
Glutathione–Hemin/Hematin Adduct Formation to Disintegrate Cytotoxic Oxidant Hemin/Hematin in Human K562 Cells and Red Blood Cells’ Hemolysates: Impact of Glutathione on the Hemolytic Disorders and Homeostasis

 , , ,
, , ,
Abstract
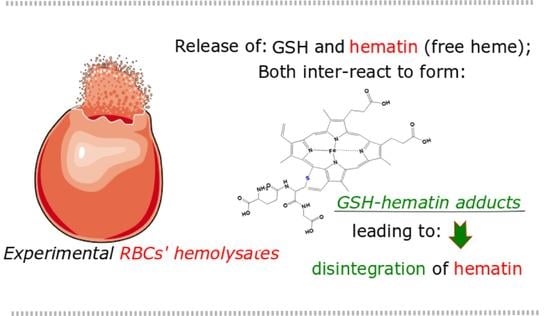
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Reagents
2.2. Assessment of Cell Growth and Viability
2.3. Determination of GSH and GSSG Levels
2.4. Spectrophotometric Analysis of Hemin
2.5. Quantitation of the Intracellular Hemin Level
2.6. Electrospray (ESI)-LC-MS/MS Analysis
2.6.1. In Vitro Reaction
2.6.2. LC-MS/MS Analysis
2.6.3. Quantitation of molecular Ions
2.7. Isolation of RBCs
2.8. Extraction of Cellular Low Molecular Weight (LMW) Metabolites
2.9. Statistical Analysis
3. Results
3.1. Endogenous GSH Served as a Defensive Barrier to HIC in Default Cell Culture Conditions in K562 Cells
3.2. The Exogenous Addition of GSH Rescued K562 Cells from HIC: Analysis of the Molecular Mechanism
3.2.1. GSH Biosynthesis Increased in Thiols (NAC and GSH)-Induced Prevention of HIC
3.2.2. GSH Biosynthesis Was Not a Key Mediator of Thiols (NAC and GSH)-Induced Prevention of HIC
3.2.3. Management of HIC, a Common Property of Thiols, Involved Hemin Structural Destabilization and Inhibition in Intracellular Accumulation of Hemin

3.3. Novel Covalent Adducts between GSH and Hemin (GSH–Hemin Adducts) and Their Derivatives Were Identified in the In Vitro Reaction between GSH and Hemin
3.4. GSH–Hematin Adducts Were Identified in Traces at Healthy Human RBCs and Intensified at Experimental Hemolysates

4. Discussion
4.1. GSH Was a Fundamental Constituent in the Management of HIC in K562 Cells: Analysis of the Mechanism
4.2. Elucidation of the Chemical Reaction between GSH and Hemin
4.3. GSH–Hematin Adducts Were Identified as Novel Metabolites in Experimental Hemolysates
4.4. Future Studies Regarding the Possible Biological Significance of GSH–Hematin Adducts Formation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tsiftsoglou, A.S.; Tsamadou, A.I.; Papadopoulou, L.C. Heme as key regulator of major mammalian cellular functions: Molecular, cellular, and pharmacological aspects. Pharmacol. Ther. 2006, 111, 327–345. [Google Scholar] [CrossRef]
- Mairbaurl, H.; Weber, R.E. Oxygen transport by hemoglobin. Compr. Physiol. 2012, 2, 1463–1489. [Google Scholar]
- Chiabrando, D.; Mercurio, S.; Tolosano, E. Heme and erythropoieis: More than a structural role. Haematologica 2014, 99, 973–983. [Google Scholar] [CrossRef]
- Chen, L.T.; Weiss, L. The role of the sinus wall in the passage of erythrocytes through the spleen. Blood 1973, 41, 529–537. [Google Scholar] [CrossRef]
- de Back, D.Z.; Kostova, E.B.; van Kraaij, M.; van den Berg, T.K.; van Bruggen, R. Of macrophages and red blood cells; a complex love story. Front. Physiol. 2014, 5, 9. [Google Scholar] [CrossRef]
- Gouveia, Z.; Carlos, A.R.; Yuan, X.; Aires-da-Silva, F.; Stocker, R.; Maghzal, G.J.; Leal, S.S.; Gomes, C.M.; Todorovic, S.; Iranzo, O.; et al. Characterization of plasma labile heme in hemolytic conditions. FEBS J. 2017, 284, 3278–3301. [Google Scholar] [CrossRef]
- Liu, S.C.; Zhai, S.; Palek, J. Detection of hemin release during hemoglobin S denaturation. Blood 1988, 71, 1755–1758. [Google Scholar] [CrossRef]
