
Adaptative Up-Regulation of PRX2 and PRX5 Expression Characterizes Brain from a Mouse Model of Chorea-Acanthocytosis

 , , ,
, , ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mouse Strains and Design of the Study
2.2. Patient Characteristics
2.3. Western-Blot Analysis and Immunoprecipitation Assay
2.4. Statistical Analysis
3. Results
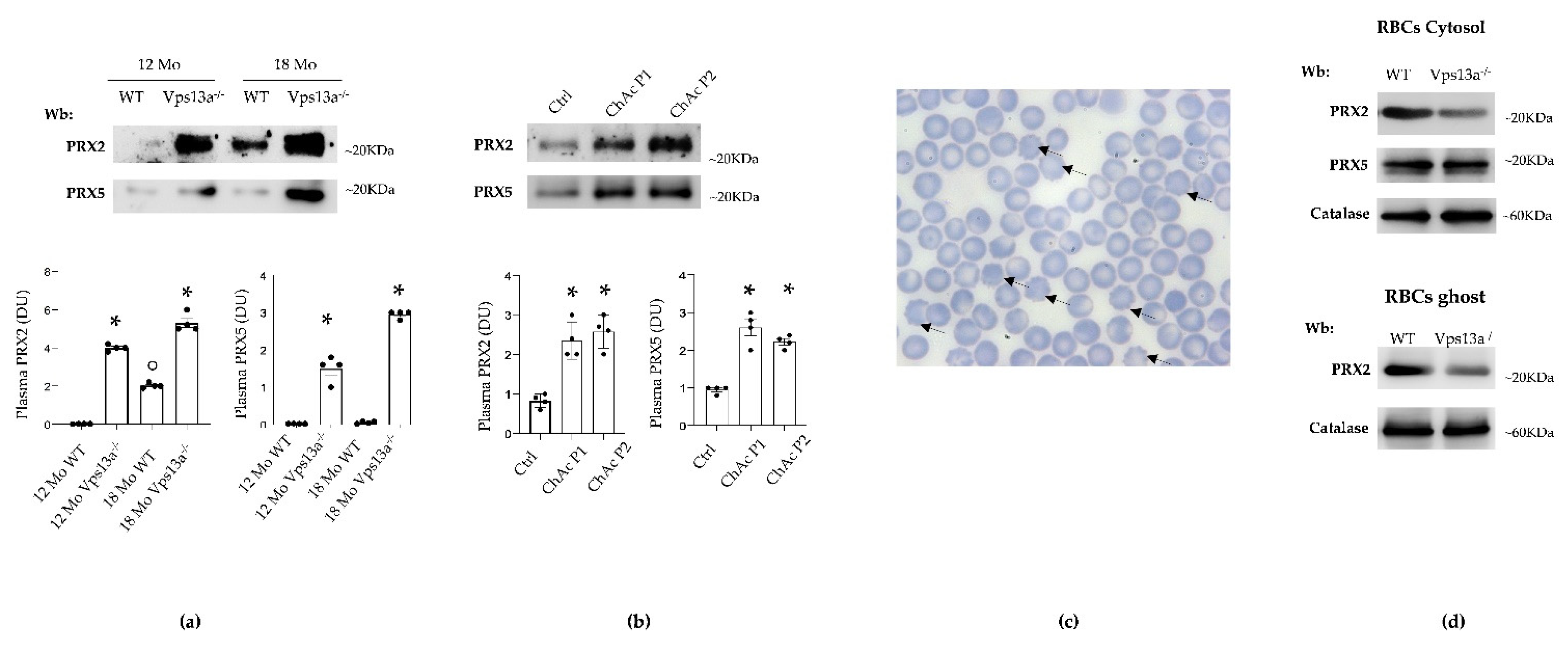
3.1. Plasma PRX2 and PRX5 Are Increased in Both Vps13a−/− Mice and Patients with ChAc
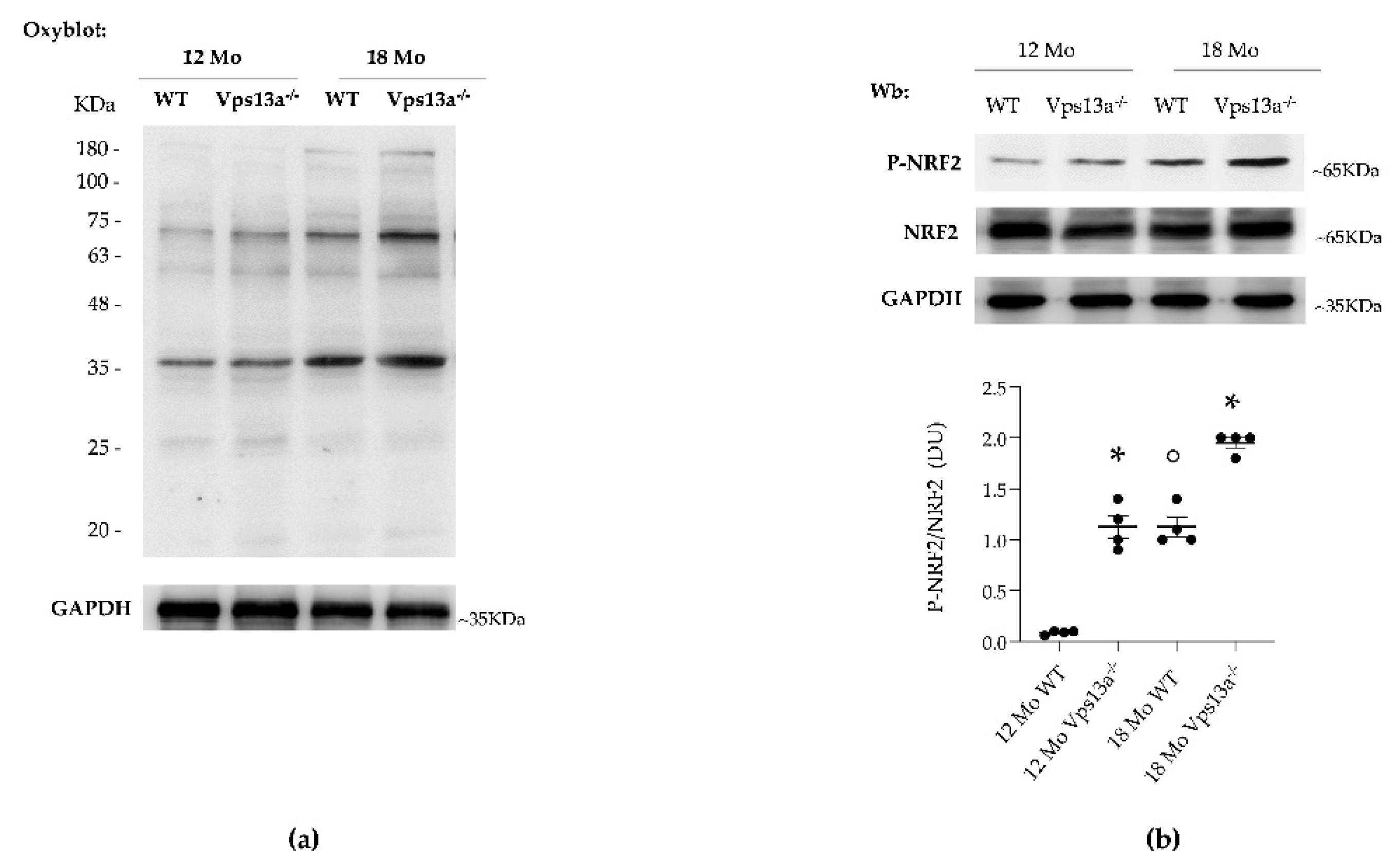
3.2. Activation of NRF2 and Up-Regulation of PRX2/5 Characterizes Basal Ganglia from Vps13a−/− Mice
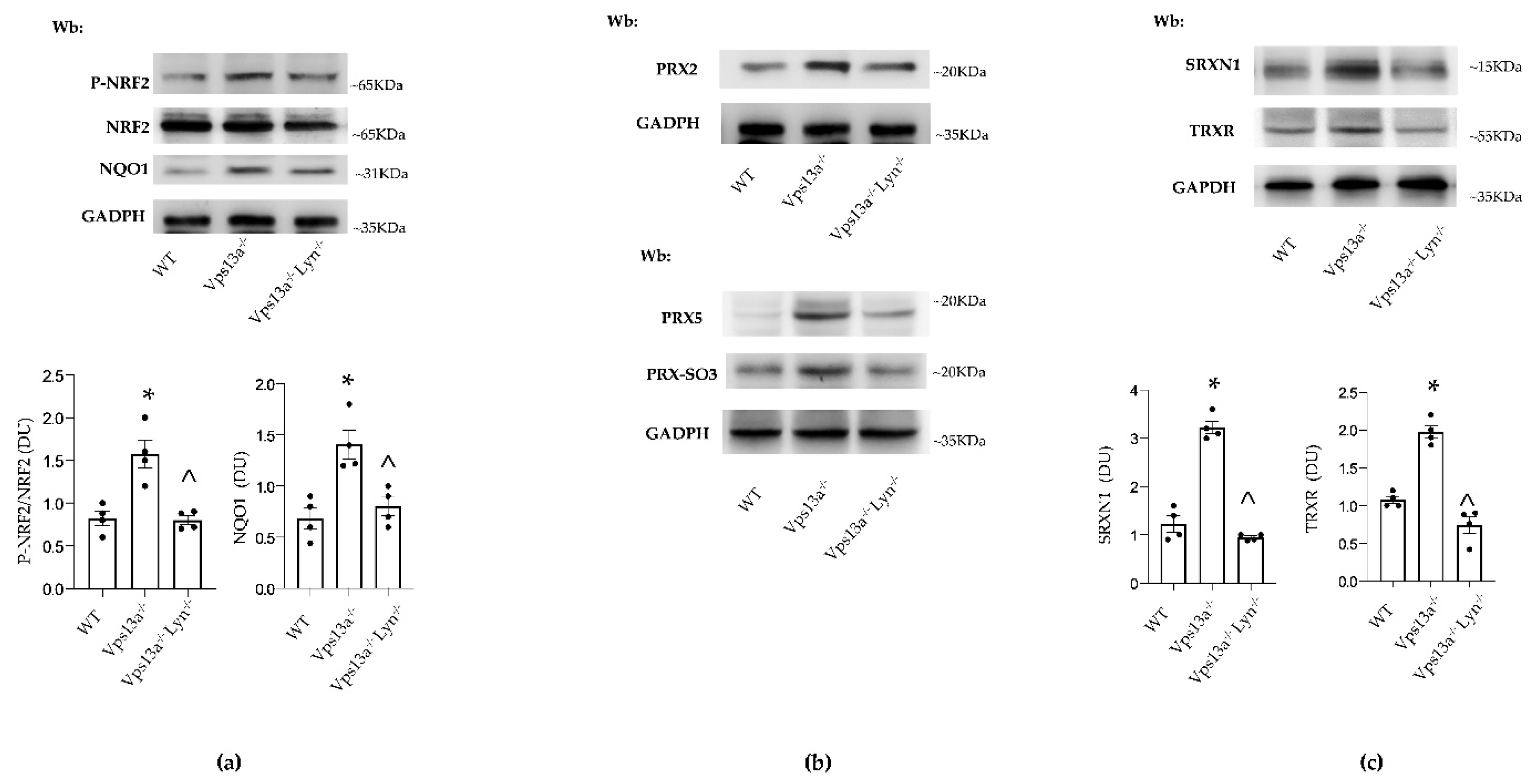
3.3. Improvement of Autophagy by Inhibition of Active Lyn Prevents NRF2 Activation and Down-Regulates PRX2/5 Expression in Vps13a−/− Basal Ganglia
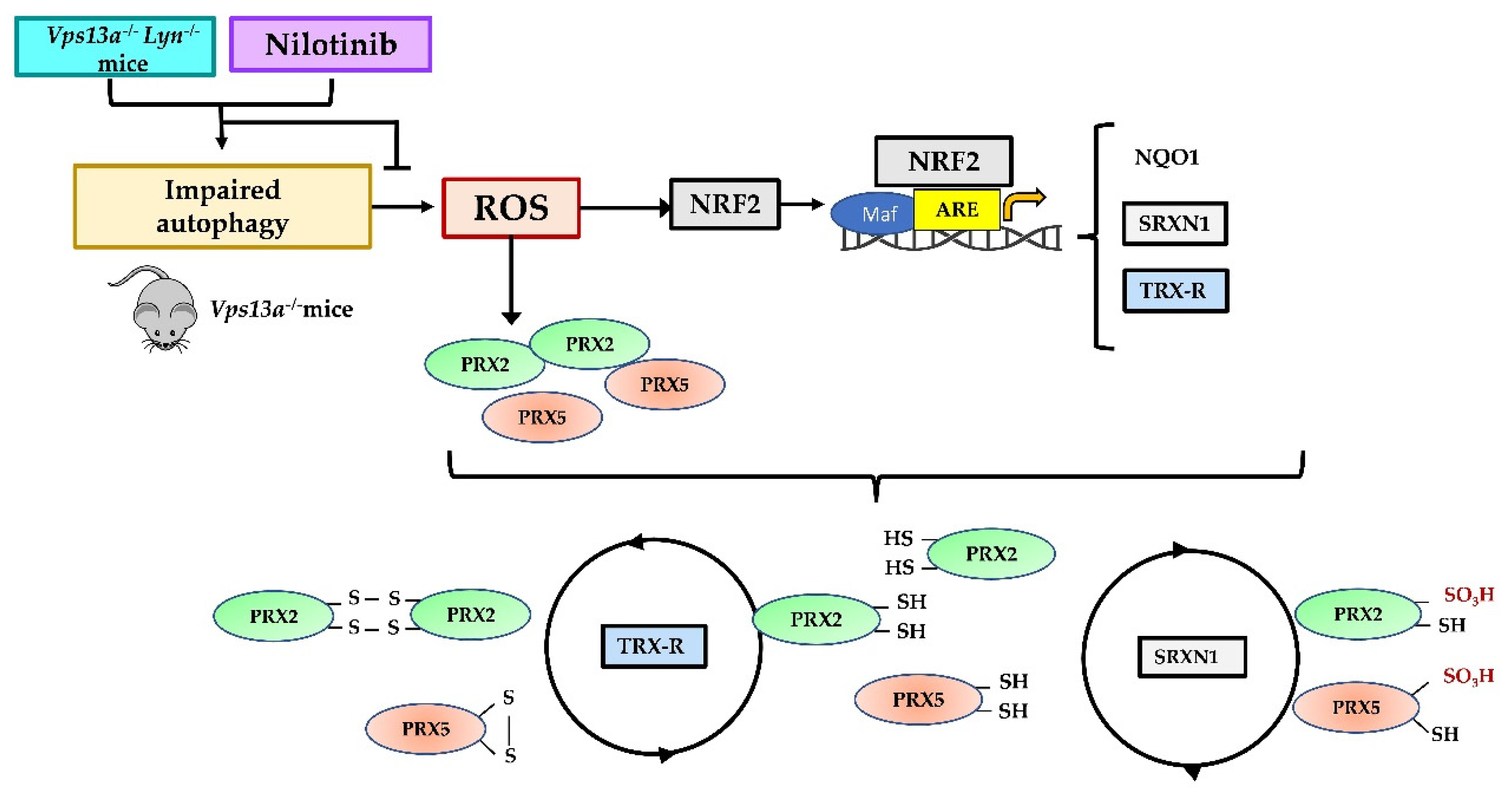
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wood, Z.A.; Schröder, E.; Harris, J.R.; Poole, L.B. Structure, mechanism and regulation of peroxiredoxins. Trends Biochem. Sci. 2003, 28, 32–40. [Google Scholar] [CrossRef]
- Wood, Z.A.; Poole, L.B.; Hantgan, R.R.; Karplus, P.A. Dimers to doughnuts: Redox-sensitive oligomerization of 2-cysteine peroxiredoxins. Biochemistry 2002, 41, 5493–5504. [Google Scholar] [CrossRef] [PubMed]
- Low, F.M.; Hampton, M.B.; Winterbourn, C.C. Peroxiredoxin 2 and Peroxide Metabolism in the Erythrocyte. Antioxid. Redox Signal. 2008, 10, 1621–1630. [Google Scholar] [CrossRef]
- Manta, B.; Hugo, M.; Ortiz, C.; Ferrer-Sueta, G.; Trujillo, M.; Denicola, A. The peroxidase and peroxynitrite reductase activity of human erythrocyte peroxiredoxin 2. Arch. Biochem. Biophys. 2009, 484, 146–154. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.G. Overview on Peroxiredoxin. Mol. Cells 2016, 39, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Rhee, S.G.; Kang, S.W.; Chang, T.S.; Jeong, W.; Kim, K. Peroxiredoxin, a novel family of peroxidases. IUBMB Life 2001, 52, 35–41. [Google Scholar] [CrossRef]
- Chae, H.Z.; Uhm, T.B.; Rhee, S.G. Dimerization of thiol-specific antioxidant and the essential role of cysteine 47. Proc. Natl. Acad. Sci. USA 1994, 91, 7022–7026. [Google Scholar] [CrossRef] [Green Version]
- Matte, A.; Pantaleo, A.; Ferru, E.; Turrini, F.; Bertoldi, M.; Lupo, F.; Siciliano, A.; Ho Zoon, C.; De Franceschi, L. The novel role of peroxiredoxin-2 in red cell membrane protein homeostasis and senescence. Free Radic. Biol. Med. 2014, 76, 80–88. [Google Scholar] [CrossRef]
