohn’s disease: A 6-month follow-up pilot study. Neurogastroenterol. Motil. 2016, 28, 948–953. [Google Scholar] [CrossRef] [PubMed]
- Jaglin, M.; Rhimi, M.; Philippe, C.; Pons, N.; Bruneau, A.; Goustard, B.; Daugé, V.; Maguin, E.; Naudon, L.; Rabot, S. Indole, a Signaling Molecule Produced by the Gut Microbiota, Negatively Impacts Emotional Behaviors in Rats. Front. Neurosci. 2018, 12, 216. [Google Scholar] [CrossRef]
- Woźniak, D.; Cichy, W.; Przysławski, J.; Drzymała-Czyż, S. The role of microbiota and enteroendocrine cells in maintaining homeostasis in the human digestive tract. Adv. Med. Sci. 2021, 66, 284–292. [Google Scholar] [CrossRef]
- Latorre, R.; Sternini, C.; de Giorgio, R.; Meerveld, B.G.-V. Enteroendocrine cells: A review of their role in brain-gut communication. Neurogastroenterol. Motil. 2015, 28, 620–630. [Google Scholar] [CrossRef] [Green Version]
- Chimerel, C.; Emery, E.; Summers, D.K.; Keyser, U.; Gribble, F.M.; Reimann, F. Bacterial Metabolite Indole Modulates Incretin Secretion from Intestinal Enteroendocrine L Cells. Cell Rep. 2014, 9, 1202–1208. [Google Scholar] [CrossRef] [Green Version]
- Psichas, A.; Sleeth, M.L.; Murphy, K.G.; Brooks, L.; Bewick, G.A.; Hanyaloglu, A.C.; Ghatei, M.A.; Bloom, S.R.; Frost, G. The short chain fatty acid propionate stimulates GLP-1 and PYY secretion via free fatty acid receptor 2 in rodents. Int. J. Obes. 2015, 39, 424–429. [Google Scholar] [CrossRef] [Green Version]
- Lund, M.L.; Sorrentino, G.; Egerod, K.L.; Kroone, C.; Mortensen, B.; Knop, F.K.; Reimann, F.; Gribble, F.M.; Drucker, D.J.; de Koning, E.J.P.; et al. L-Cell Differentiation Is Induced by Bile Acids Through GPBAR1 and Paracrine GLP-1 and Serotonin Signaling. Diabetes 2020, 69, 614–623. [Google Scholar] [CrossRef]
- Panwar, H.; Calderwood, D.; Gillespie, A.L.; Wylie, A.R.; Graham, S.F.; Grant, I.R.; Grover, S.; Green, B.D. Identification of lactic acid bacteria strains modulating incretin hormone secretion and gene expression in enteroendocrine cells. J. Funct. Foods 2016, 23, 348–358. [Google Scholar] [CrossRef] [Green Version]
- Ticho, A.L.; Malhotra, P.; Dudeja, P.K.; Gill, R.K.; Alrefai, W.A. Bile acid receptors and gastrointestinal functions. Liver Res. 2019, 3, 31–39. [Google Scholar] [CrossRef]
- Alemi, F.; Poole, D.P.; Chiu, J.; Schoonjans, K.; Cattaruzza, F.; Grider, J.R.; Bunnett, N.W.; Corvera, C.U. The receptor TGR5 mediates the prokinetic actions of intestinal bile acids and is required for normal defecation in mice. Gastroenterology 2013, 144, 145–154. [Google Scholar] [CrossRef]
- Silva, Y.P.; Bernardi, A.; Frozza, R.L. The Role of Short-Chain Fatty Acids From Gut Microbiota in Gut-Brain Communication. Front. Endocrinol. (Lausanne) 2020, 11, 25. [Google Scholar] [CrossRef] [Green Version]
- Covasa, M.; Stephens, R.W.; Toderean, R.; Cobuz, C. Intestinal sensing by gut microbiota: Targeting gut peptides. Front. Endocrinol. (Lausanne) 2019, 10, 82. [Google Scholar] [CrossRef] [Green Version]
- Mishra, S.P.; Karunakar, P.; Taraphder, S.; Yadav, H. Free fatty acid receptors 2 and 3 as microbial metabolite sensors to shape host health: Pharmacophysiological view. Biomedicines 2020, 8, 154. [Google Scholar] [CrossRef]
- Venegas, D.P.; De La Fuente, M.K.; Landskron, G.; González, M.J.; Quera, R.; Dijkstra, G.; Harmsen, H.J.M.; Faber, K.N.; Hermoso, M.A. Short chain fatty acids (SCFAs)mediated gut epithelial and immune regulation and its relevance for inflammatory bowel diseases. Front. Immunol. 2019, 10, 277. [Google Scholar] [CrossRef] [Green Version]
- Overby, H.B.; Ferguson, J.F. Gut Microbiota-Derived Short-Chain Fatty Acids Facilitate Microbiota: Host Cross talk and Modulate Obesity and Hypertension. Curr. Hypertens. Rep. 2021, 23, 8. [Google Scholar] [CrossRef]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [Green Version]
- Litvak, Y.; Byndloss, M.X.; Bäumler, A.J. Colonocyte metabolism shapes the gut microbiota. Science 2018, 362, eaat9076. [Google Scholar] [CrossRef] [Green Version]
- Gershon, M.D.; Tack, J. The Serotonin Signaling System: From Basic Understanding To Drug Development for Functional GI Disorders. Gastroenterology 2007, 132, 397–414. [Google Scholar] [CrossRef]
- Mawe, G.M.; Hoffman, J.M. Serotonin Signaling in the Gastrointestinal Tract: Functions, dysfunctions, and therapeutic targets. Nat. Rev. Gastroenterol. Hepatol. 2013, 22, 313–333. [Google Scholar] [CrossRef]
- Yano, J.M.; Yu, K.; Donaldson, G.P.; Shastri, G.G.; Ann, P.; Ma, L.; Nagler, C.R.; Ismagilov, R.F.; Mazmanian, S.K.; Hsiao, E.Y. Indigenous Bacteria from the Gut Microbiota Regulate Host Serotonin Biosynthesis. Cell 2015, 161, 264–276. [Google Scholar] [CrossRef] [Green Version]
- Ozogul, F.; Kuley, E.; Ozogul, Y.; Özoğul, I. The Function of Lactic Acid Bacteria on Biogenic Amines Production by Food-Borne Pathogens in Arginine Decarboxylase Broth. Food Sci. Technol. Res. 2012, 18, 795–804. [Google Scholar] [CrossRef] [Green Version]
- Terry, N.; Margolis, K.G. Serotonergic Mechanisms Regulating the GI Tract: Experimental Evidence and Therapeutic Relevance. Gastrointest. Pharmacol. 2016, 239, 319–342. [Google Scholar] [CrossRef] [Green Version]
- De Vadder, F.; Grasset, E.; Holm, L.M.; Karsenty, G.; MacPherson, A.J.; Olofsson, L.E.; Bäckhed, F. Gut microbiota regulates maturation of the adult enteric nervous system via enteric serotonin networks. Proc. Natl. Acad. Sci. USA 2018, 115, 6458–6463. [Google Scholar] [CrossRef] [Green Version]
- Kelly, J.R.; Kennedy, P.J.; Cryan, J.F.; Dinan, T.G.; Clarke, G.; Hyland, N.P. Breaking down the barriers: The gut microbiome, intestinal permeability and stress-related psychiatric disorders. Front. Cell. Neurosci. 2015, 9, 392. [Google Scholar] [CrossRef] [Green Version]
- Golubeva, A.V.; Joyce, S.A.; Moloney, G.; Burokas, A.; Sherwin, E.; Arboleya, S.; Flynn, I.; Khochanskiy, D.; Moya-Pérez, A.; Peterson, V.; et al. Microbiota-related Changes in Bile Acid & Tryptophan Metabolism are Associated with Gastrointestinal Dysfunction in a Mouse Model of Autism. EBioMedicine 2017, 24, 166–178. [Google Scholar] [CrossRef] [Green Version]
- Wikoff, W.R.; Anfora, A.T.; Liu, J.; Schultz, P.G.; Lesley, S.A.; Peters, E.C.; Siuzdak, G. Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc. Natl. Acad. Sci. USA 2009, 106, 3698–3703. [Google Scholar] [CrossRef] [Green Version]
- Sjögren, K.; Engdahl, C.; Henning, P.; Lerner, U.H.; Tremaroli, V.; Lagerquist, M.K.; Bäckhed, F.; Ohlsson, C. The gut microbiota regulates bone mass in mice. J. Bone Miner. Res. 2012, 27, 1357–1367. [Google Scholar] [CrossRef] [Green Version]
- Mandić, A.D.; Woting, A.; Jaenicke, T.; Sander, A.; Sabrowski, W.; Rolle-Kampcyk, U.; Von Bergen, M.; Blaut, M. Clostridium ramosum regulates enterochromaffin cell development and serotonin release. Sci. Rep. 2019, 9, 1177. [Google Scholar] [CrossRef] [Green Version]
- Fond, G.; Loundou, A.; Hamdani, N.; Boukouaci, W.; Dargel, A.; Oliveira, J.; Roger, M.; Tamouza, R.; Leboyer, M.; Boyer, L. Anxiety and depression comorbidities in irritable bowel syndrome (IBS): A systematic review and meta-analysis. Eur. Arch. Psychiatry Clin. Neurosci. 2014, 264, 651–660. [Google Scholar] [CrossRef]
- Fung, T.C. The microbiota-immune axis as a central mediator of gut-brain communication. Neurobiol. Dis. 2019, 136, 104714. [Google Scholar] [CrossRef]
- Dumitrescu, L.; Popescu-Olaru, I.; Cozma, L.; Tulbă, D.; Hinescu, M.E.; Ceafalan, L.C.; Gherghiceanu, M.; Popescu, B.O. Oxidative Stress and the Microbiota-Gut-Brain Axis. Oxidative Med. Cell. Longev. 2018, 2018, 2406594. [Google Scholar] [CrossRef] [Green Version]
- MacDonald, T.T.; Monteleone, G. Immunity, Inflammation, and Allergy in the Gut. Science 2005, 307, 1920–1925. [Google Scholar] [CrossRef] [Green Version]
- Hooper, L.V.; Littman, D.R.; MacPherson, A.J. Interactions between the Microbiota and the Immune System. Science 2012, 336, 1268–1273. [Google Scholar] [CrossRef] [Green Version]
- Powell, N.; Walker, M.M.; Talley, N.J. The mucosal immune system: Master regulator of bidirectional gut–brain communications. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 143–159. [Google Scholar] [CrossRef]
- Sommer, F.; Bäckhed, F. The gut microbiota—Masters of host development and physiology. Nat. Rev. Microbiol. 2013, 11, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Hall, J.A.; Bouladoux, N.; Sun, C.M.; Wohlfert, E.A.; Blank, R.B.; Zhu, Q.; Grigg, M.E.; Berzofsky, J.A.; Belkaid, Y. Commensal DNA Limits Regulatory T Cell Conversion and Is a Natural Adjuvant of Intestinal Immune Responses. Immunity 2008, 29, 637–649. [Google Scholar] [CrossRef] [Green Version]
- Cheng, H.; Guan, X.; Chen, D.; Ma, W. The th17/treg cell balance: A gut microbiota-modulated story. Microorganisms 2019, 7, 583. [Google Scholar] [CrossRef] [Green Version]
- Karimi, K.; Inman, M.D.; Bienenstock, J.; Forsythe, P. Lactobacillus reuteri–induced Regulatory T cells Protect against an Allergic Airway Response in Mice. Am. J. Respir. Crit. Care Med. 2009, 179, 186–193. [Google Scholar] [CrossRef] [PubMed]
- Lyons, A.; O’Mahony, D.; O’Brien, F.; Mac Sharry, J.; Sheil, B.; Ceddia, M.; Russell, W.M.; Forsythe, P.; Bienenstock, J.; Kiely, B.; et al. Bacterial strain-specific induction of Foxp3+T regulatory cells is protective in murine allergy models. Clin. Exp. Allergy 2010, 40, 811–819. [Google Scholar] [CrossRef]
- Atarashi, K.; Tanoue, T.; Shima, T.; Imaoka, A.; Kuwahara, T.; Momose, Y.; Cheng, G.; Yamasaki, S.; Saito, T.; Ohba, Y.; et al. Induction of colonic regulatory T cells by indigenous clostridium species. Science 2011, 331, 337–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, L.; Barbi, J.; Pan, F. The regulation of immune tolerance by FOXP3. Nat. Rev. Immunol. 2017, 17, 703–717. [Google Scholar] [CrossRef] [PubMed]
- Bain, C.C.; Schridde, A. Origin, differentiation, and function of intestinal macrophages. Front. Immunol. 2018. [Google Scholar] [CrossRef]
- Jacobson, A.; Yang, D.; Vella, M.; Chiu, I.M. The intestinal neuro-immune axis: Crosstalk between neurons, immune cells, and microbes. Mucosal Immunol. 2021, 14, 555–565. [Google Scholar] [CrossRef]
- Chang, P.V.; Hao, L.; Offermanns, S.; Medzhitov, R. The microbial metabolite butyrate regulates intestinal macrophage function via histone deacetylase inhibition. Proc. Natl. Acad. Sci. USA 2014, 111, 2247–2252. [Google Scholar] [CrossRef] [Green Version]
- Muller, P.A.; Koscsó, B.; Rajani, G.M.; Stevanovic, K.; Berres, M.L.; Hashimoto, D.; Mortha, A.; Leboeuf, M.; Li, X.M.; Mucida, D.; et al. Crosstalk between muscularis macrophages and enteric neurons regulates gastrointestinal motility. Cell 2014, 158, 300–313. [Google Scholar] [CrossRef] [Green Version]
- Obata, Y.; Pachnis, V. The Effect of Microbiota and the Immune System on the Development and Organization of the Enteric Nervous System. Gastroenterology 2016, 151, 836–844. [Google Scholar] [CrossRef] [Green Version]
- Gabanyi, I.; Muller, P.A.; Feighery, L.; Oliveira, T.Y.; Costa-Pinto, F.A.; Mucida, D. Neuro-immune Interactions Drive Tissue Programming in Intestinal Macrophages. Cell 2016, 164, 378–391. [Google Scholar] [CrossRef] [Green Version]
- Lycke, N.Y.; Bemark, M. The regulation of gut mucosal IgA B-cell responses: Recent developments. Mucosal Immunol. 2017, 10, 1361–1374. [Google Scholar] [CrossRef]
- Zheng, W.; He, R.; Yan, Z.; Huang, Y.; Huang, W.; Cai, Z.; Su, Y.; Liu, S.; Deng, Y.; Wang, Q.; et al. Regulation of immune-driven pathogenesis in Parkinson’s disease by gut microbiota. Brain. Behav. Immun. 2020, 87, 890–897. [Google Scholar] [CrossRef]
- Chen, Q.-Q.; Haikal, C.; Li, W.; Li, J.-Y. Gut Inflammation in Association with Pathogenesis of Parkinson’s Disease. Front. Mol. Neurosci. 2019, 12, 218. [Google Scholar] [CrossRef] [Green Version]
- Keshavarzian, A.; Green, S.J.; Engen, P.A.; Voigt, R.M.; Naqib, A.; Forsyth, C.B.; Mutlu, E.; Shannon, K.M. Colonic bacterial composition in Parkinson’s disease. Mov. Disord. 2015, 30, 1351–1360. [Google Scholar] [CrossRef]
- Scheperjans, F.; Aho, V.; Pereira, P.A.; Koskinen, K.; Paulin, L.; Pekkonen, E.; Haapaniemi, E.; Kaakkola, S.; Eerola-Rautio, J.; Pohja, M.; et al. Gut microbiota are related to Parkinson’s disease and clinical phenotype. Mov. Disord. 2015, 30, 350–358. [Google Scholar] [CrossRef]
- Unger, M.M.; Spiegel, J.; Dillmann, K.-U.; Grundmann, D.; Philippeit, H.; Bürmann, J.; Faßbender, K.; Schwiertz, A.; Schäfer, K.H. Short chain fatty acids and gut microbiota differ between patients with Parkinson’s disease and age-matched controls. Parkinsonism Relat. Disord. 2016, 32, 66–72. [Google Scholar] [CrossRef]
- Li, W.; Wu, X.L.; Hu, X.; Wang, T.; Liang, S.; Duan, Y.F.; Jin, F.; Qin, B. Structural changes of gut microbiota in Parkinson’s disease and its correlation with clinical features. Sci. China Life Sci. 2017, 60, 1223–1233. [Google Scholar] [CrossRef]
- Barichella, M.; Severgnini, M.; Cilia, R.; Cassani, E.; Bolliri, C.; Caronni, S.; Ferri, V.; Cancello, R.; Ceccarani, C.; Faierman, S.; et al. Unraveling gut microbiota in Parkinson’s disease and atypical parkinsonism. Mov. Disord. 2018, 34, 396–405. [Google Scholar] [CrossRef] [PubMed]
- Campos-Acuña, J.; Elgueta, D.; Pacheco, R. T-Cell-Driven Inflammation as a Mediator of the Gut-Brain Axis Involved in Parkinson’s Disease. Front. Immunol. 2019, 10, 239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosley, R.L.; Hutter-Saunders, J.A.; Stone, D.K.; Gendelman, H.E. Inflammation and Adaptive Immunity in Parkinson’s Disease. Cold Spring Harb. Perspect. Med. 2011, 2, a009381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jahromi, M.F.; Liang, J.B.; Ebrahimi, R.; Soleimani, A.F.; Rezaeizadeh, A.; Abdullah, N.; Shokryazdan, P. Protective potential of Lactobacillus species in lead toxicity model in broiler chickens. Animal 2017, 11, 755–761. [Google Scholar] [CrossRef] [Green Version]
- Bedarf, J.R.; Hildebrand, F.; Coelho, L.P.; Sunagawa, S.; Bahram, M.; Goeser, F.; Bork, P.; Wüllner, U. Functional implications of microbial and viral gut metagenome changes in early stage L-DOPA-naïve Parkinson’s disease patients. Genome Med. 2017, 9, 39. [Google Scholar] [CrossRef]
- Devos, D.; Lebouvier, T.; Lardeux, B.; Biraud, M.; Rouaud, T.; Pouclet, H.; Coron, E.; Varannes, S.B.D.; Naveilhan, P.; Nguyen, J.-M.; et al. Colonic inflammation in Parkinson’s disease. Neurobiol. Dis. 2013, 50, 42–48. [Google Scholar] [CrossRef]