- Larsen, R.; Gozzelino, R.; Jeney, V.; Tokaji, L.; Bozza, F.A.; Japiassu, A.M.; Bonaparte, D.; Cavalcante, M.M.; Chora, A.; Ferreira, A.; et al. A central role for free heme in the pathogenesis of severe sepsis. Sci. Transl. Med. 2010, 2, 51ra71. [Google Scholar] [CrossRef]
- Kai, O.K.; Roberts, D.J. The pathophysiology of malarial anaemia: Where have all the red cells gone? BMC Med. 2008, 6, 24. [Google Scholar] [CrossRef]
- Kassa, T.; Jana, S.; Meng, F.; Alayash, A.I. Differential heme release from various hemoglobin redox states and the upregulation of cellular heme oxygenase-1. FEBS Open Bio 2016, 6, 876–884. [Google Scholar] [CrossRef]
- Wagener, F.A.; Eggert, A.; Boerman, O.C.; Oyen, W.J.; Verhofstad, A.; Abraham, N.G.; Adema, G.; van Kooyk, Y.; de Witte, T.; Figdor, C.G. Heme is a potent inducer of inflammation in mice and is counteracted by heme oxygenase. Blood 2001, 98, 1802–1811. [Google Scholar] [CrossRef]
- Wagener, F.A.; Abraham, N.G.; van Kooyk, Y.; de Witte, T.; Figdor, C.G. Heme-induced cell adhesion in the pathogenesis of sickle-cell disease and inflammation. Trends Pharmacol. Sci. 2001, 22, 52–54. [Google Scholar] [CrossRef]
- Voskou, S.; Aslan, M.; Fanis, P.; Phylactides, M.; Kleanthous, M. Oxidative stress in beta-thalassaemia and sickle cell disease. Redox Biol. 2015, 6, 226–239. [Google Scholar] [CrossRef]
- Balla, G.; Jacob, H.S.; Eaton, J.W.; Belcher, J.D.; Vercellotti, G.M. Hemin: A possible physiological mediator of low density lipoprotein oxidation and endothelial injury. Arterioscler. Thromb. J. Vasc. Biol. 1991, 11, 1700–1711. [Google Scholar] [CrossRef]
- Deuel, J.W.; Schaer, C.A.; Boretti, F.S.; Opitz, L.; Garcia-Rubio, I.; Baek, J.H.; Spahn, D.R.; Buehler, P.W.; Schaer, D.J. Hemoglobinuria-related acute kidney injury is driven by intrarenal oxidative reactions triggering a heme toxicity response. Cell Death Dis. 2016, 7, e2064. [Google Scholar] [CrossRef] [PubMed]
- Franco, R.; Navarro, G.; Martinez-Pinilla, E. Antioxidant Defense Mechanisms in Erythrocytes and in the Central Nervous System. Antioxidants 2019, 8, 46. [Google Scholar] [CrossRef]
- van ‘t Erve, T.J.; Wagner, B.A.; Ryckman, K.K.; Raife, T.J.; Buettner, G.R. The concentration of glutathione in human erythrocytes is a heritable trait. Free Radic. Biol. Med. 2013, 65, 742–749. [Google Scholar] [CrossRef]
- Szeinberg, A.; Asher, Y.; Sheba, C. Studies on glutathione stability in erythrocytes of cases with past history of favism or sulfa-drug-induced hemolysis. Blood 1958, 13, 348–358. [Google Scholar] [CrossRef]
- Richie, J.P., Jr.; Abraham, P.; Leutzinger, Y. Long-term stability of blood glutathione and cysteine in humans. Clin. Chem. 1996, 42, 1100–1105. [Google Scholar] [CrossRef]
- Richie, J.P., Jr.; Skowronski, L.; Abraham, P.; Leutzinger, Y. Blood glutathione concentrations in a large-scale human study. Clin. Chem. 1996, 42, 64–70. [Google Scholar] [CrossRef]
- Forman, H.J.; Zhang, H.; Rinna, A. Glutathione: Overview of its protective roles, measurement, and biosynthesis. Mol. Asp. Med. 2009, 30, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franco, R.; Cidlowski, J.A. Apoptosis and glutathione: Beyond an antioxidant. Cell Death Differ. 2009, 16, 1303–1314. [Google Scholar] [CrossRef] [PubMed]
- Lushchak, V.I. Glutathione homeostasis and functions: Potential targets for medical interventions. J. Amino Acids 2012, 2012, 736837. [Google Scholar] [CrossRef] [PubMed]
