Abstract
Nonalcoholic fatty liver disease (NAFLD) is now the leading cause of chronic liver disease in western countries. The molecular mechanisms leading to NAFLD are only partially understood, and effective therapeutic interventions are clearly needed. Therefore, preclinical research is required to improve knowledge about NAFLD physiopathology and to identify new therapeutic targets. Primary human hepatocytes, human hepatic cell lines, and human stem cell-derived hepatocyte-like cells exhibit different hepatic phenotypes and have been widely used for studying NAFLD pathogenesis. In this paper, apart from employing the different in vitro cell models for the in vitro assessment of NAFLD, we also reviewed other approaches (metabolomics, transcriptomics, and high-content screening). We aimed to summarize the characteristics of different cell types and methods and to discuss their major advantages and disadvantages for NAFLD modeling.
1. Introduction
Nonalcoholic fatty liver disease (NAFLD) is the most common liver disorder in western countries, affecting 17–46% of adults and characterized by increased fat accumulation [1]. NAFLD comprises two different histological forms: nonalcoholic fatty liver (or simple steatosis) and nonalcoholic steatohepatitis (NASH) [2]. Steatosis can be defined as the presence of increased fat accumulation in the liver without hepatocellular necrosis and no, or minimal, inflammation. NASH is characterized by steatosis, liver inflammation, and hepatocyte ballooning with or without fibrosis. The progression of NASH underlies cirrhosis and hepatocellular carcinoma [2].
Evidence for hepatic steatosis in the absence of other causes of liver fat accumulation (i.e., significant alcohol consumption or hereditary disorders) is required to diagnose NAFLD, which is often asymptomatic and usually discovered during routine laboratory examinations with high transaminase levels. Most of these patients may have normal ALT levels, which tend to remain low as the disease progresses [3]. NAFLD is characterized by the presence of steatosis in >5% of the hepatocytes assessed by either histological analysis or a proton density fat fraction, determined by proton magnetic resonance imaging or magnetic resonance imaging (MRI) [2]. However, a multicenter study with adults demonstrated that MRI proton density fat fraction showed greater practicality and narrower sampling variability [4,5]. Additionally, European NAFLD management guidelines recommend ultrasonography to diagnose simple steatosis, but predictive serum biomarkers of disease risk and progression are urgently needed [3]. In fact, the international consortia LITMUS, by the European Union, and NIMBLE, by the United States of America, focus on finding reliable NAFLD and NASH biomarkers [6]. The definitive NASH diagnosis and its differentiation from simple steatosis always requires liver biopsy [2].
NAFLD is the result of multiple molecular and cellular changes and involves genetic, dietary, metabolic, and immunological factors [7]. The most common risk factors for NAFLD include obesity, type 2 diabetes, and metabolic syndrome features [8]. Hence, global NAFLD prevalence is expected to increase in forthcoming years in parallel to a rise in insulin resistance and obesity [8], which spells a growing economic challenge. Furthermore, it is widely accepted that there are evident sex differences in NAFLD [9]. The prevalence of the disease and its fibrotic progression follow a sexually dimorphic pattern [10]. Fertile women have been found to be at lower risk of NAFLD than men [11], but post-menopausal women lose the protection conferred by estrogens [9,10,11].
Drug-induced steatosis has the potential to directly produce NAFLD and, although drugs are not normally a primary cause of NAFLD, it has been shown that they may underlie, mimic, or aggravate NAFLD [12]. There are reports that drugs often used in clinical practice, such as amiodarone, methotrexate, tamoxifen, and some chemotherapeutic agents, may lead to hepatic steatosis [12,13].
Although NAFLD is the most common liver disease, no pharmacological treatment is presently approved, and the first line of treatment is lifestyle modification [14]. Even so, more than 200 active trials of NAFLD treatments are presently underway [15]. The multidimensional NAFLD etiopathogenesis is also reflected in drug development, as treatments focus on controlling the different pathways implicated in disease progression, such as chronic inflammation, insulin resistance, and fibrogenesis [7].
The need for safe, effective treatments has urged the development of both in vivo and in vitro models of NAFLD to better understand its pathogenesis, to identify potential targets, and to assess the therapeutic potential of drugs before transferring them to clinics [16]. In vitro models of NAFLD comprise simple cell-based models incubated with different lipids to more complex three-dimensional (3D) organoids. These liver cell-based NAFLD models include conventionally cultured hepatic cell lines, primary human hepatocytes (PHH), cocultures of different liver cells, and engineered liver platforms, such as perfusion systems [17]. Disease heterogeneity is relevant in clinical practice for using more personalized diagnostic and treatment approaches [18], and also in the preclinical setting to utilize in vitro models that better reflect population variability (including sex differences in NAFLD and differential drug responses). To achieve more patient-specific human cell-based models that accurately mimic pathologies in the liver, new in vitro systems have recently been developed, of which induced pluripotent stem cells (iPSCs) differentiated into hepatocyte-like cells (HLCs) are a promising option for liver disease modeling in vitro by allowing the study of the mechanisms implicated in the development and progression of this disease [19].
Despite the clear need for robust in vitro systems that mimic NAFLD, the development and employment of predictive assays are also essential, such as studying transcriptional expression, identifying potential protein and metabolite biomarkers, and running cytomic assays. The combination of results from well-validated in vitro models and new omic approaches will result in more predictive assays that can help to understand NAFLD and its progression, and that will also be useful during drug development.
Although clear progress in clarifying NAFLD etiopathogenesis and developing pharmacological treatments has been described, there are still many challenges to elucidate the mechanisms implicated in this disease and its progression, and to find new therapies. Therefore, basic preclinical research is essential. Here we review the major mechanisms of liver injury in NAFLD, and the advances made in the in vitro models and tools currently used for studying NAFLD, by focusing on employing iPSCs as a personalized medicine approach to study this disease.
2. Mechanisms of Liver Injury in NAFLD: Mitochondrial Dysfunction, Oxidative Stress, and Lipotoxicity
Uptake of non-esterified fatty acids (NEFA) from the diet, circulating NEFA released from lipolysis in adipose tissue, and de novo synthesis in the cytosol of hepatocytes are the major origins of NEFA present in the liver. The two major pathways to remove NEFA from hepatocytes are fatty acid oxidation in mitochondria (β-oxidation) and their export to blood as VLDL (very low-density lipoproteins). NEFA are stored mainly as triacylglycerides (TAG) in small intracellular fat droplets. TAG accumulation in hepatocytes is not necessarily a pathological condition, but is considered a protective mechanism to avoid the potential toxic effects induced by high NEFA levels, particularly saturated long-chain fatty acids [20].
A very close association between insulin resistance and NAFLD is well accepted [21]. Insulin resistance favors lipolysis in white adipose tissue, which leads to more NEFA being released to the liver. In the liver, hyperinsulinemia and/or insulin resistance result in hepatic lipid overaccumulation by down-regulating triglyceride export from the liver as VLDL, and by promoting the use of glucose (and fructose) for the de novo synthesis of lipids (lipogenesis) in the cytosol of hepatocytes [22,23]. Insulin-mediated effects on lipogenesis and lipid accumulation in the liver are strictly regulated by several nuclear receptors and transcription factors (e.g., SREBP1c, ChREBP, PPARs, and LXR) [24,25].
Mitochondria play a key role in the homeostasis of lipid metabolism. Under normal physiological conditions, most NEFA enter mitochondria via carnitine palmitoyl transferase, where they are converted into acetyl-CoA by the β-oxidation pathway, with subsequent ATP generation via the tricarboxylic acid cycle and oxidative phosphorylation. With increased NEFA flux in hepatocytes, the upregulation of fatty acid oxidation in mitochondria is one of the first metabolic adaptations that is triggered in the liver to prevent fat overaccumulation [26,27]. Thus, increased electron flux to the mitochondrial respiratory chain favors the formation of reactive oxygen species (ROS), mainly superoxide anion, that can contribute to altering mitochondria function and integrity (reduced ATP synthesis, mitochondrial membrane depolarization, depletion of mitochondrial DNA, release of cytochrome c and pro-apoptotic factors, and impaired mitochondrial biogenesis) (Figure 1). In turn, these mitochondrial alterations can favor ROS overproduction and, thus, contribute to exacerbating mitochondrial damage and oxidative stress [28,29].
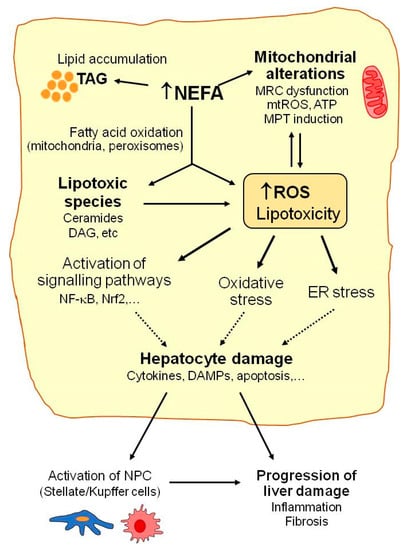
Figure 1.
Mechanisms implicated in NAFLD. Many molecular pathways contribute to NAFLD development. When the liver’s capacity to control energy substrates is overwhelmed, toxic lipid species may accumulate. This can be associated with increased reactive oxygen species (ROS) production, mitochondrial injury (mitochondrial respiratory chain (MRC) alterations, increased mitochondrial ROS (mtROS), or mitochondrial membrane permeability transition (MPT) induction), hepatocellular stress, and liver injury. Excessively high non esterified fatty acid (NEFA) levels enhance triacylglycerides’ (TAG) accumulation and fatty acid oxidation in hepatocytes and favor ROS generation, which may contribute to alterations in mitochondrial function and metabolic energy homeostasis in hepatocytes. Accumulation of lipotoxic species (e.g., ceramides, diacylglycerol (DAG)) may aggravate mitochondrial dysfunction and contribute to ROS overproduction, oxidative stress, endoplasmic reticulum (ER) stress, and activation of the signaling pathways involved in hepatocyte injury, including the release of cytokines or damage-associated molecular patterns (DAMPs). Finally, activation of non-parenchymal cells (NPC) by oxidative stress or by the signals released by damaged hepatocytes may contribute to NAFLD progression.
Moreover, with marked lipid metabolism dysregulation, which occurs in NAFLD, mitochondrial β-oxidation is insufficient to metabolize excess NEFAs, which accumulate in hepatocytes and may lead to more lipotoxic species forming (e.g., ceramides, diacylglycerols, lysophosphatidylcholines, or other lysophosphatidic acid derivatives, saturated fatty acids, and oxidized low-density lipoproteins) [20,30,31]. This excessive accumulation of fat and lipotoxic intermediates has deleterious effects on diverse cell organelles and functions, which are generally known as lipotoxicity (Figure 1). Mitochondrial dysfunction, endoplasmic reticulum (ER) stress, and the activation of signaling pathways related to inflammation and cell death are among the most prominent consequences of these deleterious effects [30].
Lipotoxic species such as ceramides can inhibit mitochondrial respiratory chain complexes and lead to ATP depletion, increased ROS production, the deregulation of the mitochondrial antioxidant defense system, and other mitochondrial disturbances and, thus, contribute to the aggravation of mitochondrial dysfunction and oxidative stress [28,32]. ER stress induced by lipotoxic agents has been related to ROS overproduction and exacerbated oxidative stress in NAFLD [33]. ER stress also contributes to reduced triglyceride synthesis and VLDL export, which increase intracellular fat accumulation in the liver. Lipotoxicity also up-regulates the signaling pathways (e.g., NF-κB, JNK, and NLRP3) that promote the release of the pro-inflammatory and pro-apoptotic mediators that actively participate in liver cell injury progression [28,34]. Not only hepatocytes but also non parenchymal liver cells (NPC, Kupffer, stellate, or sinusoidal endothelial cells) are susceptible to oxidative stress and lipotoxic species by playing important roles in NAFLD. The cytokines and DAMPs (damage-associated molecular patterns) released by damaged or dead hepatocytes may induce the activation of innate immune cells, the recruitment of macrophages, and the activation of Kupffer cells (KC), which trigger an inflammatory response in the liver. Oxidative species may also induce the activation of stellate cells and stimulate their collagen production to, thus, promote liver fibrosis [28]. KC and infiltrating monocyte-derived macrophages perform an important dual function in the development and progression of NAFLD [35]. They may release pro-inflammatory cytokines and promote liver injury or produce anti-inflammatory cytokines and limit disease progression [35]. It has been reported that exposure to saturated fatty acids (e.g., PA) favors the polarization of liver macrophages to the pro-inflammatory M1 phenotype, while unsaturated fatty acids (e.g., OA) promote KC differentiation to the anti-inflammatory M2 phenotype [36]. Further research is necessary to better understand the role of KC polarization in NAFLD and to clarify its potential utility as a future therapeutic target in NAFLD.