- Osburg, B.; Peiser, C.; Dömling, D.; Schomburg, L.; Ko, Y.T.; Voigt, K.; Bickel, U. Effect of endotoxin on expression of TNF receptors and transport of TNF-α at the blood-brain barrier of the rat. Am. J. Physiol. Metab. 2002, 283, E899–E908. [Google Scholar] [CrossRef] [Green Version]
- Benner, E.J.; Banerjee, R.; Reynolds, A.D.; Sherman, S.; Pisarev, V.M.; Tsiperson, V.; Nemachek, C.; Ciborowski, P.; Przedborski, S.; Mosley, R.L.; et al. Nitrated α–Synuclein Immunity Accelerates Degeneration of Nigral Dopaminergic Neurons. PLoS ONE 2008, 3, e1376. [Google Scholar] [CrossRef]
- Choi, J.G.; Kim, N.; Ju, I.G.; Eo, H.; Lim, S.-M.; Jang, S.-E.; Kim, D.-H.; Oh, M.S. Oral administration of Proteus mirabilis damages dopaminergic neurons and motor functions in mice. Sci. Rep. 2018, 8, 1275. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Chen, Y.; Zhang, Y.; Wang, F.; Yu, H.; Zhang, C.; Jiang, Z.; Luo, W. Iron deposition in Parkinson’s disease by quantitative susceptibility mapping. BMC Neurosci. 2019, 20, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miranda, M.; López-Alonso, M.; Castillo, C.; Hernández, J.L.; Benedito, J. Effects of moderate pollution on toxic and trace metal levels in calves from a polluted area of northern Spain. Environ. Int. 2005, 31, 543–548. [Google Scholar] [CrossRef]
- McConnell, J.R.; Edwards, R. Coal burning leaves toxic heavy metal legacy in the Arctic. Proc. Natl. Acad. Sci. USA 2008, 105, 12140–12144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petersen, M.S.; Halling, J.; Bech, S.; Wermuth, L.; Weihe, P.; Nielsen, F.; Jørgensen, P.J.; Budtz-Jørgensen, E.; Grandjean, P. Impact of dietary exposure to food contaminants on the risk of Parkinson’s disease. Neurotoxicology 2008, 29, 584–590. [Google Scholar] [CrossRef]
- Hadayat, N.; De Oliveira, L.M.; Da Silva, E.; Han, L.; Hussain, M.; Liu, X.; Ma, L.Q. Assessment of trace metals in five most-consumed vegetables in the US: Conventional vs. organic. Environ. Pollut. 2018, 243, 292–300. [Google Scholar] [CrossRef]
- Fu, L.; Lu, X.; Niu, K.; Tan, J.; Chen, J. Bioaccumulation and human health implications of essential and toxic metals in freshwater products of Northeast China. Sci. Total Environ. 2019, 673, 768–776. [Google Scholar] [CrossRef]
- Miclean, M.; Cadar, O.; Levei, E.A.; Roman, R.; Ozunu, A.; Levei, L. Metal (Pb, Cu, Cd, and Zn) Transfer along Food Chain and Health Risk Assessment through Raw Milk Consumption from Free-Range Cows. Int. J. Environ. Res. Public Health 2019, 16, 4064. [Google Scholar] [CrossRef] [Green Version]
- Tshala-Katumbay, D.; Mwanza, J.-C.; Rohlman, D.S.; Maestre, G.E.; Oriá, R. A global perspective on the influence of environmental exposures on the nervous system. Nature 2015, 527, S187–S192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, H.; Yu, L.; Tian, F.; Zhai, Q.; Fan, L.; Chen, W. Gut microbiota: A target for heavy metal toxicity and a probiotic protective strategy. Sci. Total Environ. 2020, 742, 140429. [Google Scholar] [CrossRef]
- Giambò, F.; Italia, S.; Teodoro, M.; Briguglio, G.; Furnari, N.; Catanoso, R.; Costa, C.; Fenga, C. Influence of toxic metal exposure on the gut microbiota (Review). World Acad. Sci. J. 2021, 3, 1. [Google Scholar] [CrossRef]
- Rosenfeld, C.S. Gut Dysbiosis in Animals Due to Environmental Chemical Exposures. Front. Cell. Infect. Microbiol. 2017, 7, 396. [Google Scholar] [CrossRef] [Green Version]
- Priyadarshi, A.; Khuder, S.A.; Schaub, E.A.; Priyadarshi, S.S. Environmental Risk Factors and Parkinson’s Disease: A Metaanalysis. Environ. Res. 2001, 86, 122–127. [Google Scholar] [CrossRef] [Green Version]
- Dick, F.D.; De Palma, G.; Ahmadi, A.; Scott, N.W.; Prescott, G.J.; Bennett, J.; Semple, S.; Dick, S.; Counsell, C.; Mozzoni, P.; et al. Environmental risk factors for Parkinson’s disease and parkinsonism: The Geoparkinson study. Occup. Environ. Med. 2007, 64, 666–672. [Google Scholar] [CrossRef] [Green Version]
- Seidler, A.; Hellenbrand, W.; Robra, B.-P.; Vieregge, P.; Nischan, P.; Joerg, J.; Oertel, W.H.; Ulm, G.; Schneider, E. Possible environmental, occupational, and other etiologic factors for Parkinson’s disease: A case-control study in Germany. Neurology 1996, 46, 1275. [Google Scholar] [CrossRef]
- Jinsmaa, Y.; Sullivan, P.; Gross, D.; Cooney, A.; Sharabi, Y.; Goldstein, D.S. Divalent metal ions enhance DOPAL-induced oligomerization of alpha-synuclein. Neurosci. Lett. 2014, 569, 27–32. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Cai, T.; Zhao, F.; Yao, T.; Chen, Y.; Liu, X.; Luo, W.; Chen, J. The Role of α-synuclein and Tau Hyperphosphorylation-Mediated Autophagy and Apoptosis in Lead-induced Learning and Memory Injury. Int. J. Biol. Sci. 2012, 8, 935–944. [Google Scholar] [CrossRef]
- Kwakye, G.F.; Paoliello, M.M.B.; Mukhopadhyay, S.; Bowman, A.B.; Aschner, M. Manganese-induced Parkinsonism and Parkinson’s disease: Shared and distinguishable features. Int. J. Environ. Res. Public Health 2015, 12, 7519–7540. [Google Scholar] [CrossRef] [Green Version]
- Miller, K.; Ochudło, S.; Opala, G.; Smolicha, W.; Siuda, J. Parkinsonism in chronic occupational metallic mercury intoxication. Neurol. Neurochir. Polska 2003, 37, 31–38. [Google Scholar]
- Bridges, C.C.; Zalups, R.K. Molecular and ionic mimicry and the transport of toxic metals. Toxicol. Appl. Pharmacol. 2005, 204, 274–308. [Google Scholar] [CrossRef] [Green Version]
- Fujishiro, H.; Okugaki, S.; Kubota, K.; Fujiyama, T.; Miyataka, H.; Himeno, S. The role of ZIP8 down-regulation in cadmium-resistant metallothionein-null cells. J. Appl. Toxicol. 2009, 29, 367–373. [Google Scholar] [CrossRef]
- Vesey, D.A. Transport pathways for cadmium in the intestine and kidney proximal tubule: Focus on the interaction with essential metals. Toxicol. Lett. 2010, 198, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Caito, S.; Aschner, M. Neurotoxicity of metals. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2015. [Google Scholar]
- Roth, J.; Ponzoni, S.; Aschner, M. Manganese Homeostasis and Transport. Met. Cell 2012, 12, 169–201. [Google Scholar] [CrossRef]
- Ballatori, N. Transport of toxic metals by molecular mimicry. Environ. Health Perspect. 2002, 110, 689–694. [Google Scholar] [CrossRef] [Green Version]
- Bressler, J.P.; Olivi, L.; Cheong, J.H.; Kim, Y.; Maerten, A.; Bannon, D. Metal transporters in intestine and brain: Their involvement in metal-associated neurotoxicities. Hum. Exp. Toxicol. 2007, 26, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Dubey, V.; Mishra, A.K.; Ghosh, A.R.; Mandal, B.K. Probiotic Pediococcus pentosaceus GS4 shields brush border membrane and alleviates liver toxicity imposed by chronic cadmium exposure in Swiss albino mice. J. Appl. Microbiol. 2019, 126, 1233–1244. [Google Scholar] [CrossRef]
- Tulpule, K.; Robinson, S.R.; Bishop, G.M.; Dringen, R. Uptake of ferrous iron by cultured rat astrocytes. J. Neurosci. Res. 2010, 88, 563–571. [Google Scholar] [CrossRef] [PubMed]
- Bannon, D.I.; Abounader, R.; Lees, P.S.J.; Bressler, J.P. Effect of DMT1 knockdown on iron, cadmium, and lead uptake in Caco-2 cells. Am. J. Physiol. Cell Physiol. 2003, 284, C44–C50. [Google Scholar] [CrossRef] [Green Version]
- Singh, N. The Role of Iron in Prion Disease and Other Neurodegenerative Diseases. PLoS Pathog. 2014, 10, e1004335. [Google Scholar] [CrossRef]
- Chiacchia, M.; Cerutti, C.; Gromnicova, R.; Rietdorf, K.; Romero, I.A.; Bradshaw, D. Zinc-imidazolate polymers (ZIPs) as a potential carrier to brain capillary endothelial cells. J. Mater. Chem. B 2015, 3, 9053–9059. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Wang, Y.; Song, N.; Wang, J.; Jiang, H.; Xie, J. New Progress on the Role of Glia in Iron Metabolism and Iron-Induced Degeneration of Dopamine Neurons in Parkinson’s Disease. Front. Mol. Neurosci. 2018, 10, 455. [Google Scholar] [CrossRef]
- Hoepken, H.H.; Korten, T.; Robinson, S.R.; Dringen, R. Iron accumulation, iron-mediated toxicity and altered levels of ferritin and transferrin receptor in cultured astrocytes during incubation with ferric ammonium citrate. J. Neurochem. 2004, 88, 1194–1202. [Google Scholar] [CrossRef]
- Steimle, B.L.; Smith, F.M.; Kosman, D.J. The solute carriers ZIP8 and ZIP14 regulate manganese accumulation in brain microvascular endothelial cells and control brain manganese levels. J. Biol. Chem. 2019, 294, 19197–19208. [Google Scholar] [CrossRef]
- Kumar, V. Mechanism of Microbial Heavy Metal Accumulation from a Polluted Environment and Bioremediation. In Microbial Cell Factories; Deepansh Sharma, B.S.S., Ed.; CRC Press: Boca Raton, FL, USA, 2018; pp. 156–158. [Google Scholar]
- Yokooji, T.; Murakami, T.; Yumoto, R.; Nagai, J.; Takano, M. Site-specific bidirectional efflux of 2,4-dinitrophenyl-S-glutathione, a substrate of multidrug resistance-associated proteins, in rat intestine and Caco-2 cells. J. Pharm. Pharmacol. 2007, 59, 513–520. [Google Scholar] [CrossRef]
- Calatayud, M.; Barrios, J.A.; Vélez, D.; Devesa, V. In Vitro Study of Transporters Involved in Intestinal Absorption of Inorganic Arsenic. Chem. Res. Toxicol. 2012, 25, 446–453. [Google Scholar] [CrossRef]
- Maciaszczyk-Dziubinska, E.; Wawrzycka, D.; Wysocki, R. Arsenic and Antimony Transporters in Eukaryotes. Int. J. Mol. Sci. 2012, 13, 3527–3548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nico, B.; Frigeri, A.; Nicchia, G.P.; Corsi, P.; Ribatti, D.; Quondamatteo, F.; Herken, R.; Girolamo, F.; Marzullo, A.; Svelto, M.; et al. Severe alterations of endothelial and glial cells in the blood-brain barrier of dystrophic mdx mice. Glia 2003, 42, 235–251. [Google Scholar] [CrossRef]
- Zheng, W.; Aschner, M.; Ghersi-Egea, J.-F. Brain barrier systems: A new frontier in metal neurotoxicological research. Toxicol. Appl. Pharmacol. 2003, 192, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, T.; Shimohata, T. Vascular Dysfunction Induced by Mercury Exposure. Int. J. Mol. Sci. 2019, 20, 2435. [Google Scholar] [CrossRef] [Green Version]
- Cragg, R.A.; Christie, G.R.; Phillips, S.R.; Russi, R.M.; Küry, S.; Mathers, J.C.; Taylor, P.M.; Ford, D. A Novel Zinc-regulated Human Zinc Transporter, hZTL1, Is Localized to the Enterocyte Apical Membrane. J. Biol. Chem. 2002, 277, 22789–22797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garza-Lombó, C.; Posadas, Y.; Quintanar, L.; Gonsebatt, M.E.; Franco, R. Neurotoxicity Linked to Dysfunctional Metal Ion Homeostasis and Xenobiotic Metal Exposure: Redox Signaling and Oxidative Stress. Antioxid. Redox Signal. 2018, 28, 1669–1703. [Google Scholar] [CrossRef] [Green Version]
- La-Llave-León, O.; Méndez-Hernández, E.M.; Castellanos-Juárez, F.X.; Esquivel-Rodríguez, E.; Vázquez-Alaniz, F.; Sandoval-Carrillo, A.; García-Vargas, G.; Duarte-Sustaita, J.; Candelas-Rangel, J.L.; Salas-Pacheco, J.M. Association between Blood Lead Levels and Delta-Aminolevulinic Acid Dehydratase in Pregnant Women. Int. J. Environ. Res. Public Health 2017, 14, 432. [Google Scholar] [CrossRef] [Green Version]
- Dallas, S.; Zhu, X.; Baruchel, S.; Schlichter, L.; Bendayan, R. Functional Expression of the Multidrug Resistance Protein 1 in Microglia. J. Pharmacol. Exp. Ther. 2003, 307, 282–290. [Google Scholar] [CrossRef] [Green Version]
- Meyer, N.; Koehler, Y.; Tulpule, K.; Dringen, R. Arsenate accumulation and arsenate-induced glutathione export in astrocyte-rich primary cultures. Neurochem. Int. 2013, 62, 1012–1019. [Google Scholar] [CrossRef]
- Su, W.; Pasternak, G.W. The role of multidrug resistance-associated protein in the blood-brain barrier and opioid analgesia. Synapse 2013, 67, 609–619. [Google Scholar] [CrossRef] [Green Version]
- Sabath, E.; Robles-Osorio, M.L. Medio ambiente y riñón: Nefrotoxicidad por metales pesados. Nefrologia 2012, 32, 279286. [Google Scholar]
- Zalups, R.K.; Koropatnick, J. Cellular and Molecular Biology of Metals; CRC Press: Boca Raton, FL, USA, 2010; ISBN 9781420059984. [Google Scholar]
- Bridges, C.C.; Zalups, R.K. Mechanisms involved in the transport of mercuric ions in target tissues. Arch. Toxicol. 2016, 91, 63–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.-T.; An, J.-X.; Xu, J.-Y.; Tuo, B.-G. Overview of organic anion transporters and organic anion transporter polypeptides and their roles in the liver. World J. Clin. Cases 2019, 7, 3915–3933. [Google Scholar] [CrossRef]
- Menon, A.V.; Chang, J.O.; Kim, J. Mechanisms of divalent metal toxicity in affective disorders. Toxicology 2015, 339, 58–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peer, W.A.; Baxter, I.R.; Richards, E.L.; Freeman, J.L.; Murphy, A.S. Molecular Biology of Metal Homeostasis and Detoxification; Springer: Berlin/Heidelberg, Germany; New York, NY, USA, 2006; ISBN 3-540-22175-1. [Google Scholar]
- Wu, L.J.-C.; Leenders, A.G.M.; Cooperman, S.; Meyron-Holtz, E.; Smith, S.; Land, W.; Tsai, R.Y.L.; Berger, U.V.; Sheng, Z.-H.; Rouault, T.A. Expression of the iron transporter ferroportin in synaptic vesicles and the blood–brain barrier. Brain Res. 2004, 1001, 108–117. [Google Scholar] [CrossRef]
- Villa-Bellosta, R.; Sorribas, V. Arsenate transport by sodium/phosphate cotransporter type IIb. Toxicol. Appl. Pharmacol. 2010. [Google Scholar] [CrossRef]
- Miyamoto, K.I.; Haito-Sugino, S.; Kuwahara, S.; Ohi, A.; Nomura, K.; Ito, M.; Kuwahata, M.; Kido, S.; Tatsumi, S.; Kaneko, I.; et al. Sodium-dependent phosphate cotransporters: Lessons from gene knockout and mutation studies. J. Pharm. Sci. 2011, 100, 3719–3730. [Google Scholar]
- Mukhopadhyay, R.; Bhattacharjee, H.; Rosen, B.P. Aquaglyceroporins: Generalized metalloid channels. Biochim. Biophys. Acta Gen. Subj. 2014, 1840, 1583–1591. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharjee, H.; Rosen, B.P.; Mukhopadhyay, R. Aquaglyceroporins and Metalloid Transport: Implications in Human Diseases. Aquaporins 2009, 190, 309–325. [Google Scholar] [CrossRef] [Green Version]
- Ximenes-Da-Silva, A. Metal Ion Toxins and Brain Aquaporin-4 Expression: An Overview. Front. Neurosci. 2016, 10, 233. [Google Scholar] [CrossRef] [Green Version]
- Sabolić, I.; Breljak, D.; Škarica, M.; Herak-Kramberger, C.M. Role of metallothionein in cadmium traffic and toxicity in kidneys and other mammalian organs. Biometals 2010, 23, 897–926. [Google Scholar] [CrossRef]
- Ruttkay-Nedecky, B.; Nejdl, L.; Gumulec, J.; Zitka, O.; Masarik, M.; Eckschlager, T.; Stiborova, M.; Adam, V.; Kizek, R. The Role of Metallothionein in Oxidative Stress. Int. J. Mol. Sci. 2013, 14, 6044–6066. [Google Scholar] [CrossRef] [Green Version]