- Raftos, J.E.; Whillier, S.; Kuchel, P.W. Glutathione synthesis and turnover in the human erythrocyte: Alignment of a model based on detailed enzyme kinetics with experimental data. J. Biol. Chem. 2010, 285, 23557–23567. [Google Scholar] [CrossRef] [PubMed]
- Chou, A.C.; Fitch, C.D. Mechanism of hemolysis induced by ferriprotoporphyrin IX. J. Clin. Investig. 1981, 68, 672–677. [Google Scholar] [CrossRef]
- Sahini, V.E.; Dumitrescu, M.; Volanschi, E.; Birla, L.; Diaconu, C. Spectral and interferometrical study of the interaction of haemin with glutathione. Biophys. Chem. 1996, 58, 245–253. [Google Scholar] [CrossRef]
- Shviro, Y.; Shaklai, N. Glutathione as a scavenger of free hemin. A mechanism of preventing red cell membrane damage. Biochem. Pharmacol. 1987, 36, 3801–3807. [Google Scholar] [CrossRef]
- Atamna, H.; Ginsburg, H. Heme degradation in the presence of glutathione. A proposed mechanism to account for the high levels of non-heme iron found in the membranes of hemoglobinopathic red blood cells. J. Biol. Chem. 1995, 270, 24876–24883. [Google Scholar] [CrossRef]
- Georgiou-Siafis, S.K.; Tsiftsoglou, A.S. Activation of KEAP1/NRF2 stress signaling involved in the molecular basis of hemin-induced cytotoxicity in human pro-erythroid K562 cells. Biochem. Pharmacol. 2020, 175, 113900. [Google Scholar] [CrossRef]
- Tsolaki, V.C.; Georgiou-Siafis, S.K.; Tsamadou, A.I.; Tsiftsoglou, S.A.; Samiotaki, M.; Panayotou, G.; Tsiftsoglou, A.S. Hemin accumulation and identification of a heme-binding protein clan in K562 cells by proteomic and computational analysis. J. Cell. Physiol. 2022, 237, 1315–1340. [Google Scholar] [CrossRef]
- Georgiou-Siafis, S.K.; Samiotaki, M.K.; Demopoulos, V.J.; Panayotou, G.; Tsiftsoglou, A.S. Formation of novel N-acetylcysteine-hemin adducts abrogates hemin-induced cytotoxicity and suppresses the NRF2-driven stress response in human pro-erythroid K562 cells. Eur. J. Pharmacol. 2020, 880, 173077. [Google Scholar] [CrossRef] [PubMed]
- Tsiftsoglou, A.S.; Wong, W.; Robinson, S.H.; Hensold, J. Hemin increase production of beta-like globin RNA transcripts in human erythroleukemia K-562 cells. Dev. Genet. 1989, 10, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Gusella, J.F.; Tsiftsoglou, A.S.; Volloch, V.; Weil, S.C.; Neumann, J.; Housman, D.E. Dissociation of hemoglobin accumulation and commitment during murine erythroleukemia cell differentiation by treatment with imidazole. J. Cell. Physiol. 1982, 113, 179–185. [Google Scholar] [CrossRef]
- Strober, W. Trypan blue exclusion test of cell viability. Curr. Protoc. Immunol. 2001. Appendix 3. [Google Scholar]
- Rahman, I.; Kode, A.; Biswas, S.K. Assay for quantitative determination of glutathione and glutathione disulfide levels using enzymatic recycling method. Nat. Protoc. 2006, 1, 3159–3165. [Google Scholar] [CrossRef]
- Gallagher, W.A.; Elliott, W.B. The formation of pyridine haemochromogen. Biochem. J. 1965, 97, 187–193. [Google Scholar] [CrossRef]
- Sinclair, P.R.; Gorman, N.; Jacobs, J.M. Measurement of heme concentration. Curr. Protoc. Toxicol. 2001, 8, 8.3.1–8.3.7. [Google Scholar] [CrossRef]
- Bilia, A.R.; Lazari, D.; Messori, L.; Taglioli, V.; Temperini, C.; Vincieri, F.F. Simple and rapid physico-chemical methods to examine action of antimalarial drugs with hemin: Its application to Artemisia annua constituents. Life Sci. 2002, 70, 769–778. [Google Scholar] [CrossRef]
