

Oxidative Stress-Induced Cellular Senescence in Aging Retina and Age-Related Macular Degeneration


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
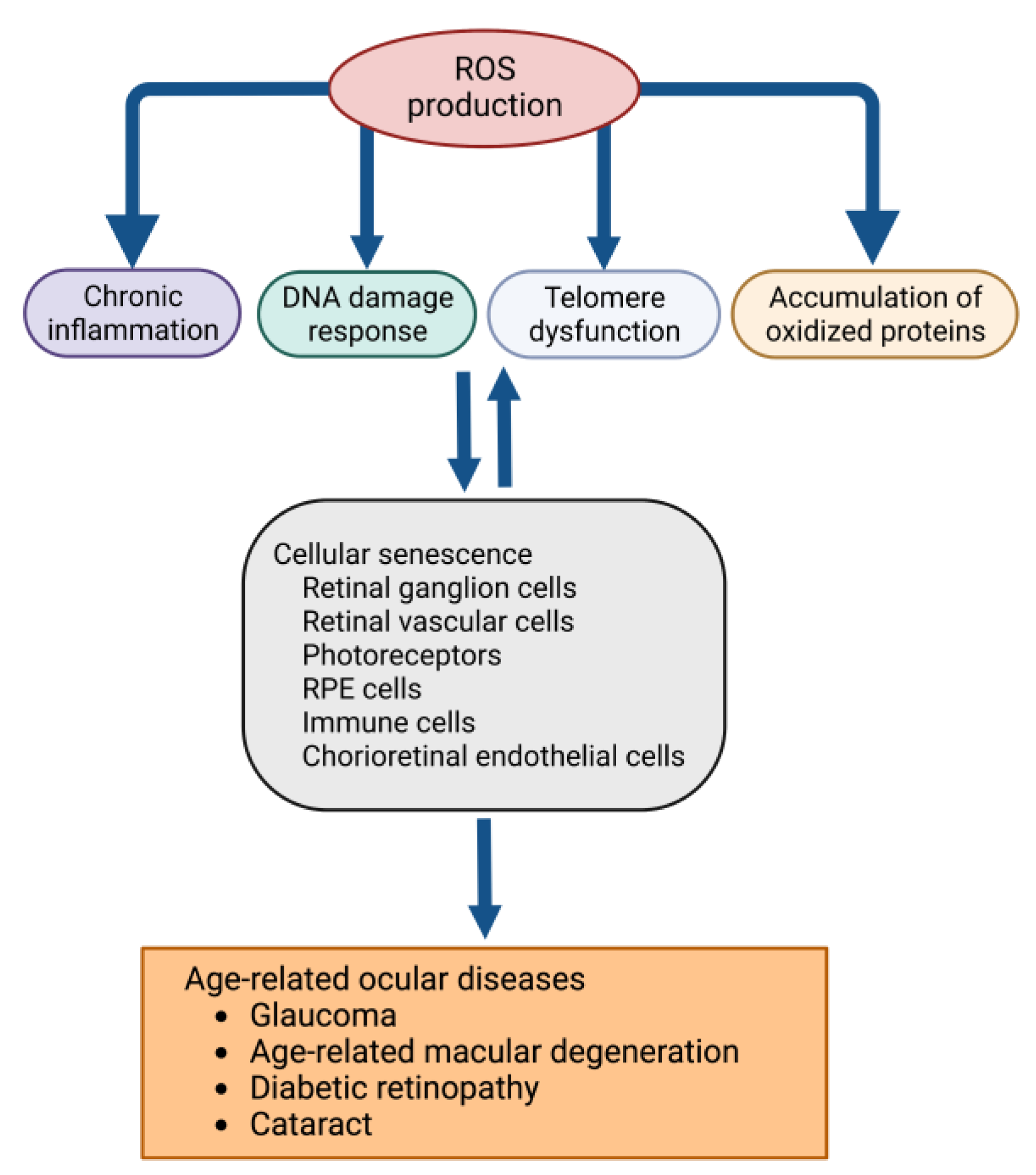
2. Oxidative Stress and Cellular Dysfunction in the Eye
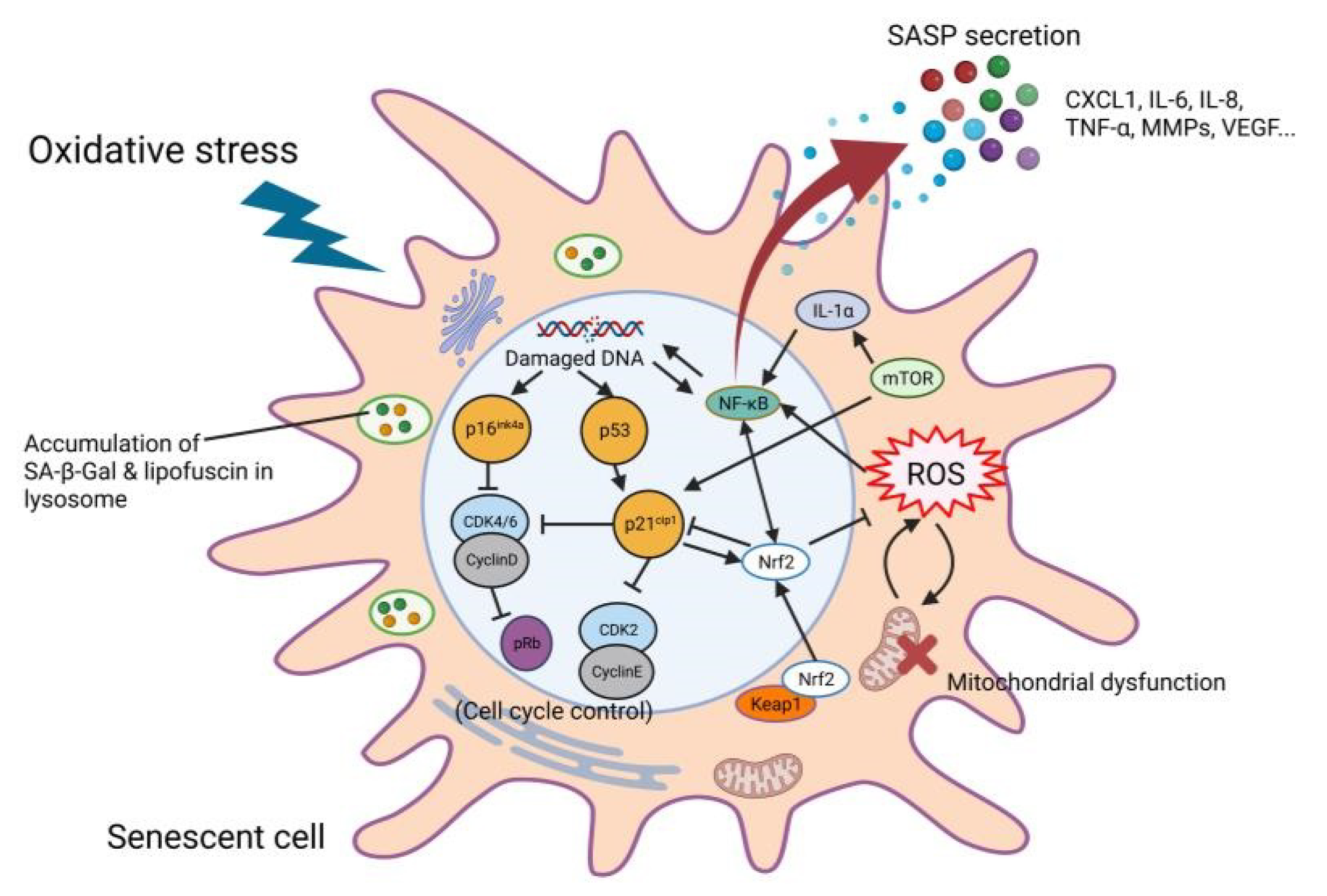
3. Molecular Biological Relationships between Oxidative Stress and Cellular Senescence
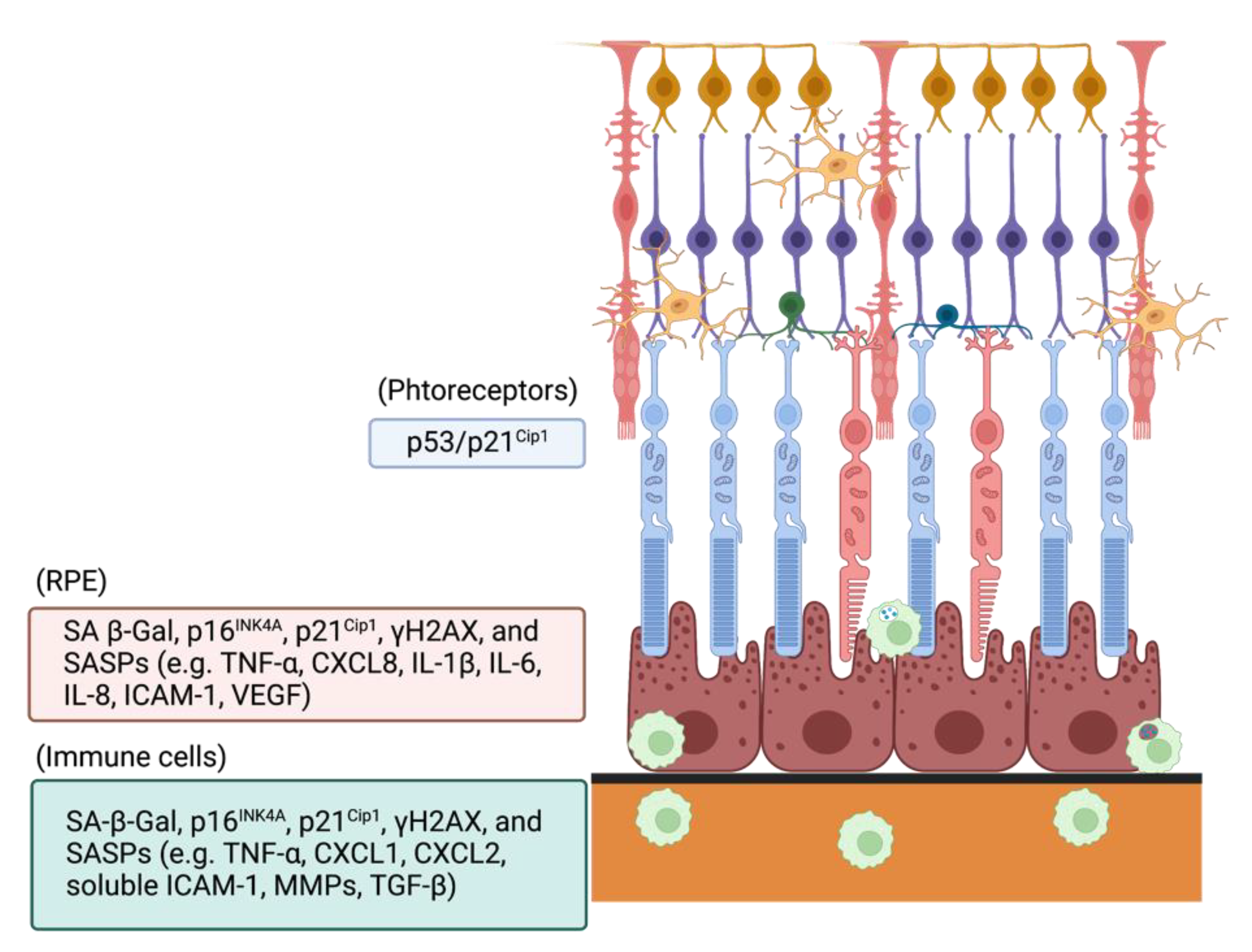
4. Roles of Oxidative Stress-Induced Cellular Senescence in Retina and Age-Related Macular Degeneration
5. Senolytics Targeting Oxidative Stress-Induced Cellular Senescence
6. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Flatt, T. A new definition of aging? Front. Genet. 2012, 3, 148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ni, Y.Q.; Liu, Y.S. New Insights into the Roles and Mechanisms of Spermidine in Aging and Age-Related Diseases. Aging Dis. 2021, 12, 1948–1963. [Google Scholar] [CrossRef] [PubMed]
- Wong, T.Y.; Chakravarthy, U.; Klein, R.; Mitchell, P.; Zlateva, G.; Buggage, R.; Fahrbach, K.; Probst, C.; Sledge, I. The natural history and prognosis of neovascular age-related macular degeneration: A systematic review of the literature and meta-analysis. Ophthalmology 2008, 115, 116–126. [Google Scholar] [CrossRef] [PubMed]
- Wong, W.L.; Su, X.; Li, X.; Cheung, C.M.; Klein, R.; Cheng, C.Y.; Wong, T.Y. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: A systematic review and meta-analysis. Lancet Glob. Health 2014, 2, e106–e116. [Google Scholar] [CrossRef] [Green Version]