- Petering, D.H.; Krezoski, S.; Tabatabai, N.M. 12 Metallothionein Toxicology: Metal Ion Trafficking and Cellular Protection. In Metallothioneins and Related Chelators; De Gruyter: Berlin, Germany, 2015. [Google Scholar]
- Anderson, G.J.; Vulpe, C.D. Mammalian iron transport. Cell. Mol. Life Sci. 2009, 66, 3241–3261. [Google Scholar] [CrossRef]
- Gonick, H.C. Lead-Binding Proteins: A Review. J. Toxicol. 2011, 2011, 686050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fanali, G.; Cao, Y.; Ascenzi, P.; Fasano, M. Mn(II) binding to human serum albumin: A 1H-NMR relaxometric study. J. Inorg. Biochem. 2012, 117, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Yun, Z.; Li, L.; Liu, L.; He, B.; Zhao, X.; Jiang, G. Characterization of mercury-containing protein in human plasma. Metallomics 2013, 5, 821–827. [Google Scholar] [CrossRef] [PubMed]
- Briner, W. The Role of Metal Regulatory Proteins in Brain Oxidative Stress: A Tutorial. Oxidative Med. Cell. Longev. 2012, 2012, 981561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pacini, A.; Branca, J.J.V.; Morucci, G. Cadmium-induced neurotoxicity: Still much ado. Neural Regen. Res. 2018, 13, 1879–1882. [Google Scholar] [CrossRef] [PubMed]
- Hussain, A.; Raveendran, V.A.; Kundu, S.; Samanta, T.; Shunmugam, R.; Pal, D.; Sarma, J. Das Mechanisms of Arsenic-Induced Toxicity with Special Emphasis on Arsenic-Binding Proteins. In Arsenic—Analytical and Toxicological Studies; IntechOpen: London, UK, 2018; Available online: https://www.intechopen.com/chapters/60425 (accessed on 3 November 2021). [CrossRef] [Green Version]
- Ball, N.; Teo, W.-P.; Chandra, S.; Chapman, J. Parkinson’s Disease and the Environment. Front. Neurol. 2019, 10, 218. [Google Scholar] [CrossRef] [Green Version]
- Prabu, S.M.; Shagirtha, K.; Renugadevi, J. Amelioration of Cadmium-Induced Oxidative Stress, Impairment in Lipids and Plasma Lipoproteins by the Combined Treatment with Quercetin and α-Tocopherol in Rats. J. Food Sci. 2010, 75, T132–T140. [Google Scholar] [CrossRef]
- Obermeier, B.; Verma, A.; Ransohoff, R.M. The blood-brain barrier. Handb Clin Neurol. 2016, 133, 39–59. [Google Scholar] [CrossRef]
- Nemeth, E.; Tuttle, M.S.; Powelson, J.; Vaughn, M.B.; Donovan, A.; Ward, D.M.V.; Ganz, T.; Kaplan, J. Hepcidin Regulates Cellular Iron Efflux by Binding to Ferroportin and Inducing Its Internalization. Science 2004, 306, 2090–2093. [Google Scholar] [CrossRef] [Green Version]
- Crichton, R.; Ward, R. Role of Metal Ions in Brain Function, Metal Transport, Storage and Homoeostasis. In Metal-Based Neurodegeneration; John Wiley and Sons Ltd.: Hoboken, NJ, USA, 2013. [Google Scholar]
- Chiou, B.; Neal, E.H.; Bowman, A.B.; Lippmann, E.S.; Simpson, I.A.; Connor, J.R. Endothelial cells are critical regulators of iron transport in a model of the human blood–brain barrier. J. Cereb. Blood Flow Metab. 2018, 39, 2117–2131. [Google Scholar] [CrossRef] [PubMed]
- Crossgrove, J.S.; Allen, D.D.; Bukaveckas, B.L.; Rhineheimer, S.S.; Yokel, R.A. Manganese Distribution Across the Blood–Brain Barrier: I. Evidence for Carrier-Mediated Influx of Manganese Citrate as Well as Manganese and Manganese Transferrin. Neurotoxicology 2003, 24, 3–13. [Google Scholar] [CrossRef]
- Ye, Q.; Park, J.E.; Gugnani, K.; Betharia, S.; Pino-Figueroa, A.; Kim, J. Influence of iron metabolism on manganese transport and toxicity. Metallomics 2017, 9, 1028–1046. [Google Scholar] [CrossRef]
- Karri, V.; Schuhmacher, M.; Kumar, V. Heavy metals (Pb, Cd, As and MeHg) as risk factors for cognitive dysfunction: A general review of metal mixture mechanism in brain. Environ. Toxicol. Pharmacol. 2016, 48, 203–213. [Google Scholar] [CrossRef] [Green Version]
- Yukutake, Y.; Tsuji, S.; Hirano, Y.; Adachi, T.; Takahashi, T.; Fujihara, K.; Agre, P.; Yasui, M.; Suematsu, M. Mercury chloride decreases the water permeability of aquaporin-4-reconstituted proteoliposomes. Biol. Cell 2008, 100, 355–363. [Google Scholar] [CrossRef]
- Song, H.; Zheng, G.; Liu, Y.; Shen, X.-F.; Zhao, Z.-H.; Aschner, M.; Luo, W.-J.; Chen, J.-Y. Cellular uptake of lead in the blood-cerebrospinal fluid barrier: Novel roles of Connexin 43 hemichannel and its down-regulations via Erk phosphorylation. Toxicol. Appl. Pharmacol. 2016, 297, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Branca, J.J.V.; Maresca, M.; Morucci, G.; Mello, T.; Becatti, M.; Pazzagli, L.; Colzi, I.; Gonnelli, C.; Carrino, D.; Paternostro, F.; et al. Effects of Cadmium on ZO-1 Tight Junction Integrity of the Blood Brain Barrier. Int. J. Mol. Sci. 2019, 20, 6010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chtourou, Y.; Trabelsi, K.; Fetoui, H.; Mkannez, G.; Kallel, H.; Zeghal, N. Manganese Induces Oxidative Stress, Redox State Unbalance and Disrupts Membrane Bound ATPases on Murine Neuroblastoma Cells In Vitro: Protective Role of Silymarin. Neurochem. Res. 2011, 36, 1546–1557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trist, B.G.; Hare, D.J.; Double, K.L. Oxidative stress in the aging substantia nigra and the etiology of Parkinson’s disease. Aging Cell 2019, 18, e13031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prakash, C.; Soni, M.; Kumar, V. Mitochondrial oxidative stress and dysfunction in arsenic neurotoxicity: A review. J. Appl. Toxicol. 2015, 36, 179–188. [Google Scholar] [CrossRef]
- Schneider, C.B.; Donix, M.; Linse, K.; Werner, A.; Fauser, M.; Klingelhoefer, L.; Löhle, M.; Von Kummer, R.; Reichmann, H.; Storch, A.; et al. Accelerated Age-Dependent Hippocampal Volume Loss in Parkinson Disease with Mild Cognitive Impairment. Am. J. Alzheimer Dis. Other Dement. 2017, 32, 313–319. [Google Scholar] [CrossRef]
- Lamtai, M.; Zghari, O.; Ouakki, S.; Marmouzi, I.; Mesfioui, A.; El Hessni, A.; Ouichou, A. Chronic copper exposure leads to hippocampus oxidative stress and impaired learning and memory in male and female rats. Toxicol. Res. 2020, 36, 359–366. [Google Scholar] [CrossRef]
- Cobley, J.N.; Fiorello, M.L.; Bailey, D.M. 13 reasons why the brain is susceptible to oxidative stress. Redox Biol. 2018, 15, 490–503. [Google Scholar] [CrossRef] [PubMed]
- Rao, R.K.; Li, L.; Baker, R.D.; Baker, S.S.; Gupta, A. Glutathione oxidation and PTPase inhibition by hydrogen peroxide in Caco-2 cell monolayer. Am. J. Physiol. Gastrointest. Liver Physiol. 2000, 279, G332–G340. [Google Scholar] [CrossRef]
- Jaishankar, M.; Tseten, T.; Anbalagan, N.; Mathew, B.B.; Beeregowda, K.N. Toxicity, mechanism and health effects of some heavy metals. Interdiscip. Toxicol. 2014, 7, 60–72. [Google Scholar] [CrossRef] [Green Version]
- Ferruzza, S.; Scacchi, M.; Scarino, M.L.; Sambuy, Y. Iron and copper alter tight junction permeability in human intestinal Caco-2 cells by distinct mechanisms. Toxicol. Vitr. 2002, 16, 399–404. [Google Scholar] [CrossRef]
- Valko, M.; Morris, H.; Cronin, M.T.D. Metals, Toxicity and Oxidative Stress. Curr. Med. Chem. 2005, 12, 1161–1208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, R. Oxidative stress-induced disruption of epithelial and endothelial tight junctions. Front. Biosci. 2008, 13, 7210–7226. [Google Scholar] [CrossRef] [Green Version]
- Bonetto, J.; Villaamil-Lepori, E. Update on the oxidative stress associated with arsenic exposure. Curr. Top. Toxicol. 2014, 10, 37–48. [Google Scholar]
- Flora, G.; Gupta, D.; Tiwari, A. Toxicity of lead: A review with recent updates. Interdiscip. Toxicol. 2012, 5, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Neal, A.P.; Guilarte, T.R. Mechanisms of lead and manganese neurotoxicity. Toxicol. Res. 2013, 2, 99–114. [Google Scholar] [CrossRef]
- Nemmiche, S. Oxidative Signaling Response to Cadmium Exposure. Toxicol. Sci. 2017, 156, 4–10. [Google Scholar] [CrossRef] [Green Version]
- Patra, R.C.; Rautray, A.K.; Swarup, D. Oxidative Stress in Lead and Cadmium Toxicity and Its Amelioration. Vet. Med. Int. 2011, 2011, 457327. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Yalowich, J.C.; Hasinoff, B.B. Cadmium is a catalytic inhibitor of DNA topoisomerase II. J. Inorg. Biochem. 2011, 105, 833–838. [Google Scholar] [CrossRef] [Green Version]
- Branco, V.; Canário, J.; Lu, J.; Holmgren, A.; Carvalho, C. Mercury and selenium interaction in vivo: Effects on thioredoxin reductase and glutathione peroxidase. Free. Radic. Biol. Med. 2011, 52, 781–793. [Google Scholar] [CrossRef]
- Farina, M.; Avila, D.S.; da Rocha, J.B.T.; Aschner, M. Metals, oxidative stress and neurodegeneration: A focus on iron, manganese and mercury. Neurochem. Int. 2012, 62, 575–594. [Google Scholar] [CrossRef] [Green Version]
- Fretham, S.J.B.; Caito, S.; Martinez-Finley, E.J.; Aschner, M. Mechanisms and modifiers of methylmercury-induced neurotoxicity. Toxicol. Res. 2012, 1, 32–38. [Google Scholar] [CrossRef] [Green Version]
- Glaser, V.; Leipnitz, G.; Straliotto, M.R.; de Oliveira, J.; Dos Santos, V.V.; Wannmacher, C.M.D.; De Bem, A.F.; da Rocha, J.B.T.; Farina, M.; Latini, A. Oxidative stress-mediated inhibition of brain creatine kinase activity by methylmercury. Neurotoxicology 2010, 31, 454–460. [Google Scholar] [CrossRef]
- Mazerik, J.N.; Mikkilineni, H.; Kuppusamy, V.A.; Steinhour, E.; Peltz, A.; Marsh, C.B.; Kuppusamy, P.; Parinandi, N.L. Mercury Activates Phospholipase A2and Induces Formation of Arachidonic Acid Metabolites in Vascular Endothelial Cells. Toxicol. Mech. Methods 2007, 17, 541–557. [Google Scholar] [CrossRef] [PubMed]
- Ueda, K.; Makino, R.; Tobe, T.; Okamoto, Y.; Kojima, N. Effects of organic and inorganic mercury(II) on gene expression via DNA conformational changes. Fundam. Toxicol. Sci. 2014, 1, 73–79. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.Y.; Tsao, G.C.; Zhao, Q.; Zheng, W. Differential Cytotoxicity of Mn(II) and Mn(III): Special Reference to Mitochondrial [Fe-S] Containing Enzymes. Toxicol. Appl. Pharmacol. 2001, 175, 160–168. [Google Scholar] [CrossRef] [Green Version]
- Gunter, T.E.; Gerstner, B.; Lester, T.; Wojtovich, A.P.; Malecki, J.; Swarts, S.G.; Brookes, P.S.; Gavin, C.E.; Gunter, K.K. An analysis of the effects of Mn2+ on oxidative phosphorylation in liver, brain, and heart mitochondria using state 3 oxidation rate assays. Toxicol. Appl. Pharmacol. 2010, 249, 65–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harischandra, D.S.; Rokad, D.; Neal, M.L.; Ghaisas, S.; Manne, S.; Sarkar, S.; Panicker, N.; Zenitsky, G.; Jin, H.; Lewis, M.; et al. Manganese promotes the aggregation and prion-like cell-to-cell exosomal transmission of α-synuclein. Sci. Signal. 2019, 12, eaau4543. [Google Scholar] [CrossRef]
- Zheng, W.; Ren, S.; Graziano, J.H. Manganese inhibits mitochondrial aconitase: A mechanism of manganese neurotoxicity1Published on the World Wide Web on 3 June 1998.1. Brain Res. 1998, 799, 334–342. [Google Scholar] [CrossRef]
- Abeyawardhane, D.L.; Lucas, H.R. Iron Redox Chemistry and Implications in the Parkinson’s Disease Brain. Oxidative Med. Cell. Longev. 2019, 2019, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joppe, K.; Roser, A.-E.; Maass, F.; Lingor, P. The Contribution of Iron to Protein Aggregation Disorders in the Central Nervous System. Front. Neurosci. 2019, 13, 15. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z. Iron and oxidizing species in oxidative stress and Alzheimer’s disease. Aging Med. 2019, 2, 82–87. [Google Scholar] [CrossRef]
- Garza-Lombo, C.; Pappa, A.; Panayiotidis, M.I.; Gonsebatt, M.E.; Franco, R. Arsenic-induced neurotoxicity: A mechanistic appraisal. JBIC J. Biol. Inorg. Chem. 2019, 24, 1305–1316. [Google Scholar] [CrossRef]
- Tamás, M.J.; Sharma, S.K.; Ibstedt, S.; Jacobson, T.; Christen, P. Heavy Metals and Metalloids As a Cause for Protein Misfolding and Aggregation. Biomolecules 2014, 4, 252–267. [Google Scholar] [CrossRef] [Green Version]
- Kwon, H.S.; Koh, S.-H. Neuroinflammation in neurodegenerative disorders: The roles of microglia and astrocytes. Transl. Neurodegener. 2020, 9, 42. [Google Scholar] [CrossRef]
- Sanders, K.M.; Ward, S.M. Nitric oxide and its role as a non-adrenergic, non-cholinergic inhibitory neurotransmitter in the gastrointestinal tract. Br. J. Pharmacol. 2019, 176, 212–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedland, R.P.; Chapman, M.R. The role of microbial amyloid in neurodegeneration. PLoS Pathog. 2017, 13, e1006654. [Google Scholar] [CrossRef]
- Tschopp, J.; Schroder, K. NLRP3 inflammasome activation: The convergence of multiple signalling pathways on ROS production? Nat. Rev. Immunol. 2010, 10, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.K.; Menezes, J.S.; Umesaki, Y.; Mazmanian, S.K. Proinflammatory T-cell responses to gut microbiota promote experimental autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. USA 2010, 108, 4615–4622. [Google Scholar] [CrossRef] [Green Version]
- Kustrimovic, N.; Comi, C.; Magistrelli, L.; Rasini, E.; Legnaro, M.; Bombelli, R.; Aleksic, I.; Blandini, F.; Minafra, B.; Riboldazzi, G.; et al. Parkinson’s disease patients have a complex phenotypic and functional Th1 bias: Cross-sectional studies of CD4+ Th1/Th2/T17 and Treg in drug-naïve and drug-treated patients. J. Neuroinflammation 2018, 15, 205. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.-L.; Inserra, A.; Lewis, M.D.; Mastronardi, C.A.; Leong, L.E.X.; Choo, J.; Kentish, S.; Xie, P.; Morrison, M.; Wesselingh, S.L.; et al. Inflammasome signaling affects anxiety- and depressive-like behavior and gut microbiome composition. Mol. Psychiatry 2016, 21, 797–805. [Google Scholar] [CrossRef] [PubMed]
- Banan, A.; Fields, J.Z.; Zhang, Y.; Keshavarzian, A. iNOS upregulation mediates oxidant-induced disruption of F-actin and barrier of intestinal monolayers. Am. J. Physiol. Liver Physiol. 2001, 280, G1234–G1246. [Google Scholar] [CrossRef]
- Byndloss, M.X.; Olsan, E.E.; Rivera-Chávez, F.; Tiffany, C.R.; Cevallos, S.A.; Lokken, K.L.; Torres, T.P.; Byndloss, A.J.; Faber, F.; Gao, Y.; et al. Microbiota-activated PPAR-γ signaling inhibits dysbiotic Enterobacteriaceae expansion. Science 2017, 357, 570–575. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Chen, D.; Zheng, P.; Yu, J.; He, J.; Mao, X.; Yu, B. The Bidirectional Interactions between Resveratrol and Gut Microbiota: An Insight into Oxidative Stress and Inflammatory Bowel Disease Therapy. BioMed Res. Int. 2019, 2019, 5403761. [Google Scholar] [CrossRef]
