
Pharmacological Inhibition of Class III Alcohol Dehydrogenase 5: Turning Remote Ischemic Conditioning Effective in a Diabetic Stroke Model


Abstract
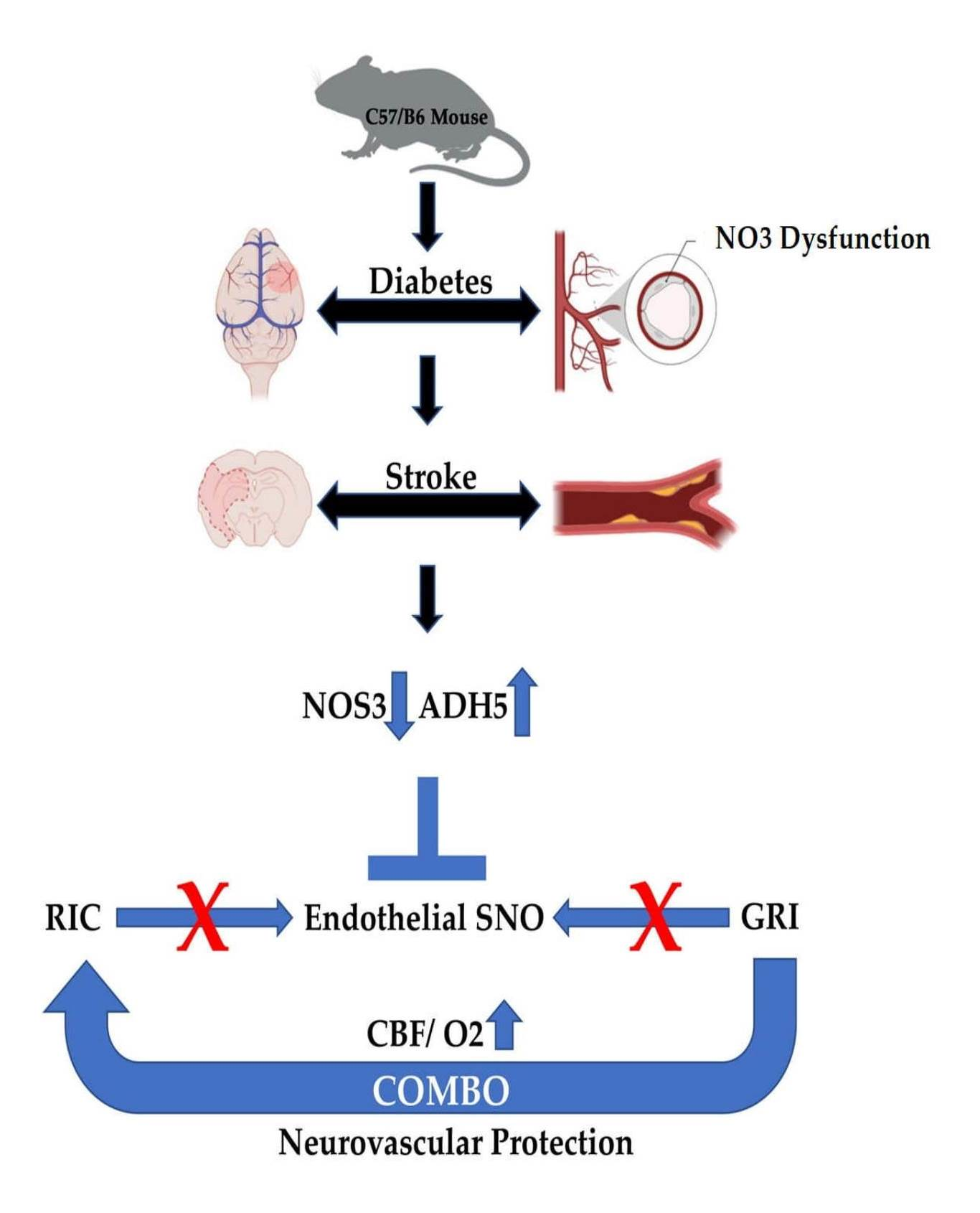
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animals, Experimental Groups, and Approaches
2.2. Streptozotocin (STZ)-Induced Model of Type 1 Diabetes
2.3. Models of Thrombotic and Reperfused Strokes
2.3.1. Photothrombotic (PT) Model of Stroke
2.3.2. Reperfused Transient MCA Occlusion (tMCAO) Model of Stroke
2.4. Treatments and Therapies after Stroke
2.5. Measurement of Relative Cerebral Blood Flow (CBF)
2.6. Quantification of Hemoglobin (Hb) Content
2.7. Analysis of Plasma Nitrite (NO2−) Level
2.8. Isolation and Enrichment of Mouse BMECs
2.9. Estimation of GSNOR Activity in Plasma and BMECs
2.10. Immunoblot of GSNOR Expression in BMECs
2.11. Behavioral Tests for Motor Function and Neurologic Deficits
2.12. Measurement of Infarct Volume, Edema, and BBB Permeability
2.13. Measurement of Brain Tissue Oxygenation (PbtO2)
2.14. Semi-Quantitative Real-Time PCR (qPCR) for Inflammatory Gene Expressions
2.15. Data Collection and Statistical Analyses
3. Results
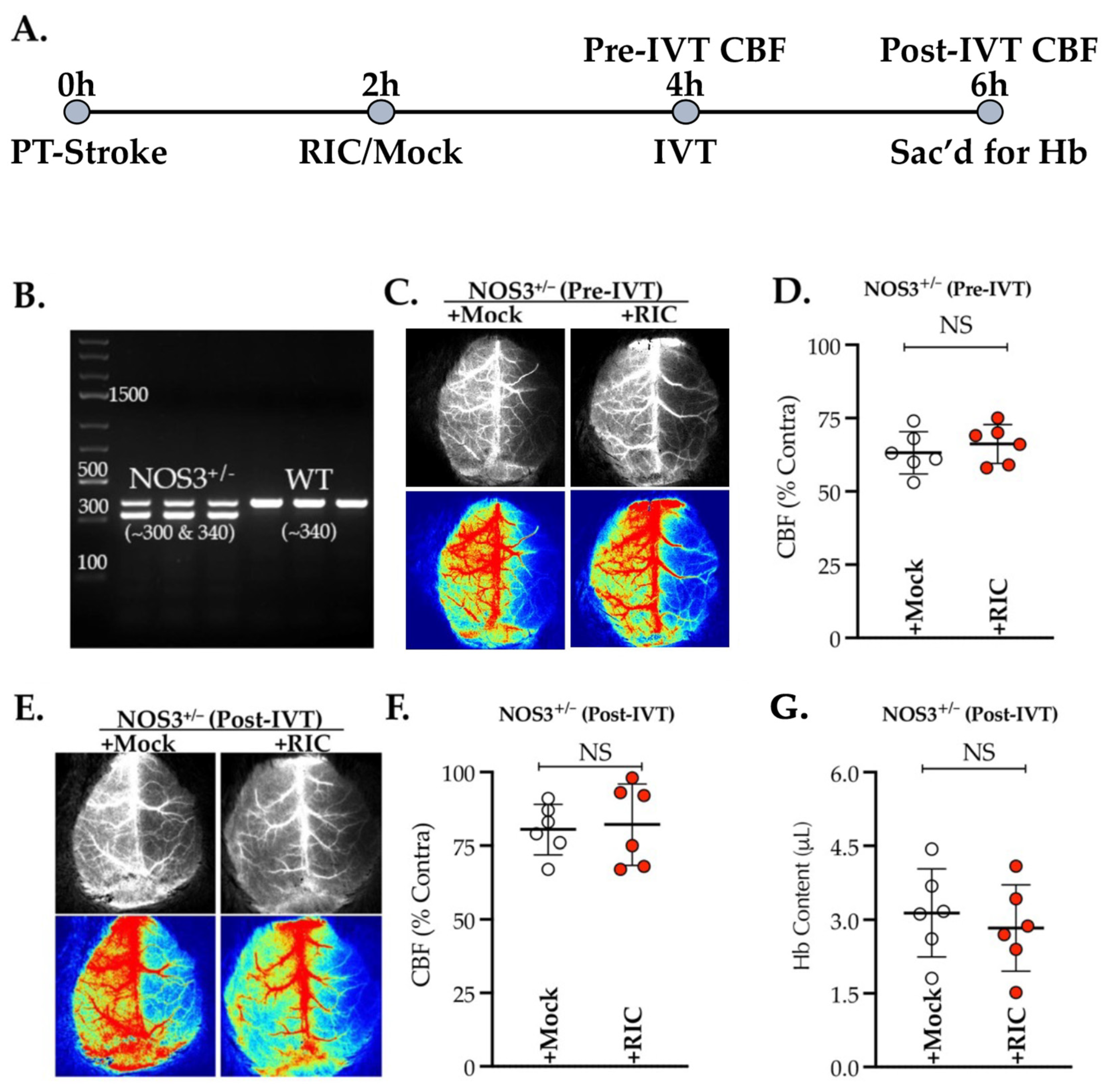
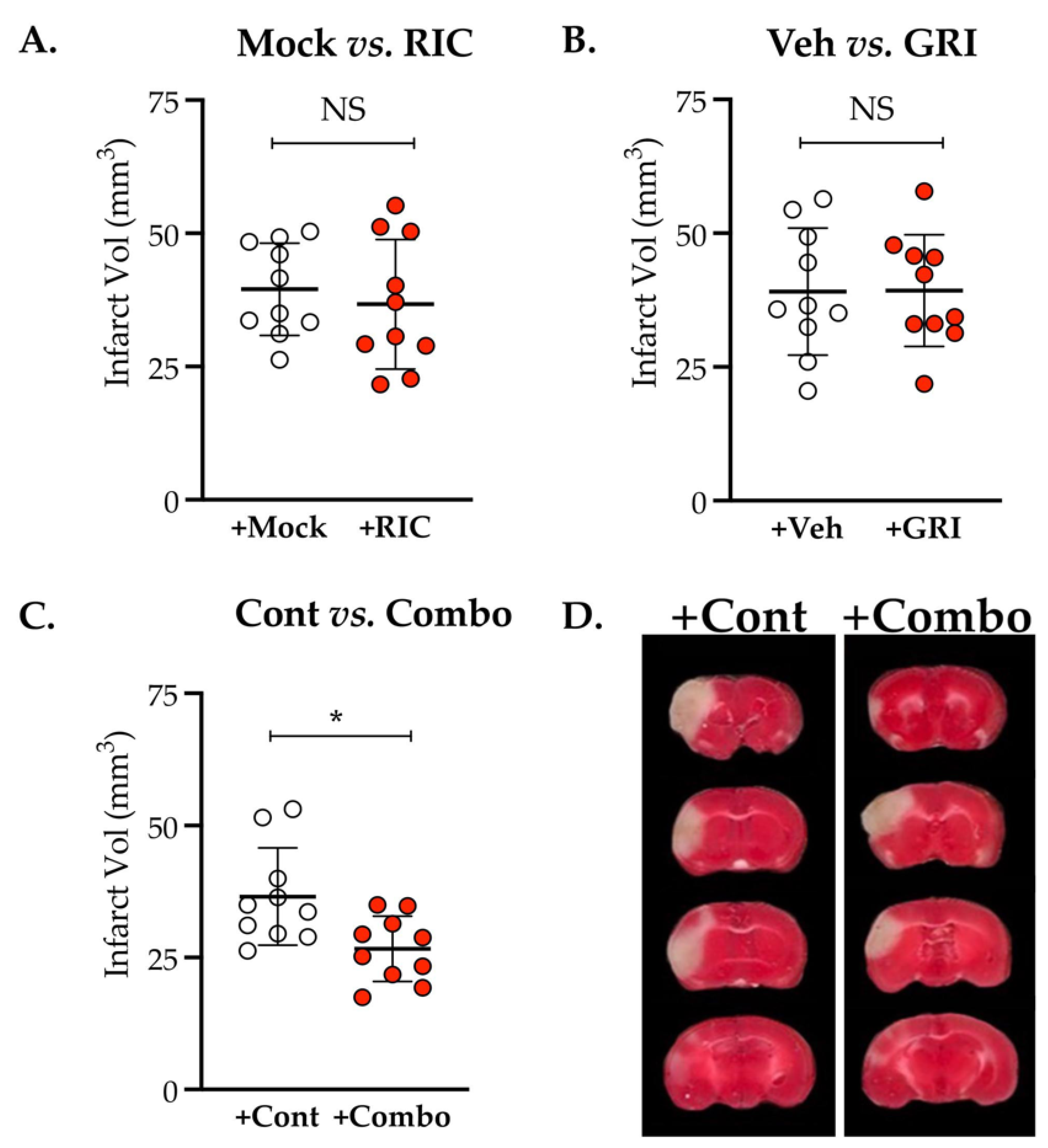
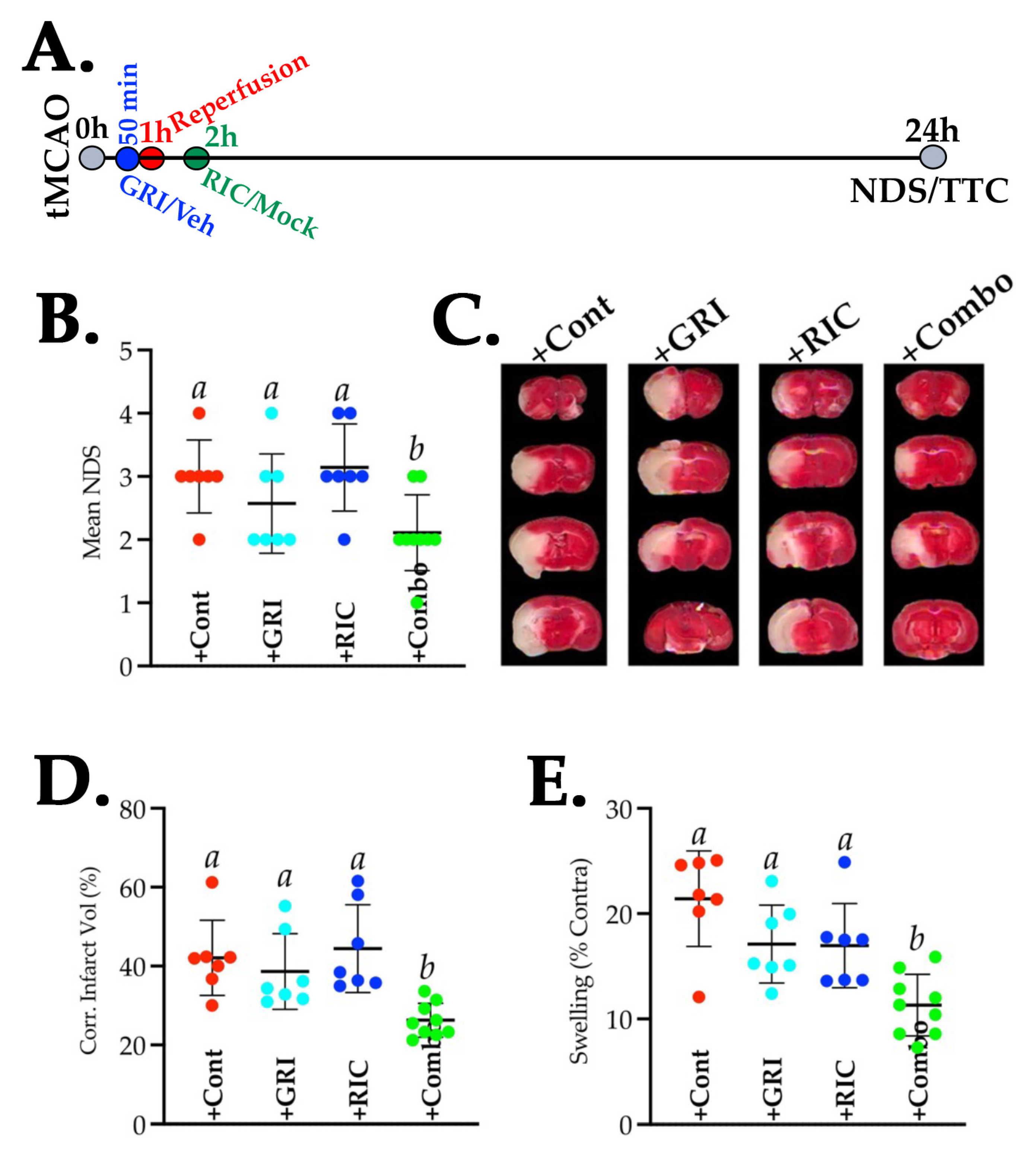
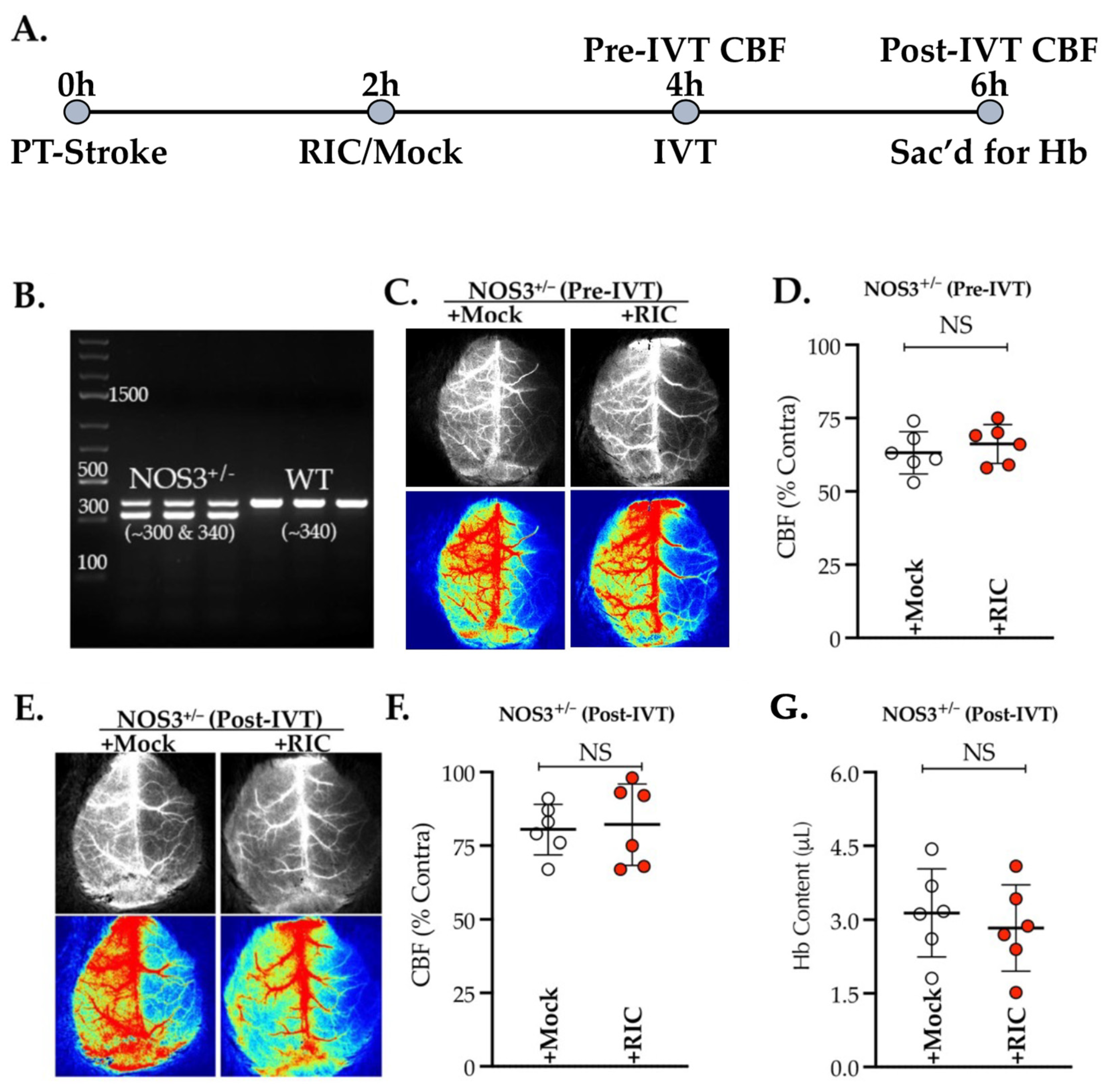
3.1. NOS3 Depletion Abolishes the Efficacy of RIC Therapy in Thrombotic Stroke
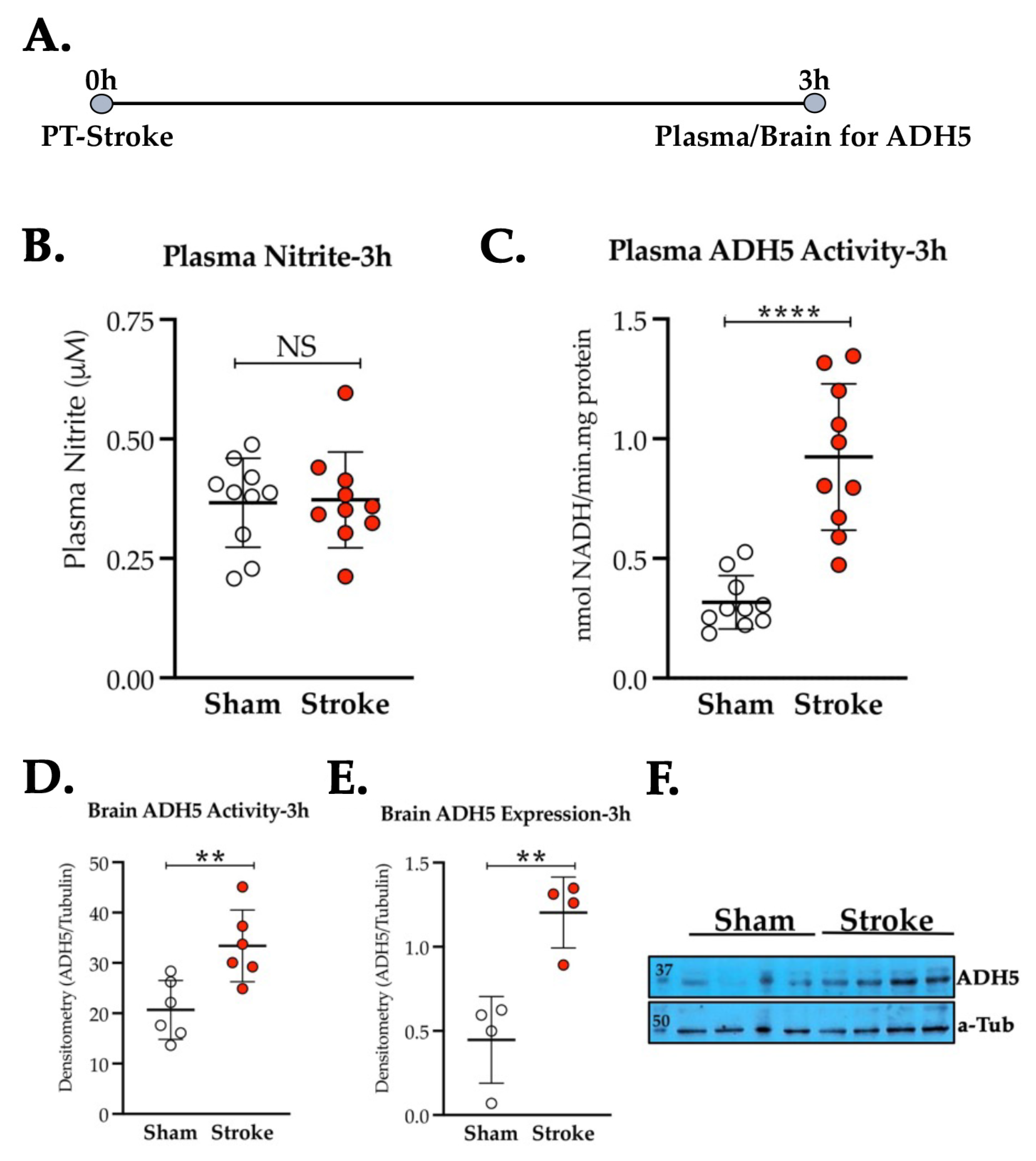
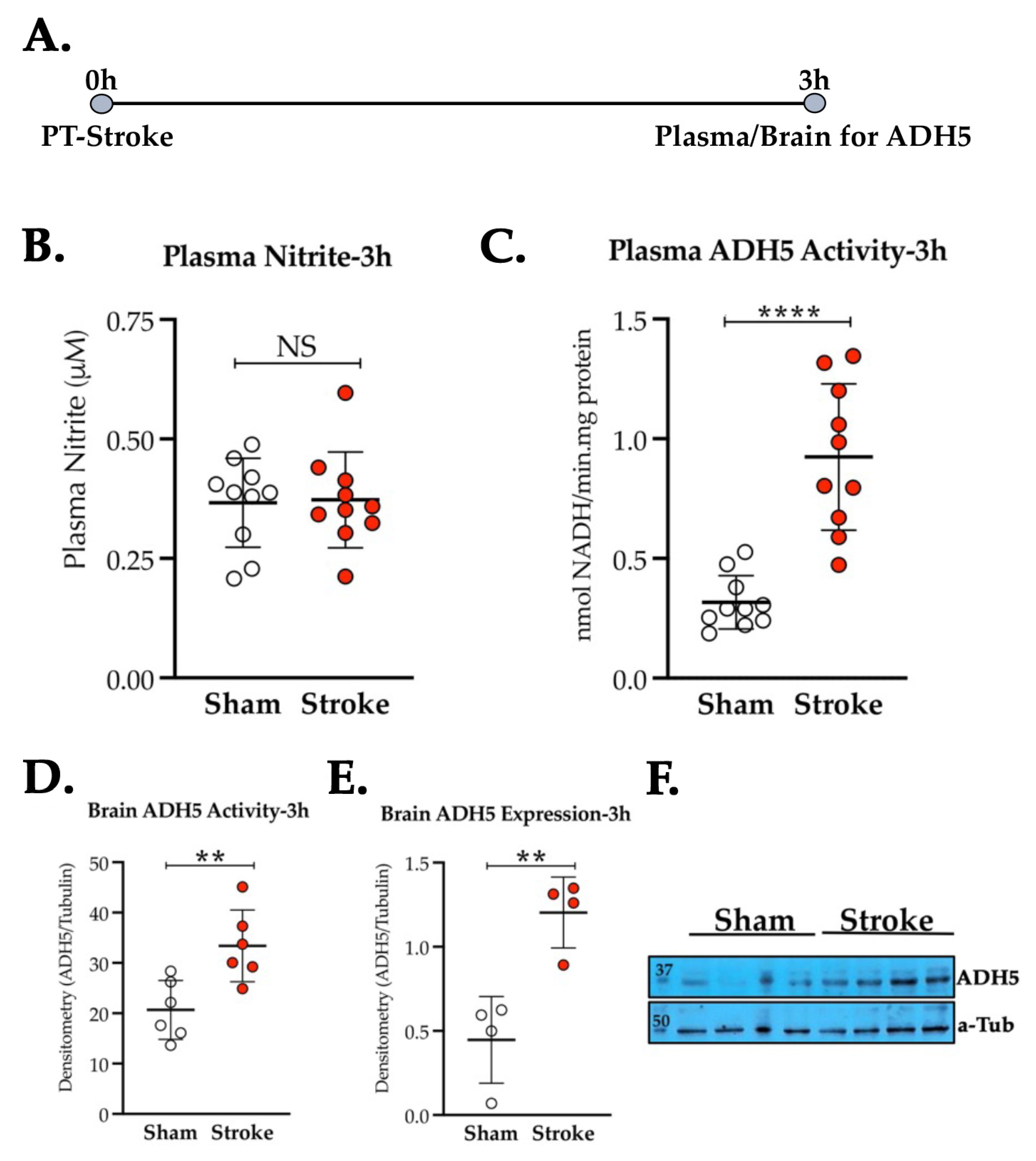
3.2. Thrombotic Stroke in Diabetic Mice Does Not Modulate the Plasma NO2− Level but Increases the Activity and Expression of ADH5/GSNOR
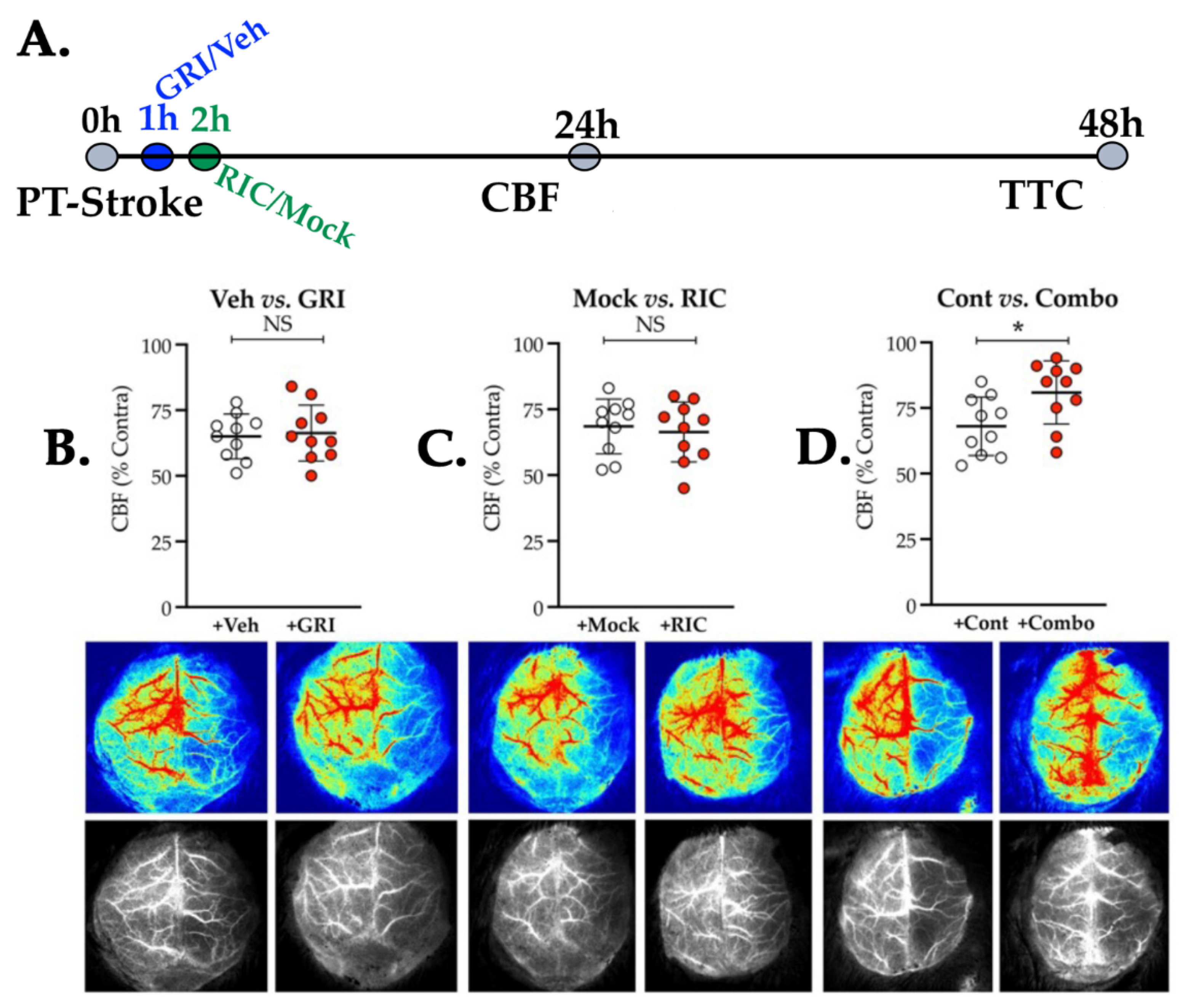
3.3. Neither GRI nor RIC Therapy Alone but a Combination of Both Enhances CBF Following Thrombotic Stroke in Diabetic Mice

3.4. Combination Therapy of GRI and RIC but Neither Therapy Alone in Diabetic Mice Is Neuroprotective against Thrombotic Stroke

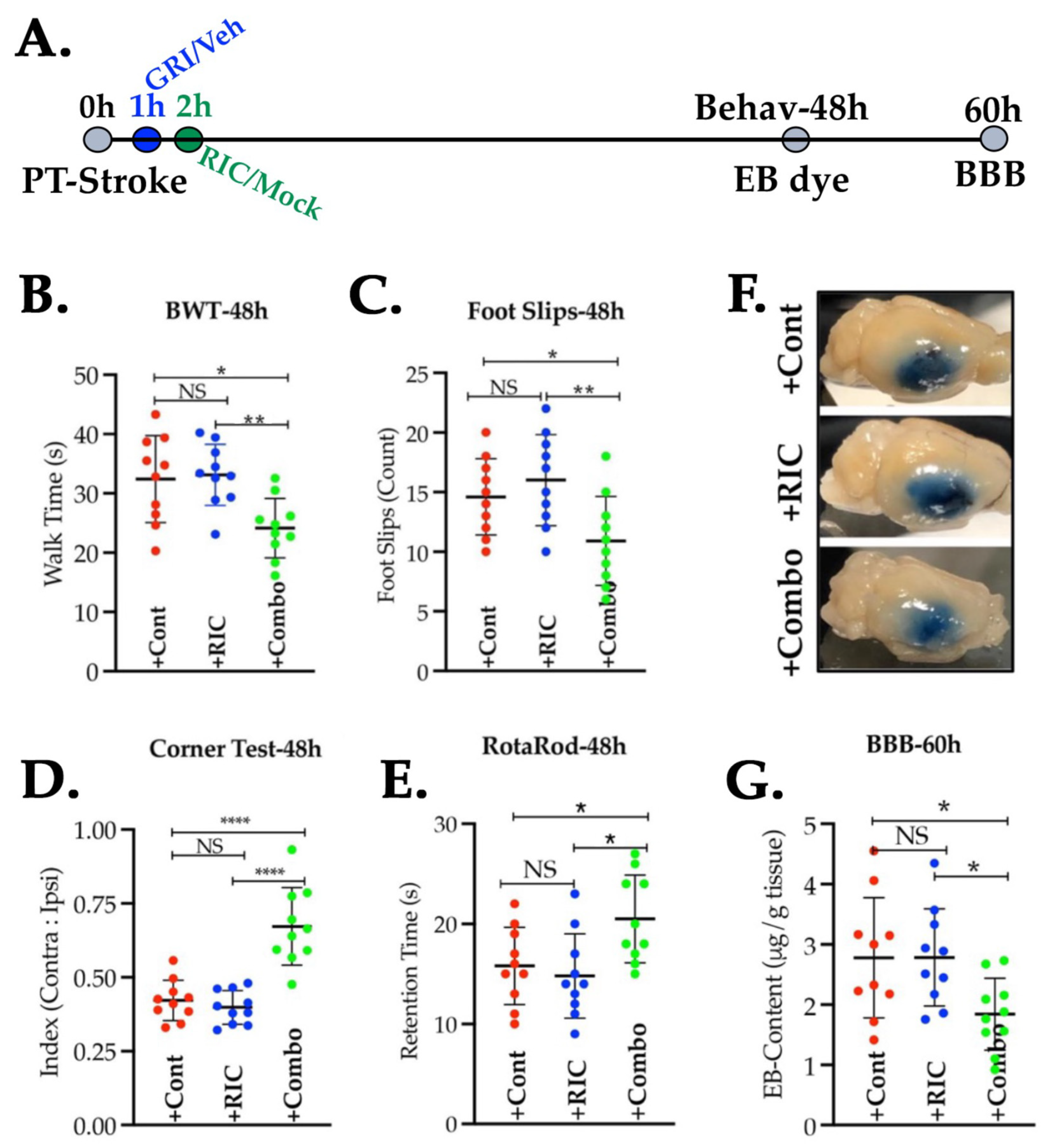
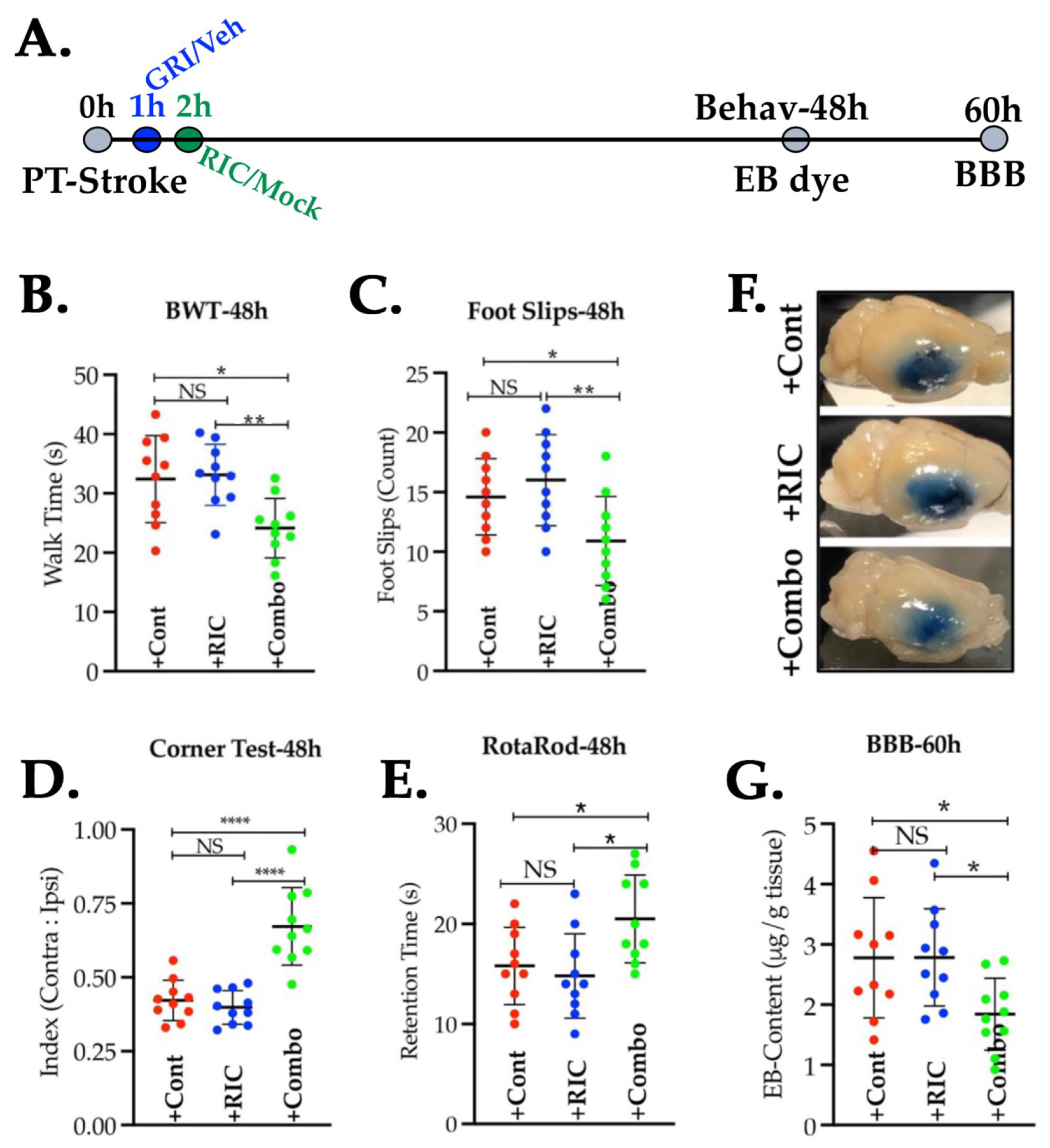
3.5. Combination of GRI and RIC Therapies but Not RIC Alone in Thrombotic Diabetic Stroke Improves Behavioral Outcomes and Attenuates BBB Disruption
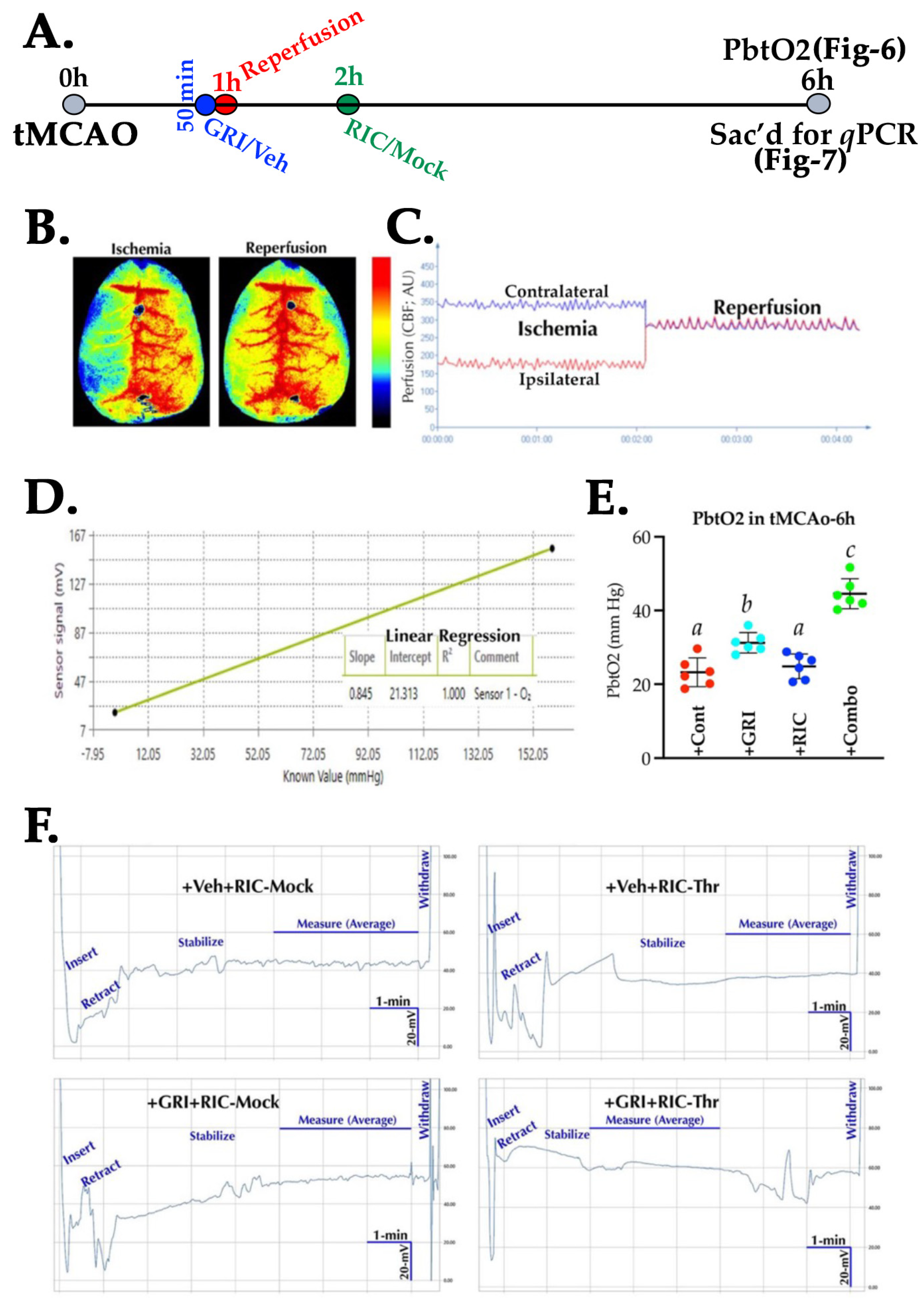
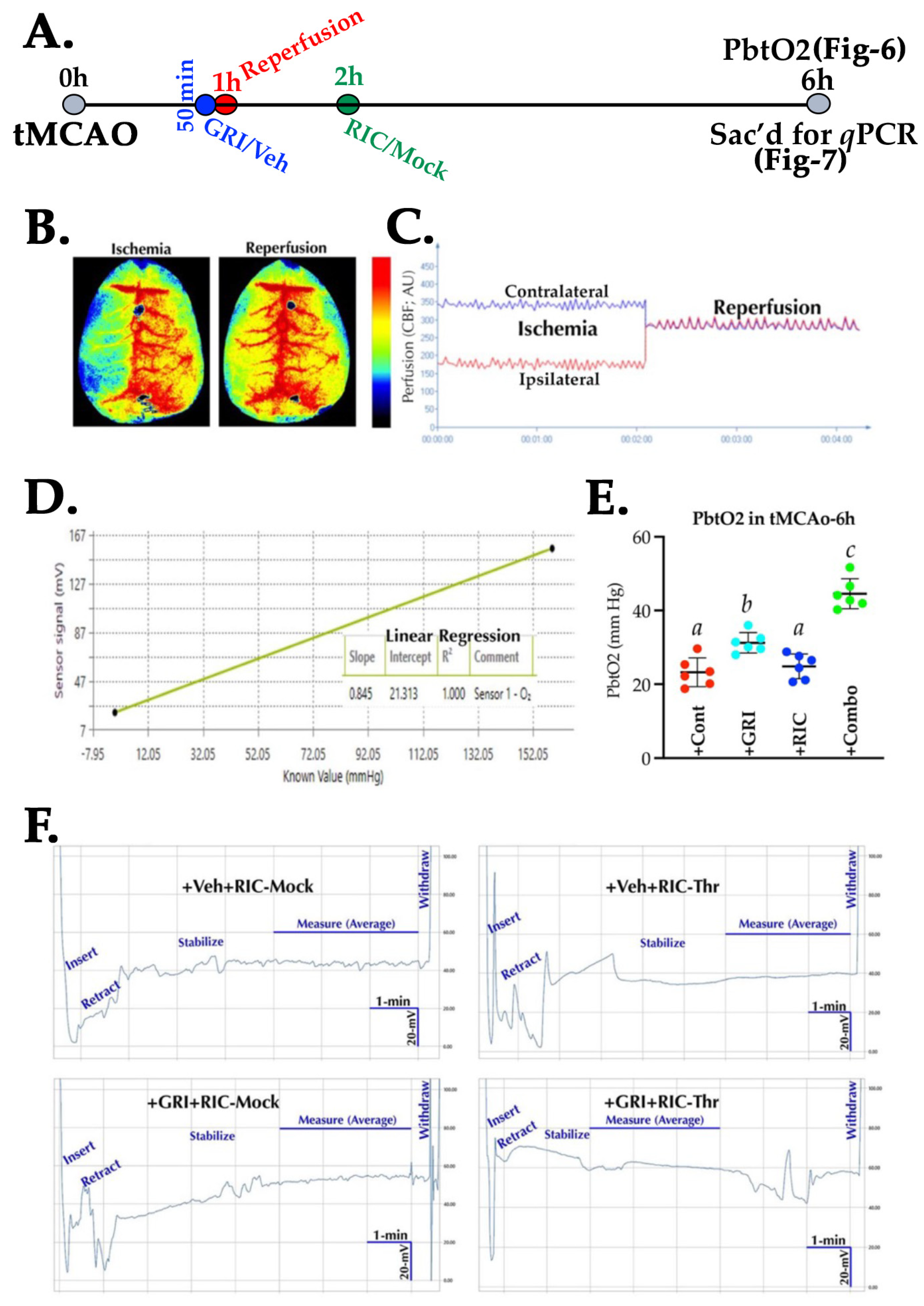
3.6. GRI and RIC Therapies Show Differential Effects on PbtO2 in Reperfused Diabetic Stroke
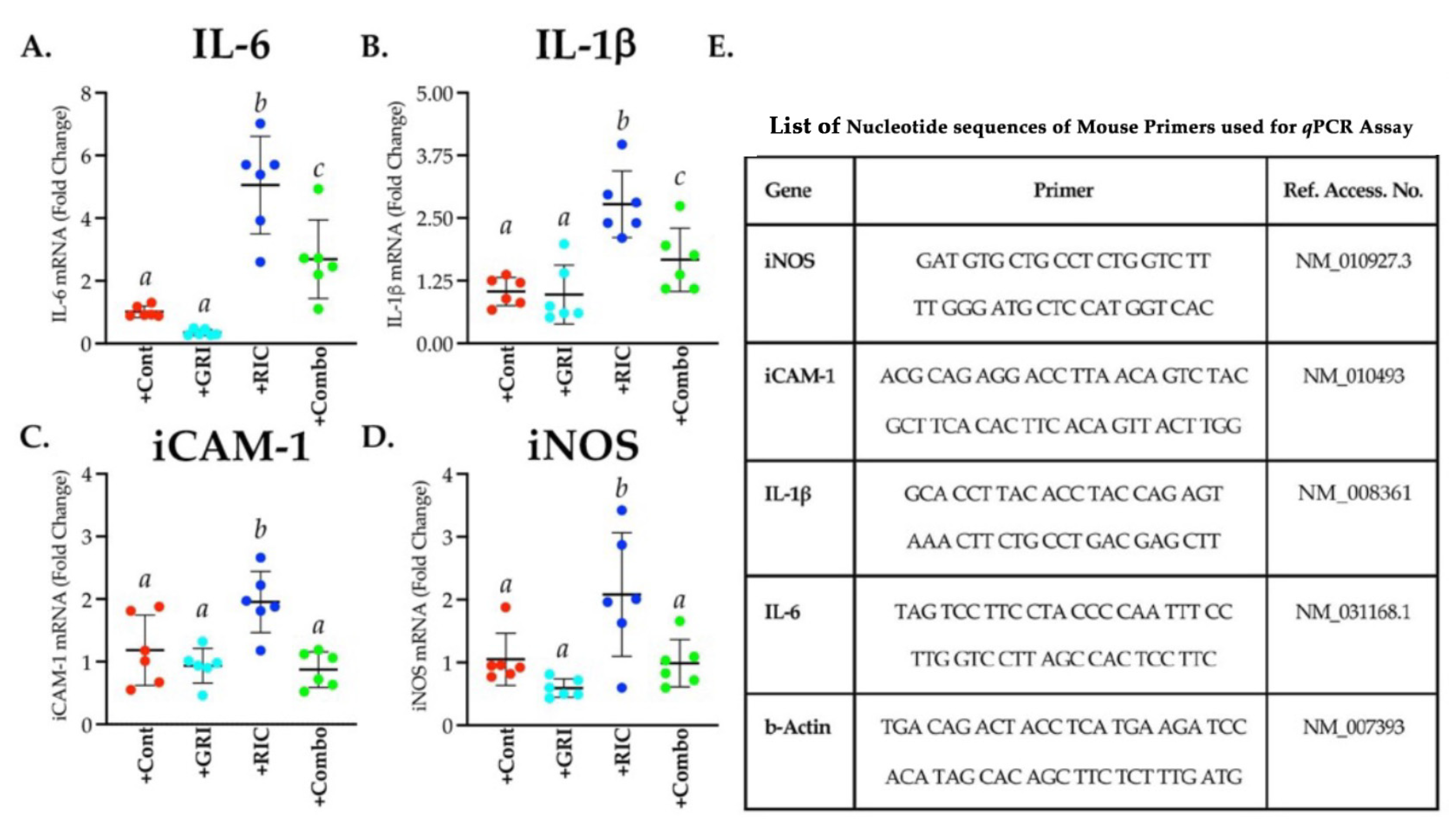
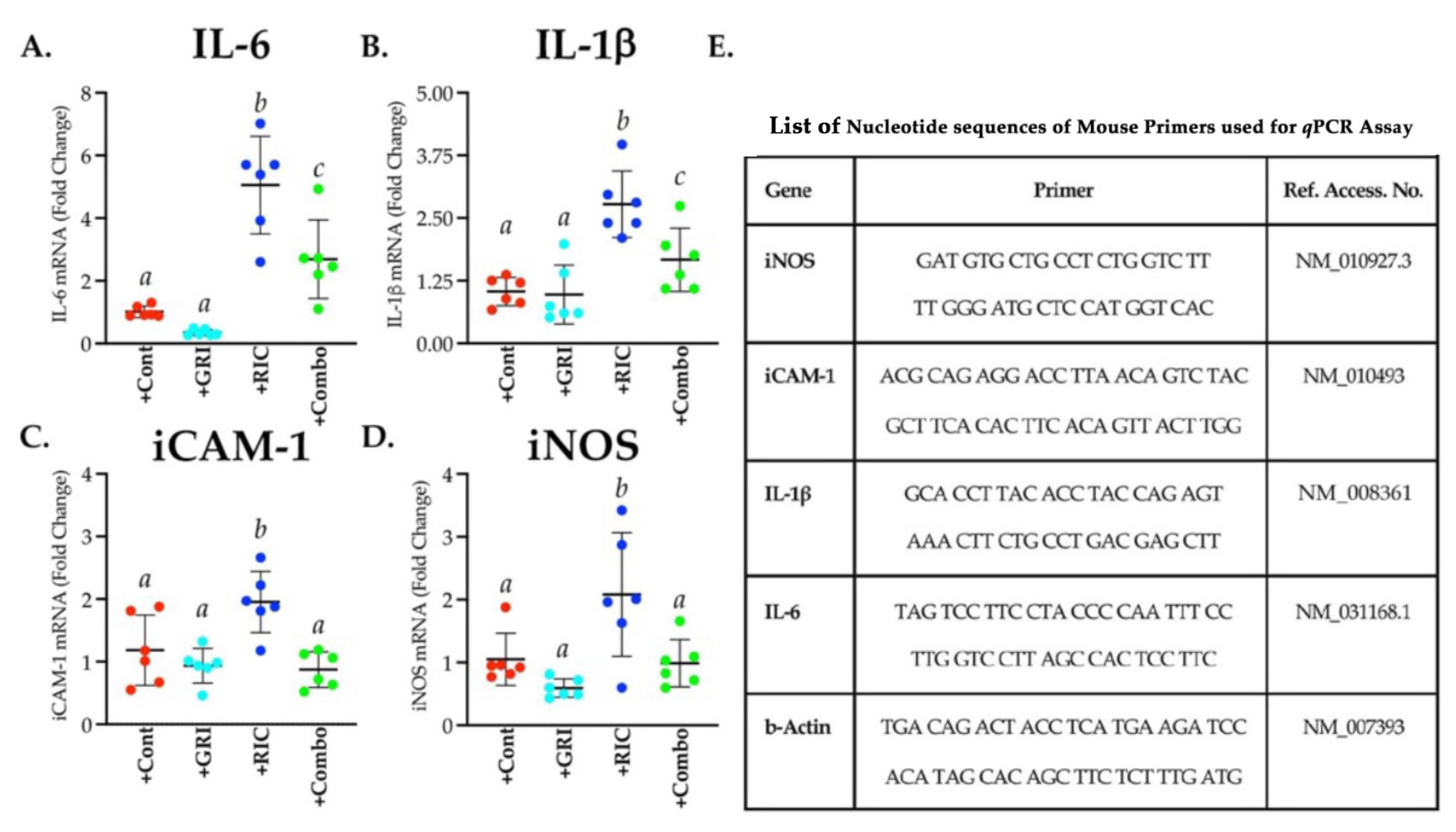
3.7. RIC Therapy Triggers Inflammatory Gene Expressions in Reperfused Diabetic Stroke which Is Attenuated in Combination with GRI Treatment