- Sarks, S.H. Ageing and degeneration in the macular region: A clinico-pathological study. Br. J. Ophthalmol. 1976, 60, 324–341. [Google Scholar] [CrossRef] [Green Version]
- Apte, R.S. Age-Related Macular Degeneration. N. Engl. J. Med. 2021, 385, 539–547. [Google Scholar] [CrossRef]
- Lim, L.S.; Mitchell, P.; Seddon, J.M.; Holz, F.G.; Wong, T.Y. Age-related macular degeneration. Lancet 2012, 379, 1728–1738. [Google Scholar] [CrossRef]
- Elfawy, H.A.; Das, B. Crosstalk between mitochondrial dysfunction, oxidative stress, and age related neurodegenerative disease: Etiologies and therapeutic strategies. Life Sci. 2019, 218, 165–184. [Google Scholar] [CrossRef]
- Luo, J.; Mills, K.; le Cessie, S.; Noordam, R.; van Heemst, D. Ageing, age-related diseases and oxidative stress: What to do next? Ageing Res. Rev. 2020, 57, 100982. [Google Scholar] [CrossRef]
- Zhang, Z.Y.; Bao, X.L.; Cong, Y.Y.; Fan, B.; Li, G.Y. Autophagy in Age-Related Macular Degeneration: A Regulatory Mechanism of Oxidative Stress. Oxid. Med. Cell. Longev. 2020, 2020, 2896036. [Google Scholar] [CrossRef]
- Chen, Q.; Fischer, A.; Reagan, J.D.; Yan, L.J.; Ames, B.N. Oxidative DNA damage and senescence of human diploid fibroblast cells. Proc. Natl. Acad. Sci. USA 1995, 92, 4337–4341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munoz-Espin, D.; Serrano, M. Cellular senescence: From physiology to pathology. Nat. Rev. Mol. Cell Biol. 2014, 15, 482–496. [Google Scholar] [CrossRef] [PubMed]