- Hawkes, C.H.; Del Tredici, K.; Braak, H. A timeline for Parkinson’s disease. Parkinsonism Relat. Disord. 2010, 16, 79–84. [Google Scholar] [CrossRef]
- Dos Santos, A.A.; Ferrer, B.; Gonçalves, F.M.; Tsatsakis, A.M.; Renieri, E.A.; Skalny, A.V.; Farina, M.; Rocha, J.B.T.; Aschner, M. Oxidative Stress in Methylmercury-Induced Cell Toxicity. Toxics 2018, 6, 47. [Google Scholar] [CrossRef] [Green Version]
- Fernandes, J.; Hao, L.; Bijli, K.M.; Chandler, J.D.; Orr, M.; Hu, X.; Jones, D.P.; Go, Y.-M. From the Cover: Manganese Stimulates Mitochondrial H2O2Production in SH-SY5Y Human Neuroblastoma Cells OVER Physiologic as well as Toxicologic Range. Toxicol. Sci. 2017, 155, 213–223. [Google Scholar] [CrossRef] [Green Version]
- Fernandes, J.; Chandler, J.D.; Lili, L.N.; Uppal, K.; Hu, X.; Hao, L.; Go, Y.-M.; Jones, D.P. Transcriptome Analysis Reveals Distinct Responses to Physiologic versus Toxic Manganese Exposure in Human Neuroblastoma Cells. Front. Genet. 2019, 10, 676. [Google Scholar] [CrossRef] [Green Version]
- Sidoryk-Wegrzynowicz, M.; Aschner, M. Manganese toxicity in the central nervous system: The glutamine/glutamate-γ-aminobutyric acid cycle. J. Intern. Med. 2013, 273, 466–477. [Google Scholar] [CrossRef] [Green Version]
- Ke, T.; Sidoryk-Wegrzynowicz, M.; Pajarillo, E.; Rizor, A.; Soares, F.A.A.; Lee, E.; Aschner, M. Role of Astrocytes in Manganese Neurotoxicity Revisited. Neurochem. Res. 2019, 44, 2449–2459. [Google Scholar] [CrossRef] [PubMed]
- Raj, K.; Kaur, P.; Gupta, G.D.; Singh, S. Metals associated neurodegeneration in Parkinson’s disease: Insight to physiological, pathological mechanisms and management. Neurosci. Lett. 2021, 753, 135873. [Google Scholar] [CrossRef]
- Azevedo, B.F.; Furieri, L.B.; Peçanha, F.M.; Wiggers, G.; Vassallo, P.F.; Simões, M.R.; Fiorim, J.; De Batista, P.R.; Fioresi, M.; Rossoni, L.; et al. Toxic Effects of Mercury on the Cardiovascular and Central Nervous Systems. J. Biomed. Biotechnol. 2012, 2012, 949048. [Google Scholar] [CrossRef]
- Segura-Aguilar, J.; Metodiewa, D.; Welch, C.J. Metabolic activation of dopamine o-quinones to o-semiquinones by NADPH cytochrome P450 reductase may play an important role in oxidative stress and apoptotic effects. Biochim. Biophys. Acta Gen. Subj. 1998, 1381, 1–6. [Google Scholar] [CrossRef]
- Desole, M.S.; Esposito, G.; Migheli, R.; Fresu, L.; Sircana, S.; Zangani, D.; Miele, M.; Miele, E. Cellular defence mechanisms in the striatum of young and aged rats subchronically exposed to manganese. Neuropharmacology 1995, 34, 289–295. [Google Scholar] [CrossRef]
- Serra, P.A.; Esposito, G.; Enrico, P.; Mura, M.A.; Migheli, R.; Delogu, M.R.; Miele, M.; Desole, M.S.; Grella, G.; Miele, E. Manganese increases L-DOPA auto-oxidation in the striatum of the freely moving rat: Potential implications to L-DOPA long-term therapy of Parkinson’s disease. Br. J. Pharmacol. 2000, 130, 937–945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desole, M.S.; Sciola, G.L.; Delogu, M.R.; Sircana, S.; Migheli, R. Manganese and 1-methyl-4-(2′-ethylphenyl)-1,2,3,6-tetrahydropyridine induce apoptosis in PC12 cells. Neurosci. Lett. 1996, 209, 193–196. [Google Scholar] [CrossRef]
- Fitsanakis, V.A.; Aschner, M. The importance of glutamate, glycine, and γ-aminobutyric acid transport and regulation in manganese, mercury and lead neurotoxicity. Toxicol. Appl. Pharmacol. 2005, 204, 343–354. [Google Scholar] [CrossRef]
- Racette, B.A.; Nielsen, S.S.; Criswell, S.R.; Sheppard, L.; Seixas, N.; Warden, M.N.; Checkoway, H. Dose-dependent progression of parkinsonism in manganese-exposed welders. Neurology 2017, 88, 344–351. [Google Scholar] [CrossRef] [Green Version]
- Block, M.L.; Zecca, L.; Hong, J.-S. Microglia-mediated neurotoxicity: Uncovering the molecular mechanisms. Nat. Rev. Neurosci. 2007, 8, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, E.C.; Hunot, S. Neuroinflammation in Parkinson’s disease: A target for neuroprotection? Lancet Neurol. 2009, 8, 382–397. [Google Scholar] [CrossRef]
- Tiwari, P.C.; Pal, R. The potential role of neuroinflammation and transcription factors in Parkinson disease. Dialogues Clin. Neurosci. 2017, 19, 71–80. [Google Scholar] [CrossRef]
- Moos, T.; Nielsen, T.R.; Skjørringe, T.; Morgan, E.H. Iron trafficking inside the brain. J. Neurochem. 2007, 103, 1730–1740. [Google Scholar] [CrossRef] [PubMed]
- Neal, A.P.; Guilarte, T.R. Molecular neurobiology of lead (Pb2+): Effects on synaptic function. Mol. Neurobiol. 2010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Florea, A.-M.; Taban, J.; Varghese, E.; Alost, B.T.; Moreno, S.; Büsselberg, D. Lead (Pb2+) neurotoxicity: Ion-mimicry with calcium (Ca2+) impairs synaptic transmission. A review with animated illustrations of the pre- and post-synaptic effects of lead. J. Local Glob. Heal. Sci. 2013. [Google Scholar] [CrossRef]
- Chater, T.E.; Goda, Y. The role of AMPA receptors in postsynaptic mechanisms of synaptic plasticity. Front. Cell. Neurosci. 2014. [Google Scholar] [CrossRef] [PubMed]
- Hanisch, U.-K.; Kettenmann, H. Microglia: Active sensor and versatile effector cells in the normal and pathologic brain. Nat. Neurosci. 2007, 10, 1387–1394. [Google Scholar] [CrossRef] [PubMed]
- Nagatsu, T.; Mogi, M.; Ichinose, H.; Togari, A.; Riederer, P. Cytokines in Parkinson’s disease. Neurosci. News 1999, 58, 143–151. [Google Scholar]
- Kebir, H.; Kreymborg, K.; Ifergan, I.; Dodelet-Devillers, A.; Cayrol, R.; Bernard, M.; Giuliani, F.; Arbour, N.; Becher, B.; Prat, A. Human TH17 lymphocytes promote blood-brain barrier disruption and central nervous system inflammation. Nat. Med. 2007, 13, 1173–1175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dardalhon, V.; Korn, T.; Kuchroo, V.K.; Anderson, A.C. Role of Th1 and Th17 cells in organ-specific autoimmunity. J. Autoimmun. 2008, 31, 252–256. [Google Scholar] [CrossRef] [Green Version]
- Barcia, C.; Ros, F.; Annese, V.; Gómez, C.M.R.; Bernal, F.R.; Aguado-Llera, D.; Pagán, M.E.M.; De Pablos, V.; Villalba, E.F.; Herrero, M.T. IFN-γ signaling, with the synergistic contribution of TNF-α, mediates cell specific microglial and astroglial activation in experimental models of Parkinson’s disease. Cell Death Dis. 2011, 2, e142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, T.; Kai, S.; Matsuyama, T.; Adachi, T.; Fukuda, K.; Hirota, K. General Anesthetics Inhibit LPS-Induced IL-1β Expression in Glial Cells. PLoS ONE 2013, 8, e82930. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Liu, Y.; Zhou, J. Neuroinflammation in Parkinson’s disease and its potential as therapeutic target. Transl. Neurodegener. 2015, 4, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Troncoso-Escudero, P.; Parra, A.; Nassif, M.; Vidal, R.L. Outside in: Unraveling the Role of Neuroinflammation in the Progression of Parkinson’s Disease. Front. Neurol. 2018, 9, 860. [Google Scholar] [CrossRef] [PubMed]
- Pajarillo, E.; Nyarko-Danquah, I.; Adinew, G.; Rizor, A.; Aschner, M.; Lee, E. Neurotoxicity Mechanisms of Manganese in the Central Nervous System; Elsevier: Amsterdam, The Netherlands, 2021. [Google Scholar]
- Li, B.; Xia, M.; Zorec, R.; Parpura, V.; Verkhratsky, A. Astrocytes in heavy metal neurotoxicity and neurodegeneration. Brain Res. 2021, 1752, 147234. [Google Scholar] [CrossRef]
- Johnson, J.; Pajarillo, E.A.B.; Taka, E.; Reams, R.; Son, D.S.; Aschner, M.; Lee, E. Valproate and sodium butyrate attenuate manganese-decreased locomotor activity and astrocytic glutamate transporters expression in mice. Neurotoxicology 2018, 64, 230–239. [Google Scholar] [CrossRef] [Green Version]
- Deng, Y.; Xu, Z.; Xu, B.; Xu, D.; Tian, Y.; Feng, W. The protective effects of riluzole on manganese-induced disruption of glutamate transporters and glutamine synthetase in the cultured astrocytes. Biol. Trace Elem. Res. 2012, 148, 242–249. [Google Scholar] [CrossRef]
- Gu, X.-L.; Long, C.-X.; Sun, L.; Xie, C.; Lin, X.; Cai, H. Astrocytic expression of Parkinson’s disease-related A53T α-synuclein causes neurodegeneration in mice. Mol. Brain 2010, 3, 12. [Google Scholar] [CrossRef] [Green Version]
- Gelders, G.; Baekelandt, V.; Van Der Perren, A. Linking Neuroinflammation and Neurodegeneration in Parkinson’s Disease. J. Immunol. Res. 2018, 2018, 4784268. [Google Scholar] [CrossRef] [Green Version]
- Clarkson, T.W. The three modern faces of mercury. Environ. Health Perspect. 2002, 110, 11–23. [Google Scholar] [CrossRef] [Green Version]
- Rooney, J.P.K. The role of thiols, dithiols, nutritional factors and interacting ligands in the toxicology of mercury. Toxicology 2007, 234, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Atchison, W.D.; Hare, M.F. Mechanisms of methylmercury-induced neurotoxicity. FASEB J. 1994, 8, 622–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olivieri, G.; Brack, C.; Müller-Spahn, F.; Stähelin, H.B.; Herrmann, M.; Renard, P.; Brockhaus, M.; Hock, C. Mercury Induces Cell Cytotoxicity and Oxidative Stress and Increases β-Amyloid Secretion and Tau Phosphorylation in SHSY5Y Neuroblastoma Cells. J. Neurochem. 2000, 74, 231–236. [Google Scholar] [CrossRef]
- Kim, S.H.; Johnson, V.J.; Sharma, R.P. Mercury inhibits nitric oxide production but activates proinflammatory cytokine expression in murine macrophage: Differential modulation of NF-κB and p38 MAPK signaling pathways. Nitric Oxide 2002, 7, 67–74. [Google Scholar] [CrossRef]
- Fonnum, F.; Lock, E.A. The contributions of excitotoxicity, glutathione depletion and DNA repair in chemically induced injury to neurones: Exemplified with toxic effects on cerebellar granule cells. J. Neurochem. 2003, 88, 513–531. [Google Scholar] [CrossRef] [Green Version]
- Castoldi, A.F.; Barni, S.; Turin, I.; Gandini, C.; Manzo, L. Early acute necrosis, delayed apoptosis and cytoskeletal breakdown in cultured cerebellar granule neurons exposed to methylmercury. J. Neurosci. Res. 2000, 59, 775–787. [Google Scholar] [CrossRef]
- Wang, X.; Yan, M.; Zhao, L.; Wu, Q.; Wu, C.; Chang, X.; Zhou, Z. Low-Dose Methylmercury-Induced Apoptosis and Mitochondrial DNA Mutation in Human Embryonic Neural Progenitor Cells. Oxidative Med. Cell. Longev. 2016, 2016, 5137042. [Google Scholar] [CrossRef] [Green Version]
- Perry, T.L.; Godin, D.V.; Hansen, S. Parkinson’s disease: A disorder due to nigral glutathione deficiency? Neurosci. Lett. 1982, 33, 305–310. [Google Scholar] [CrossRef]
- Serru, V.; Baudin, B.; Ziegler, F.; David, J.-P.; Cals, M.-J.; Vaubourdolle, M.; Mario, N. Quantification of Reduced and Oxidized Glutathione in Whole Blood Samples by Capillary Electrophoresis. Clin. Chem. 2001, 47, 1321–1324. [Google Scholar] [CrossRef]
- Hsu, M.; Srinivas, B.; Kumar, J.K.; Subramanian, R.; Andersen, J. Glutathione depletion resulting in selective mitochondrial complex I inhibition in dopaminergic cells is via an NO-mediated pathway not involving peroxynitrite: Implications for Parkinson’s disease: Glutathione Affects Mitochondrial Complex I via NO Not ONOO. J. Neurochem. 2005, 92, 1091–1103. [Google Scholar] [CrossRef]
- Ballatori, N.; Krance, S.M.; Notenboom, S.; Shi, S.; Tieu, K.; Hammond, C.L. Glutathione dysregulation and the etiology and progression of human diseases. Biol. Chem. 2009, 390, 191–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Götz, M.E.; Double, K.; Gerlach, M.; Youdim, M.B.H.; Riederere, P. The Relevance of Iron in the Pathogenesis of Parkinson’s Disease. Ann. N. Y. Acad. Sci. 2004, 1012, 193–208. [Google Scholar] [CrossRef]
- Hare, D.J.; Lei, P.; Ayton, S.; Roberts, B.R.; Grimm, R.; George, J.L.; Bishop, D.P.; Beavis, A.D.; Donovan, S.J.; McColl, G.; et al. An iron–dopamine index predicts risk of parkinsonian neurodegeneration in the substantia nigra pars compacta. Chem. Sci. 2014, 5, 2160–2169. [Google Scholar] [CrossRef] [Green Version]
- Hare, D.J.; Double, K.L. Iron and dopamine: A toxic couple. Brain 2016, 139, 1026–1035. [Google Scholar] [CrossRef] [PubMed]
- Guiney, S.J.; Adlard, P.A.; Bush, A.I.; Finkelstein, D.I.; Ayton, S. Ferroptosis and cell death mechanisms in Parkinson’s disease. Neurochem. Int. 2017, 104, 34–48. [Google Scholar] [CrossRef]
- Ma, L.; Azad, M.G.; Dharmasivam, M.; Richardson, V.; Quinn, R.J.; Feng, Y.; Pountney, D.L.; Tonissen, K.F.; Mellick, G.D.; Yanatori, I.; et al. Parkinson’s disease: Alterations in iron and redox biology as a key to unlock therapeutic strategies. Redox Biol. 2021, 41, 101896. [Google Scholar] [CrossRef]
- Collins, L.M.; Toulouse, A.; Connor, T.J.; Nolan, Y.M. Contributions of central and systemic inflammation to the pathophysiology of Parkinson’s disease. Neuropharmacology 2012, 62, 2154–2168. [Google Scholar] [CrossRef] [Green Version]
- Hare, D.J.; Ayton, S.; Bush, A.I.; Lei, P. A delicate balance: Iron metabolism and diseases of the brain. Front. Aging Neurosci. 2013, 5, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urrutia, P.; Aguirre, P.; Esparza, A.; Tapia, V.; Mena, N.P.; Arredondo, M.; González-Billault, C.; Núñez, M.T. Inflammation alters the expression of DMT1, FPN1 and hepcidin, and it causes iron accumulation in central nervous system cells. J. Neurochem. 2013, 126, 541–549. [Google Scholar] [CrossRef] [PubMed]
- Niranjan, R. The Role of Inflammatory and Oxidative Stress Mechanisms in the Pathogenesis of Parkinson’s Disease: Focus on Astrocytes. Mol. Neurobiol. 2013, 49, 28–38. [Google Scholar] [CrossRef]
- Lian, T.-H.; Guo, P.; Zuo, L.-J.; Hu, Y.; Yu, S.-Y.; Yu, Q.-J.; Jin, Z.; Wang, R.-D.; Li, L.-X.; Zhang, W. Tremor-Dominant in Parkinson Disease: The Relevance to Iron Metabolism and Inflammation. Front. Neurosci. 2019, 13, 255. [Google Scholar] [CrossRef]
- Kortekaas, R.; Leenders, K.L.; Van Oostrom, J.C.H.; Vaalburg, W.; Bart, J.; Willemsen, A.T.M.; Hendrikse, N.H. Blood-brain barrier dysfunction in parkinsonian midbrain in vivo. Ann. Neurol. 2005, 57, 176–179. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.T.; Dong, M.H.; Zhang, J.Q.; Bai, Y.; Kuang, F.; Chen, L.W. Microglia and astroglia: The role of neuroinflammation in lead toxicity and neuronal injury in the brain. Neuroimmunol. Neuroinflamm. 2015, 2, 131–137. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Su, P.; Meng, S.; Aschner, M.; Cao, Y.; Luo, W.; Zheng, G.; Liu, M. Role of matrix metalloproteinase-2/9 (MMP2/9) in lead-induced changes in an in vitro blood-brain barrier model. Int. J. Biol. Sci. 2017, 13, 1351–1360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mangia, S.; Simpson, I.A.; Vannucci, S.J.; Carruthers, A. The in vivo neuron-to-astrocyte lactate shuttle in human brain: Evidence from modeling of measured lactate levels during visual stimulation. J. Neurochem. 2009, 109, 55–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jozefczak, M.; Remans, T.; Vangronsveld, J.; Cuypers, A. Glutathione Is a Key Player in Metal-Induced Oxidative Stress Defenses. Int. J. Mol. Sci. 2012, 13, 3145–3175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabezas, R.; Fidel, M.; Torrente, D.; El-Bach, R.S.; Morales, L.; Gonzalez, J.; Barreto, G.E. Astrocytes Role in Parkinson: A Double-Edged Sword. Neurodegener. Dis. 2013. [Google Scholar] [CrossRef] [Green Version]