3. Liver Cell Models to Study NAFLD
The biological systems used for the in vitro study of NAFLD range from monolayer cell–cultures to more complex 3D cultures. The purpose is to recapitulate the biology of NAFLD and identify the specific pathways implicated in the pathogenesis of this disease, and to also find useful therapeutic targets for drug development. Apart from hepatocytes, different NPC, such as KC, liver endothelial cells (LEC), or hepatic stellate cells (HSC), may affect liver biology, and have been described to play an important role in NAFLD pathogenesis. In recent years, new in vitro models have been suggested for studying NAFLD pathogenesis. Yet, despite a majority of in vitro studies focusing on the biology of NAFLD, publications reporting compound testing have increased in the last three years [37]. Figure 2 schematically presents the major advantages and disadvantages of each described model. Table 1 summarizes the main in vitro models for studying NAFLD.
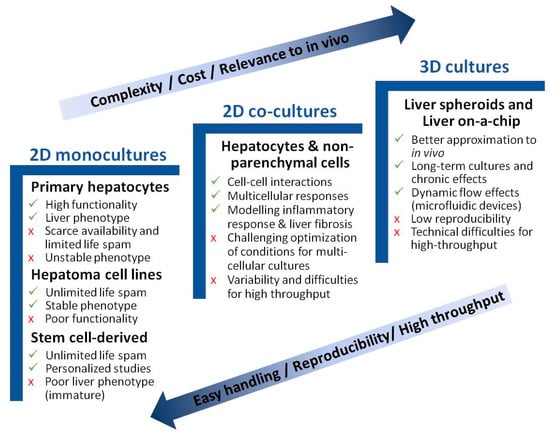
Figure 2.
Advantages and disadvantages of in vitro models for studying NAFLD. Cell culture models are listed per increasing cost, longevity, and complexity. Simpler models are better suited for high-throughput applications.

Table 1.
Cellular models to study NAFLD in vitro.
3.1. Monoculture Models
PHH are considered the closest in vitro model to the human liver. Hepatocytes are obtained by liver tissue digestion and retained in culture-specific liver functions, such as metabolic detoxification of foreign compounds, glycogen synthesis and storage, lipid metabolism, urea and albumin production, and the functional expression of membrane transport proteins [38]. Therefore, PHH have been proposed as an in vitro model for diverse hepatology fields, such as pharmacological and toxicological studies, and liver diseases modeling, including NAFLD. However, their widespread use is hindered by scarce liver tissue availability, high interdonor variability, and the short life and progressive loss of hepatocytes’ functionality during culture [39]. In contrast, immortalized hepatic cell lines offer an almost unlimited proliferation potential, high availability, easy handling, and a stable phenotype, and are widely used as alternative cell models to PHH. Their main disadvantage is the low expression of certain enzymes, mainly those involved in drug metabolism [40].
The most widespread approach to generate in vitro models of NAFLD is adding NEFA to cell culture medium. In particular, an NEFA mixture containing oleic and palmitic acids (OA and PA, at the 2:1 ratio) has been shown to induce lipid accumulation in both PHH and hepatoma cell lines (e.g., HepG2, Huh7, HepaRG, or LO2), which can be easily evidenced by Nile Red, BODIPY 493/503, or Oil Red O staining. Cells incubated with NEFA show the formation of cytosolic fat droplets, and their morphology resembles ballooned hepatocytes, which are typical of the steatosis process. The extent of lipid accumulation, mainly TAG, in PHH exposed to NEFA is similar to that observed in the liver of patients with steatosis [41]. Apart from lipid accumulation, induction of apoptosis, increased ER stress, and inhibition of protein synthesis have also been observed in NEFA-treated hepatocytes [41,42]. Cytotoxicity, increased ROS, and induction of oxidative stress, mitochondrial alterations, and production of inflammatory or fibrinogenic cytokines are other effects observed in NAFLD cell models [43,44,45,46]. The composition of the NEFA mixtures used to induce fat-overloaded cells can influence the observed effects. NEFA mixtures with a low proportion of saturated PA induced lipid levels display minor effects on cell viability and, thus, render an NAFLD cell model that mimics benign steatosis. In contrast, a high proportion of PA favors not only fat accumulation but also harmful cytotoxic and apoptotic effects [41].
In addition to NEFA, drugs or endocrine disruptors such as bisphenol A have been used to generate NAFLD cell models [47,48,49]. Drug-induced steatosis has been well-studied in vitro and associated with both increases in lipid accumulation and ROS formation in several liver cell models [50,51]. More detailed studies into HepG2 cells have provided a better understanding of the mechanisms associated with amiodarone, a well-known steatogenic compound that not only produces increased lipid accumulation but also changes in the expression of the genes related to lipid metabolism, such as SREBP1c and DGAT1, along with increased activation of ER-stress regulator IRE1α and induction of autophagy [52]. The steatogenic effects of drugs have also been assessed in Lo2 cells, where exposure to valproic acid increases cytoplasmic lipid levels and enhances oxidative stress, as reflected by the lower GSH level or the higher MDA and ROS levels. This increase has been linked with an increased expression of isoform 2E1 of cytochrome P450 (CYP), as valproic acid-induced ROS accumulation and hepatic steatosis were attenuated when CYP2E1 was inhibited using the CYP2E1 inhibitor or CYP2E1 CRISPR knockdown (CYP2E1-KD) [53].
A comparative study has revealed that HepaRG cells exhibit more sensitivity to drug-induced steatosis than HepG2 cells [54]. HepaRG cells are known to show a better expression of different drug-metabolizing enzymes and liver functions than other hepatoma cell lines, such as HepG2 [55]. Allard et al. recently studied the mechanisms by which 12 commonly used drugs produced steatosis in HepaRG, and observed different patterns [56]. For instance, a group of drugs that includes amiodarone and rifampicin caused steatosis by decreasing mitochondrial fatty acid oxidation, whereas another set of drugs (i.e., allopurinol, fluorouracil, and troglitazone) reduced the expression of the proteins involved in VLDL secretion, such as APOB and induced ER stress [56]. Regarding the antioxidant capacity of different cell lines, a comparative study of HepG2 and HepaRG cells showed a more significant decrease in GSH or increase in ROS in HepaRG than in HepG2 cells after treatment with compounds that are known to produce oxidative stress induction [57].
Single-cell in vitro models fail to accurately recapitulate the pathological mechanisms of liver diseases. Therefore, more physiologically relevant models such as cocultures or 3D systems have been developed.
3.2. Coculture Models
Although hepatocytes are the major cell type in the liver, they co-exist with other NPC, such as LEC, KC, and HSC, which play important roles in NAFLD development. Barbero-Becerra et al. studied the interaction between HuH7 and LX2 (a hepatic stellate cell line) and observed that NEFA exposure increases the α-smooth muscle actin (α-SMA) expression in LX2, regardless of the cell–cell contact with hepatocytes [58]. In a similar model, greater α-SMA production and an increase in extracellular matrix (ECM) components, such as collagen I, collagen II, collagen IV, fibronectin, and profibrotic proteins such as MMP-2 and MMP-9, have been reported and linked with higher oxidative stress [59]. Activation of cocultured HSC was related to up-regulation of antioxidant regulator Nrf2 in hepatocytes damaged by lipid accumulation, and Nrf2 was suggested as a potential therapeutic target to prevent or delay NASH progression. These studies evidenced the interplay between lipid accumulation in hepatocytes and HSC activation with a fibronogenic phenotype in co-culture models of NASH.
KC are resident macrophages in the liver responsible for detecting local damage and eliminating foreign substances. The coculture of KC and hepatocytes has been used to study not only the inflammatory process that accompanies NAFLD, but also possible therapeutic targets [60]. The coculture of mouse primary hepatocytes and KC in a transwell system incubated with PA has been used as a model of NASH and to study drug effects. The model confirmed the role of KC in NASH progression and elucidated different implicated molecular mechanisms [61]. Cocultured hepatocytes show higher sensitivity to the toxicity, due to some compounds with oxidative properties, than do hepatocyte monocultures [62]. This has been related to the generation of toxic metabolites that decrease GSH levels in hepatocytes and activate ROS formation by KC causing further GSH depletion.
3.3. 3D Models of NAFLD
By better understanding that 3D cultures more accurately reflect in vivo physiology, in the last few decades, many researchers have focused on developing and optimizing different 3D strategies to better preserve liver properties in vitro by mimicking the architecture and cell–cell interactions [63]. 3D models include scaffold-containing and scaffold-free systems.
Cell spheroids are one of the most widely used scaffold-free strategies to generate 3D NAFLD models. This approach consists of generating cell aggregates by different techniques, such as hanging drop, low-adherence substrates, microwells, bioreactors, and magnetic levitation [64]. PHH spheroids maintain their viability and functionality up to 21 days and have been used to generate an NAFLD model after exposure to high levels of insulin, NEFA, and monosaccharides [65]. After a seven-day accumulation of intracellular lipids, the development of insulin resistance was detected in spheroids on day 14, as evidenced by the increased expression of PCK1 and PDK4 and by reduced GSK3β phosphorylation [65]. The spheroid methodology was followed to coculture HepG2 and LX-2 cells [66]. This system demonstrated that hepatic stellate LX-2 cells facilitate the compactness of spheroids, which confirms the role of HSC in matrix remodeling. Treatment with a mixture of NEFA brought about an increase in cytoplasmic lipids and a higher COL1A1 expression, and allowed the effects of anti-NASH drugs such as liraglutide or elafibranor to be studied [66]. Some 3D liver in vitro tissue models are commercially available, such as 3D InSightTM Human Liver Microtissues (InSphero). The system consists of PHH, HSCs, LEC, and KC, which are organized in microspheres. It has been used to model NASH with severe fibrosis by exposing microtissues to high PA concentrations, which results in the induction of a fibrotic and proinflammatory profile in tissue with increased expressions of IL-8 and collagen 1 and 3 [67]. This model has been described as particularly useful for mechanistic exploration of inflammation-associated processes that would be of special interest for studying the antioxidant response [68].
Native tissues comprise a 3D viscoelastic milieu, the ECM, which guides cells’ development, interactions, and homeostasis. Therefore, mimicking the natural ECM is a promising approach to gain a 3D structure for a cell culture that better represents what happens in vivo. For instance, the PHH cultured in a collagen-sandwich configuration retain higher CYP expression, greater metabolic activity, and a longer life span to, thus, delay the dedifferentiation process by up to 14 days in culture [69]. This system has also been used for the coculture of PHH, macrophages, and HSC in another system that incorporates both perfusion and hemodynamic shear forces [70]. Increases in cytoplasmic lipids and the synthesis of triglycerides, diglycerides, and cholesterol esters, accompanied by the expression of the genes associated with apoptotic and oxidative stress signals, have been observed after a 10-day exposure to high concentrations of glucose, insulin, and NEFA. Greater insulin resistance (increased PCK1 expression and the generation of a proinflammatory profile) and a fibrotic profile (increased α-SMA, TGF-β, and steopontin) have also been reported [70].
Hydrogel-based systems are 3D hydrophilic polymeric networks that mimic ECM and allow the free diffusion of oxygen and nutrients. Duriez et al. developed a 3D NASH model by combining the culture of four cell types embedded in collagen hydrogel with exposure to glucose, NEFA, and TNF-α for 15 days. This model showed the accumulation of lipid droplets in the cytoplasm, the generation of a proinflammatory environment determined by increased IL6 and CCL2 expressions, and the induction of early fibrosis with the expressions of MMP2 and MMP9 [71].