3.8. GRI Treatment in Combination with RIC Therapy Attenuates Reperfusion Injury and Enhances the Neuroprotection in Diabetic Stroke

4. Discussion
5. Conclusions
6. Patents
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

References
- French, B.R.; Boddepalli, R.S.; Govindarajan, R. Acute Ischemic Stroke: Current Status and Future Directions. Mo. Med. 2016, 113, 480–486. [Google Scholar] [PubMed]
- Gursoy-Ozdemir, Y.; Yemisci, M.; Dalkara, T. Microvascular protection is essential for successful neuroprotection in stroke. J. Neurochem. 2012, 123 (Suppl. 2), 2–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singel, D.J.; Stamler, J.S. Chemical physiology of blood flow regulation by red blood cells: The role of nitric oxide and S-nitrosohemoglobin. Annu. Rev. Physiol. 2005, 67, 99–145. [Google Scholar] [CrossRef] [PubMed]
- Stamler, J.S.; Jia, L.; Eu, J.P.; McMahon, T.J.; Demchenko, I.T.; Bonaventura, J.; Gernert, K.; Piantadosi, C.A. Blood flow regulation by S-nitrosohemoglobin in the physiological oxygen gradient. Science 1997, 276, 2034–2037. [Google Scholar] [CrossRef] [Green Version]
- Stamler, J.S.; Reynolds, J.D.; Hess, D.T. Endocrine nitric oxide bioactivity and hypoxic vasodilation by inhaled nitric oxide. Circ. Res. 2012, 110, 652–654. [Google Scholar] [CrossRef] [Green Version]
- Bath, P.M.; Krishnan, K.; Appleton, J.P. Nitric oxide donors (nitrates), L-arginine, or nitric oxide synthase inhibitors for acute stroke. Cochrane Database Syst. Rev. 2017, 2017, CD000398. [Google Scholar] [CrossRef]
- Endres, M.; Laufs, U.; Liao, J.K.; Moskowitz, M.A. Targeting eNOS for stroke protection. Trends. Neurosci. 2004, 27, 283–289. [Google Scholar] [CrossRef]
- Cortese-Krott, M.M.; Kelm, M. Endothelial nitric oxide synthase in red blood cells: Key to a new erythrocrine function? Redox Biol. 2014, 2, 251–258. [Google Scholar] [CrossRef] [Green Version]
- Green, A.R. Pharmacological approaches to acute ischaemic stroke: Reperfusion certainly, neuroprotection possibly. Br. J. Pharmacol. 2008, 153, S325–S338. [Google Scholar] [CrossRef] [Green Version]
- Huang, Z.; Hunag, P.L.; Ma, J.; Meng, W.; Ayata, C.; Fishman, M.C.; Moskowitz, M.A. Enlarged infarcts in endothelial nitric oxide synthase knockout mice are attenuated by nitro-L-arginine. J. Cereb. Blood Flow Metab. 1996, 16, 981–987. [Google Scholar] [CrossRef]