- Matte, A.; Bertoldi, M.; Mohandas, N.; An, X.; Bugatti, A.; Brunati, A.M.; Rusnati, M.; Tibaldi, E.; Siciliano, A.; Turrini, F.; et al. Membrane association of peroxiredoxin-2 in red cells is mediated by the N-terminal cytoplasmic domain of band 3. Free Radic. Biol. Med. 2013, 55, 27–35. [Google Scholar] [CrossRef]
- De Franceschi, L.; Bertoldi, M.; Matte, A.; Santos Franco, S.; Pantaleo, A.; Ferru, E.; Turrini, F. Oxidative stress and beta-thalassemic erythroid cells behind the molecular defect. Oxid. Med. Cell. Longev. 2013, 2013, 985210. [Google Scholar] [CrossRef] [Green Version]
- Matte, A.; De Falco, L.; Iolascon, A.; Mohandas, N.; An, X.; Siciliano, A.; Leboeuf, C.; Janin, A.; Bruno, M.; Choi, S.Y.; et al. The Interplay Between Peroxiredoxin-2 and Nuclear Factor-Erythroid 2 Is Important in Limiting Oxidative Mediated Dysfunction in beta-Thalassemic Erythropoiesis. Antioxid. Redox Signal. 2015, 23, 1284–1297. [Google Scholar] [CrossRef] [Green Version]
- Matte, A.; De Falco, L.; Federti, E.; Cozzi, A.; Iolascon, A.; Levi, S.; Mohandas, N.; Zamo, A.; Bruno, M.; Lebouef, C.; et al. Peroxiredoxin-2: A Novel Regulator of Iron Homeostasis in Ineffective Erythropoiesis. Antioxid. Redox Signal. 2018, 28, 1–14. [Google Scholar] [CrossRef]
- Rhee, S.G.; Chae, H.Z.; Kim, K. Peroxiredoxins: A historical overview and speculative preview of novel mechanisms and emerging concepts in cell signaling. Free Radic. Biol. Med. 2005, 38, 1543–1552. [Google Scholar] [CrossRef]
- Woo, H.A.; Jeong, W.; Chang, T.S.; Park, K.J.; Park, S.J.; Yang, J.S.; Rhee, S.G. Reduction of cysteine sulfinic acid by sulfiredoxin is specific to 2-cys peroxiredoxins. J. Biol. Chem. 2005, 280, 3125–3128. [Google Scholar] [CrossRef] [Green Version]
- Hall, A.; Nelson, K.; Poole, L.B.; Karplus, P.A. Structure-based insights into the catalytic power and conformational dexterity of peroxiredoxins. Antioxid. Redox Signal. 2011, 15, 795–815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhee, S.G.; Woo, H.A. Multiple functions of 2-Cys peroxiredoxins, I and II, and their regulations via post-translational modifications. Free Radic. Biol. Med. 2020, 152, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Romero-Puertas, M.C.; Laxa, M.; Matte, A.; Zaninotto, F.; Finkemeier, I.; Jones, A.M.; Perazzolli, M.; Vandelle, E.; Dietz, K.J.; Delledonne, M. S-nitrosylation of peroxiredoxin II E promotes peroxynitrite-mediated tyrosine nitration. Plant. Cell 2007, 19, 4120–4130. [Google Scholar] [CrossRef] [Green Version]