- Kiela, P.R.; Ghishan, F.K. Physiology of Intestinal Absorption and Secretion. Best Pract. Res. Clin. Gastroenterol. 2016, 30, 145–159. [Google Scholar] [CrossRef] [Green Version]
- Onakpa, M.M.; Njan, A.A.; Kalu, O.C. A Review of Heavy Metal Contamination of Food Crops in Nigeria. Ann. Glob. Health 2018, 84, 488–494. [Google Scholar] [CrossRef] [Green Version]
- Chi, L.; Bian, X.; Gao, B.; Tu, P.; Ru, H.; Lu, K. The Effects of an Environmentally Relevant Level of Arsenic on the Gut Microbiome and Its Functional Metagenome. Toxicol. Sci. 2017, 160, 193–204. [Google Scholar] [CrossRef]
- Sears, M.E. Chelation: Harnessing and Enhancing Heavy Metal Detoxification—A Review. Sci. World J. 2013, 2013, 219840. [Google Scholar] [CrossRef] [Green Version]
- Bisanz, J.E.; Enos, M.K.; Mwanga, J.R.; Changalucha, J.P.; Burton, J.; Gloor, G.B.; Reid, G. Randomized Open-Label Pilot Study of the Influence of Probiotics and the Gut Microbiome on Toxic Metal Levels in Tanzanian Pregnant Women and School Children. mBio 2014, 5, e01580-14. [Google Scholar] [CrossRef] [Green Version]
- Astolfi, M.L.; Protano, C.; Schiavi, E.; Marconi, E.; Capobianco, D.; Massimi, L.; Ristorini, M.; Baldassarre, M.E.; Laforgia, N.; Vitali, M.; et al. A prophylactic multi-strain probiotic treatment to reduce the absorption of toxic elements: In-vitro study and biomonitoring of breast milk and infant stools. Environ. Int. 2019, 130, 104818. [Google Scholar] [CrossRef] [PubMed]
- Raghuvanshi, R.; Chaudhari, A.; Kumar, G.N. Amelioration of cadmium- and mercury-induced liver and kidney damage in rats by genetically engineered probiotic Escherichia coli Nissle 1917 producing pyrroloquinoline quinone with oral supplementation of citric acid. Nutrition 2016, 32, 1285–1294. [Google Scholar] [CrossRef] [PubMed]
- Zeitoun, M.; Al-Damegh, M.; Abdel-Salam, A. The Role of Fermented Milk Containing Probiotic, Dandelion as Prebiotic or their Combination on Serum Metabolites, Enzymes, Testosterone and Testicular Histopathology of Arsenic-Intoxicated Male Rats. J. Basic Appl. Sci. 2014, 10, 492–503. [Google Scholar] [CrossRef]
- Gerbino, E.; Carasi, P.; Tymczyszyn, E.E.; Gómez-Zavaglia, A. Removal of cadmium by Lactobacillus kefir as a protective tool against toxicity. J. Dairy Res. 2014, 81, 280–287. [Google Scholar] [CrossRef] [PubMed]
- Jama, A.M.; Ćulafić, D.M.; Kolarevic, S.; Djurasevic, S.; Knezevic-Vukcevic, J. Protective effect of probiotic bacteria against cadmium-induced genotoxicity in rat hepatocytes in vivo and in vitro. Arch. Biol. Sci. 2012, 64, 1197–1206. [Google Scholar] [CrossRef]
- Djurasevic, S.; Jama, A.; Jasnic, N.; Vujovic, P.; Jovanovic, M.; Ćulafić, D.M.; Knežević-Vukčević, J.; Cakic-Milosevic, M.; Ilijevic, K.; Djordjevic, J. The Protective Effects of Probiotic Bacteria on Cadmium Toxicity in Rats. J. Med. Food 2017, 20, 189–196. [Google Scholar] [CrossRef]
- Song, S.; Oh, S.; Lim, K.-T. Lactobacillus plantarum L67 glycoprotein protects against cadmium chloride toxicity in RAW 264.7 clls. J. Dairy Sci. 2016, 99, 1812–1821. [Google Scholar] [CrossRef] [Green Version]
- Zhai, Q.; Tian, F.; Zhao, J.; Zhang, H.; Narbad, A.; Chen, W. Oral Administration of Probiotics Inhibits Absorption of the Heavy Metal Cadmium by Protecting the Intestinal Barrier. Appl. Environ. Microbiol. 2016, 82, 4429–4440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abd-El-Moneim, O.M.; Abd El-Kader, H.A.; Abd El-Rahim, A.H.; Radwan, H.A.; Fadel, M.; Farag, I.M. Modulatory role of Saccharomyces cerevisiae against cadmium-induced genotoxicity in mice. J. Arab Soc. Med. Res. 2017, 12, 27–38. [Google Scholar] [CrossRef]
- Jafarpour, D.; Shekarforoush, S.S.; Ghaisari, H.R.; Nazifi, S.; Sajedianfard, J.; Eskandari, M.H. Protective effects of synbiotic diets of Bacillus coagulans, Lactobacillus plantarum and inulin against acute cadmium toxicity in rats. BMC Complement. Altern. Med. 2017, 17, 291. [Google Scholar] [CrossRef] [PubMed]
- Kadry, M.O.; Megeed, R.A. Probiotics as a Complementary Therapy in the Model of Cadmium Chloride Toxicity: Crosstalk of β-Catenin, BDNF, and StAR Signaling Pathways. Biol. Trace Elem. Res. 2018, 185, 404–413. [Google Scholar] [CrossRef] [PubMed]
- Allam, N.; Ali, E.; Shabanna, S.; Abd-Elrahman, E. Protective efficacy of Streptococcus thermophilus against acute cadmium toxicity in mice. Iran. J. Pharm. Res. 2018, 17, 695–707. [Google Scholar] [CrossRef]
- Wang, N.; Jiang, M.; Zhang, P.; Shu, H.; Li, Y.; Guo, Z.; Li, Y. Amelioration of Cd-induced bioaccumulation, oxidative stress and intestinal microbiota by Bacillus cereus in Carassius auratus gibelio. Chemosphere 2020, 245, 125613. [Google Scholar] [CrossRef]
- Zhai, Q.; Yu, L.; Li, T.; Zhu, J.; Zhang, C.; Zhao, J.; Zhang, H.; Chen, W. Effect of dietary probiotic supplementation on intestinal microbiota and physiological conditions of Nile tilapia (Oreochromis niloticus) under waterborne cadmium exposure. Antonie van Leeuwenhoek Int. J. Gen. Mol. Microbiol. 2017, 110, 501–513. [Google Scholar] [CrossRef] [PubMed]
- Abu-Braka, A.Z.; Zaki, M.S.; Abbas, H.H.; Ismail, N.E.D.A.; Khalil, R.; Tanekhy, M.; Saad, T. Filed studies on some probiotics to minimize hazard effects of prevailing heavy metals contamination for improving immunity and growth performance of Oreochromis niloticus. Electron. Physician 2017, 9, 4138. [Google Scholar] [CrossRef] [Green Version]
- Feng, S.; Liu, Y.; Huang, Y.; Zhao, J.; Zhang, H.; Zhai, Q.; Chen, W. Influence of oral administration of Akkermansia muciniphila on the tissue distribution and gut microbiota composition of acute and chronic cadmium exposure mice. FEMS Microbiol. Lett. 2019, 366, fnz160. [Google Scholar] [CrossRef]
- Tian, F.; Zhai, Q.; Zhao, J.; Liu, X.; Wang, G.; Zhang, H.; Zhang, H.; Chen, W. Lactobacillus plantarum CCFM8661 Alleviates Lead Toxicity in Mice. Biol. Trace Elem. Res. 2012, 150, 264–271. [Google Scholar] [CrossRef]
- Zhai, Q.; Liu, Y.; Wang, C.; Qu, D.; Zhao, J.; Zhang, H.; Tian, F.; Chen, W. Lactobacillus plantarum CCFM8661 modulates bile acid enterohepatic circulation and increases lead excretion in mice. Food Funct. 2019, 10, 1455–1464. [Google Scholar] [CrossRef]
- Zhai, Q.; Qu, D.; Feng, S.; Yu, Y.; Yu, L.; Tian, F.; Zhao, J.; Zhang, H.; Chen, W. Oral Supplementation of Lead-Intolerant Intestinal Microbes Protects Against Lead (Pb) Toxicity in Mice. Front. Microbiol. 2020, 10, 3161. [Google Scholar] [CrossRef]
- Zhai, Q.; Wang, G.; Zhao, J.; Liu, X.; Tian, F.; Zhang, H.; Chen, W. Protective Effects of Lactobacillus plantarum CCFM8610 against Acute Cadmium Toxicity in Mice. Appl. Environ. Microbiol. 2013, 79, 1508–1515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giri, S.S.; Yun, S.; Jun, J.W.; Kim, H.J.; Kim, S.G.; Kang, J.W.; Kim, S.W.; Han, S.J.; Sukumaran, V.; Park, S.C. Therapeutic Effect of Intestinal Autochthonous Lactobacillus reuteri P16 Against Waterborne Lead Toxicity in Cyprinus carpio. Front. Immunol. 2018, 9, 1824. [Google Scholar] [CrossRef]
- Ghenioa, A.; Okle, O.; Nazem, A.; Ashry, K.M. Protective Effect of Probiotic Bactosac Against Induced Sub Chronic Lead Toxicity in Broiler Chicks. Alex. J. Vet. Sci. 2015, 47, 53–64. [Google Scholar] [CrossRef]
- Li, B.; Jin, D.; Yu, S.; Evivie, S.E.; Muhammad, Z.; Huo, G.; Liu, F. In Vitro and In Vivo Evaluation of Lactobacillus delbrueckii subsp. bulgaricus KLDS1.0207 for the Alleviative Effect on Lead Toxicity. Nutrients 2017, 9, 845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, X.; Gu, S.; Liu, D.; Zhao, L.; Xia, S.; He, X.; Chen, H.; Ge, J. Lactobacillus brevis 23017 Relieves Mercury Toxicity in the Colon by Modulation of Oxidative Stress and Inflammation Through the Interplay of MAPK and NF-κB Signaling Cascades. Front. Microbiol. 2018, 9, 2425. [Google Scholar] [CrossRef] [PubMed]
- Majlesi, M.; Shekarforoush, S.S.; Ghaisari, H.R.; Nazifi, S.; Sajedianfard, J.; Eskandari, M.H. Effect of Probiotic Bacillus Coagulans and Lactobacillus Plantarum on Alleviation of Mercury Toxicity in Rat. Probiotics Antimicrob. Proteins 2017, 9, 300–309. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Salam, A.M.; Al-Dekheil, A.; Babkr, A.; Farahna, M.; Mousa, H.M. High fiber probiotic fermented mare’s milk reduces the toxic effects of mercury in rats. N. Am. J. Med Sci. 2010, 2, 569–575. [Google Scholar] [CrossRef] [PubMed]
- Shao, M.; Zhu, Y. Long-term metal exposure changes gut microbiota of residents surrounding a mining and smelting area. Sci. Rep. 2020, 10, 4453. [Google Scholar] [CrossRef] [PubMed]
- Hattori, Y.; Naito, M.; Satoh, M.; Nakatochi, M.; Naito, H.; Kato, M.; Takagi, S.; Matsunaga, T.; Seiki, T.; Sasakabe, T.; et al. MetallothioneinMT2AA-5G Polymorphism as a Risk Factor for Chronic Kidney Disease and Diabetes: Cross-Sectional and Cohort Studies. Toxicol. Sci. 2016, 152, 181–193. [Google Scholar] [CrossRef] [Green Version]
- Nair, A.R.; Lee, W.-K.; Smeets, K.; Swennen, Q.; Sanchez, A.; Thévenod, F.; Cuypers, A. Glutathione and mitochondria determine acute defense responses and adaptive processes in cadmium-induced oxidative stress and toxicity of the kidney. Arch. Toxicol. 2014, 89, 2273–2289. [Google Scholar] [CrossRef]
- Mozzi, F.; Raya, R.R.; Vignolo, G.M. Biotechnology of Lactic Acid Bacteria: Novel Applications; John Wiley & Sons: Hoboken, NJ, USA, 2010; ISBN 9780813815831. [Google Scholar]
- Wedajo, B. Lactic Acid Bacteria: Benefits, Selection Criteria and Probiotic Potential in Fermented Food. J. Probiotics Health 2015, 3, 1–9. [Google Scholar] [CrossRef]
- Ameen, F.A.; Hamdan, A.M.; El-Naggar, M.Y. Assessment of the heavy metal bioremediation efficiency of the novel marine lactic acid bacterium, Lactobacillus plantarum MF042018. Sci. Rep. 2020, 10, 314. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Xiao, X.; Feng, P.; Xie, F.; Yu, Z.; Yuan, W.; Liu, P.; Li, X. Gut remediation: A potential approach to reducing chromium accumulation using Lactobacillus plantarum TW1-1. Sci. Rep. 2017, 7, 15000. [Google Scholar] [CrossRef] [Green Version]
- Chandran, H.; Meena, M.; Sharma, K. Microbial Biodiversity and Bioremediation Assessment through Omics Approaches. Front. Environ. Chem. 2020, 1, 9. [Google Scholar] [CrossRef]
- Cassani, E.; Privitera, G.; Pezzoli, G.; Pusani, C.; Madio, C.; Iorio, L.; Barichella, M. Use of probiotics for the treatment of constipation in Parkinson’s disease patients. Minerva Gastroenterol. Dietol. 2011, 57, 117–121. [Google Scholar] [PubMed]
- Tamtaji, O.R.; Taghizadeh, M.; Kakhaki, R.D.; Kouchaki, E.; Bahmani, F.; Borzabadi, S.; Oryan, S.; Mafi, A.; Asemi, Z. Clinical and metabolic response to probiotic administration in people with Parkinson’s disease: A randomized, double-blind, placebo-controlled trial. Clin. Nutr. 2019, 38, 1031–1035. [Google Scholar] [CrossRef]
- Tamtaji, O.R.; Kouchaki, E.; Salami, M.; Aghadavod, E.; Akbari, E.; Tajabadi-Ebrahimi, M.; Asemi, Z. The Effects of Probiotic Supplementation on Gene Expression Related to Inflammation, Insulin, and Lipids in Patients With Multiple Sclerosis: A Randomized, Double-Blind, Placebo-Controlled Trial. J. Am. Coll. Nutr. 2017, 36, 660–665. [Google Scholar] [CrossRef]
- Georgescu, D.; Ancusa, O.E.; Georgescu, L.A.; Ionita, I.; Reisz, D. Nonmotor gastrointestinal disorders in older patients with Parkinson’s disease: Is there hope? Clin. Interv. Aging 2016, 11, 1601–1608. [Google Scholar] [CrossRef] [Green Version]
- Barichella, M.; Pacchetti, C.; Bolliri, C.; Cassani, E.; Iorio, L.; Pusani, C.; Pinelli, G.; Privitera, G.; Cesari, I.; Faierman, S.A.; et al. Probiotics and prebiotic fiber for constipation associated with Parkinson disease. Neurology 2016, 87, 1274–1280. [Google Scholar] [CrossRef]
- Magistrelli, L.; Amoruso, A.; Mogna, L.; Graziano, T.; Cantello, R.; Pane, M.; Comi, C. Probiotics May Have Beneficial Effects in Parkinson’s Disease: In vitro Evidence. Front. Immunol. 2019, 10, 969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srivastav, S.; Neupane, S.; Bhurtel, S.; Katila, N.; Maharjan, S.; Choi, H.; Hong, J.T.; Choi, D.-Y. Probiotics mixture increases butyrate, and subsequently rescues the nigral dopaminergic neurons from MPTP and rotenone-induced neurotoxicity. J. Nutr. Biochem. 2019, 69, 73–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsieh, T.-H.; Kuo, C.-W.; Hsieh, K.-H.; Shieh, M.-J.; Peng, C.-W.; Chen, Y.-C.; Chang, Y.-L.; Huang, Y.-Z.; Chen, C.-C.; Chang, P.-K.; et al. Probiotics Alleviate the Progressive Deterioration of Motor Functions in a Mouse Model of Parkinson’s Disease. Brain Sci. 2020, 10, 206. [Google Scholar] [CrossRef] [Green Version]
- Sjögren, Y.M.; Tomicic, S.; Lundberg, A.; Böttcher, M.F.; Björkstén, B.; Sverremark-Ekström, E.; Jenmalm, M.C. Influence of early gut microbiota on the maturation of childhood mucosal and systemic immune responses. Gut microbiota and immune responses. Clin. Exp. Allergy 2009, 39, 1842–1851. [Google Scholar] [CrossRef] [Green Version]
- Fujimura, K.E.; Lynch, S.V. Microbiota in Allergy and Asthma and the Emerging Relationship with the Gut Microbiome. Cell Host Microbe 2015, 17, 592–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zoghi, A.; Khosravi-Darani, K.; Sohrabvandi, S. Surface Binding of Toxins and Heavy Metals by Probiotics. Mini-Rev. Med. Chem. 2014, 14, 84–98. [Google Scholar] [CrossRef] [PubMed]
- Pieper, D.H.; Reineke, W. Engineering bacteria for bioremediation. Curr. Opin. Biotechnol. 2000, 11, 262–270. [Google Scholar] [CrossRef]
- Bhakta, J.N.; Ohnishi, K.; Munekage, Y.; Iwasaki, K.; Wei, M.Q. Characterization of lactic acid bacteria-based probiotics as potential heavy metal sorbents. J. Appl. Microbiol. 2012, 112, 1193–1206. [Google Scholar] [CrossRef]
- Patel, A.; SV, A.; Shah, N.; Verma, D.K. Lactic Acid Bacteria As Metal Quenchers To Improve Food Safety and Quality. AgroLife 2017.