3.4. Liver-on-a-Chip
The liver-on-a-chip methodology is based on combining different structures that recreate the conditions and dynamics of a small-scale liver. On these platforms, several polymeric chambers act as supports to generate cell cultures which are connected by channels that distribute microfluids with nutrients and oxygen [72]. This technology allows the generation of cell–cell interactions and a dynamic flow to increase cell survival and functionality. Liver-on-a-chip has emerged as a very powerful tool to not only study pathologies but to also preclinically study potential therapies [73,74]. The ability to connect different chambers also allows the combination of distinct tissues that may be important in disease development. Despite its advantages, this model is not widely used because of its high cost and considerable complexity.
The liver-on-a-chip technology has been utilized to model NAFLD by employing simple cell systems, such as HepG2 cells, and ranges to more complex ones that combine different cell types. The culture of HepG2 cells in an NEFA perfused device for 24 h and 48 h allows more gradual triglyceride deposition and increased cell survival which, thus, resemble chronic steatosis that develops in vivo [75]. This model confirmed that PA is more cytotoxic than OA and revealed a direct contribution of PA overload to induce oxidative stress (production of total ROS and superoxide) in liver cells [76].
Besides steatosis, inflammation and fibrosis are two important events in NAFLD development. The use of liver-on-a-chip that combines several liver cells (hepatocytes, HSC, LEC, and KC) and stimulation with NEFA and LPS results in increased cytoplasmic lipid accumulation, ballooned hepatocytes, and higher a-SMA, collagen 1A1, and TIMP-1 production, which are indicators of liver fibrosis [77]. The system recapitulates NASH endpoints and also allows testing of anti-NASH drugs, with a very high potential for further drug testing [77].
The organ-on-a-chip technology has been used to assess the role of other tissues in NAFLD development. Lee et al. constructed a gut–liver microfluidic chip and showed that most of the lipids that accumulated in hepatocytes while this disease developed were obtained from exogenous input and transport through the gut, and were key. Additionally, the efficacy of antisteatotic compounds, whose mechanism of action is based on altering lipid absorption at the intestinal level, has been evidenced [78]. More recently, the influence of lipolysis and insulin resistance on NAFLD development has been explored by a model of adipocyte and hepatocyte chambers. With this model, in a lipotoxic environment, adipocytes modulated adipokine secretion and lipolysis to increase hepatic steatosis [79].
4. Human Hepatocyte-like Cells Deriving from Pluripotent Stem Cells for NAFLD Modeling
Since their discovery, stem cells have been a proven potential tool for developmental biology, drug toxicity, or regenerative medicine thanks to their capability to proliferate and differentiate into somatic cells. The possibility of using stem cells to recreate some hallmarks of different diseases has also been explored in vitro, and has been demonstrated as a promising approach for disease modeling and a competent alternative to PHH and immortalized cell lines [82,83].
Human adult stem cells reside in specific niches inside tissues, and their multipotency allows them to differentiate into different tissue-specific somatic cells to contribute to tissue regeneration and renewal [84]. In fact, stem cells from different tissues, such as bone marrow, adipose tissue, or umbilical cord blood, have been differentiated into cells with the hepatic phenotype [85]. Human skin-derived precursors have been shown to differentiate into hepatic progenitor cells in vitro with suitable properties for drug hepatotoxicity studies [86], NAFLD modeling [87], and efficacy assessments of potential anti-NASH compounds [88,89]. Nonetheless, adult stem cells are rare, their isolation may prove difficult given tissue accessibility restrictions, and long times are required to extend the obtained cells in vitro to generate sufficient stock [90].
Human pluripotent stem cells (PSCs), such as embryonic stem cells (ESCs) and iPSCs, indefinitely proliferate and are capable of forming cells of all germ layers. PSCs can be differentiated into hepatocyte-like cells (HLCs) that are morphologically and functionally similar to PHH [91] and provide an unlimited source of HLCs that can be cultured longer before loss functionality.
PSC-derived HLCs have been used to generate in vitro models that recapitulate some NAFLD features (Table 2). Lipid accumulation has been accurately recreated in HLC-based models upon lipid overloading [19,92,93,94,95,96], high-energy substrate induction [97], or steatogenic drug exposure [98]. Figure 3 depicts a representative example of increased lipid accumulation induced by drugs in an NAFLD model of HLCs deriving from iPSCs.
Table 2.
Pluripotent stem cell-derived cell models to study NAFLD.
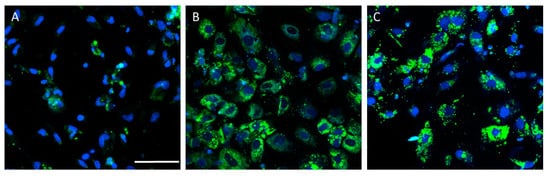
Figure 3.
Drug-induced lipid accumulation in the hepatocyte-like cells (HLCs) derived from induced-pluripotent stem cells (iPSCs) from a healthy donor. Representative images of untreated HLCs (A), and HLCs treated with valproate (B) or amiodarone (C), are also shown. Nuclei were stained with Hoechst 33342 (blue) and neutral lipids with BODIPY 493 (green). Scale bar (50 µm) applies to all images.
Other NAFLD hallmarks, such as mitochondrial dysfunction and oxidative stress, have been recreated in different HLC models [95,97]. Some cell stressors, such as ER stressor thapsigargin, are useful for potentiating the steatosis phenotype upon NEFA overload in iPSC-derived HLCs, which confirms the link between ER homeostasis and lipid metabolism. This exacerbated phenotype has allowed the effects of the therapeutic drug tauroursodeoxycholic acid and experimental compound obeticholic acid to be evaluated, which reduced the steatosic phenotype in iPSC-derived-HLCs [94]. This scenario suggests that HLC-based NAFLD modeling is a promising tool for testing potential anti-NASH compounds. HLCs have also been described as a potential platform for assessing drug-induced steatosis and as a similar cell model to HepG2 [98].
One of the major advantages of iPSCs is that they can be obtained by a well-standardized reprogramming process using somatic cells, which can be obtained by minimal invasive methods (blood or urine collection). The iPSCs from patient somatic cells can be employed to generate HLCs, which maintain the patient-specific genotype and metabolic variations [99,100] to allow the study of the interindividual variability associated with disease development and progression, as in NAFLD [83]. In fact, the iPSC-derived HLCs from NAFLD patients have been developed to obtain a potential NAFLD model to study the idiosyncratic nature of this disease [101,102]. An HLC-derived NAFLD cell model from different NAFLD patients has revealed some differences in the steatotic phenotype among donors, including differences in drug response. By analyzing all the transcriptome data from a single donor, a different response to AdipoRon (an adiponectin-like small molecule that can revert the steatotic phenotype) treatment has been observed, which could be partly due to variations in donors’ individual genetic backgrounds [103].
Specific genetic alterations related to increased NAFLD susceptibility can be modeled with HLCs derived from NAFLD patients. For instance, mutations in patatin-like phospholipase domain-containing protein 3 (PNPLA3), such as variant I148M (responsible for loss of enzymatic activity [104]), have been successfully recreated in iPSC-derived HLCs by the CRISPR/Cas9 technology for either knockout PNPLA3 or the knockin I148M variant [96]. PNPLA3I148M–HLCs exhibit a similar lipidomic profile to liver biopsies from donors with the I148M variant and show more susceptibility to other toxicity types, due to the down-regulation in the detoxification pathways related to variant I148M [96].
Nevertheless, 2D monocellular HLC models failed to recapitulate some NAFLD hallmarks because the cytoarchitecture was lacking, which mimics liver parenchyma and critical hepatocyte–NPC interactions [93]. Liver organoids have been developed with PSCs, adult stem cells, or fetal/adult liver cells. Under specific culture conditions (including growth factors and cytokines), stem cells differentiate and progress, like liver development, to generate adult somatic cells in a 3D environment and to give organoids that mimic structural and functional liver parameters [105]. These hepatic organoids not only recreate NAFLD hallmarks (e.g., lipid accumulation, increased lipid peroxidation, and higher ROS levels), but also show bile canaliculi network disruption upon lipid induction [95], as described in NAFLD-patient samples [106]. Lipid droplet accumulation in HLCs has been observed in the organoids formed by HLCs from NAFLD patient-derived iPSCs and NPC lineages (macrophages, mesenchymal stem cells, and endothelial cells) when exposed to an NEFA mixture. Interestingly, Gurevich et al. noted how these NAFLD patient-derived HLCs organoids displayed spontaneous lipid accumulation without lipid overloading [93].
The bioengineered tissue formed by iPSC-derived HLCs with an inducible knockdown expression of deacetylase sirtuin-1 (SIRT1), cultured in combination with macrophages, has been reported to mimic the pro-inflammatory phenotype upon lipid overloading and revealed the major implication of sirt1 in de novo lipogenesis and β-oxidation regulation, along with their pivotal role in NAFLD development and progression [92]. Thus, 3D models are powerful tools to elucidate the mechanisms underlying NAFLD development with a view to study cell interactions in the progression of this disease. However, these cell culture models are still expensive and need further technological improvements.
HLCs are a proven competent tool for mimicking several NAFLD hallmarks in vitro by studying the cellular mechanisms implicated in the disease and their progression, and the genetic alterations related to NAFLD development, by recreating the NAFLD patient-specific genotype and learning about the genetic predisposition to NAFLD and its idiosyncrasy. This personalized in vitro approach would better reflect specific pathobiological features of disease among NAFLD patients, such as metabolic status or sex differences. Table 2 summarizes the different PSC-cell models used to study NAFLD. Three-dimensional cultures of HLCs more closely recreate the liver’s cytoarchitecture (including the bile network) than 2D cultures, while 3D cocultures with NPCs have revealed the possibility of more accurately mimicking other NAFLD hallmarks, such as the proinflammatory phenotype. Nevertheless, more studies are required, and other approaches should be considered, such as including other NPCs in HLCs cocultures, like HSCs, which play an important role in fibrotic NAFLD progression to NASH [58]. Disease modeling based on HLCs has to deal with other drawbacks, such as the inability to obtain a fully mature phenotype in HLCs, because they exhibit characteristics of immature hepatocytes [91,99,107]. Therefore, differentiation protocols are constantly improving to obtain mature HLCs, which have become the best starting point for disease modeling [108]. Although stem cell-based NAFLD modeling is a promising tool in the research and clinical fields, it cannot be denied that further research is needed to standardize culture conditions.
5. New Approaches and Tools for the In Vitro Assessment of NAFLD
NAFLD is characterized by alterations in a wide range of molecules, such as proteins, NEFA and other lipids, and metabolites [109], and is also related to changes in expression patterns [110]. Therefore, a wide spectrum of techniques (staining lipids with specific dyes, coupled-reaction assays for metabolites, enzyme activity measurements, analysis of protein expression by immunochemistry or Western blot analyses, or analysis of mRNA by RT-PCR) has been routinely followed to identify and analyze cellular, biochemical, and functional changes in NAFLD models. By applying these traditional methodologies, only a single parameter (or a few parameters) is evaluated in each assay, which is time-consuming and seriously limits the full characterization of NAFLD-related events. In contrast, omics-based technologies allow the simultaneous analysis of many parameters in the same cell system and offer the possibility of performing more comprehensive mechanistic studies about the global events involved in NAFLD.
Many omics studies have been performed to analyze patients’ samples (plasma, liver tissue, faeces) or samples from in vivo experimental models of NAFLD, to find new biomarkers for the prognosis, diagnosis, or monitoring of the disease [111,112,113,114,115]. Omics studies have also been applied to NAFLD in vitro models as novel high-throughput strategies to search and identify new NAFLD biomarkers or therapeutic targets, and to test new molecules as potential drugs to treat NAFLD (Table 3). In many studies, the in vitro results obtained in NAFLD cell models have been validated with, or compared to, data from in vivo serum-based studies.
Table 3.
Omics approaches for the in vitro modeling of NAFLD.