- Caceres-Velez, P.R.; Hui, F.; Hercus, J.; Bui, B.; Jusuf, P.R. Restoring the oxidative balance in age-related diseases—An approach in glaucoma. Ageing Res. Rev. 2022, 75, 101572. [Google Scholar] [CrossRef] [PubMed]
- Moldogazieva, N.T.; Mokhosoev, I.M.; Mel’nikova, T.I.; Porozov, Y.B.; Terentiev, A.A. Oxidative Stress and Advanced Lipoxidation and Glycation End Products (ALEs and AGEs) in Aging and Age-Related Diseases. Oxid. Med. Cell. Longev. 2019, 2019, 3085756. [Google Scholar] [CrossRef] [Green Version]
- Brand, M.D.; Orr, A.L.; Perevoshchikova, I.V.; Quinlan, C.L. The role of mitochondrial function and cellular bioenergetics in ageing and disease. Br. J. Dermatol. 2013, 169 (Suppl. 2), 1–8. [Google Scholar] [CrossRef] [Green Version]
- Kauppinen, A.; Paterno, J.J.; Blasiak, J.; Salminen, A.; Kaarniranta, K. Inflammation and its role in age-related macular degeneration. Cell. Mol. Life Sci. 2016, 73, 1765–1786. [Google Scholar] [CrossRef] [Green Version]
- Neofytou, E.; Tzortzaki, E.G.; Chatziantoniou, A.; Siafakas, N.M. DNA damage due to oxidative stress in Chronic Obstructive Pulmonary Disease (COPD). Int. J Mol. Sci. 2012, 13, 16853–16864. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Dong, Q.; Liu, X.; Wei, L.; Liu, L.; Li, Y.; Wang, X. Dynamic transcriptome profiling in DNA damage-induced cellular senescence and transient cell-cycle arrest. Genomics 2020, 112, 1309–1317. [Google Scholar] [CrossRef]