- Szeliga, M. Peroxiredoxins in Neurodegenerative Diseases. Antioxidants 2020, 9, 1203. [Google Scholar] [CrossRef]
- Stepler, K.E.; Mahoney, E.R.; Kofler, J.; Hohman, T.J.; Lopez, O.L.; Robinson, R.A.S. Inclusion of African American/Black adults in a pilot brain proteomics study of Alzheimer’s disease. Neurobiol. Dis. 2020, 146, 105129. [Google Scholar] [CrossRef]
- Fang, J.; Nakamura, T.; Cho, D.H.; Gu, Z.; Lipton, S.A. S-nitrosylation of peroxiredoxin 2 promotes oxidative stress-induced neuronal cell death in Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2007, 104, 18742–18747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorolla, M.A.; Reverter-Branchat, G.; Tamarit, J.; Ferrer, I.; Ros, J.; Cabiscol, E. Proteomic and oxidative stress analysis in human brain samples of Huntington disease. Free Radic. Biol. Med. 2008, 45, 667–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basso, M.; Giraudo, S.; Corpillo, D.; Bergamasco, B.; Lopiano, L.; Fasano, M. Proteome analysis of human substantia nigra in Parkinson’s disease. Proteomics 2004, 4, 3943–3952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shichita, T.; Hasegawa, E.; Kimura, A.; Morita, R.; Sakaguchi, R.; Takada, I.; Sekiya, T.; Ooboshi, H.; Kitazono, T.; Yanagawa, T.; et al. Peroxiredoxin family proteins are key initiators of post-ischemic inflammation in the brain. Nat. Med. 2012, 18, 911–917. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.H.; Lee, Y.H.; Kim, J.M.; Sun, H.N.; Moon, E.Y.; Shong, M.H.; Kim, S.U.; Lee, S.H.; Lee, T.H.; Yu, D.Y.; et al. Characterization of neural cell types expressing peroxiredoxins in mouse brain. Neurosci. Lett. 2005, 381, 252–257. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.; Park, J.; Chang, K.T.; Lee, D.S. Peroxiredoxin 5 prevents amyloid-beta oligomer-induced neuronal cell death by inhibiting ERK-Drp1-mediated mitochondrial fragmentation. Free Radic. Biol. Med. 2016, 90, 184–194. [Google Scholar] [CrossRef]
- Kunze, A.; Zierath, D.; Tanzi, P.; Cain, K.; Becker, K. Peroxiredoxin 5 (PRX5) is correlated inversely to systemic markers of inflammation in acute stroke. Stroke 2014, 45, 608–610. [Google Scholar] [CrossRef]