- Halttunen, T.; Collado, M.C.; El-Nezami, H.; Meriluoto, J.; Salminen, S. Combining strains of lactic acid bacteria may reduce their toxin and heavy metal removal efficiency from aqueous solution. Lett. Appl. Microbiol. 2008, 46, 160–165. [Google Scholar] [CrossRef]
- Monachese, M.; Burton, J.P.; Reid, G. Bioremediation and tolerance of humans to heavy metals through microbial processes: A potential role for probiotics? Appl. Environ. Microbiol. 2012, 78, 6397–6404. [Google Scholar] [CrossRef] [Green Version]
- Beeler, E.; Singh, O.V. Extremophiles as sources of inorganic bio-nanoparticles. World J. Microbiol. Biotechnol. 2016, 32, 156. [Google Scholar] [CrossRef]
- De Vadder, F.; Kovatcheva-Datchary, P.; Goncalves, D.; Vinera, J.; Zitoun, C.; Duchampt, A.; Bäckhed, F.; Mithieux, G. Microbiota-Generated Metabolites Promote Metabolic Benefits via Gut-Brain Neural Circuits. Cell 2014, 156, 84–96. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Li, Y.; Liu, K.; Shen, J. Exposing to Cadmium Stress Cause Profound Toxic Effect on Microbiota of the Mice Intestinal Tract. PLoS ONE 2014, 9, e85323. [Google Scholar] [CrossRef]
- Roberfroid, M.; Gibson, G.R.; Hoyles, L.; McCartney, A.L.; Rastall, R.; Rowland, I.; Wolvers, D.; Watzl, B.; Szajewska, H.; Stahl, B.; et al. Prebiotic effects: Metabolic and health benefits. Br. J. Nutr. 2010, 104, S1–S63. [Google Scholar] [CrossRef] [Green Version]
- Tian, F.; Xiao, Y.; Li, X.; Zhai, Q.; Wang, G.; Zhang, Q.; Zhang, H.; Chen, W. Protective Effects of Lactobacillus plantarum CCFM8246 against Copper Toxicity in Mice. PLoS ONE 2015, 10, e0143318. [Google Scholar] [CrossRef]
- Zhai, Q.; Wang, G.; Zhao, J.; Liu, X.; Narbad, A.; Chen, Y.Q.; Zhang, H.; Tian, F.; Chen, W. Protective Effects of Lactobacillus plantarum CCFM8610 against Chronic Cadmium Toxicity in Mice Indicate Routes of Protection besides Intestinal Sequestration. Appl. Environ. Microbiol. 2014, 80, 4063–4071. [Google Scholar] [CrossRef] [Green Version]
- Walter, J. Ecological Role of Lactobacilli in the Gastrointestinal Tract: Implications for Fundamental and Biomedical Research. Appl. Environ. Microbiol. 2008, 74, 4985–4996. [Google Scholar] [CrossRef] [Green Version]
- D’Antona, G.; Ragni, M.; Cardile, A.; Tedesco, L.; Dossena, M.; Bruttini, F.; Caliaro, F.; Corsetti, G.; Bottinelli, R.; Carruba, M.O.; et al. Branched-Chain Amino Acid Supplementation Promotes Survival and Supports Cardiac and Skeletal Muscle Mitochondrial Biogenesis in Middle-Aged Mice. Cell Metab. 2010, 12, 362–372. [Google Scholar] [CrossRef] [Green Version]
- Valerio, A.; D’Antona, G.; Nisoli, E. Branched-chain amino acids, mitochondrial biogenesis, and healthspan: An evolutionary perspective. Aging 2011, 3, 464–478. [Google Scholar] [CrossRef] [Green Version]
- Sridharan, G.V.; Choi, K.; Klemashevich, C.; Wu, C.; Prabakaran, D.; Bin Pan, L.; Steinmeyer, S.; Mueller, C.; Yousofshahi, M.; Alaniz, R.C.; et al. Prediction and quantification of bioactive microbiota metabolites in the mouse gut. Nat. Commun. 2014, 5, 5492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliphant, K.; Allen-Vercoe, E. Macronutrient metabolism by the human gut microbiome: Major fermentation by-products and their impact on host health. Microbiome 2019, 7, 91. [Google Scholar] [CrossRef] [PubMed]
- Novarino, G.; El-Fishawy, P.; Kayserili, H.; Meguid, N.A.; Scott, E.M.; Schroth, J.; Silhavy, J.L.; Kara, M.; Khalil, R.O.; Ben-Omran, T.; et al. Mutations in BCKD-kinase Lead to a Potentially Treatable Form of Autism with Epilepsy. Science 2012, 338, 394–397. [Google Scholar] [CrossRef] [Green Version]
- Mally, J.; Szalai, G.; Stone, T.W. Changes in the concentration of amino acids in serum and cerebrospinal fluid of patients with Parkinson’s disease. J. Neurol. Sci. 1997, 151, 159–162. [Google Scholar] [CrossRef]
- Molina, J.A.; Jiménez-Jiménez, F.J.; Gómez, P.; Vargas, C.; Navarro, J.A.; Ortí-Pareja, M.; Gasalla, T.; Benito-León, J.; Bermejo, F.; Arenas, J. Decreased cerebrospinal fluid levels of neutral and basic amino acids in patients with Parkinson’s disease. J. Neurol. Sci. 1997, 150, 123–127. [Google Scholar] [CrossRef]
- Yan, Z.; Yang, F.; Cao, J.; Ding, W.; Yan, S.; Shi, W.; Wen, S.; Yao, L. Alterations of gut microbiota and metabolome with Parkinson’s disease. Microb. Pathog. 2021, 160, 105187. [Google Scholar] [CrossRef] [PubMed]
- Wuolikainen, A.; Jonsson, P.; Ahnlund, M.; Antti, H.; Marklund, S.L.; Moritz, T.; Forsgren, L.; Andersen, P.M.; Trupp, M. Multi-platform mass spectrometry analysis of the CSF and plasma metabolomes of rigorously matched amyotrophic lateral sclerosis, Parkinson’s disease and control subjects. Mol. BioSyst. 2016, 12, 1287–1298. [Google Scholar] [CrossRef]
- Bravo, J.A.; Forsythe, P.; Chew, M.V.; Escaravage, E.; Savignac, H.M.; Dinan, T.G.; Bienenstock, J.; Cryan, J.F. Ingestion of Lactobacillus strain regulates emotional behavior and central GABA receptor expression in a mouse via the vagus nerve. Proc. Natl. Acad. Sci. USA 2011, 108, 16050–16055. [Google Scholar] [CrossRef] [Green Version]
- Zheng, H.; Xu, P.; Jiang, Q.; Xu, Q.; Zheng, Y.; Yan, J.; Ji, H.; Ning, J.; Zhang, X.; Li, C.; et al. Correction to: Depletion of acetate-producing bacteria from the gut microbiota facilitates cognitive impairment through the gut-brain neural mechanism in diabetic mice. Microbiome 2021, 9, 164. [Google Scholar] [CrossRef]
- Shehata, M.G.; El Sohaimy, S.A.; El-Sahn, M.A.; Youssef, M.M. Screening of isolated potential probiotic lactic acid bacteria for cholesterol lowering property and bile salt hydrolase activity. Ann. Agric. Sci. 2016, 61, 65–75. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Marcelin, G.; Blouet, C.; Jeong, J.H.; Jo, Y.-H.; Schwartz, G.J.; Chua, S. A gut–brain axis regulating glucose metabolism mediated by bile acids and competitive fibroblast growth factor actions at the hypothalamus. Mol. Metab. 2017, 8, 37–50. [Google Scholar] [CrossRef]
- Lyte, M. Host-microbiota neuroendocrine interactions influencing brain and behavior. Gut Microbes 2014, 5, 381–389. [Google Scholar] [CrossRef] [Green Version]
- Erikson, K.M.; Aschner, M. Manganese neurotoxicity and glutamate-GABA interaction. Neurochem. Int. 2003, 43, 475–480. [Google Scholar] [CrossRef]
- Nyarko-Danquah, I.; Pajarillo, E.; Digman, A.; Soliman, K.F.A.; Aschner, M.; Lee, E. Manganese Accumulation in the Brain via Various Transporters and Its Neurotoxicity Mechanisms. Molecules 2020, 25, 5880. [Google Scholar] [CrossRef]
- Gros, D.F.; Antony, M.M.; McCabe, R.E.; Swinson, R.P. Frequency and severity of the symptoms of irritable bowel syndrome across the anxiety disorders and depression. J. Anxiety Disord. 2009, 23, 290–296. [Google Scholar] [CrossRef]
- Kim, S.-H.; Field, K.G.; Morrissey, M.T.; Price, R.J.; Wei, C.-I.; An, H. Source and Identification of Histamine-Producing Bacteria from Fresh and Temperature-Abused Albacore. J. Food Prot. 2001, 64, 1035–1044. [Google Scholar] [CrossRef]
- Smolinska, S.; Jutel, M.; Crameri, R.; O’Mahony, L. Histamine and gut mucosal immune regulation. Allergy 2013, 69, 273–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferstl, R.; Frei, R.; Schiavi, E.; Konieczna, P.; Barcik, W.; Ziegler, M.; Lauener, R.P.; Chassard, C.; Lacroix, C.; Akdis, C.A.; et al. Histamine receptor 2 is a key influence in immune responses to intestinal histamine-secreting microbes. J. Allergy Clin. Immunol. 2014, 134, 744–746. [Google Scholar] [CrossRef] [PubMed]
- Karri, V.; Schuhmacher, M.; Kumar, V. A systems toxicology approach to compare the heavy metal mixtures (Pb, As, MeHg) impact in neurodegenerative diseases. Food Chem. Toxicol. 2020, 139, 111257. [Google Scholar] [CrossRef]
- Stilling, R.M.; Moloney, G.M.; Ryan, F.J.; Hoban, A.E.; Bastiaanssen, T.F.S.; Shanahan, F.; Clarke, G.; Claesson, M.J.; Dinan, T.G.; Cryan, J.F. Social interaction-induced activation of RNA splicing in the amygdala of microbiome-deficient mice. eLife 2018, 7, e33070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tilocca, B.; Pieroni, L.; Soggiu, A.; Britti, D.; Bonizzi, L.; Roncada, P.; Greco, V. Gut–Brain Axis and Neurodegeneration: State-of-the-Art of Meta-Omics Sciences for Microbiota Characterization. Int. J. Mol. Sci. 2020, 21, 4045. [Google Scholar] [CrossRef]
- Wanichthanarak, K.; Fahrmann, J.F.; Grapov, D. Genomic, Proteomic, and Metabolomic Data Integration Strategies. Biomark. Insights 2015, 10s4, BMI.S29511–BMI.S29516. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Shimoji, M.; Thomas, B.; Moore, D.J.; Yu, S.-W.; Marupudi, N.I.; Torp, R.; Torgner, I.A.; Ottersen, O.P.; Dawson, T.M.; et al. Mitochondrial localization of the Parkinson’s disease related protein DJ-1: Implications for pathogenesis. Hum. Mol. Genet. 2005, 14, 2063–2073. [Google Scholar] [CrossRef] [Green Version]