- Green, L.S.; Chun, E.; Patton, A.K.; Sun, X.; Rosenthal, G.J.; Richards, J.P. Mechanism of inhibition for N6022, a first-in-class drug targeting s-nitrosoglutathione reductase. Biochemistry 2012, 51, 2157–2168. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, S.K.; Ahmed, F.; Alkhatabi, H.; Hoda, M.N.; Qhatani, M.A. Nebulization of Low-Dose s-Nitrosoglutathione in Diabetic Stroke Enhances Benefits of Rep. erfusion and Prevents Post-Thrombolysis Hemorrhage. Biomolecules 2021, 11, 1587. [Google Scholar] [CrossRef] [PubMed]
- Lima, B.; Lam, G.K.W.; Xie, L.; Diesen, D.L.; Villamizar, N.; Nienaber, J.; Messina, E.; Bowles, D.; Konstos, C.D.; Hare, J.M.; et al. Endogenous S-nitrosothiols protect against myocardial injury. Proc. Natl. Acad. Sci. USA 2009, 106, 6297–6302. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.; Dhammu, T.S.; Qiao, F.; Singh, A.K.; Sing, I. s-Nitrosoglutathione Mimics the Beneficial Activity of Endothelial Nitric Oxide Synthase-Derived Nitric Oxide in a Mouse Model of Stroke. J. Stroke Cerebrovasc. Dis. 2019, 28, 104470. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Sievers, R.E.; Varga, M.; Kharait, S.; Haddad, D.J.; Patton, A.K.; Delany, C.S.; Mutka, S.C.; Blonder, J.P.; Dube, J.P.; et al. Pharmacological inhibition of s-nitrosoglutathione reductase improves endothelial vasodilatory function in rats in vivo. J. Appl. Physiol. (1985) 2013, 114, 752–760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hess, D.C.; Hoda, M.N.; Khan, M.B. Humoral Mediators of Remote Ischemic Conditioning: Important Role of eNOS/NO/Nitrite. Acta Neurochir. Suppl. 2016, 121, 45–48. [Google Scholar]
- Schulz, R.; Kelm, M.; Heusch, G. Nitric oxide in myocardial ischemia/reperfusion injury. Cardiovasc. Res. 2004, 61, 402–413. [Google Scholar] [CrossRef] [Green Version]
- Jung, H.; Choi, E.U.; Baek, S.I.; Cho, C.; Jin, Y.; Kwak, K.H.; Jeon, Y.; Park, S.S.; Kim, S.; Lim, D.G. The Effect of Nitric Oxide on Remote Ischemic Preconditioning in Renal Ischemia Reperfusion Injury in Rats. Dose Response 2019, 17, 1559325819853651. [Google Scholar] [CrossRef]
- Jankovic, A.; Zakic, T.; Milicic, M.; Unic-Stojanovic, D.; Kalezic, A.; Korac, A.; Jovic, M.; Korac, B. Effects of Remote Ischaemic Preconditioning on the Internal Thoracic Artery Nitric Oxide Synthase Isoforms in Patients Undergoing Coronary Artery Bypass Grafting. Antioxidants 2021, 10, 1910. [Google Scholar] [CrossRef]
- Hoda, M.N.; Bhatia, K.; Sharef, S.S.; Johnson, M.H.; Siddiqui, S.; Ergul, A.; Zaidi, S.K.; Fagan, S.C.; Hess, D.C. Remote ischemic perconditioning is effective after embolic stroke in ovariectomized female mice. Transl. Stroke Res. 2014, 5, 484–490. [Google Scholar] [CrossRef] [Green Version]