- Abruzzo, P.M.; Matte, A.; Bolotta, A.; Federti, E.; Ghezzo, A.; Guarnieri, T.; Marini, M.; Posar, A.; Siciliano, A.; De Franceschi, L.; et al. Plasma peroxiredoxin changes and inflammatory cytokines support the involvement of neuro-inflammation and oxidative stress in Autism Spectrum Disorder. J. Transl. Med. 2019, 17, 332. [Google Scholar] [CrossRef]
- Wu, J.; Chen, Y.; Yu, S.; Li, L.; Zhao, X.; Li, Q.; Zhao, J.; Zhao, Y. Neuroprotective effects of sulfiredoxin-1 during cerebral ischemia/reperfusion oxidative stress injury in rats. Brain Res. Bull. 2017, 132, 99–108. [Google Scholar] [CrossRef]
- Peikert, K.; Danek, A.; Hermann, A. Current state of knowledge in Chorea-Acanthocytosis as core Neuroacanthocytosis syndrome. Eur. J. Med. Genet. 2018, 61, 699–705. [Google Scholar] [CrossRef]
- Dobson-Stone, C.; Danek, A.; Rampoldi, L.; Hardie, R.J.; Chalmers, R.M.; Wood, N.W.; Bohlega, S.; Dotti, M.T.; Federico, A.; Shizuka, M.; et al. Mutational spectrum of the CHAC gene in patients with chorea-acanthocytosis. Eur. J. Hum. Genet. 2002, 10, 773–781. [Google Scholar] [CrossRef] [Green Version]
- Rampoldi, L.; Dobson-Stone, C.; Rubio, J.P.; Danek, A.; Chalmers, R.M.; Wood, N.W.; Verellen, C.; Ferrer, X.; Malandrini, A.; Fabrizi, G.M.; et al. A conserved sorting-associated protein is mutant in chorea-acanthocytosis. Nat. Genet. 2001, 28, 119–120. [Google Scholar] [CrossRef] [PubMed]
- Ueno, S.; Maruki, Y.; Nakamura, M.; Tomemori, Y.; Kamae, K.; Tanabe, H.; Yamashita, Y.; Matsuda, S.; Kaneko, S.; Sano, A. The gene encoding a newly discovered protein, chorein, is mutated in chorea-acanthocytosis. Nat. Genet. 2001, 28, 121–122. [Google Scholar] [CrossRef] [PubMed]
- Leonzino, M.; Reinisch, K.M.; De Camilli, P. Insights into VPS13 properties and function reveal a new mechanism of eukaryotic lipid transport. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2021, 1866, 159003. [Google Scholar] [CrossRef] [PubMed]
- Dziurdzik, S.K.; Conibear, E. The Vps13 Family of Lipid Transporters and Its Role at Membrane Contact Sites. Int. J. Mol. Sci. 2021, 22, 2905. [Google Scholar] [CrossRef]
- Lupo, F.; Tibaldi, E.; Matte, A.; Sharma, A.K.; Brunati, A.M.; Alper, S.L.; Zancanaro, C.; Benati, D.; Siciliano, A.; Bertoldi, M.; et al. A new molecular link between defective autophagy and erythroid abnormalities in chorea-acanthocytosis. Blood 2016, 128, 2976–2987. [Google Scholar] [CrossRef] [Green Version]