- Fyrestam, J.; Ostman, C. Determination of heme in microorganisms using HPLC-MS/MS and cobalt(III) protoporphyrin IX inhibition of heme acquisition in Escherichia coli. Anal. Bioanal. Chem. 2017, 409, 6999–7010. [Google Scholar] [CrossRef]
- Crook, T.R.; Souhami, R.L.; Whyman, G.D.; McLean, A.E. Glutathione depletion as a determinant of sensitivity of human leukemia cells to cyclophosphamide. Cancer Res. 1986, 46, 5035–5038. [Google Scholar] [PubMed]
- Shrieve, D.C.; Denekamp, J.; Minchinton, A.I. Effects of glutathione depletion by buthionine sulfoximine on radiosensitization by oxygen and misonidazole in vitro. Radiat. Res. 1985, 102, 283–294. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, L.; Teng, Z.P.; Zeserson, E.; Patel, M.; Regan, R.F. Hemin induces an iron-dependent, oxidative injury to human neuron-like cells. J. Neurosci. Res. 2003, 73, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Whillier, S.; Raftos, J.E.; Chapman, B.; Kuchel, P.W. Role of N-acetylcysteine and cystine in glutathione synthesis in human erythrocytes. Redox Rep. Commun. Free Radic. Res. 2009, 14, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Raftos, J.E.; Whillier, S.; Chapman, B.E.; Kuchel, P.W. Kinetics of uptake and deacetylation of N-acetylcysteine by human erythrocytes. Int. J. Biochem. Cell Biol. 2007, 39, 1698–1706. [Google Scholar] [CrossRef] [PubMed]
- Rushworth, G.F.; Megson, I.L. Existing and potential therapeutic uses for N-acetylcysteine: The need for conversion to intracellular glutathione for antioxidant benefits. Pharmacol. Ther. 2014, 141, 150–159. [Google Scholar] [CrossRef]
- Mlejnek, P.; Dolezel, P.; Kriegova, E.; Pastvova, N. N-acetylcysteine Can Induce Massive Oxidative Stress, Resulting in Cell Death with Apoptotic Features in Human Leukemia Cells. Int. J. Mol. Sci. 2021, 22, 12635. [Google Scholar] [CrossRef]
- Bachhawat, A.K.; Thakur, A.; Kaur, J.; Zulkifli, M. Glutathione transporters. Biochim. Biophys. Acta 2013, 1830, 3154–3164. [Google Scholar] [CrossRef]
- Jian, W.; Yao, M.; Zhang, D.; Zhu, M. Rapid detection and characterization of in vitro and urinary N-acetyl-L-cysteine conjugates using quadrupole-linear ion trap mass spectrometry and polarity switching. Chem. Res. Toxicol. 2009, 22, 1246–1255. [Google Scholar] [CrossRef]
- Sweeny, E.A.; Singh, A.B.; Chakravarti, R.; Martinez-Guzman, O.; Saini, A.; Haque, M.M.; Garee, G.; Dans, P.D.; Hannibal, L.; Reddi, A.R.; et al. Glyceraldehyde-3-phosphate dehydrogenase is a chaperone that allocates labile heme in cells. J. Biol. Chem. 2018, 293, 14557–14568. [Google Scholar] [CrossRef]
- Aich, A.; Freundlich, M.; Vekilov, P.G. The free heme concentration in healthy human erythrocytes. Blood Cells Mol. Dis. 2015, 55, 402–409. [Google Scholar] [CrossRef]
- Boys, B.L.; Konermann, L. Folding and assembly of hemoglobin monitored by electrospray mass spectrometry using an on-line dialysis system. J. Am. Soc. Mass Spectrom. 2007, 18, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Light, W.R., 3rd; Olson, J.S. Transmembrane movement of heme. J. Biol. Chem. 1990, 265, 15623–15631. [Google Scholar] [CrossRef]