- Hoda, M.N.; Siddiqui, S.; Herberg, S.; Periyasamy-Thandavan, S.; Bhatia, K.; Sharef, S.S.; Johnson, M.H.; Hill, W.D.; Ergul, A.; Fagan, S.C.; et al. Remote ischemic perconditioning is effective alone and in combination with intravenous tissue-type plasminogen activator in murine model of embolic stroke. Stroke 2012, 43, 2794–2799. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Ma, Y.; Shuaib, A.; Winship, I.R. Improved collateral flow and reduced damage after remote ischemic perconditioning during distal middle cerebral artery occlusion in aged rats. Sci. Rep. 2020, 10, 12392–12406. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Nguyen, T.; Aponte, A.M.; Menazza, S.; Kohr, M.J.; Roth, D.M.; Patel, H.H.; Murphy, E.; Steenbergen, C. Ischaemic preconditioning preferentially increases protein s-nitrosylation in subsarcolemmal mitochondria. Cardiovasc. Res. 2015, 106, 227–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schenning, K.J.; Anderson, S.; Alkayed, N.J.; Hutchens, M.P. Hyperglycemia abolishes the protective effect of ischemic preconditioning in glomerular endothelial cells in vitro. Physiol. Rep. 2015, 3, e12346. [Google Scholar] [CrossRef]
- Kersten, J.R.; Schmeling, T.J.; Orth, K.G.; Pagel, P.S.; Warltier, D.C. Acute hyperglycemia abolishes ischemic preconditioning in vivo. Am. J. Physiol. 1998, 275, H721–H725. [Google Scholar] [CrossRef]
- Baranyai, T.; Nagy, C.T.; Koncsos, G.; Onodi, Z.; Karolyi-Szabo, M.; Makkos, A.; Varga, Z.V.; Ferdinandy, P.; Giricz, Z. Acute hyperglycemia abolishes cardioprotection by remote ischemic perconditioning. Cardiovasc. Diabetol. 2015, 14, 151–160. [Google Scholar] [CrossRef] [Green Version]
- Lau, L.H.; Lew, J.; Borschmann, K.; Thijs, V.; Ekinci, E.I. Prevalence of diabetes and its effects on stroke outcomes: A meta-analysis and literature review. J. Diabetes Investig. 2019, 10, 780–792. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Forstermann, U. Pharmacological prevention of eNOS uncoupling. Curr. Pharm. Des. 2014, 20, 3595–3606. [Google Scholar] [CrossRef]
- Thum, T.; Fraccarollo, D.; Schultheiss, M.; Froese, S.; Galuppo, P.; Widder, J.D.; Tsikas, D.; Ertk, G.; Bauersachs, J. Endothelial nitric oxide synthase uncoupling impairs endothelial progenitor cell mobilization and function in diabetes. Diabetes 2007, 56, 666–674. [Google Scholar] [CrossRef] [Green Version]