- Peikert, K.; Federti, E.; Matte, A.; Constantin, G.; Pietronigro, E.C.; Fabene, P.F.; Defilippi, P.; Turco, E.; Del Gallo, F.; Pucci, P.; et al. Therapeutic targeting of Lyn kinase to treat chorea-acanthocytosis. Acta Neuropathol. Commun. 2021, 9, 81. [Google Scholar] [CrossRef] [PubMed]
- Peikert, K.; Glass, H.; Federti, E.; Matte, A.; Pelzl, L.; Akgun, K.; Ziemssen, T.; Ordemann, R.; Lang, F.; Patients, T.; et al. Targeting Lyn Kinase in Chorea-Acanthocytosis: A Translational Treatment Approach in a Rare Disease. J. Pers. Med. 2021, 11, 392. [Google Scholar] [CrossRef]
- De Franceschi, L.; Tomelleri, C.; Matte, A.; Brunati, A.M.; Bovee-Geurts, P.H.; Bertoldi, M.; Lasonder, E.; Tibaldi, E.; Danek, A.; Walker, R.H.; et al. Erythrocyte membrane changes of chorea-acanthocytosis are the result of altered Lyn kinase activity. Blood 2011, 118, 5652–5663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Franceschi, L.; Olivieri, O.; Miraglia del Giudice, E.; Perrotta, S.; Sabato, V.; Corrocher, R.; Iolascon, A. Membrane cation and anion transport activities in erythrocytes of hereditary spherocytosis: Effects of different membrane protein defects. Am. J. Hematol. 1997, 55, 121–128. [Google Scholar] [CrossRef]
- Brugnara, C.; de Franceschi, L. Effect of cell age and phenylhydrazine on the cation transport properties of rabbit erythrocytes. J. Cell Physiol. 1993, 154, 271–280. [Google Scholar] [CrossRef]
- Kalish, B.T.; Matte, A.; Andolfo, I.; Iolascon, A.; Weinberg, O.; Ghigo, A.; Cimino, J.; Siciliano, A.; Hirsch, E.; Federti, E.; et al. Dietary omega-3 fatty acids protect against vasculopathy in a transgenic mouse model of sickle cell disease. Haematologica 2015, 100, 870–880. [Google Scholar] [CrossRef] [Green Version]
- Matte, A.; Federti, E.; Kung, C.; Kosinski, P.A.; Narayanaswamy, R.; Russo, R.; Federico, G.; Carlomagno, F.; Desbats, M.A.; Salviati, L.; et al. The pyruvate kinase activator mitapivat reduces hemolysis and improves anemia in a beta-thalassemia mouse model. J. Clin. Investig. 2021, 131, e144206. [Google Scholar] [CrossRef]
- Matte, A.; Lupo, F.; Tibaldi, E.; Di Paolo, M.L.; Federti, E.; Carpentieri, A.; Pucci, P.; Brunati, A.M.; Cesaro, L.; Turrini, F.; et al. Fyn specifically regulates the activity of red cell glucose-6-phosphate-dehydrogenase. Redox Biol. 2020, 36, 101639–101651. [Google Scholar] [CrossRef] [PubMed]