- Ponka, P.; Sheftel, A.D.; English, A.M.; Scott Bohle, D.; Garcia-Santos, D. Do Mammalian Cells Really Need to Export and Import Heme? Trends Biochem. Sci. 2017, 42, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Chiu, D.T.; Huang, T.Y.; Hung, I.J.; Wei, J.S.; Liu, T.Z.; Stern, A. Hemin-induced membrane sulfhydryl oxidation: Possible involvement of thiyl radicals. Free Radic. Res. 1997, 27, 55–62. [Google Scholar] [CrossRef]
- Fortes, G.B.; Alves, L.S.; de Oliveira, R.; Dutra, F.F.; Rodrigues, D.; Fernandez, P.L.; Souto-Padron, T.; De Rosa, M.J.; Kelliher, M.; Golenbock, D.; et al. Heme induces programmed necrosis on macrophages through autocrine TNF and ROS production. Blood 2012, 119, 2368–2375. [Google Scholar] [CrossRef]
- Higdon, A.N.; Benavides, G.A.; Chacko, B.K.; Ouyang, X.; Johnson, M.S.; Landar, A.; Zhang, J.; Darley-Usmar, V.M. Hemin causes mitochondrial dysfunction in endothelial cells through promoting lipid peroxidation: The protective role of autophagy. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H1394–H1409. [Google Scholar] [CrossRef]
- Gibson, K.R.; Neilson, I.L.; Barrett, F.; Winterburn, T.J.; Sharma, S.; MacRury, S.M.; Megson, I.L. Evaluation of the antioxidant properties of N-acetylcysteine in human platelets: Prerequisite for bioconversion to glutathione for antioxidant and antiplatelet activity. J. Cardiovasc. Pharmacol. 2009, 54, 319–326. [Google Scholar] [CrossRef]
- NaveenKumar, S.K.; Hemshekhar, M.; Kemparaju, K.; Girish, K.S. Hemin-induced platelet activation and ferroptosis is mediated through ROS-driven proteasomal activity and inflammasome activation: Protection by Melatonin. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 2303–2316. [Google Scholar] [CrossRef]
- Haenen, H.E.; Rogmans, P.; Temmink, J.H.; van Bladeren, P.J. Differential detoxification of two thioether conjugates of menadione in confluent monolayers of rat renal proximal tubular cells. Toxicol. Vitr. 1994, 8, 207–214. [Google Scholar] [CrossRef]
- Haenen, H.E.; Spenkelink, A.; Teunissen, C.; Temmink, J.H.; Koeman, J.H.; van Bladeren, P.J. Transport and metabolism of glutathione conjugates of menadione and ethacrynic acid in confluent monolayers of rat renal proximal tubular cells. Toxicology 1996, 112, 117–130. [Google Scholar] [CrossRef]
- Chakravarthi, S.; Jessop, C.E.; Bulleid, N.J. The role of glutathione in disulphide bond formation and endoplasmic-reticulum-generated oxidative stress. EMBO Rep. 2006, 7, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Vallelian, F.; Deuel, J.W.; Opitz, L.; Schaer, C.A.; Puglia, M.; Lonn, M.; Engelsberger, W.; Schauer, S.; Karnaukhova, E.; Spahn, D.R.; et al. Proteasome inhibition and oxidative reactions disrupt cellular homeostasis during heme stress. Cell Death Differ. 2015, 22, 597–611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michelet, F.; Gueguen, R.; Leroy, P.; Wellman, M.; Nicolas, A.; Siest, G. Blood and plasma glutathione measured in healthy subjects by HPLC: Relation to sex, aging, biological variables, and life habits. Clin. Chem. 1995, 41, 1509–1517. [Google Scholar] [CrossRef] [PubMed]
- Mlejnek, P. Direct Interaction between N-Acetylcysteine and Cytotoxic Electrophile—An Overlooked In Vitro Mechanism of Protection. Antioxidants 2022, 11, 1485. [Google Scholar] [CrossRef]
- Toader, A.M.; Diaconu, C.; Volanschi, E. Spectroelectrochemical Study of the Hemin-Glutathione Interaction in the Absence and Presence of Surfactants. ECS Trans. 2007, 3, 155–166. [Google Scholar] [CrossRef]