- Padron, J.; Peiro, C.; Cercas, E.; Llegro, J.L.; Sanchez-Ferrer, C.F. Enhancement of s-nitrosylation in glycosylated hemoglobin. Biochem. Biophys. Res. Commun. 2000, 271, 217–221. [Google Scholar] [CrossRef]
- Sun, Y.Y.; Kuo, Y.M.; Chen, H.R.; Short-Miller, J.C.; Smucker, M.R.; Kuan, C.Y. A murine photothrombotic stroke model with an increased fibrin content and improved responses to tPA-lytic treatment. Blood Adv. 2020, 4, 1222–1231. [Google Scholar] [CrossRef] [PubMed]
- Hoda, M.N.; Singh, I.; Singh, A.K.; Khan, M. Reduction of lipoxidative load by secretory phospholipase A2 inhibition protects against neurovascular injury following experimental stroke in rat. J. Neuroinflamm. 2009, 6, 21–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoda, M.N.; Li, W.; Ahmad, A.; Ogbi, S.; Zemskova, M.A.; Johnson, M.H.; Ergul, A.; Hill, W.D.; Hess, D.C.; Sazonova, I.Y. Sex-independent neuroprotection with minocycline after experimental thromboembolic stroke. Exp. Transl. Stroke Med. 2011, 3, 16–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoda, M.N.; Fagan, S.C.; Khan, M.B.; Vaibhav, K.; Chaudhary, A.; Wang, P.; Dhandapani, K.M.; Waller, J.L.; Hess, D.C. A 2 × 2 factorial design for the combination therapy of minocycline and remote ischemic perconditioning: Efficacy in a preclinical trial in murine thromboembolic stroke model. Exp. Transl. Stroke Med. 2014, 6, 10–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruck, T.; Bittner, S.; Epping, L.; Herrmann, A.M.; Meuth, S.G. Isolation of primary murine brain microvascular endothelial cells. J. Vis. Exp. 2014, 14, e52204. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Zeng, X.; Li, G.; Yang, Q.; Xu, J.; Zhang, M.; Mao, X.; Cao, Y.; Wang, L.; Xu, Y.; et al. Inactivation of endothelial adenosine A2A receptors protects mice from cerebral ischaemia-induced brain injury. Br. J. Pharmacol. 2019, 176, 2250–2263. [Google Scholar]
- Brown-Steinke, K.; deRonde, K.; Yemen, S.; Palmer, L.A. Gender differences in s-nitrosoglutathione reductase activity in the lung. PLoS ONE 2010, 5, e14007. [Google Scholar] [CrossRef] [Green Version]
- Hayashida, K.; Bagchi, A.; Miyazaki, Y.; Hirai, S.; Seth, D.; Silverman, M.G.; Rezoagli, E.; Marutani, E.; Mori, N.; Magliocca, A.; et al. Improvement in Outcomes After Cardiac Arrest and Resuscitation by Inhibition of s-Nitrosoglutathione Reductase. Circulation 2019, 139, 815–827. [Google Scholar] [CrossRef]
- Khan, M.B.; Hafez, S.S.; Hoda, M.N.; Baban, B.; Wagner, J.; Awad, M.E.; Sangabathula, H.; Haigh, S.; Elsalanty, M.; Waller, J.L.; et al. Chronic Remote Ischemic Conditioning Is Cerebroprotective and Induces Vascular Remodeling in a VCID Model. Transl. Stroke Res. 2018, 9, 51–63. [Google Scholar] [CrossRef] [Green Version]