- El Eter, E.; Al Masri, A.; Habib, S.; Al Zamil, H.; Al Hersi, A.; Al Hussein, F.; Al Omran, M. Novel links among peroxiredoxins, endothelial dysfunction, and severity of atherosclerosis in type 2 diabetic patients with peripheral atherosclerotic disease. Cell Stress Chaperones 2014, 19, 173–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pantaleo, A.; Ferru, E.; Pau, M.C.; Khadjavi, A.; Mandili, G.; Matte, A.; Spano, A.; De Franceschi, L.; Pippia, P.; Turrini, F. Band 3 Erythrocyte Membrane Protein Acts as Redox Stress Sensor Leading to Its Phosphorylation by p (72) Syk. Oxid. Med. Cell. Longev. 2016, 2016, 6051093. [Google Scholar] [CrossRef] [Green Version]
- Jazvinscak Jembrek, M.; Orsolic, N.; Mandic, L.; Sadzak, A.; Segota, S. Anti-Oxidative, Anti-Inflammatory and Anti-Apoptotic Effects of Flavonols: Targeting Nrf2, NF-kappaB and p53 Pathways in Neurodegeneration. Antioxidants 2021, 10, 1628. [Google Scholar] [CrossRef] [PubMed]
- Bolduc, J.; Koruza, K.; Luo, T.; Malo Pueyo, J.; Vo, T.N.; Ezerina, D.; Messens, J. Peroxiredoxins wear many hats: Factors that fashion their peroxide sensing personalities. Redox Biol. 2021, 42, 101959. [Google Scholar] [CrossRef]
- Bi, M.; Du, X.; Xiao, X.; Dai, Y.; Jiao, Q.; Chen, X.; Zhang, L.; Jiang, H. Deficient immunoproteasome assembly drives gain of alpha-synuclein pathology in Parkinson’s disease. Redox Biol. 2021, 47, 102167. [Google Scholar] [CrossRef]
- Al-Mubarak, B.R.; Bell, K.F.S.; Chowdhry, S.; Meakin, P.J.; Baxter, P.S.; McKay, S.; Dando, O.; Ashford, M.L.J.; Gazaryan, I.; Hayes, J.D.; et al. Non-canonical Keap1-independent activation of Nrf2 in astrocytes by mild oxidative stress. Redox Biol. 2021, 47, 102158. [Google Scholar] [CrossRef]
- Villavicencio Tejo, F.; Quintanilla, R.A. Contribution of the Nrf2 Pathway on Oxidative Damage and Mitochondrial Failure in Parkinson and Alzheimer’s Disease. Antioxidants 2021, 10, 1069. [Google Scholar] [CrossRef]
- Davies, D.A.; Adlimoghaddam, A.; Albensi, B.C. Role of Nrf2 in Synaptic Plasticity and Memory in Alzheimer’s Disease. Cells 2021, 10, 1884. [Google Scholar] [CrossRef] [PubMed]