- Aiken, C.T.; Kaake, R.M.; Wang, X.; Huang, L. Oxidative stress-mediated regulation of proteasome complexes. Mol. Cell. Proteom. 2011, 10, R110.006924. [Google Scholar] [CrossRef] [Green Version]
- Jarrett, S.G.; Lin, H.; Godley, B.F.; Boulton, M.E. Mitochondrial DNA damage and its potential role in retinal degeneration. Prog. Retin. Eye Res. 2008, 27, 596–607. [Google Scholar] [CrossRef]
- Balistreri, C.R.; Candore, G.; Accardi, G.; Colonna-Romano, G.; Lio, D. NF-kappaB pathway activators as potential ageing biomarkers: Targets for new therapeutic strategies. Immun. Ageing 2013, 10, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, M.M.; Liu, N.; Qin, Z.H.; Wang, Y. Mitochondrial-derived damage-associated molecular patterns amplify neuroinflammation in neurodegenerative diseases. Acta Pharmacol. Sin. 2022, 43, 2439–2447. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-kappaB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [Green Version]
- Lingappan, K. NF-kappaB in Oxidative Stress. Curr. Opin. Toxicol. 2018, 7, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Xiao, J.H. The Keap1-Nrf2 System: A Mediator between Oxidative Stress and Aging. Oxid. Med. Cell. Longev. 2021, 2021, 6635460. [Google Scholar] [CrossRef] [PubMed]