- Schellenberger, J.; Que, R.; Fleming, R.M.T.; Thiele, I.; Orth, J.D.; Feist, A.M.; Zielinski, D.C.; Bordbar, A.; Lewis, N.E.; Rahmanian, S.; et al. Quantitative prediction of cellular metabolism with constraint-based models: The COBRA Toolbox v2.0. Nat. Protoc. 2011, 6, 1290–1307. [Google Scholar] [CrossRef] [Green Version]
- Mo, M.L.; Jamshidi, N.; Palsson, B. A genome-scale, constraint-based approach to systems biology of human metabolism. Mol. BioSyst. 2007, 3, 598–603. [Google Scholar] [CrossRef]
- Orth, J.D.; Thiele, I.; Palsson, B.Ø. What is flux balance analysis? Nat. Biotechnol. 2010, 28, 245–248. [Google Scholar] [CrossRef]
- Thiele, I.; Sahoo, S.; Heinken, A.; Hertel, J.; Heirendt, L.; Aurich, M.K.; Fleming, R.M. Personalized whole-body models integrate metabolism, physiology, and the gut microbiome. Mol. Syst. Biol. 2020, 16, e8982. [Google Scholar] [CrossRef]
- Lu, K.; Abo, R.P.; Schlieper, K.A.; Graffam, M.E.; Levine, S.; Wishnok, J.S.; Swenberg, J.A.; Tannenbaum, S.R.; Fox, J.G. Arsenic Exposure Perturbs the Gut Microbiome and Its Metabolic Profile in Mice: An Integrated Metagenomics and Metabolomics Analysis. Environ. Health Perspect. 2014, 122, 284–291. [Google Scholar] [CrossRef]
- Richardson, J.B.; Dancy, B.C.R.; Horton, C.L.; Lee, Y.S.; Madejczyk, M.S.; Xu, Z.Z.; Ackermann, G.; Humphrey, G.; Palacios, G.; Knight, R.; et al. Exposure to toxic metals triggers unique responses from the rat gut microbiota. Sci. Rep. 2018, 8, 6578. [Google Scholar] [CrossRef]
- Latha, S.; Vinothini, G.; Dhanasekaran, D. Chromium [Cr(VI)] biosorption property of the newly isolated actinobacterial probiont Streptomyces werraensis LD22. 3 Biotech 2014, 5, 423–432. [Google Scholar] [CrossRef] [Green Version]
- Chi, L.; Gao, B.; Bian, X.; Tu, P.; Ru, H.; Lu, K. Manganese-induced sex-specific gut microbiome perturbations in C57BL/6 mice. Toxicol. Appl. Pharmacol. 2017, 331, 142–153. [Google Scholar] [CrossRef] [Green Version]
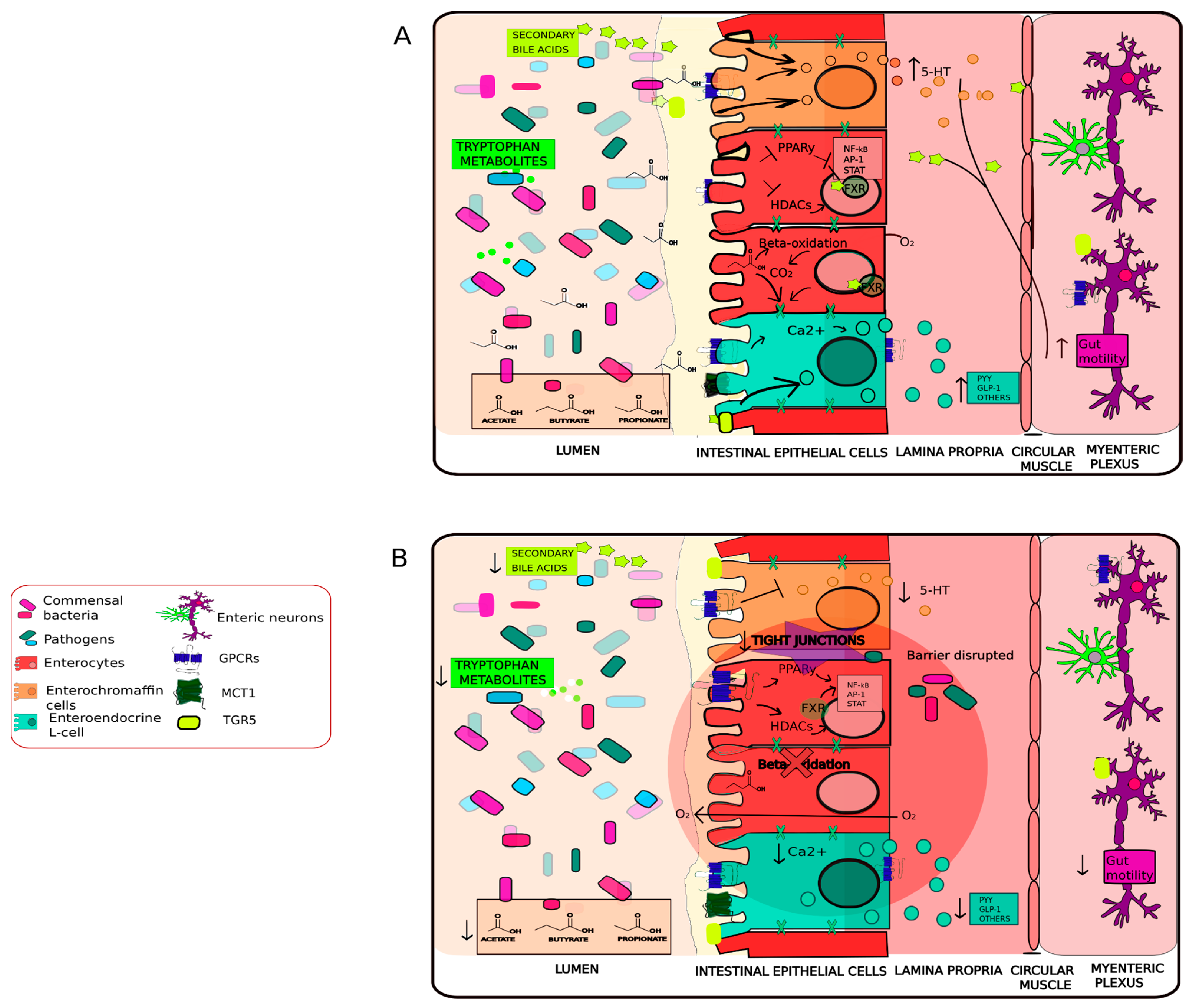
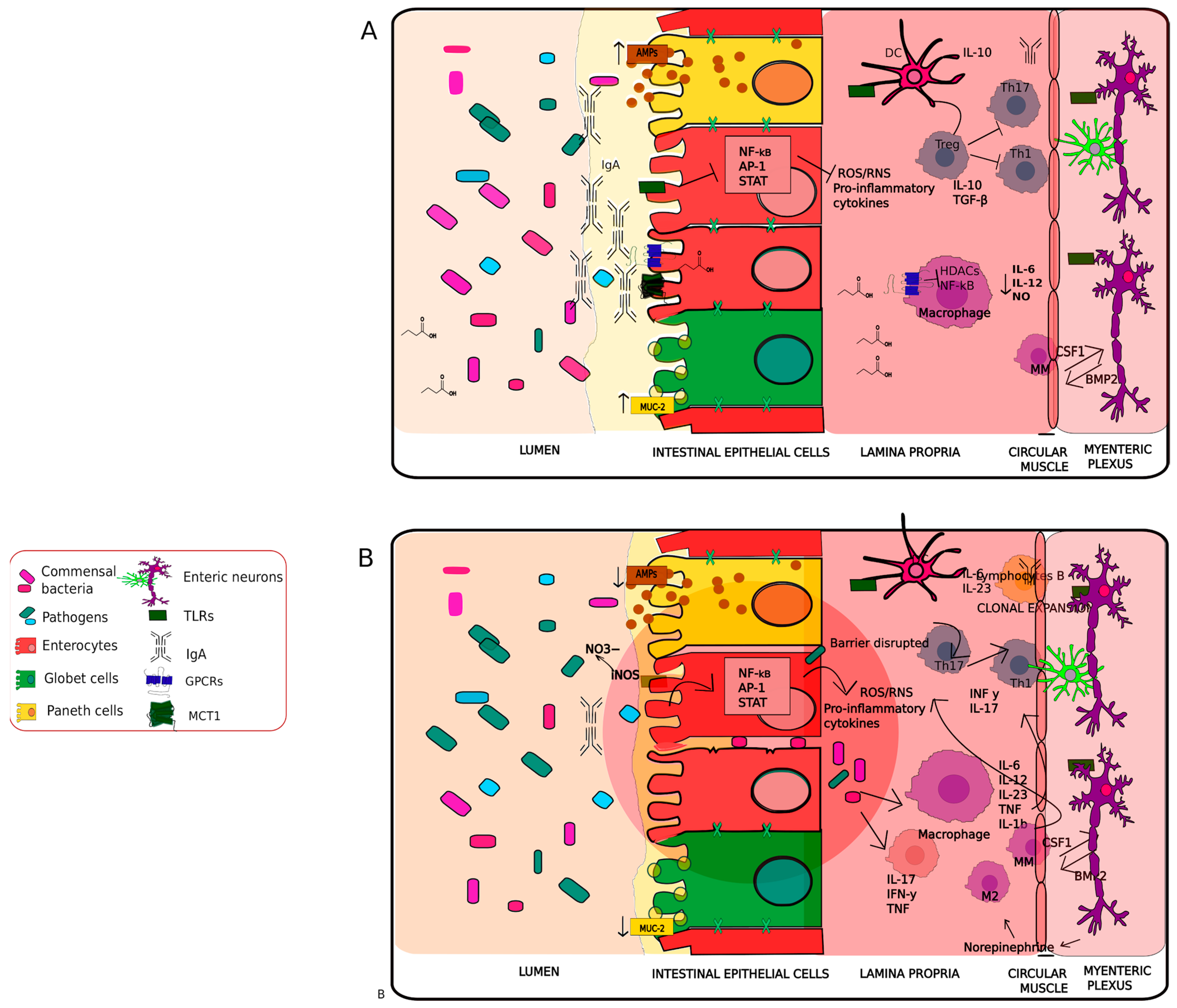
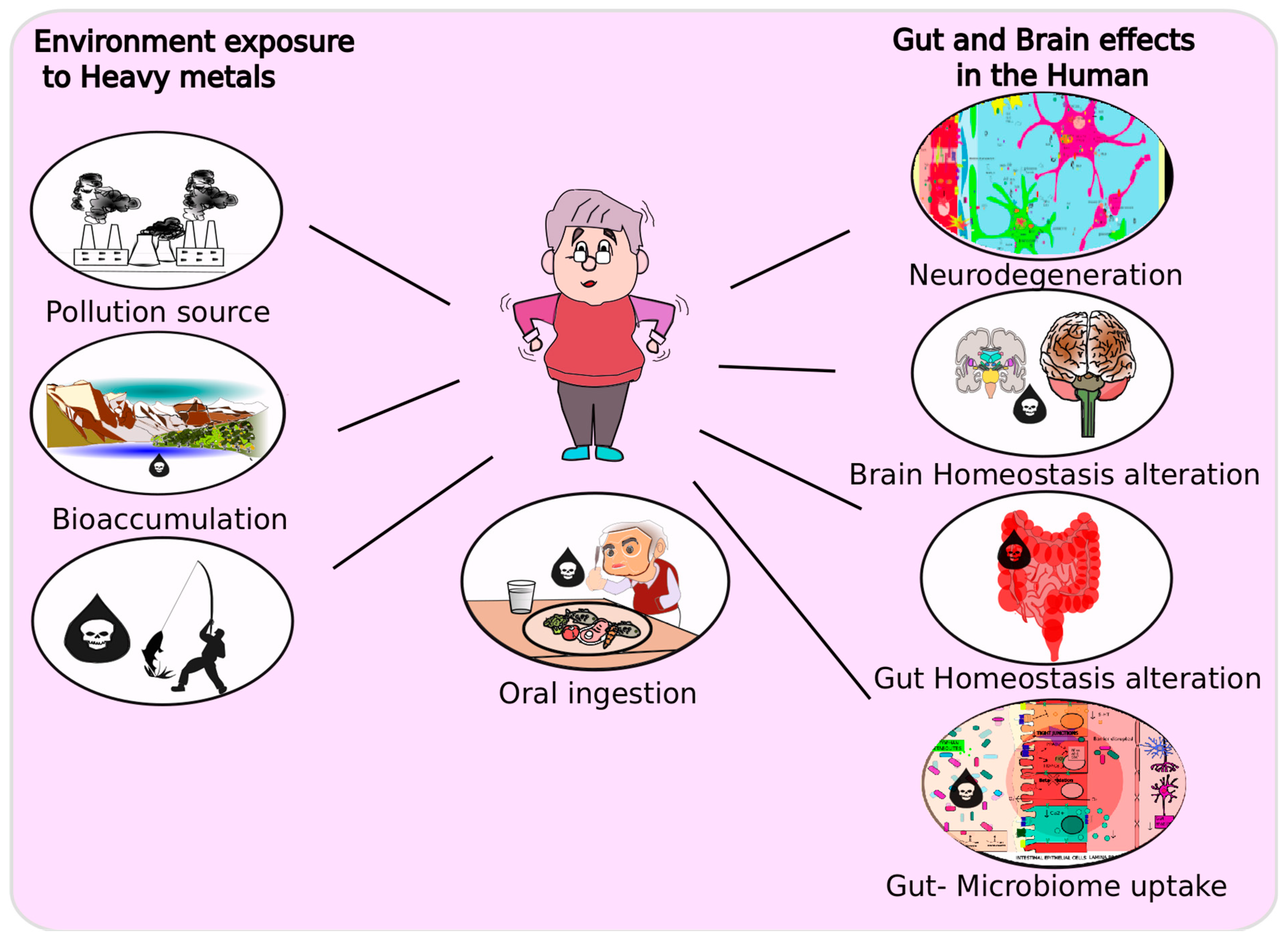

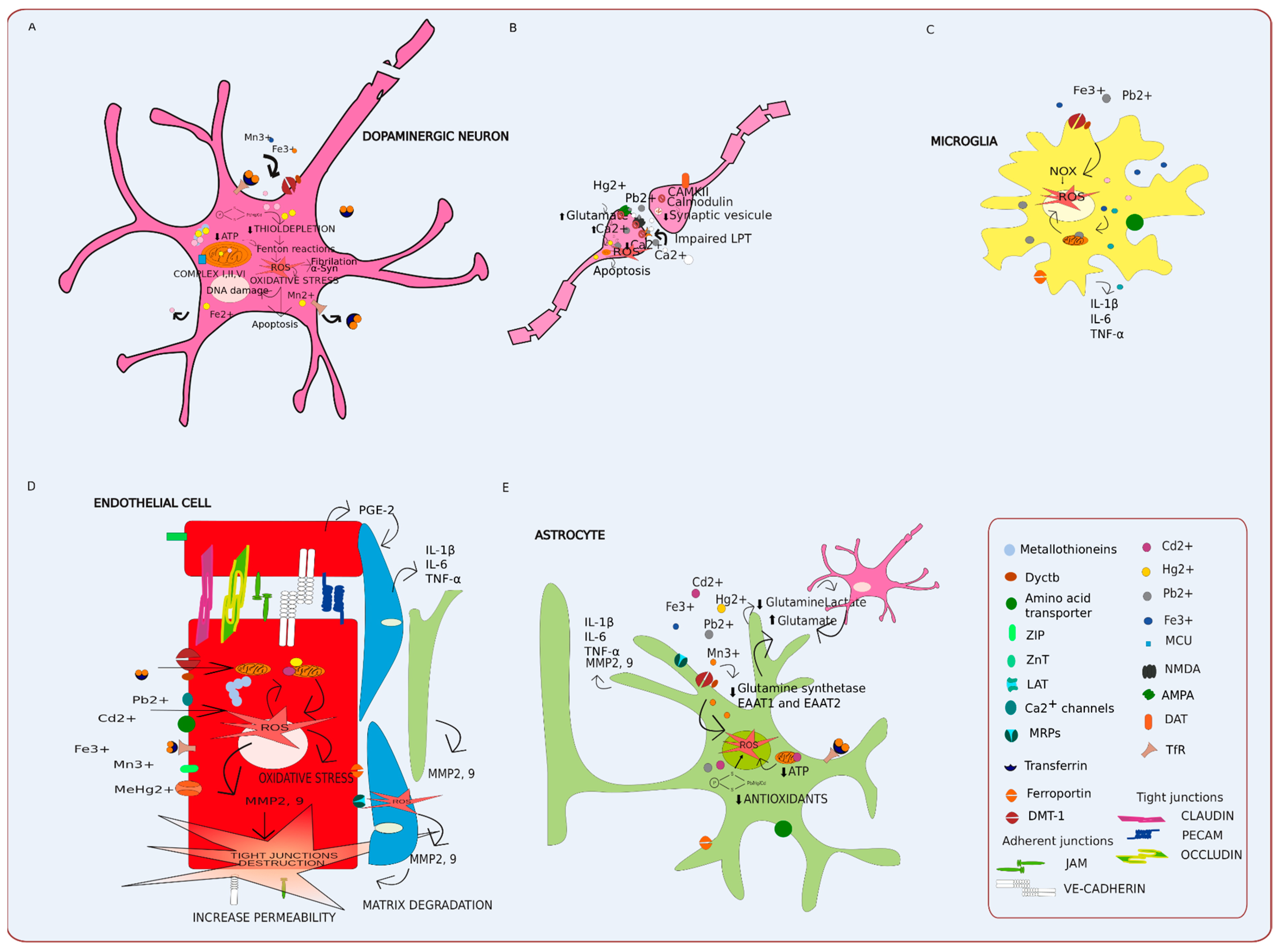

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Transporters | Molecule/Ion Metal Being Mimicked | Metal Replacement | Cells Containing the Transporters of the Transporter | Citation |
|---|---|---|---|---|
| Organic anion transporters: | CH3Hg+ | Endothelial cells/Glial cells/Enterocytes | [201,230,231] | |
| OAT1 | GSH | |||
| Cysteine | GS-Cd-S-G | |||
| OAT3 | ||||
| CH3Hg-S-Cys | ||||
| Zinc-regulated zinc transporter 1 (hZTL1) | Zn 2+ | Cd2+ | Enterocytes/Neurons | [218,219,220] |
| Ca2+ channels | Ca2+ | Cd2+ | Neurons/Endothelial cells/glial cells | [201,223,224] |
| Pb2+ | ||||
| Mn2+ | ||||
| Divalent metal transporter 1 | Fe2+ | Pb2+ | Enterocytes/Endothelial cells/Neurons/Glial cells | [217] |
| DMT-1 | Mn2+ | |||
| Cd2+ | ||||
| Zinc-imidazolate polymers (ZIPs) | Zn 2+ | Pb2+ | Enterocytes/Endothelial/Astrocytes | [201,221,222] |
| 1.2 | Mn2+ | |||
| 8, 14 | Cd2+ | |||
| Hg2+ | ||||
| Transferrin receptor | Fe2+ | Mn2+ | Neurons/Endothelial cells/Glial cells | [229] |
| TfR | ||||
| Amino acid transporters (system b0,+, system L) | Cysteine | CH3Hg-S-Cys | Enterocytes/Endothelial cells/Glial cells | [201,226,227,228] |
| Methionine | Cys-S-Hg-S-Cys | |||
| Cys-S-Cd-S-Cys | ||||
| CH3Hg-S-CysGly | ||||
| Multidrug resistance-associated proteins | CH3Hg+ | Enterocytes/Endothelial cells/glial cells | [201,232] | |
| MRP 1, 2, 3,4 | GSSG | G-S-Cd-S-G | ||
| GSH | CH3Hg-S-G | |||
| As (III) | ||||
| As–GSH | ||||
| Ferroportin | Fe2+ | Cd2+ | Enterocytes/Endothelial cells/Neurons/Oligodendrocytes/Astrocytes | [201,233,234,235] |
| Mn2+ | ||||
| Glucose permeases | Glucose | As (III) | Enterocytes/Endothelial cells/Astrocytes | [210] |
| GLUT 1, 2, 5 | ||||
| Sodium-dependent phosphate transporters | Phosphate | As(V) | Enterocytes | [218,219,220,236,237] |
| NaPiIIb | ||||
| Aquaporins | Glycerol | As(III) | [218,219,220,238,239,240] | |
| AQP 3, 10 | Water | Hg2+ | Enterocytes/Enteric neurons/Endothelial cells/Astrocytes | |
| AQP 4 | Pb2+ | |||
| Fe2+ | ||||
| Mn2+ | ||||
| Organic anion transporting polypeptides | Amphipathic organic compounds | As(III) | Enterocytes/Astrocytes/Endothelial | [218,219,220] |
| OATPB |
| Metal Ion | Reactive Species | Oxidized Molecules | Adverse Outcomes | Cite |
|---|---|---|---|---|
| Lead | Pb2+ | δ-aminolevulinic acid dehydratase (ALAD) | Reduces the antioxidant (glutathione) levels | [275,276] |
| Hydrogen peroxide Superoxide radical | Superoxide dismutase (SOD) | Oxidative stress | ||
| Hydroxyl radicals | Catalase | Alteration in Ca2+ influx | ||
| Glucose-6-phosphate dehydrogenase (G6PD) | Apoptosis | |||
| GSH | ||||
| Glutathione reductase | ||||
| Glutathione peroxidase | ||||
| Glutathione s-transferase | ||||
| Voltage-gated calcium (Ca2+) channels | ||||
| N-methyl-d-aspartate (NMDA) | ||||
| Cadmium | Cd2+ | Thioredoxin | Depresses antioxidants (glutathione) levels | [258,274,277,278,279] |
| Superoxide anion | Cysteine | Oxidative stress | ||
| Hydrogen peroxide | Ubiquitin enzymes | Damage in the electron transport chain | ||
| Hydroxyl radicals | Mitochondrial Complex II III | Apoptosis | ||
| Topoisomerase II | Lipid peroxidation | |||
| DNA methyltransferases | Alteration in maintaining genomic integrity | |||
| GSH | ||||
| Glutathione reductase | ||||
| Glutathione peroxidase | ||||
| Glutathione s-transferase | ||||
| Mercury | MeHg | DNA | Depresses antioxidants (glutathione) levels | [280,281,282,283,284,285] |
| Hg2+ | Thioredoxin reductase | Oxidative stress | ||
| Hydrogen peroxide | Nitric oxide synthase | Lipid peroxidation | ||
| Nitric oxide | Monoamine oxidase | Mitochondrial function | ||
| Glutathione reductase | Decreases GABA signaling | |||
| Glutathione peroxidase | Neurotransmitter metabolism | |||
| Astrocytic glutamine transporter | Glutamine uptake | |||
| Choline acetyltransferase | Acetylcholine synthesis | |||
| Creatine kinase | Decrease ATP production | |||
| Cytosolic phospholipase A2 | Membrane damage in Endothelial cells | |||
| Enolase | ||||
| Glutamate transporters | ||||
| Ca2+ ATP | ||||
| Mn | Mn2+ | Dopamine | Impairment of oxidative phosphorylation | [276,281,286,287,288,289] |
| Mn3+ | SOD | Decrease ATP synthesis | ||
| DA-o-quinone | Complex I, II | Disruption of mitochondrial energy production | ||
| Aminochrome | Aconitase | Alteration in Ca2+ influx | ||
| Adenylate cyclase | ||||
| Succinate | ||||
| Malate | ||||
| Glutamate | ||||
| N-methyl-d-aspartate (NMDA) | ||||
| ATP synthase | ||||
| Iron | Fe2+ | Complex I III | Lipid peroxidation | [281,290,291,292] |
| Fe3+ | SOD | Dopamine metabolism | ||
| Hydrogen peroxide Superoxide radical | α-synuclein | Mitochondrial functions disruption in ATP synthesis | ||
| Hydroxyl radicals | Tyrosine hydroxylase | Apoptosis | ||
| 3,4- dihydroxyphenylacetaldehyde DA-o-quinone | Hydrogen peroxide | DNA/protein degradation | ||
| 6-hydroxydopamine | Lipid peroxide | |||
| Aminochrome | DNA/RNA | |||
| Creatine kinase BB | ||||
| Cytochrome c oxidase | ||||
| Ketoglutarate dehydrogenase | ||||
| Arsenic | iAsV | ATP synthase | Uncouples oxidative phosphorylation | [250,293,294] |
| iAsIII | β-tubulin | Decrease ATP formation in the mitochondria | ||
| Monomethyl arsonous acid (MMAIII), | Glucose 6-arsenate | Inhibition of the hexokinase | ||
| Monomethylarsonic acid (MMAV) | Peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α) | Decrease in mitochondrial biogenesis | ||
| Dimethylarsinous acid (DMAIII), and | Mitochondrial transcription factor A (TFAM) | |||
| Dimethylarsinic acid (DMAV) | Pyruvate dehydrogenase | |||
| Arsenic triglutathione |
| Probiotic Microorganism | Toxic Element | Model/Cell Line | Treatment Effects | Citation |
|---|---|---|---|---|
| Lactobacillus rhamnosus GR-1 | Arsenic, Cadmium, Mercury, Lead | Humans | Reduction in toxic levels in blood and microbiota changes (Succinivibrionaceae and Gammaproteobacteria families) | [372] |
| Mix of microorganisms: L. plantarum DSM 24730, L. acidophilus DSM 24735, L. paracasei DSM 24733, B. infantis DSM 24737, B. longum DSM 24736, L. delbrueckii subsp. bulgaricus DSM 24734, B. breve DSM 24732 and Streptococcus thermophilus DSM 24731. | Arsenic, Cadmium, Mercury, Lead | Humans | Concentration of Cd, Hg, and Pb in breast milk, lower concentration of Cd in stools from newborns treated with the probiotics | [373] |
| Lactobacillus rhamnosus GR-1 | Cadmium, Lead | Caco-2 cells | Reduce translocation of toxics | [24] |