- Brandes, M.S.; Gray, N.E. NRF2 as a Therapeutic Target in Neurodegenerative Diseases. ASN Neuro 2020, 12, 1759091419899782. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.A.; Johnson, J.A. Nrf2--a therapeutic target for the treatment of neurodegenerative diseases. Free Radic. Biol. Med. 2015, 88, 253–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, H.; Santo, A.; Li, Y. The antioxidant enzyme peroxiredoxin and its protective role in neurological disorders. Exp. Biol. Med. 2012, 237, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; An, C.; Gao, Y.; Leak, R.K.; Chen, J.; Zhang, F. Emerging roles of Nrf2 and phase II antioxidant enzymes in neuroprotection. Prog. Neurobiol. 2013, 100, 30–47. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Yu, S.; Wu, J.; Zou, Y.; Zhao, Y. Sulfiredoxin-1 protects PC12 cells against oxidative stress induced by hydrogen peroxide. J. Neurosci. Res. 2013, 91, 861–870. [Google Scholar] [CrossRef]
- Zhou, Y.; Duan, S.; Zhou, Y.; Yu, S.; Wu, J.; Wu, X.; Zhao, J.; Zhao, Y. Sulfiredoxin-1 attenuates oxidative stress via Nrf2/ARE pathway and 2-Cys Prdxs after oxygen-glucose deprivation in astrocytes. J. Mol. Neurosci. 2015, 55, 941–950. [Google Scholar] [CrossRef]
- de Franceschi, L.; Turrini, F.; Honczarenko, M.; Ayi, K.; Rivera, A.; Fleming, M.D.; Law, T.; Mannu, F.; Kuypers, F.A.; Bast, A.; et al. In vivo reduction of erythrocyte oxidant stress in a murine model of beta-thalassemia. Haematologica 2004, 89, 1287–1298. [Google Scholar]
- Hommen, F.; Bilican, S.; Vilchez, D. Protein clearance strategies for disease intervention. J. Neural Transm. 2021, 1–32. [Google Scholar] [CrossRef]
- Moors, T.E.; Hoozemans, J.J.; Ingrassia, A.; Beccari, T.; Parnetti, L.; Chartier-Harlin, M.C.; van de Berg, W.D. Therapeutic potential of autophagy-enhancing agents in Parkinson’s disease. Mol. Neurodegener. 2017, 12, 11. [Google Scholar] [CrossRef] [Green Version]
- Turner, R.S.; Hebron, M.L.; Lawler, A.; Mundel, E.E.; Yusuf, N.; Starr, J.N.; Anjum, M.; Pagan, F.; Torres-Yaghi, Y.; Shi, W.; et al. Nilotinib Effects on Safety, Tolerability, and Biomarkers in Alzheimer’s Disease. Ann. Neurol. 2020, 88, 183–194. [Google Scholar] [CrossRef]
- Simuni, T.; Fiske, B.; Merchant, K.; Coffey, C.S.; Klingner, E.; Caspell-Garcia, C.; Lafontant, D.E.; Matthews, H.; Wyse, R.K.; Brundin, P.; et al. Efficacy of Nilotinib in Patients With Moderately Advanced Parkinson Disease: A Randomized Clinical Trial. JAMA Neurol. 2021, 78, 312–320. [Google Scholar] [CrossRef]