Transcriptomics is probably the most widely used omics for the in vitro study of NAFLD. Both microarray and RNA sequencing (RNASeq) technologies have been followed to identify and quantify RNA expression profiles in in vitro NAFLD models (Table 3). For instance, untargeted microarray analyses in steatotic and non steatotic Huh7 cells [116] have revealed 88 differentially expressed genes, including cytokine CXCL10, which was previously identified by in vivo studies as a powerful biomarker for NASH [127,128]. A transcriptomic analysis in an NAFLD model in PSC-derived HLCs has identified a steatotic profile, which includes numerous genes related to the PPAR pathway [19], as previously demonstrated in vivo [129]. The transcriptomics analysis has also been applied to evaluate drug candidates to treat NAFLD in vitro [88,100]. Some studies have also focused on the RNA-based regulatory mechanisms that underlie NAFLD, because transcriptomics has allowed the identification and quantification of non-coding RNAs, such as microRNAs (miRNAs) [19] or long non-coding RNAs (lncRNAs) [119,120].
Proteomics, metabolomics, and lipidomics (the branch of metabolomics that centers on studying lipidic perturbations that is especially relevant in steatosis studies) have also been useful in the in vitro study of NAFLD. The ever-growing use of these three analytical tools for research purposes has been driven by the recent methodological advances made in chromatography, either gas (GC) or liquid (LC), and mass spectrometry (MS). The analysis of the proteomic, metabolomic, and lipidomic profiles in in vitro NAFLD models may provide additional global insights into the mechanisms involved in NAFLD to identify new specific biomarkers for the clinical diagnosis of NAFLD or to discover novel drugs to treat NAFLD patients. By way of example, an untargeted proteomics study in an octanoate-based model of steatosis with C3A hepatoblastoma cells has demonstrated that lipid metabolism-related proteins, such as serum albumin, Perilipin-2, APOAI, AKR1C1, or FABP1, are the most altered proteins [121]. Metabolomics studies with a model of steatosis using OA/PA-treated HepaRG cells have shown significant changes in lipids, glutathione, carnitine, and tricarboxilic acid cycle intermediates, and metabolites related to oxidative stress, energy metabolism, and insulin resistance [43]. Similar results have been reported by Cuykx et al. in a metabolomics study with a valproate-induced HepaRG model of steatosis [123]. They found perturbations in the levels of lipids, such as ceramides, tryglicerides, and carnitine, among others. An untargeted lipidomics study with an OA/PA-treated HepG2 model of steatosis has demonstrated that levels of phospholipids, triglycerides, ceramides, and sphingomyelins alter under steatogenic conditions [124]. All these studies sustain the suitability of proteomics, metabolomics, and lipidomics studies for assessing NAFLD in vitro.
The application of combined strategies with two omics technologies or more will lead to a more exhaustive and global NAFLD characterization and will contribute to a better understanding of all the involved mechanisms. Along these lines, a few reports illustrate the application of this integrative strategy to study NAFLD in diverse cell liver models, such as PHH, hepatoma cell lines, or HLCs [70,97,125,126]. However, the interpretation and integration of data from multiple omics are difficult because alterations in gene or protein expression and metabolite levels occur on different time scales.
Finally, cytomics, the comprehensive, structural, and functional study of the cytome at a level of individual cells, has been proposed as a multiparametric tool to examine the complex and dynamics biology of in vitro cell models. Cytomics is based on using multiplexed cell staining assays to analyze alterations in cell structures or functions. Flow cytometry is the main technique followed in cytomics studies, but some microscopy-based technologies such as high-content screening (HCS) have also been applied to study NAFLD in vitro. For instance, different fluorescent probes can be used to stain and quantify lipids [126]. Flow cytometry has been applied and/or HCS assays have been run to study drug-induced steatosis in HepG2, HepaRG, or Upcyte cells [50,51,54,81]. This technique also allows the combination of several fluorescent probes to identify steatogenic drugs with high sensitivity and to analyze the key mitochondrial alterations and oxidative stress associated with increased intracellular lipid levels [130].
6. Conclusions
NAFLD is a complex disease that can be triggered by a combination of different genetic, metabolic, immunological, and dietary factors. Moreover, there is compelling evidence for sex differences and the effect of reproductive status on NAFLD that remain largely unexplored [9]. Experimental methodologies in NAFLD should allow us to understand the pathogenesis of this disease, to evaluate the response to drugs, and to assess differential responses and pathogenesis due to gender. In the past few years, the diversity and complexity of cellular models used to study NAFLD have increased. However, these in vitro test systems still have many limitations and need further technological developments to provide a more in-depth mechanistic understanding of the disease and the implicated therapeutic mechanisms. The use of iPSCs deriving from patients and differentiated into HLCs may recapitulate many human NAFLD features and allow the effects of one drug, or more, on the key relevant pathways for disease progression to be assessed. The use of IPSCs deriving from donors with different NAFLD disease grades provides a new valuable tool to study whether differential responses to treatments are partially due to variations in individual genetic backgrounds. As traditional 2D cultures do not recapitulate native 3D spatial organization and intercellular interactions, new in vitro models (3D organoids, hydrogels, or liver-on-a-chip systems) attempt to mimic the real microenvironment of liver cells to provide more valuable and predictive models. Finally, the application of readout technologies that offer an exhaustive analysis of alterations at different levels by transcriptomics, metabolomics, or proteomics provides powerful mechanistic information that can even be applied to clinical practice, after an appropriate validation process. Although there is still much work to be done, the combination of new in vitro models that better recapitulate NAFLD and the use of new methodological approaches are expected to help us understand the disease and to extend these preclinical findings to humans.
Author Contributions
Writing—original draft preparation, M.P., E.V.-B., E.T.-L., M.T.D. and L.T.; writing—review and editing, M.T.D. and L.T. All authors have read and agreed to the published version of the manuscript.
Funding
This work has been supported by the Carlos III Health Institute (ISCIII, Plan Estatal de I+D+i 2013–2016) and co-financed by the European Regional Development Fund “A way to achieve Europe” (FEDER) through Grants PI18/00993 and CP16/00097, by the Spanish Ministry of Science and Innovation “Agencia Estatal de Investigación” through Grant PID2019-106000RB-C22 funded by MCIN/AEI/10.13039/501100011033, and by the Generalitat Valenciana (PROMETEO/2019/060). M.P. was supported by ACIF/2018/0226 (Generalitat Valenciana), L.T. by ISCIII CP16/00097, and E.T. by FPU19/02240 (Spanish Ministry of Universities).
Acknowledgments
The authors acknowledge Helen Warburton for her correction and support in linguistic matters.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
3D: three dimensional; α-SMA: α-smooth muscle actin; CYP cytochrome P450; DAMPs: damage-associated molecular patterns; ECM: extracellular matrix; ER: endoplasmic reticulum; ESC: embryonic stem cells; MRI: magnetic resonance imaging; NEFA: non esterified fatty acids; GC: gas chromatography; HCS: high-content screening; HLCs: hepatocyte-like cells; HSC: hepatic stellate cells; iPSCs: induced-pluripotent stem cells; KC: Kupffer cells; LC: liquid chromatography; LEC: liver endothelial cells; MS: mass spectrometry; NAFLD: nonalcoholic fatty liver disease; NASH: nonalcoholic steatohepatitis; NPC: non parenchymal cells; OA: oleic acid; PA: palmitic acid; PHH: primary human hepatocytes; PSC: pluripotent stem cells; ROS: reactive oxygen species; SIRT1: deacetylase sirtuin-1; TAG: triacylglyceride; VLDL: very low-density lipoproteins.
References