- Webb, R.L.; Kaiser, E.E.; Scoville, S.L.; Thompson, T.A.; Fatima, S.; Pandya, C.; Sriram, K.; Swetenburg, R.L.; Vaibhav, K.; Arbab, A.S.; et al. Human Neural Stem Cell Extracellular Vesicles Improve Tissue and Functional Recovery in the Murine Thromboembolic Stroke Model. Transl. Stroke Res. 2018, 9, 530–539. [Google Scholar] [CrossRef] [Green Version]
- Balkaya, M.; Krober, J.M.; Rex, A.; Endres, M. Assessing post-stroke behavior in mouse models of focal ischemia. J. Cereb. Blood Flow Metab. 2013, 33, 330–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almendros, I.; Farre, R.; Planas, A.M.; Torres, M.; Bonsignore, M.R.; Navajas, D.; Montserrat, J.M. Tissue oxygenation in brain, muscle, and fat in a rat model of sleep apnea: Differential effect of obstructive apneas and intermittent hypoxia. Sleep 2011, 34, 1127–1133. [Google Scholar] [CrossRef] [PubMed]
- Geneslaw, A.S.; Zhao, M.; Mao, H.; Schwartz, T.H. Tissue hypoxia correlates with intensity of interictal spikes. J. Cereb. Blood Flow Metab. 2011, 31, 1394–1402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huchzermeyer, C.; Berndt, N.; Holzhutter, H.G.; Kann, O. Oxygen consumption rates during three different neuronal activity states in the hippocampal CA3 network. J. Cereb. Blood Flow Metab. 2013, 33, 263–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abu-Amara, M.; Yang, S.Y.; Quaglia, A.; Rowley, P.; Mel, A.D.; Tapuria, N.; Seifalian, A.; Davidson, B.; Fuller, B. Nitric oxide is an essential mediator of the protective effects of remote ischaemic preconditioning in a mouse model of liver ischaemia/reperfusion injury. Clin. Sci. 2011, 121, 257–266. [Google Scholar] [CrossRef] [Green Version]
- Abu-Amara, M.; Yang, S.Y.; Quaglia, A.; Rowley, P.; Fuller, B.; Seifalian, A.; Davidson, B. Role of endothelial nitric oxide synthase in remote ischemic preconditioning of the mouse liver. Liver Transpl. 2011, 17, 610–619. [Google Scholar] [CrossRef]
- Pico, F.; Lapergue, B.; Ferrigno, M.; Rosso, C.; Meseguer, E.; Chadenat, M.L.; Bourdain, F.; Obadia, M.; Hirel, C.; Duong, D.L.; et al. Effect of In-Hospital Remote Ischemic Perconditioning on Brain Infarction Growth and Clinical Outcomes in Patients With Acute Ischemic Stroke: The RESCUE BRAIN Randomized Clinical Trial. JAMA Neurol. 2020, 77, 725–734. [Google Scholar] [CrossRef]