- Pagan, F.L.; Wilmarth, B.; Torres-Yaghi, Y.; Hebron, M.L.; Mulki, S.; Ferrante, D.; Matar, S.; Ahn, J.; Moussa, C. Long-Term Safety and Clinical Effects of Nilotinib in Parkinson’s Disease. Mov. Disord. 2021, 36, 740–749. [Google Scholar] [CrossRef]
- Pagan, F.; Hebron, M.; Valadez, E.H.; Torres-Yaghi, Y.; Huang, X.; Mills, R.R.; Wilmarth, B.M.; Howard, H.; Dunn, C.; Carlson, A.; et al. Nilotinib Effects in Parkinson’s disease and Dementia with Lewy bodies. J. Parkinsons Dis. 2016, 6, 503–517. [Google Scholar] [CrossRef] [Green Version]
- Chou, J.L.; Wu, C.H.; Tsai, C.Y.; Chang, A.Y.; Chan, S.H. Proteomic investigation of a neural substrate intimately related to brain death. Proteomics 2011, 11, 239–248. [Google Scholar] [CrossRef]
- Dayon, L.; Turck, N.; Garci-Berrocoso, T.; Walter, N.; Burkhard, P.R.; Vilalta, A.; Sahuquillo, J.; Montaner, J.; Sanchez, J.C. Brain extracellular fluid protein changes in acute stroke patients. J. Proteome Res. 2011, 10, 1043–1051. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Federti, E.; Matte, A.; Riccardi, V.; Peikert, K.; Alper, S.L.; Danek, A.; Walker, R.H.; Siciliano, A.; Iatcenko, I.; Hermann, A.; et al. Adaptative Up-Regulation of PRX2 and PRX5 Expression Characterizes Brain from a Mouse Model of Chorea-Acanthocytosis. Antioxidants 2022, 11, 76. https://doi.org/10.3390/antiox11010076
Federti E, Matte A, Riccardi V, Peikert K, Alper SL, Danek A, Walker RH, Siciliano A, Iatcenko I, Hermann A, et al. Adaptative Up-Regulation of PRX2 and PRX5 Expression Characterizes Brain from a Mouse Model of Chorea-Acanthocytosis. Antioxidants. 2022; 11(1):76. https://doi.org/10.3390/antiox11010076
Chicago/Turabian StyleFederti, Enrica, Alessandro Matte, Veronica Riccardi, Kevin Peikert, Seth L. Alper, Adrian Danek, Ruth H. Walker, Angela Siciliano, Iana Iatcenko, Andreas Hermann, and et al. 2022. "Adaptative Up-Regulation of PRX2 and PRX5 Expression Characterizes Brain from a Mouse Model of Chorea-Acanthocytosis" Antioxidants 11, no. 1: 76. https://doi.org/10.3390/antiox11010076
APA StyleFederti, E., Matte, A., Riccardi, V., Peikert, K., Alper, S. L., Danek, A., Walker, R. H., Siciliano, A., Iatcenko, I., Hermann, A., & De Franceschi, L. (2022). Adaptative Up-Regulation of PRX2 and PRX5 Expression Characterizes Brain from a Mouse Model of Chorea-Acanthocytosis. Antioxidants, 11(1), 76. https://doi.org/10.3390/antiox11010076