- Vernon, G.; Baranova, A.; Younossi, Z.M. Systematic review: The epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults. Aliment. Pharmacol. Ther. 2011, 34, 274–285. [Google Scholar] [CrossRef] [PubMed]
- European Association for the Study of the Liver; European Association for the Study of Diabetes; European Association for the Study of Obesity. EASL-EASD-EASO Clinical Practice Guidelines for the management of non-alcoholic fatty liver disease. J. Hepatol. 2016, 64, 1388–1402. [Google Scholar] [CrossRef]
- Berardo, C.; Di Pasqua, L.G.; Cagna, M.; Richelmi, P.; Vairetti, M.; Ferrigno, A. Nonalcoholic Fatty Liver Disease and Non-Alcoholic Steatohepatitis: Current Issues and Future Perspectives in Preclinical and Clinical Research. Int. J. Mol. Sci. 2020, 21, 9646. [Google Scholar] [CrossRef]
- Middleton, M.S.; Heba, E.R.; Hooker, C.A.; Bashir, M.R.; Fowler, K.J.; Sandrasegaran, K.; Brunt, E.M.; Kleiner, D.E.; Doo, E.; Van Natta, M.L.; et al. Agreement between Magnetic Resonance Imaging Proton Density Fat Fraction Measurements and Pathologist-Assigned Steatosis Grades of Liver Biopsies From Adults With Nonalcoholic Steatohepatitis. Gastroenterology 2017, 153, 753–761. [Google Scholar] [CrossRef] [Green Version]
- Tang, A.; Tan, J.; Sun, M.; Hamilton, G.; Bydder, M.; Wolfson, T.; Gamst, A.C.; Middleton, M.; Brunt, E.M.; Loomba, R.; et al. Nonalcoholic fatty liver disease: MR imaging of liver proton density fat fraction to assess hepatic steatosis. Radiology 2013, 267, 422–431. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Sanchez, G.N.; Dominguez-Perez, M.; Uribe, M.; Chavez-Tapia, N.C.; Nuno-Lambarri, N. Non-alcoholic fatty liver disease and microRNAs expression, how it affects the development and progression of the disease. Ann. Hepatol. 2021, 21, 100212. [Google Scholar] [CrossRef]
- Thiagarajan, P.; Aithal, G.P. Drug Development for Nonalcoholic Fatty Liver Disease: Landscape and Challenges. J. Clin. Exp. Hepatol. 2019, 9, 515–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Estes, C.; Razavi, H.; Loomba, R.; Younossi, Z.; Sanyal, A.J. Modeling the epidemic of nonalcoholic fatty liver disease demonstrates an exponential increase in burden of disease. Hepatology 2018, 67, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Lonardo, A.; Suzuki, A. Sexual Dimorphism of NAFLD in Adults. Focus on Clinical Aspects and Implications for Practice and Translational Research. J. Clin. Med. 2020, 9, 1278. [Google Scholar] [CrossRef]
- Lonardo, A.; Arab, J.P.; Arrese, M. Perspectives on Precision Medicine Approaches to NAFLD Diagnosis and Management. Adv. Ther. 2021, 38, 2130–2158. [Google Scholar] [CrossRef]
- Balakrishnan, M.; Patel, P.; Dunn-Valadez, S.; Dao, C.; Khan, V.; Ali, H.; El-Serag, L.; Hernaez, R.; Sisson, A.; Thrift, A.P.; et al. Women Have a Lower Risk of Nonalcoholic Fatty Liver Disease but a Higher Risk of Progression vs Men: A Systematic Review and Meta-analysis. Clin. Gastroenterol. Hepatol. 2021, 19, 61–71.e15. [Google Scholar] [CrossRef]
- Liebe, R.; Esposito, I.; Bock, H.H.; Vom Dahl, S.; Stindt, J.; Baumann, U.; Luedde, T.; Keitel, V. Diagnosis and management of secondary causes of steatohepatitis. J. Hepatol. 2021, 74, 1455–1471. [Google Scholar] [CrossRef]
- Miele, L.; Liguori, A.; Marrone, G.; Biolato, M.; Araneo, C.; Vaccaro, F.G.; Gasbarrini, A.; Grieco, A. Fatty liver and drugs: The two sides of the same coin. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 86–94. [Google Scholar] [PubMed]
- Zelber-Sagi, S.; Ratziu, V.; Oren, R. Nutrition and physical activity in NAFLD: An overview of the epidemiological evidence. World J. Gastroenterol. 2011, 17, 3377–3389. [Google Scholar] [CrossRef] [PubMed]
- Drew, L. Drug development: Sprint finish. Nature 2017, 551, S86–S89. [Google Scholar] [CrossRef]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef] [PubMed]
- Oseini, A.M.; Cole, B.K.; Issa, D.; Feaver, R.E.; Sanyal, A.J. Translating scientific discovery: The need for preclinical models of nonalcoholic steatohepatitis. Hepatol. Int. 2018, 12, 6–16. [Google Scholar] [CrossRef] [Green Version]
- Arrese, M.; Arab, J.P.; Barrera, F.; Kaufmann, B.; Valenti, L.; Feldstein, A.E. Insights into Nonalcoholic Fatty-Liver Disease Heterogeneity. Semin. Liver Dis. 2021, 41, 421–434. [Google Scholar] [CrossRef]
- Graffmann, N.; Ring, S.; Kawala, M.A.; Wruck, W.; Ncube, A.; Trompeter, H.I.; Adjaye, J. Modeling Nonalcoholic Fatty Liver Disease with Human Pluripotent Stem Cell-Derived Immature Hepatocyte-Like Cells Reveals Activation of PLIN2 and Confirms Regulatory Functions of Peroxisome Proliferator-Activated Receptor Alpha. Stem Cells Dev. 2016, 25, 1119–1133. [Google Scholar] [CrossRef] [Green Version]
- Listenberger, L.L.; Han, X.; Lewis, S.E.; Cases, S.; Farese, R.V., Jr.; Ory, D.S.; Schaffer, J.E. Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc. Natl. Acad. Sci. USA 2003, 100, 3077–3082. [Google Scholar] [CrossRef] [Green Version]
- Marchesini, G.; Brizi, M.; Bianchi, G.; Tomassetti, S.; Bugianesi, E.; Lenzi, M.; McCullough, A.J.; Natale, S.; Forlani, G.; Melchionda, N. Nonalcoholic fatty liver disease: A feature of the metabolic syndrome. Diabetes 2001, 50, 1844–1850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Ciaula, A.; Passarella, S.; Shanmugam, H.; Noviello, M.; Bonfrate, L.; Wang, D.Q.; Portincasa, P. Nonalcoholic Fatty Liver Disease (NAFLD). Mitochondria as Players and Targets of Therapies? Int. J. Mol. Sci. 2021, 22, 5375. [Google Scholar] [CrossRef] [PubMed]
- Vatner, D.F.; Majumdar, S.K.; Kumashiro, N.; Petersen, M.C.; Rahimi, Y.; Gattu, A.K.; Bears, M.; Camporez, J.P.; Cline, G.W.; Jurczak, M.J.; et al. Insulin-independent regulation of hepatic triglyceride synthesis by fatty acids. Proc. Natl. Acad. Sci. USA 2015, 112, 1143–1148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massart, J.; Begriche, K.; Moreau, C.; Fromenty, B. Role of nonalcoholic fatty liver disease as risk factor for drug-induced hepatotoxicity. J. Clin. Transl. Res. 2017, 3, 212–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mellor, C.L.; Steinmetz, F.P.; Cronin, M.T. The identification of nuclear receptors associated with hepatic steatosis to develop and extend adverse outcome pathways. Crit. Rev. Toxicol. 2016, 46, 138–152. [Google Scholar] [CrossRef]
- Hodson, L.; McQuaid, S.E.; Humphreys, S.M.; Milne, R.; Fielding, B.A.; Frayn, K.N.; Karpe, F. Greater dietary fat oxidation in obese compared with lean men: An adaptive mechanism to prevent liver fat accumulation? Am. J. Physiol. Endocrinol. Metab. 2010, 299, E584–E592. [Google Scholar] [CrossRef] [Green Version]
- Serviddio, G.; Bellanti, F.; Tamborra, R.; Rollo, T.; Romano, A.D.; Giudetti, A.M.; Capitanio, N.; Petrella, A.; Vendemiale, G.; Altomare, E. Alterations of hepatic ATP homeostasis and respiratory chain during development of non-alcoholic steatohepatitis in a rodent model. Eur. J. Clin. Investig. 2008, 38, 245–252. [Google Scholar] [CrossRef]
- Arroyave-Ospina, J.C.; Wu, Z.; Geng, Y.; Moshage, H. Role of Oxidative Stress in the Pathogenesis of Non-Alcoholic Fatty Liver Disease: Implications for Prevention and Therapy. Antioxidants 2021, 10, 174. [Google Scholar] [CrossRef]
- Serviddio, G.; Sastre, J.; Bellanti, F.; Vina, J.; Vendemiale, G.; Altomare, E. Mitochondrial involvement in non-alcoholic steatohepatitis. Mol. Aspects Med. 2008, 29, 22–35. [Google Scholar] [CrossRef]
- Gan, L.T.; Van Rooyen, D.M.; Koina, M.E.; McCuskey, R.S.; Teoh, N.C.; Farrell, G.C. Hepatocyte free cholesterol lipotoxicity results from JNK1-mediated mitochondrial injury and is HMGB1 and TLR4-dependent. J. Hepatol. 2014, 61, 1376–1384. [Google Scholar] [CrossRef] [PubMed]
- Wasilewska, N.; Lebensztejn, D.M. Non-alcoholic fatty liver disease and lipotoxicity. Clin. Exp. Hepatol. 2021, 7, 1–6. [Google Scholar] [CrossRef]
- Neuschwander-Tetri, B.A. Hepatic lipotoxicity and the pathogenesis of nonalcoholic steatohepatitis: The central role of nontriglyceride fatty acid metabolites. Hepatology 2010, 52, 774–788. [Google Scholar] [CrossRef]
- Lake, A.D.; Novak, P.; Hardwick, R.N.; Flores-Keown, B.; Zhao, F.; Klimecki, W.T.; Cherrington, N.J. The adaptive endoplasmic reticulum stress response to lipotoxicity in progressive human nonalcoholic fatty liver disease. Toxicol. Sci. 2014, 137, 26–35. [Google Scholar] [CrossRef] [Green Version]