| Escherichia coli Nissle 1917 (EcN-20) and (EcN-21) | Cadmium, Mercury | Rats | Increasing GSH levels, SOD activity and catalase, decrease lipid peroxidation in liver and kidney and decrease ALT, AST and ALP activities, bilirubin, creatinine and urea levels in serum | [374] |
| Streptococcus thermophilus, Lactobacillus acidophilus and Bifidobacterium bifidum | Arsenic | Rats | Ameliorating the toxic effects on testis and blood metabolites, increase antioxidant enzyme activity (glutathione-s-transferase) | [375] |
| Lactobacillus kefir CIDCA 8348 and JCM 5818 | Cadmium | Human hepatoma cell line | Increase cell viability by binding to the toxic metal | [376] |
| Lactobacillus rhamnosus Rosell-11, Lactobacillus acidophilus Rosell-52 and Bifidobacterium longum Rosell-175 | Cadmium | Rat hepatocytes, Rats | Decrease genotoxicity through binding to toxic metal | [377] |
| Lactobacillus rhamnosus Rosell-11, Lactobacillus acidophilus Rosell-52 and Bifidobacterium longum Rosell-175 | Cadmium | Rats | Increase in toxic metal secretion by feces and decrease in concentration in the blood. Normalized ALT and AST activities | [378] |
| Lactobacillus plantarum L67 | Cadmium | Murine RAW 264.7 cells | Inhibits cytotoxicity and intracellular Ca2+ mobilization; suppressed the expression of AP-1 and MAPK protecting against inflammation | [379] |
| Lactobacillus plantarum CCFM8610 | Cadmium | Human intestinal cell line HT-29 and mice | Protection against cell damage, reversed the disruption of tight junctions, protected against inflammation, and decreased the intestinal permeability | [380] |
| Saccharomyces cerevisiae | Cadmium | Mice | Protected against genotoxic and spermatotoxic effects | [381] |
| Lactobacillus plantarum and Bacillus coagulans | Cadmium | Rats | Decreased AST, ALT, BUN, bilirubin (increased by toxic exposition) and metal accumulation in the liver and kidney | [382] |
| Lactobacillus delbrueckii, Lactobacillus fermentum, Lactobacillus acidophilus, Bifidobacterium and Lactobacillus bulgaricus | Cadmium | Mice | Increased toxic excretion in feces, and increasing β-catenin and BDNF in brain tissue | [383] |
| Streptococcus thermophilus | Cadmium | Mice | Through toxic metal binding, decreased levels of toxic metal in blood and attenuation levels of MDA and GSH | [384] |
| Pediococcus pentosaceus GS4 | Cadmium | Mice | Reduced tissue deposition, increased fecal secretion of toxic, and increased enzymatic activity | [208] |
| Bacillus cereus | Cadmium | Fish | Reduced Cd accumulation in organs, modulate antioxidant activity and intestinal microbial composition | [385] |
| Lactobacillus plantarum CCFM8610 | Cadmium | HT-29 cells, mice | Inhibition of metal absorption, alleviated cytotoxicity, reversal of the disruption of tight junctions and inhibition of inflammation | [380] |
| Lactobacillus plantarum CCFM8610 | Cadmium | Fish | Mitigation of oxidative stress in tissues and reversed alterations in hematological and biochemical parameters | [386] |
| Lactobacillus acidophilus | Cadmium | Fish | Decreased number of micronucleus formation in erythrocytes and improvement of animal survival rate. | [387] |
| Akkermansia muciniphila | Cadmium | Mice | Modulation of gut microbiota composition | [388] |
| Lactobacillus plantarum CCFM8661 | Lead | Mice | Decreased toxic levels in blood and tissues, recover blood δ-aminolevulinic acid dehydratase activity, avoidance of alterations in glutathione, glutathione peroxidase, superoxide dismutase, malondialdehyde, and reactive oxygen species | [389] |
| Lactobacillus plantarum CCFM8662 | Lead | Mice | Induced fecal metal excretion through hepatic bile acids synthesis, enhanced biliary glutathione and increased fecal bile acid excretion. | [390] |
| Faecalibacterium prausnitzii and Oscillibacter ruminantium | Lead | Mice | Increase in the expression of TJ proteins (ZO-1, occludin and claudin-1), increased fecal Pb excretion, increase SCFAs, pH and oxidative reduction in the intestinal lumen | [391] |
| Lactobacillus plantarum CCFM8610 | Cadmium | Mice | Decrease intestinal metal absorption, tissue accumulation, oxidative stress and ameliorate hepatic histopathological changes | [392] |
| Lactobacillus reuteri | Lead | Fish | Decreased oxidative stress, reversed alterations in hemato-biochemical parameters, and restored intestinal enzymatic activities | [393] |
| Lactobacillus plantarum, Lactobacillus acidophilus, Bacillus subtilis, Pediococcus pentosaceus, Bacillus licheniformis and Saccharomyces cerevisiae | Lead | Broiler Chicks | Improved antioxidant parameters, liver transaminases, decreased accumulation of metals and morphological alterations | [394] |
| Lactobacillus delbrueckii subsp. bulgaricus KLDS1.0207 | Lead | Mice | Increased fecal excretion, decreased oxidative stress, lipid peroxide by decreasing MDA concentration, and improved antioxidant production | [395] |
| Lactobacillus pentosus ITA23 and Lactobacillus acidipiscis ITA44 | Lead | Broiler Chicks | Reduced metal tissue accumulation, decreased lipid peroxidation, and normalized antioxidant activity | [179] |
| Lactobacillus brevis 23017 | Mercury | Mice | Reduced intestinal inflammation and decreased oxidative stress | [396] |
| and Lactobacillus plantarum | Mercury | Rats | Decreased tissue accumulation of toxic metal, avoided antioxidant alterations, normalized creatinine, bilirubin, urea AST, and ALT levels | [397] |
| Streptococcus thermophilus, Lactobacillus acidophilus and Bifidobacterium bifidum | Mercury | Rats | Protection against the adverse effects in the brain and kidney. Increased activity of glutathione-S-transferase, lactate dehydrogenase, normalized creatinine, triglycerides levels and modulate histopathological changes in the brain. | [398] |
| Probiotic Microorganism | Model | Outcome | Citation |
|---|---|---|---|
| Lactobacillus casei Shirota | Humans | Improves stool consistency and bowel habits | [407] |
| Lactobacillus acidophilus, Lactobacillus reuteri, Lactobacillus fermentum and Bifidobacterium bifidum | Humans | Decreases The Movement Disorders Society-Unified Parkinson’s Disease Rating Scale (MDS-UPDRS), biomarkers of inflammation and oxidative stress (high-sensitivity C-reactive protein (hs-CRP), Malondialdehyde (MDA), Glutathione) and insulin metabolism. | [408] |
| Lactobacillus acidophilus, Bifidobacterium bifidum, L. reuteri and Lactobacillus fermentum | Humans | Downregulates gene expression levels of IL-1, IL-8 and TNF-α; increases the gene expression of TGF-β and PPAR-γ | [409] |
| Lactobacillus acidophilus and Bifidobacterium infantis | Humans | Improves abdominal pain, bloating and constipation with incomplete evacuation | [410] |
| Streptococcus salivarius subsp thermophilus, Enterococcus faecium, Lactobacillus rhamnosus GG, Lactobacillus acidophilus, Lactobacillus plantarum, Lactobacillus paracasei, Lactobacillus delbrueckii subsp bulgaricus, and Bifidobacterium (breve and animalis subsp lactis) | Humans | Improves constipation in PD patients | [411] |
| Lactobacillus salivarius LS01 and Lactobacillus acidophilus | Peripheral blood mononuclear cells from PD patients and Caco-2 cells | Reduces proinflammatory cytokines, oxidative stress and increased the anti-inflammatory response and protection of the epithelium from gut permeability | [412] |
| Lactobacillus acidophilus, Lactobacillus rhamnosus GG and Bifidobacterium animalis lactis | Mice | Increases levels of BDNF, GDNF and dopamine, and decreases levels of MAO-B in the brain. | [413] |
| Bifidobacterium bifidum, Bifidobacterium longum, Lactobacillus rhamnosus, Lactobacillus rhamnosus GG, Lactobacillus plantarum LP28 and Lactococcus lactis subsp. Lactis | Mice | Preserves dopamine neurons, reduces the motor impairments, and maintains tyrosine hydroxylase in neurons | [414] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Forero-Rodríguez, L.J.; Josephs-Spaulding, J.; Flor, S.; Pinzón, A.; Kaleta, C. Parkinson’s Disease and the Metal–Microbiome–Gut–Brain Axis: A Systems Toxicology Approach. Antioxidants 2022, 11, 71. https://doi.org/10.3390/antiox11010071
Forero-Rodríguez LJ, Josephs-Spaulding J, Flor S, Pinzón A, Kaleta C. Parkinson’s Disease and the Metal–Microbiome–Gut–Brain Axis: A Systems Toxicology Approach. Antioxidants. 2022; 11(1):71. https://doi.org/10.3390/antiox11010071
Chicago/Turabian StyleForero-Rodríguez, Lady Johanna, Jonathan Josephs-Spaulding, Stefano Flor, Andrés Pinzón, and Christoph Kaleta. 2022. "Parkinson’s Disease and the Metal–Microbiome–Gut–Brain Axis: A Systems Toxicology Approach" Antioxidants 11, no. 1: 71. https://doi.org/10.3390/antiox11010071
APA StyleForero-Rodríguez, L. J., Josephs-Spaulding, J., Flor, S., Pinzón, A., & Kaleta, C. (2022). Parkinson’s Disease and the Metal–Microbiome–Gut–Brain Axis: A Systems Toxicology Approach. Antioxidants, 11(1), 71. https://doi.org/10.3390/antiox11010071