- Hougaard, K.D.; Hjort, N.; Zeidler, D.; Sorensen, L.; Norgaard, A.; Hansen, T.M.; Weitzel-Mudersbach, P.V.; Simonsen, C.Z.; Damgaard, D.; Gottrup, H.; et al. Remote ischemic perconditioning as an adjunct therapy to thrombolysis in patients with acute ischemic stroke: A randomized trial. Stroke 2014, 45, 159–167. [Google Scholar] [CrossRef] [Green Version]
- Hess, D.C.; Blauenfeldt, R.A.; Andersen, G.; Hougaard, K.D.; Hoda, M.N.; Ding, Y.; Ji, X. Remote ischaemic conditioning-a new paradigm of self-protection in the brain. Nat. Rev. Neurol. 2015, 11, 698–710. [Google Scholar] [CrossRef]
- Hess, D.C.; Hoda, M.N.; Bhatia, K. Remote limb perconditioning [corrected] and postconditioning: Will it translate into a promising treatment for acute stroke? Stroke 2013, 44, 1191–1197. [Google Scholar] [CrossRef]
- Mozaffarian, D.; Benjamin, E.J.; Go, A.S.; Arnett, D.K.; Blaha, M.J.; Cushman, M.; De Ferranti, S.; Després, J.P.; Fullerton, H.J.; Howard, V.J.; et al. Heart disease and stroke statistics—2015 update: A report from the American Heart Association. Circulation 2015, 131, e29–e322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basri, R.; Issrani, R.; Gan, S.H.; Prabhu, N.; Alam, M.K. Burden of stroke in the Kingdom of Saudi Arabia: A soaring epidemic. Saudi Pharm. J. 2021, 29, 264–268. [Google Scholar] [CrossRef] [PubMed]
- Al Dawish, M.A.; Robert, A.A.; Braham, R.; Al Hayek, A.A.; Al Saeed, A.; Ahmed, R.A.; Al Shabaan, F.S. Diabetes Mellitus in Saudi Arabia: A Review of the Recent Literature. Curr. Diabetes Rev. 2016, 12, 359–368. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, E.J.; Blaha, M.J.; Chiuve, S.E.; Cushman, M.; Das, S.R.; Deo, R.; de Ferranti, S.D.; Floyd, J.; Fornage, M.; Gillespie, C. Heart Disease and Stroke Statistics-2018 Update: A Report from the American Heart Association. Circulation 2018, 137, e67–e492. [Google Scholar] [PubMed]
- Huang, P.L. eNOS, metabolic syndrome and cardiovascular disease. Trends. Endocrinol. Metab. 2009, 20, 295–302. [Google Scholar] [CrossRef] [Green Version]
- Rashid, P.A.; Whitehurst, A.; Lawson, N.; Bath, P.M. Plasma nitric oxide (nitrate/nitrite) levels in acute stroke and their relationship with severity and outcome. J. Stroke Cerebrovasc. Dis. 2003, 12, 82–87. [Google Scholar] [CrossRef]
- Fan, X.; Qiu, J.; Dai, H.; Singhal, A.B.; Lo, E.H.; Wang, X. A rat model of studying tissue-type plasminogen activator thrombolysis in ischemic stroke with diabetes. Stroke 2012, 43, 567–570. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.; Jatana, M.; Elango, C.; Paintlia, A.S.; Singh, A.K.; Singh, I. Cerebrovascular protection by various nitric oxide donors in rats after experimental stroke. Nitric Oxide 2006, 15, 114–124. [Google Scholar] [CrossRef]