- Brown, S.B.; Hatzikonstantinou, H.; Herries, D.G. The role of peroxide in haem degradation. A study of the oxidation of ferrihaems by hydrogen peroxide. Biochem. J. 1978, 174, 901–907. [Google Scholar] [CrossRef]
- Harvey, J.W.; Beutler, E. Binding of heme by glutathione S-transferase: A possible role of the erythrocyte enzyme. Blood 1982, 60, 1227–1230. [Google Scholar] [CrossRef]
- Li, X. Glutathione and Glutathione-S-Transferase in Detoxification Mechanisms. In General, Applied and Systems Toxicology; Wiley: Hoboken, NJ, USA, 2009. [Google Scholar]
- Tsuneshige, A.; Imai, K.; Tyuma, I. The Binding of Hemoglobin to Red Cell Membrane Lowers Its Oxygen Affinity. J. Biochem. 1987, 101, 695–704. [Google Scholar] [CrossRef]
- Gatidis, S.; Föller, M.; Lang, F. Hemin-induced suicidal erythrocyte death. Ann. Hematol. 2009, 88, 721–726. [Google Scholar] [CrossRef]
- Das, D.; Patra, M.; Chakrabarti, A. Binding of hemin, hematoporphyrin, and protoporphyrin with erythroid spectrin: Fluorescence and molecular docking studies. Eur. Biophys. J. 2015, 44, 171–182. [Google Scholar] [CrossRef]
- Kozlova, E.; Chernysh, A.; Moroz, V.; Gudkova, O.; Sergunova, V.; Kuzovlev, A. Transformation of membrane nanosurface of red blood cells under hemin action. Sci. Rep. 2014, 4, 6033. [Google Scholar] [CrossRef] [PubMed]
- Shaklai, N.; Shviro, Y.; Rabizadeh, E.; Kirschner-Zilber, I. Accumulation and drainage of hemin in the red cell membrane. Biochim. Biophys. Acta 1985, 821, 355–366. [Google Scholar] [CrossRef]
- Buehler, P.W.; Karnaukhova, E.; Gelderman, M.P.; Alayash, A.I. Blood Aging, Safety, and Transfusion: Capturing the “Radical” Menace. Antioxid. Redox Signal. 2011, 14, 1713–1728. [Google Scholar] [CrossRef] [PubMed]
- Darghouth, D.; Koehl, B.; Madalinski, G.; Heilier, J.F.; Bovee, P.; Xu, Y.; Olivier, M.F.; Bartolucci, P.; Benkerrou, M.; Pissard, S.; et al. Pathophysiology of sickle cell disease is mirrored by the red blood cell metabolome. Blood 2011, 117, e57–e66. [Google Scholar] [CrossRef] [PubMed]
- Uzunova, V.V.; Pan, W.; Galkin, O.; Vekilov, P.G. Free heme and the polymerization of sickle cell hemoglobin. Biophys. J. 2010, 99, 1976–1985. [Google Scholar] [CrossRef] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Georgiou-Siafis, S.K.; Samiotaki, M.K.; Demopoulos, V.J.; Panayotou, G.; Tsiftsoglou, A.S. Glutathione–Hemin/Hematin Adduct Formation to Disintegrate Cytotoxic Oxidant Hemin/Hematin in Human K562 Cells and Red Blood Cells’ Hemolysates: Impact of Glutathione on the Hemolytic Disorders and Homeostasis. Antioxidants 2022, 11, 1959. https://doi.org/10.3390/antiox11101959
Georgiou-Siafis SK, Samiotaki MK, Demopoulos VJ, Panayotou G, Tsiftsoglou AS. Glutathione–Hemin/Hematin Adduct Formation to Disintegrate Cytotoxic Oxidant Hemin/Hematin in Human K562 Cells and Red Blood Cells’ Hemolysates: Impact of Glutathione on the Hemolytic Disorders and Homeostasis. Antioxidants. 2022; 11(10):1959. https://doi.org/10.3390/antiox11101959
Chicago/Turabian StyleGeorgiou-Siafis, Sofia K., Martina K. Samiotaki, Vassilis J. Demopoulos, George Panayotou, and Asterios S. Tsiftsoglou. 2022. "Glutathione–Hemin/Hematin Adduct Formation to Disintegrate Cytotoxic Oxidant Hemin/Hematin in Human K562 Cells and Red Blood Cells’ Hemolysates: Impact of Glutathione on the Hemolytic Disorders and Homeostasis" Antioxidants 11, no. 10: 1959. https://doi.org/10.3390/antiox11101959