- Meakin, P.J.; Chowdhry, S.; Sharma, R.S.; Ashford, F.B.; Walsh, S.V.; McCrimmon, R.J.; Dinkova-Kostova, A.T.; Dillon, J.F.; Hayes, J.D.; Ashford, M.L. Susceptibility of Nrf2-null mice to steatohepatitis and cirrhosis upon consumption of a high-fat diet is associated with oxidative stress, perturbation of the unfolded protein response, and disturbance in the expression of metabolic enzymes but not with insulin resistance. Mol. Cell. Biol. 2014, 34, 3305–3320. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Deng, X.; Liu, Y.; Tan, Q.; Huang, G.; Che, Q.; Guo, J.; Su, Z. Kupffer Cells in Non-alcoholic Fatty Liver Disease: Friend or Foe? Int. J. Biol. Sci. 2020, 16, 2367–2378. [Google Scholar] [CrossRef]
- Luo, W.; Xu, Q.; Wang, Q.; Wu, H.; Hua, J. Effect of modulation of PPAR-gamma activity on Kupffer cells M1/M2 polarization in the development of non-alcoholic fatty liver disease. Sci. Rep. 2017, 7, 44612. [Google Scholar] [CrossRef] [Green Version]
- Jimenez Ramos, M.; Bandiera, L.; Menolascina, F.; Fallofield, J.A. In vitro models for non-alcoholic fatty liver disease: Emerging platforms and their applications. iScience 2022, 25, 103549. [Google Scholar] [CrossRef]
- Godoy, P.; Hewitt, N.J.; Albrecht, U.; Andersen, M.E.; Ansari, N.; Bhattacharya, S.; Bode, J.G.; Bolleyn, J.; Borner, C.; Bottger, J.; et al. Recent advances in 2D and 3D in vitro systems using primary hepatocytes, alternative hepatocyte sources and non-parenchymal liver cells and their use in investigating mechanisms of hepatotoxicity, cell signaling and ADME. Arch. Toxicol. 2013, 87, 1315–1530. [Google Scholar] [CrossRef] [Green Version]
- Heslop, J.A.; Rowe, C.; Walsh, J.; Sison-Young, R.; Jenkins, R.; Kamalian, L.; Kia, R.; Hay, D.; Jones, R.P.; Malik, H.Z.; et al. Mechanistic evaluation of primary human hepatocyte culture using global proteomic analysis reveals a selective dedifferentiation profile. Arch. Toxicol. 2017, 91, 439–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez-Lechon, M.J.; Donato, M.T.; Castell, J.V.; Jover, R. Human hepatocytes as a tool for studying toxicity and drug metabolism. Curr. Drug Metab. 2003, 4, 292–312. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Lechon, M.J.; Donato, M.T.; Martinez-Romero, A.; Jimenez, N.; Castell, J.V.; O’Connor, J.E. A human hepatocellular in vitro model to investigate steatosis. Chem. Biol. Interact 2007, 165, 106–116. [Google Scholar] [CrossRef]
- Rennert, C.; Heil, T.; Schicht, G.; Stilkerich, A.; Seidemann, L.; Kegel-Hubner, V.; Seehofer, D.; Damm, G. Prolonged Lipid Accumulation in Cultured Primary Human Hepatocytes Rather Leads to ER Stress than Oxidative Stress. Int. J. Mol. Sci. 2020, 21, 7097. [Google Scholar] [CrossRef]
- Brown, M.V.; Compton, S.A.; Milburn, M.V.; Lawton, K.A.; Cheatham, B. Metabolomic signatures in lipid-loaded HepaRGs reveal pathways involved in steatotic progression. Obesity 2013, 21, E561–E570. [Google Scholar] [CrossRef]
- Chavez-Tapia, N.C.; Rosso, N.; Tiribelli, C. Effect of intracellular lipid accumulation in a new model of non-alcoholic fatty liver disease. BMC Gastroenterol. 2012, 12, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Jiang, Y.; Wang, Y.; An, W. Suppression of ABCA1 by unsaturated fatty acids leads to lipid accumulation in HepG2 cells. Biochimie 2010, 92, 958–963. [Google Scholar] [CrossRef]
- Zhang, Y.; Jiang, L.; Hu, W.; Zheng, Q.; Xiang, W. Mitochondrial dysfunction during in vitro hepatocyte steatosis is reversed by omega-3 fatty acid-induced up-regulation of mitofusin 2. Metabolism 2011, 60, 767–775. [Google Scholar] [CrossRef]
- Antherieu, S.; Rogue, A.; Fromenty, B.; Guillouzo, A.; Robin, M.A. Induction of vesicular steatosis by amiodarone and tetracycline is associated with up-regulation of lipogenic genes in HepaRG cells. Hepatology 2011, 53, 1895–1905. [Google Scholar] [CrossRef]
- Dallio, M.; Masarone, M.; Errico, S.; Gravina, A.G.; Nicolucci, C.; Di Sarno, R.; Gionti, L.; Tuccillo, C.; Persico, M.; Stiuso, P.; et al. Role of bisphenol A as environmental factor in the promotion of non-alcoholic fatty liver disease: In vitro and clinical study. Aliment Pharmacol. Ther. 2018, 47, 826–837. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Yu, P.; Xie, X.; Wu, L.; Zhou, M.; Huan, F.; Jiang, L.; Gao, R. Bisphenol F induces nonalcoholic fatty liver disease-like changes: Involvement of lysosome disorder in lipid droplet deposition. Environ. Pollut. 2021, 271, 116304. [Google Scholar] [CrossRef]
- Donato, M.T.; Martinez-Romero, A.; Jimenez, N.; Negro, A.; Herrera, G.; Castell, J.V.; O’Connor, J.E.; Gomez-Lechon, M.J. Cytometric analysis for drug-induced steatosis in HepG2 cells. Chem. Biol. Interact 2009, 181, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Donato, M.T.; Tolosa, L.; Jimenez, N.; Castell, J.V.; Gomez-Lechon, M.J. High-content imaging technology for the evaluation of drug-induced steatosis using a multiparametric cell-based assay. J. Biomol. Screen 2012, 17, 394–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wandrer, F.; Frangez, Z.; Liebig, S.; John, K.; Vondran, F.; Wedemeyer, H.; Veltmann, C.; Pfeffer, T.J.; Shibolet, O.; Schulze-Osthoff, K.; et al. Autophagy alleviates amiodarone-induced hepatotoxicity. Arch. Toxicol. 2020, 94, 3527–3539. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Wang, Y.; Chen, X.; Zhao, L.; Guo, Y. Involvement of CYP2E1-ROS-CD36/DGAT2 axis in the pathogenesis of VPA-induced hepatic steatosis in vivo and in vitro. Toxicology 2020, 445, 152585. [Google Scholar] [CrossRef]
- Tolosa, L.; Gomez-Lechon, M.J.; Jimenez, N.; Hervas, D.; Jover, R.; Donato, M.T. Advantageous use of HepaRG cells for the screening and mechanistic study of drug-induced steatosis. Toxicol. Appl. Pharmacol. 2016, 302, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kanebratt, K.P.; Andersson, T.B. HepaRG cells as an in vitro model for evaluation of cytochrome P450 induction in humans. Drug Metab. Dispos. 2008, 36, 137–145. [Google Scholar] [CrossRef] [Green Version]
- Allard, J.; Bucher, S.; Massart, J.; Ferron, P.J.; Le Guillou, D.; Loyant, R.; Daniel, Y.; Launay, Y.; Buron, N.; Begriche, K.; et al. Drug-induced hepatic steatosis in absence of severe mitochondrial dysfunction in HepaRG cells: Proof of multiple mechanism-based toxicity. Cell Biol. Toxicol. 2021, 37, 151–175. [Google Scholar] [CrossRef]
- Saito, J.; Okamura, A.; Takeuchi, K.; Hanioka, K.; Okada, A.; Ohata, T. High content analysis assay for prediction of human hepatotoxicity in HepaRG and HepG2 cells. Toxicol. Vitro 2016, 33, 63–70. [Google Scholar] [CrossRef]
- Barbero-Becerra, V.J.; Giraudi, P.J.; Chavez-Tapia, N.C.; Uribe, M.; Tiribelli, C.; Rosso, N. The interplay between hepatic stellate cells and hepatocytes in an in vitro model of NASH. Toxicol. In Vitro 2015, 29, 1753–1758. [Google Scholar] [CrossRef]
- Yu, H.; Jiang, X.; Dong, F.; Zhang, F.; Ji, X.; Xue, M.; Yang, F.; Chen, J.; Hu, X.; Bao, Z. Lipid accumulation-induced hepatocyte senescence regulates the activation of hepatic stellate cells through the Nrf2-antioxidant response element pathway. Exp. Cell Res. 2021, 405, 112689. [Google Scholar] [CrossRef]
- Cole, B.K.; Feaver, R.E.; Wamhoff, B.R.; Dash, A. Non-alcoholic fatty liver disease (NAFLD) models in drug discovery. Expert Opin. Drug Discov. 2018, 13, 193–205. [Google Scholar] [CrossRef]
- Zhou, Z.; Qi, J.; Lim, C.W.; Kim, J.W.; Kim, B. Dual TBK1/IKKepsilon inhibitor amlexanox mitigates palmitic acid-induced hepatotoxicity and lipoapoptosis in vitro. Toxicology 2020, 444, 152579. [Google Scholar] [CrossRef]
- Orbach, S.M.; Ehrich, M.F.; Rajagopalan, P. High-throughput toxicity testing of chemicals and mixtures in organotypic multi-cellular cultures of primary human hepatic cells. Toxicol. In Vitro 2018, 51, 83–94. [Google Scholar] [CrossRef]
- Lauschke, V.M.; Hendriks, D.F.; Bell, C.C.; Andersson, T.B.; Ingelman-Sundberg, M. Novel 3D Culture Systems for Studies of Human Liver Function and Assessments of the Hepatotoxicity of Drugs and Drug Candidates. Chem. Res. Toxicol. 2016, 29, 1936–1955. [Google Scholar] [CrossRef]
- Liu, D.; Chen, S.; Win Naing, M. A review of manufacturing capabilities of cell spheroid generation technologies and future development. Biotechnol. Bioeng. 2021, 118, 542–554. [Google Scholar] [CrossRef] [PubMed]
- Kozyra, M.; Johansson, I.; Nordling, A.; Ullah, S.; Lauschke, V.M.; Ingelman-Sundberg, M. Human hepatic 3D spheroids as a model for steatosis and insulin resistance. Sci. Rep. 2018, 8, 14297. [Google Scholar] [CrossRef] [Green Version]
- Pingitore, P.; Sasidharan, K.; Ekstrand, M.; Prill, S.; Linden, D.; Romeo, S. Human Multilineage 3D Spheroids as a Model of Liver Steatosis and Fibrosis. Int. J. Mol. Sci. 2019, 20, 1629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukherjee, S.; Zhelnin, L.; Sanfiz, A.; Pan, J.; Li, Z.; Yarde, M.; McCarty, J.; Jarai, G. Development and validation of an in vitro 3D model of NASH with severe fibrotic phenotype. Am. J. Transl. Res. 2019, 11, 1531–1540. [Google Scholar] [PubMed]