- Molloy, J.; Martin, J.F.; Bakersville, P.A.; Fraser, S.C.; Markus, H.S. S-nitrosoglutathione reduces the rate of embolization in humans. Circulation 1998, 98, 1372–1375. [Google Scholar] [CrossRef] [Green Version]
- Sehba, F.A.; Ding, W.H.; Chereshnev, I.; Bederson, J.B. Effects of s-nitrosoglutathione on acute vasoconstriction and glutamate release after subarachnoid hemorrhage. Stroke 1999, 30, 1955–1961. [Google Scholar] [CrossRef] [Green Version]
- Rassaf, T.; Totzeck, M.; Hendgen-Cotta, U.B.; Shiva, S.; Heusch, G.; Kelm, M. Circulating nitrite contributes to cardioprotection by remote ischemic preconditioning. Circ. Res. 2014, 114, 1601–1610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hossmann, K.A. The two pathophysiologies of focal brain ischemia: Implications for translational stroke research. J. Cereb. Blood Flow Metab. 2012, 32, 1310–1316. [Google Scholar] [CrossRef] [PubMed]
- Yemisci, M.; Gursoy-Ozdemir, Y.; Vural, A.; Can, A.; Topalkara, K.; Dalkara, T. Pericyte contraction induced by oxidative-nitrative stress impairs capillary reflow despite successful opening of an occluded cerebral artery. Nat. Med. 2009, 15, 1031–1037. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zaidi, S.K.; Hoda, M.N.; Tabrez, S.; Khan, M.I. Pharmacological Inhibition of Class III Alcohol Dehydrogenase 5: Turning Remote Ischemic Conditioning Effective in a Diabetic Stroke Model. Antioxidants 2022, 11, 2051. https://doi.org/10.3390/antiox11102051
Zaidi SK, Hoda MN, Tabrez S, Khan MI. Pharmacological Inhibition of Class III Alcohol Dehydrogenase 5: Turning Remote Ischemic Conditioning Effective in a Diabetic Stroke Model. Antioxidants. 2022; 11(10):2051. https://doi.org/10.3390/antiox11102051
Chicago/Turabian StyleZaidi, Syed Kashif, Md Nasrul Hoda, Shams Tabrez, and Mohammad Imran Khan. 2022. "Pharmacological Inhibition of Class III Alcohol Dehydrogenase 5: Turning Remote Ischemic Conditioning Effective in a Diabetic Stroke Model" Antioxidants 11, no. 10: 2051. https://doi.org/10.3390/antiox11102051
APA StyleZaidi, S. K., Hoda, M. N., Tabrez, S., & Khan, M. I. (2022). Pharmacological Inhibition of Class III Alcohol Dehydrogenase 5: Turning Remote Ischemic Conditioning Effective in a Diabetic Stroke Model. Antioxidants, 11(10), 2051. https://doi.org/10.3390/antiox11102051