- Jiang, J.; Messner, S.; Kelm, J.M.; van Herwijnen, M.; Jennen, D.G.J.; Kleinjans, J.C.; de Kok, T.M. Human 3D multicellular microtissues: An upgraded model for the in vitro mechanistic investigation of inflammation-associated drug toxicity. Toxicol. Lett. 2019, 312, 34–44. [Google Scholar] [CrossRef]
- Rowe, C.; Gerrard, D.T.; Jenkins, R.; Berry, A.; Durkin, K.; Sundstrom, L.; Goldring, C.E.; Park, B.K.; Kitteringham, N.R.; Hanley, K.P.; et al. Proteome-wide analyses of human hepatocytes during differentiation and dedifferentiation. Hepatology 2013, 58, 799–809. [Google Scholar] [CrossRef] [Green Version]
- Feaver, R.E.; Cole, B.K.; Lawson, M.J.; Hoang, S.A.; Marukian, S.; Blackman, B.R.; Figler, R.A.; Sanyal, A.J.; Wamhoff, B.R.; Dash, A. Development of an in vitro human liver system for interrogating nonalcoholic steatohepatitis. JCI Insight 2016, 1, e90954. [Google Scholar] [CrossRef]
- Duriez, M.; Jacquet, A.; Hoet, L.; Roche, S.; Bock, M.D.; Rocher, C.; Haussy, G.; Vige, X.; Bocskei, Z.; Slavnic, T.; et al. A 3D Human Liver Model of Nonalcoholic Steatohepatitis. J. Clin. Transl. Hepatol. 2020, 8, 359–370. [Google Scholar] [CrossRef]
- Banaeiyan, A.A.; Theobald, J.; Paukstyte, J.; Wolfl, S.; Adiels, C.B.; Goksor, M. Design and fabrication of a scalable liver-lobule-on-a-chip microphysiological platform. Biofabrication 2017, 9, 015014. [Google Scholar] [CrossRef]
- Knowlton, S.; Tasoglu, S. A Bioprinted Liver-on-a-Chip for Drug Screening Applications. Trends Biotechnol. 2016, 34, 681–682. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, H.; Deng, P.; Tao, T.; Liu, H.; Wu, S.; Chen, W.; Qin, J. Modeling Human Nonalcoholic Fatty Liver Disease (NAFLD) with an Organoids-on-a-Chip System. ACS Biomater. Sci. Eng. 2020, 6, 5734–5743. [Google Scholar] [CrossRef]
- Gori, M.; Simonelli, M.C.; Giannitelli, S.M.; Businaro, L.; Trombetta, M.; Rainer, A. Investigating Nonalcoholic Fatty Liver Disease in a Liver-on-a-Chip Microfluidic Device. PLoS ONE 2016, 11, e0159729. [Google Scholar] [CrossRef]
- Gori, M.; Giannitelli, S.M.; Zancla, A.; Mozetic, P.; Trombetta, M.; Merendino, N.; Rainer, A. Quercetin and hydroxytyrosol as modulators of hepatic steatosis: A NAFLD-on-a-chip study. Biotechnol. Bioeng. 2021, 118, 142–152. [Google Scholar] [CrossRef]
- Freag, M.S.; Namgung, B.; Reyna Fernandez, M.E.; Gherardi, E.; Sengupta, S.; Jang, H.L. Human Nonalcoholic Steatohepatitis on a Chip. Hepatol. Commun. 2021, 5, 217–233. [Google Scholar] [CrossRef]
- Lee, S.Y.; Sung, J.H. Gut-liver on a chip toward an in vitro model of hepatic steatosis. Biotechnol. Bioeng. 2018, 115, 2817–2827. [Google Scholar] [CrossRef]
- Slaughter, V.L.; Rumsey, J.W.; Boone, R.; Malik, D.; Cai, Y.; Sriram, N.N.; Long, C.J.; McAleer, C.W.; Lambert, S.; Shuler, M.L.; et al. Validation of an adipose-liver human-on-a-chip model of NAFLD for preclinical therapeutic efficacy evaluation. Sci. Rep. 2021, 11, 13159. [Google Scholar] [CrossRef]
- Wang, X.H.; Tian, Y.; Guo, Z.J.; Fan, Z.P.; Qiu, D.K.; Zeng, M.D. Cholesterol metabolism and expression of its relevant genes in cultured steatotic hepatocytes. J. Dig. Dis. 2009, 10, 310–314. [Google Scholar] [CrossRef]
- Tolosa, L.; Gomez-Lechon, M.J.; Lopez, S.; Guzman, C.; Castell, J.V.; Donato, M.T.; Jover, R. Human Upcyte Hepatocytes: Characterization of the Hepatic Phenotype and Evaluation for Acute and Long-Term Hepatotoxicity Routine Testing. Toxicol. Sci. 2016, 152, 214–229. [Google Scholar] [CrossRef] [Green Version]
- Chagastelles, P.C.; Nardi, N.B. Biology of stem cells: An overview. Kidney Int. Suppl. 2011, 1, 63–67. [Google Scholar] [CrossRef] [Green Version]
- Larsen, L.E.; Smith, M.A.; Abbey, D.; Korn, A.; Reeskamp, L.F.; Hand, N.J.; Holleboom, A.G. Hepatocyte-like cells derived from induced pluripotent stem cells: A versatile tool to understand lipid disorders. Atherosclerosis 2020, 303, 8–14. [Google Scholar] [CrossRef]
- Mannino, G.; Russo, C.; Maugeri, G.; Musumeci, G.; Vicario, N.; Tibullo, D.; Giuffrida, R.; Parenti, R.; Lo Furno, D. Adult stem cell niches for tissue homeostasis. J. Cell. Physiol. 2021. [Google Scholar] [CrossRef]
- Lee, K.D.; Kuo, T.K.; Whang-Peng, J.; Chung, Y.F.; Lin, C.T.; Chou, S.H.; Chen, J.R.; Chen, Y.P.; Lee, O.K. In vitro hepatic differentiation of human mesenchymal stem cells. Hepatology 2004, 40, 1275–1284. [Google Scholar] [CrossRef]
- Rodrigues, R.M.; De Kock, J.; Branson, S.; Vinken, M.; Meganathan, K.; Chaudhari, U.; Sachinidis, A.; Govaere, O.; Roskams, T.; De Boe, V.; et al. Human skin-derived stem cells as a novel cell source for in vitro hepatotoxicity screening of pharmaceuticals. Stem Cells Dev. 2014, 23, 44–55. [Google Scholar] [CrossRef]
- Rodrigues, R.M.; Branson, S.; De Boe, V.; Sachinidis, A.; Rogiers, V.; De Kock, J.; Vanhaecke, T. In vitro assessment of drug-induced liver steatosis based on human dermal stem cell-derived hepatic cells. Arch. Toxicol. 2016, 90, 677–689. [Google Scholar] [CrossRef]
- Boeckmans, J.; Buyl, K.; Natale, A.; Vandenbempt, V.; Branson, S.; De Boe, V.; Rogiers, V.; De Kock, J.; Rodrigues, R.M.; Vanhaecke, T. Elafibranor restricts lipogenic and inflammatory responses in a human skin stem cell-derived model of NASH. Pharmacol. Res. 2019, 144, 377–389. [Google Scholar] [CrossRef]
- Boeckmans, J.; Natale, A.; Rombaut, M.; Buyl, K.; Vanhaecke, T.; Rogiers, V.; Rodrigues, R.M.; De Kock, J. Flow cytometric quantification of neutral lipids in a human skin stem cell-derived model of NASH. MethodsX 2020, 7, 101068. [Google Scholar] [CrossRef]
- Roh, J.K.; Jung, K.H.; Chu, K. Adult stem cell transplantation in stroke: Its limitations and prospects. Curr. Stem Cell Res. Ther. 2008, 3, 185–196. [Google Scholar] [CrossRef]
- Saito, Y.; Ikemoto, T.; Morine, Y.; Shimada, M. Current status of hepatocyte-like cell therapy from stem cells. Surg. Today 2021, 51, 340–349. [Google Scholar] [CrossRef] [PubMed]
- Collin de l’Hortet, A.; Takeishi, K.; Guzman-Lepe, J.; Morita, K.; Achreja, A.; Popovic, B.; Wang, Y.; Handa, K.; Mittal, A.; Meurs, N.; et al. Generation of Human Fatty Livers Using Custom-Engineered Induced Pluripotent Stem Cells with Modifiable SIRT1 Metabolism. Cell Metab. 2019, 30, 385–401.e9. [Google Scholar] [CrossRef]
- Gurevich, I.; Burton, S.A.; Munn, C.; Ohshima, M.; Goedland, M.E.; Czysz, K.; Rajesh, D. iPSC-derived hepatocytes generated from NASH donors provide a valuable platform for disease modeling and drug discovery. Biol. Open 2020, 9, bio055087. [Google Scholar] [CrossRef] [PubMed]
- Parafati, M.; Kirby, R.J.; Khorasanizadeh, S.; Rastinejad, F.; Malany, S. A nonalcoholic fatty liver disease model in human induced pluripotent stem cell-derived hepatocytes, created by endoplasmic reticulum stress-induced steatosis. Dis. Model Mech. 2018, 11, dmm033530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramli, M.N.B.; Lim, Y.S.; Koe, C.T.; Demircioglu, D.; Tng, W.; Gonzales, K.A.U.; Tan, C.P.; Szczerbinska, I.; Liang, H.; Soe, E.L.; et al. Human Pluripotent Stem Cell-Derived Organoids as Models of Liver Disease. Gastroenterology 2020, 159, 1471–1486.e12. [Google Scholar] [CrossRef] [PubMed]
- Tilson, S.G.; Morell, C.M.; Lenaerts, A.S.; Park, S.B.; Hu, Z.; Jenkins, B.; Koulman, A.; Liang, T.J.; Vallier, L. Modeling PNPLA3-Associated NAFLD Using Human-Induced Pluripotent Stem Cells. Hepatology 2021, 74, 2998–3017. [Google Scholar] [CrossRef]
- Lyall, M.J.; Cartier, J.; Thomson, J.P.; Cameron, K.; Meseguer-Ripolles, J.; O’Duibhir, E.; Szkolnicka, D.; Villarin, B.L.; Wang, Y.; Blanco, G.R.; et al. Modelling non-alcoholic fatty liver disease in human hepatocyte-like cells. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2018, 373, 20170362. [Google Scholar] [CrossRef] [Green Version]
- Pradip, A.; Steel, D.; Jacobsson, S.; Holmgren, G.; Ingelman-Sundberg, M.; Sartipy, P.; Bjorquist, P.; Johansson, I.; Edsbagge, J. High Content Analysis of Human Pluripotent Stem Cell Derived Hepatocytes Reveals Drug Induced Steatosis and Phospholipidosis. Stem Cells Int. 2016, 2016, 2475631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donato, M.T.; Tolosa, L. Stem-cell derived hepatocyte-like cells for the assessment of drug-induced liver injury. Differentiation 2019, 106, 15–22. [Google Scholar] [CrossRef]
- Shi, Y.; Inoue, H.; Wu, J.C.; Yamanaka, S. Induced pluripotent stem cell technology: A decade of progress. Nat. Rev. Drug Discov. 2017, 16, 115–130. [Google Scholar] [CrossRef]
- Graffmann, N.; Bohndorf, M.; Ncube, A.; Kawala, M.A.; Wruck, W.; Kashofer, K.; Zatloukal, K.; Adjaye, J. Establishment and characterization of an iPSC line from a 35 years old high grade patient with nonalcoholic fatty liver disease (30–40% steatosis) with homozygous wildtype PNPLA3 genotype. Stem Cell Res. 2018, 31, 113–116. [Google Scholar] [CrossRef]
- Graffmann, N.; Bohndorf, M.; Ncube, A.; Wruck, W.; Kashofer, K.; Zatloukal, K.; Adjaye, J. Establishment and characterization of an iPSC line from a 58 years old high grade patient with nonalcoholic fatty liver disease (70% steatosis) with homozygous wildtype PNPLA3 genotype. Stem Cell Res. 2018, 31, 131–134. [Google Scholar] [CrossRef]
- Graffmann, N.; Ncube, A.; Martins, S.; Fiszl, A.R.; Reuther, P.; Bohndorf, M.; Wruck, W.; Beller, M.; Czekelius, C.; Adjaye, J. A stem cell based in vitro model of NAFLD enables the analysis of patient specific individual metabolic adaptations in response to a high fat diet and AdipoRon interference. Biol. Open 2021, 10, bio054189. [Google Scholar] [CrossRef]
- He, S.; McPhaul, C.; Li, J.Z.; Garuti, R.; Kinch, L.; Grishin, N.V.; Cohen, J.C.; Hobbs, H.H. A sequence variation (I148M) in PNPLA3 associated with nonalcoholic fatty liver disease disrupts triglyceride hydrolysis. J. Biol. Chem. 2010, 285, 6706–6715. [Google Scholar] [CrossRef] [Green Version]
- Kammerer, S. Three-Dimensional Liver Culture Systems to Maintain Primary Hepatic Properties for Toxicological Analysis In Vitro. Int. J. Mol. Sci. 2021, 22, 214. [Google Scholar] [CrossRef] [PubMed]
- Segovia-Miranda, F.; Morales-Navarrete, H.; Kucken, M.; Moser, V.; Seifert, S.; Repnik, U.; Rost, F.; Brosch, M.; Hendricks, A.; Hinz, S.; et al. Three-dimensional spatially resolved geometrical and functional models of human liver tissue reveal new aspects of NAFLD progression. Nat. Med. 2019, 25, 1885–1893. [Google Scholar] [CrossRef] [PubMed]
- Baxter, M.; Withey, S.; Harrison, S.; Segeritz, C.P.; Zhang, F.; Atkinson-Dell, R.; Rowe, C.; Gerrard, D.T.; Sison-Young, R.; Jenkins, R.; et al. Phenotypic and functional analyses show stem cell-derived hepatocyte-like cells better mimic fetal rather than adult hepatocytes. J. Hepatol. 2015, 62, 581–589. [Google Scholar] [CrossRef]
- Schwartz, R.E.; Fleming, H.E.; Khetani, S.R.; Bhatia, S.N. Pluripotent stem cell-derived hepatocyte-like cells. Biotechnol. Adv. 2014, 32, 504–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parlati, L.; Regnier, M.; Guillou, H.; Postic, C. New targets for NAFLD. JHEP Rep. 2021, 3, 100346. [Google Scholar] [CrossRef]
- Taliento, A.E.; Dallio, M.; Federico, A.; Prati, D.; Valenti, L. Novel Insights into the Genetic Landscape of Nonalcoholic Fatty Liver Disease. Int. J. Environ. Res. Public Health 2019, 16, 2755. [Google Scholar] [CrossRef] [Green Version]
- Bell, L.N.; Theodorakis, J.L.; Vuppalanchi, R.; Saxena, R.; Bemis, K.G.; Wang, M.; Chalasani, N. Serum proteomics and biomarker discovery across the spectrum of nonalcoholic fatty liver disease. Hepatology 2010, 51, 111–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barr, J.; Caballeria, J.; Martinez-Arranz, I.; Dominguez-Diez, A.; Alonso, C.; Muntane, J.; Perez-Cormenzana, M.; Garcia-Monzon, C.; Mayo, R.; Martin-Duce, A.; et al. Obesity-dependent metabolic signatures associated with nonalcoholic fatty liver disease progression. J. Proteome Res. 2012, 11, 2521–2532. [Google Scholar] [CrossRef]
- Garcia-Canaveras, J.C.; Donato, M.T.; Castell, J.V.; Lahoz, A. A comprehensive untargeted metabonomic analysis of human steatotic liver tissue by RP and HILIC chromatography coupled to mass spectrometry reveals important metabolic alterations. J. Proteome Res. 2011, 10, 4825–4834. [Google Scholar] [CrossRef] [PubMed]
- Seko, Y.; Yamaguchi, K.; Itoh, Y. The genetic backgrounds in nonalcoholic fatty liver disease. Clin. J. Gastroenterol. 2018, 11, 97–102. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Liu, Z.; Zou, W.; Hong, H.; Fang, H.; Tong, W. Molecular regulation of miRNAs and potential biomarkers in the progression of hepatic steatosis to NASH. Biomark. Med. 2015, 9, 1189–1200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tse, E.; Helbig, K.J.; Van der Hoek, K.; McCartney, E.M.; Van der Hoek, M.; George, J.; Beard, M.R. Fatty Acids Induce a Pro-Inflammatory Gene Expression Profile in Huh-7 Cells That Attenuates the Anti-HCV Action of Interferon. J. Interferon Cytokine Res. 2015, 35, 392–400. [Google Scholar] [CrossRef]
- Hetherington, A.M.; Sawyez, C.G.; Zilberman, E.; Stoianov, A.M.; Robson, D.L.; Borradaile, N.M. Differential Lipotoxic Effects of Palmitate and Oleate in Activated Human Hepatic Stellate Cells and Epithelial Hepatoma Cells. Cell Physiol. Biochem. 2016, 39, 1648–1662. [Google Scholar] [CrossRef] [PubMed]
- Breher-Esch, S.; Sahini, N.; Trincone, A.; Wallstab, C.; Borlak, J. Genomics of lipid-laden human hepatocyte cultures enables drug target screening for the treatment of non-alcoholic fatty liver disease. BMC Med. Genom. 2018, 11, 111. [Google Scholar] [CrossRef] [Green Version]
- Atanasovska, B.; Rensen, S.S.; Marsman, G.; Shiri-Sverdlov, R.; Withoff, S.; Kuipers, F.; Wijmenga, C.; van de Sluis, B.; Fu, J. Long Non-Coding RNAs Involved in Progression of Non-Alcoholic Fatty Liver Disease to Steatohepatitis. Cells 2021, 10, 1883. [Google Scholar] [CrossRef]
- Errafii, K.; Al-Akl, N.S.; Khalifa, O.; Arredouani, A. Comprehensive analysis of LncRNAs expression profiles in an in vitro model of steatosis treated with Exendin-4. J. Transl. Med. 2021, 19, 235. [Google Scholar] [CrossRef]
- Lockman, K.A.; Htun, V.; Sinha, R.; Treskes, P.; Nelson, L.J.; Martin, S.F.; Rogers, S.M.; Le Bihan, T.; Hayes, P.C.; Plevris, J.N. Proteomic profiling of cellular steatosis with concomitant oxidative stress in vitro. Lipids Health Dis. 2016, 15, 114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chienwichai, P.; Reamtong, O.; Boonyuen, U.; Pisitkun, T.; Somparn, P.; Tharnpoophasiam, P.; Worakhunpiset, S.; Topanurak, S. Hepatic protein Carbonylation profiles induced by lipid accumulation and oxidative stress for investigating cellular response to non-alcoholic fatty liver disease in vitro. Proteome Sci. 2019, 17, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuykx, M.; Claes, L.; Rodrigues, R.M.; Vanhaecke, T.; Covaci, A. Metabolomics profiling of steatosis progression in HepaRG. Toxicol. Lett. 2018, 286, 22–30. [Google Scholar] [CrossRef] [PubMed]
- García-Cañaveras, J.C.; Peris-Díaz, M.D.; Alcoriza-Balaguer, M.I.; Cerdán-Calero, M.; Donato, M.T.; Lahoz, A. A lipidomic cell-based assay for studying drug-induced phospholipidosis and steatosis. Electrophoresis 2017, 38, 2331–2340. [Google Scholar] [CrossRef]
- Yeung, E.N.; Treskes, P.; Martin, S.F.; Manning, J.R.; Dunbar, D.R.; Rogers, S.M.; Le Bihan, T.; Lockman, K.A.; Morley, S.D.; Hayes, P.C.; et al. Fibrinogen production is enhanced in an in-vitro model of non-alcoholic fatty liver disease: An isolated risk factor for cardiovascular events? Lipids Health Dis. 2015, 14, 86. [Google Scholar] [CrossRef] [Green Version]
- Gunn, P.J.; Pramfalk, C.; Millar, V.; Cornfield, T.; Hutchinson, M.; Johnson, E.M.; Nagarajan, S.R.; Troncoso-Rey, P.; Mithen, R.F.; Pinnick, K.E.; et al. Modifying nutritional substrates induces macrovesicular lipid droplet accumulation and metabolic alterations in a cellular model of hepatic steatosis. Physiol. Rep. 2020, 8, e14482. [Google Scholar] [CrossRef] [PubMed]
- Tomita, K.; Freeman, B.L.; Bronk, S.F.; LeBrasseur, N.K.; White, T.A.; Hirsova, P.; Ibrahim, S.H. CXCL10-Mediates Macrophage, but not Other Innate Immune Cells-Associated Inflammation in Murine Nonalcoholic Steatohepatitis. Sci. Rep. 2016, 6, 28786. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Shen, J.; Man, K.; Chu, E.S.; Yau, T.O.; Sung, J.C.; Go, M.Y.; Deng, J.; Lu, L.; Wong, V.W.; et al. CXCL10 plays a key role as an inflammatory mediator and a non-invasive biomarker of non-alcoholic steatohepatitis. J. Hepatol. 2014, 61, 1365–1375. [Google Scholar] [CrossRef] [Green Version]
- Kohjima, M.; Enjoji, M.; Higuchi, N.; Kato, M.; Kotoh, K.; Yoshimoto, T.; Fujino, T.; Yada, M.; Yada, R.; Harada, N.; et al. Re-evaluation of fatty acid metabolism-related gene expression in nonalcoholic fatty liver disease. Int. J. Mol. Med. 2007, 20, 351–358. [Google Scholar] [CrossRef] [Green Version]
- Donato, M.T.; Gomez-Lechon, M.J. Drug-induced liver steatosis and phospholipidosis: Cell-based assays for early screening of drug candidates. Curr. Drug Metab. 2012, 13, 1160–1173. [Google Scholar] [CrossRef]
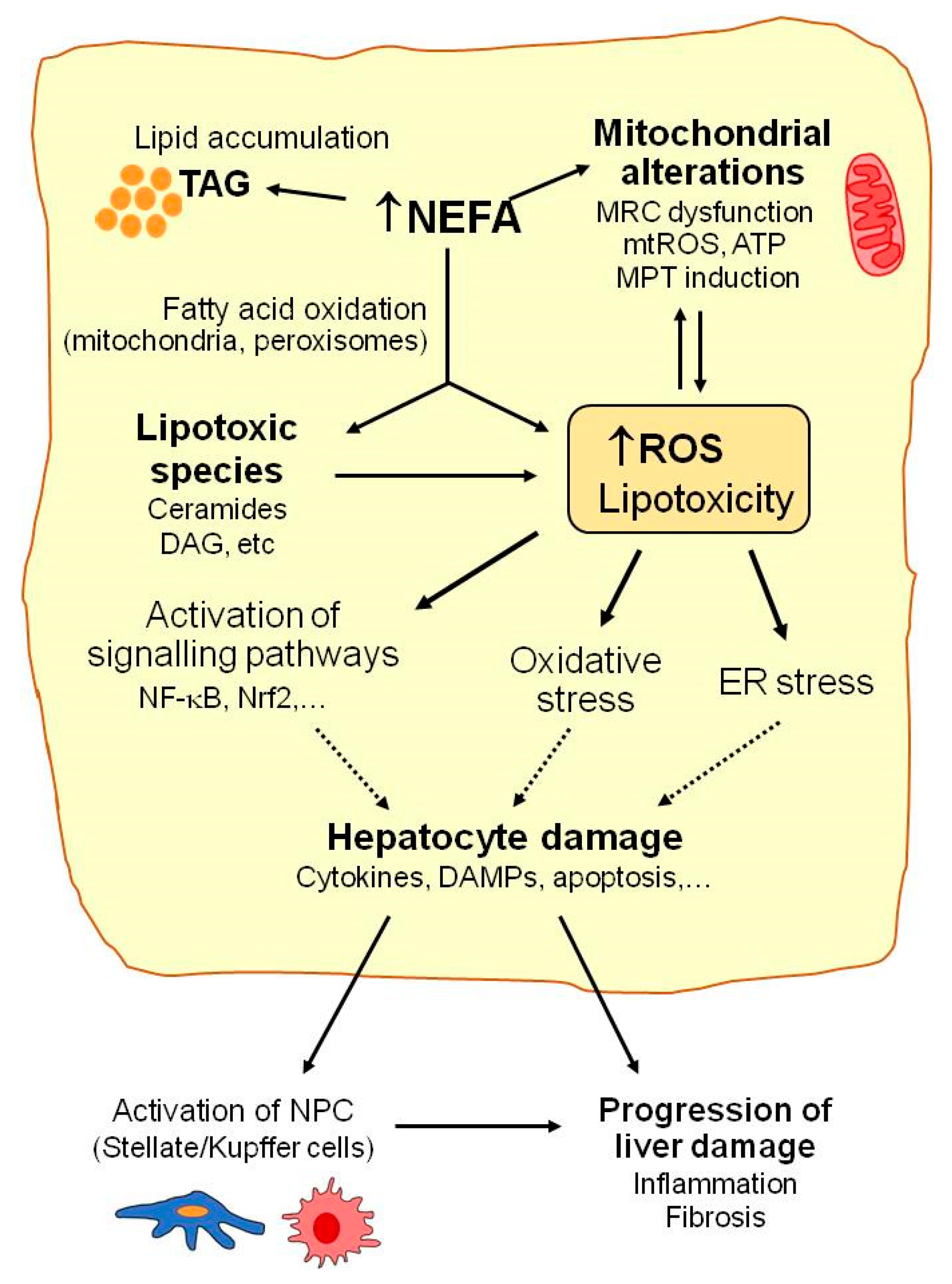
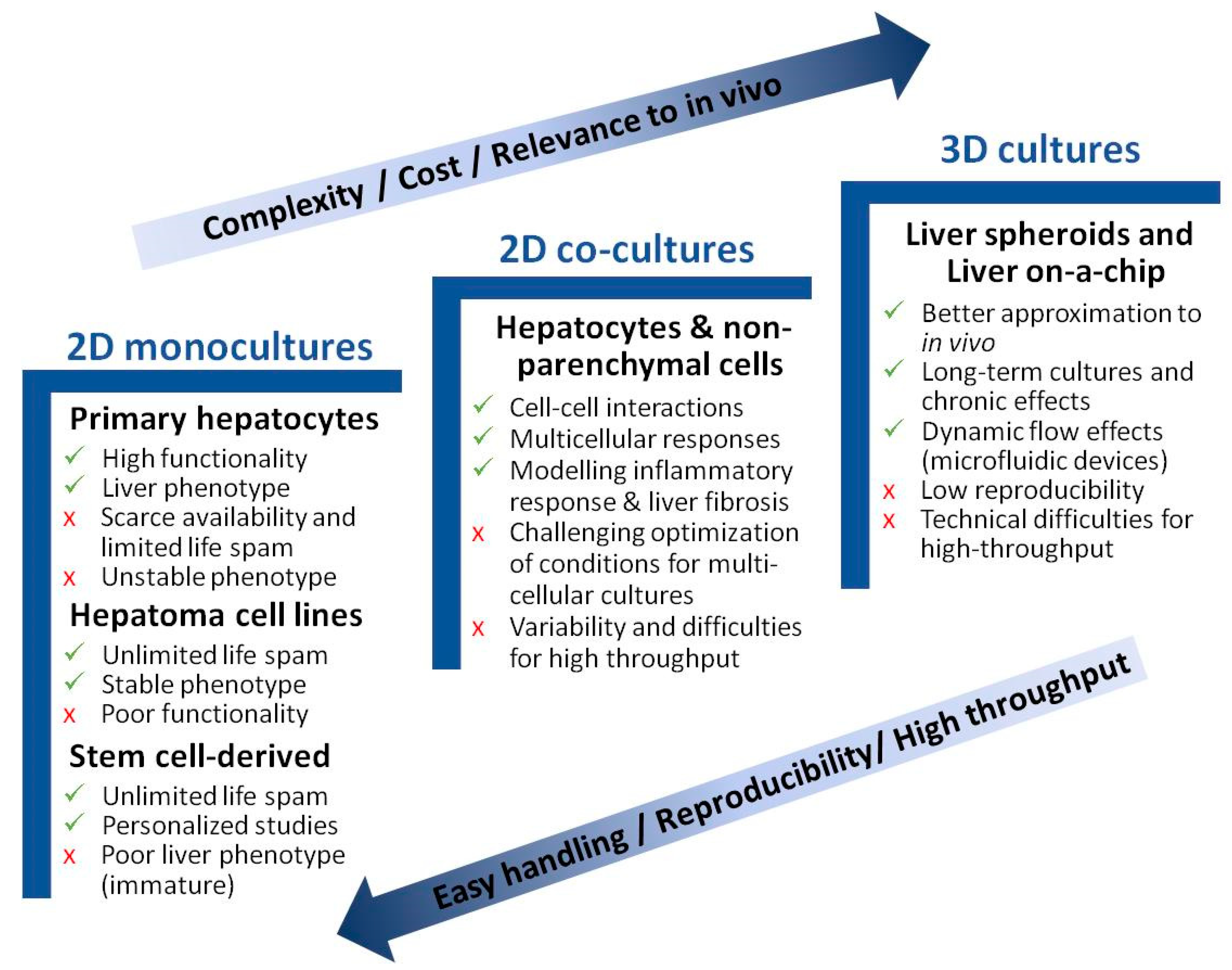
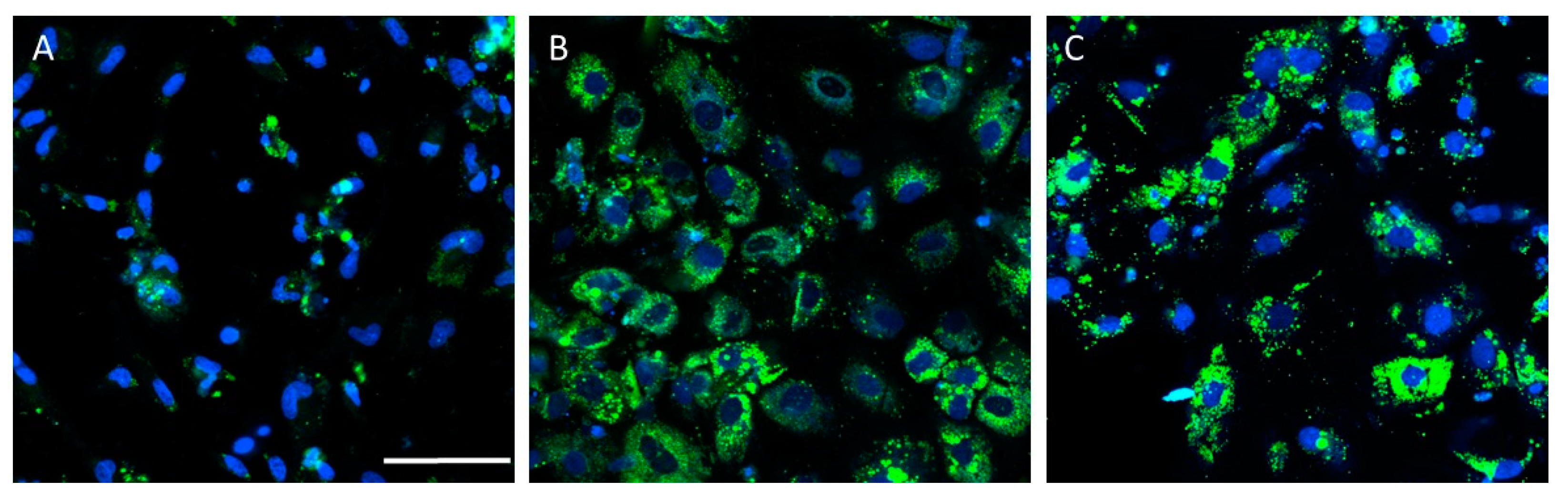
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).