- Hellyer, J.A.; Padda, S.K.; Diehn, M.; Wakelee, H.A. Clinical Implications of KEAP1-NFE2L2 Mutations in NSCLC. J. Thorac. Oncol. 2021, 16, 395–403. [Google Scholar] [CrossRef]
- Sajadimajd, S.; Khazaei, M. Oxidative Stress and Cancer: The Role of Nrf2. Curr. Cancer Drug Targets 2018, 18, 538–557. [Google Scholar] [CrossRef]
- Bellezza, I.; Giambanco, I.; Minelli, A.; Donato, R. Nrf2-Keap1 signaling in oxidative and reductive stress. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 721–733. [Google Scholar] [CrossRef]
- Lee, K.S.; Lin, S.; Copland, D.A.; Dick, A.D.; Liu, J. Cellular senescence in the aging retina and developments of senotherapies for age-related macular degeneration. J. Neuroinflamm. 2021, 18, 32. [Google Scholar] [CrossRef]
- Mitter, S.K.; Song, C.; Qi, X.; Mao, H.; Rao, H.; Akin, D.; Lewin, A.; Grant, M.; Dunn, W., Jr.; Ding, J.; et al. Dysregulated autophagy in the RPE is associated with increased susceptibility to oxidative stress and AMD. Autophagy 2014, 10, 1989–2005. [Google Scholar] [CrossRef]
- Kaarniranta, K.; Tokarz, P.; Koskela, A.; Paterno, J.; Blasiak, J. Autophagy regulates death of retinal pigment epithelium cells in age-related macular degeneration. Cell Biol. Toxicol. 2017, 33, 113–128. [Google Scholar] [CrossRef] [Green Version]
- Ballinger, S.W.; Van Houten, B.; Jin, G.F.; Conklin, C.A.; Godley, B.F. Hydrogen peroxide causes significant mitochondrial DNA damage in human RPE cells. Exp. Eye Res. 1999, 68, 765–772. [Google Scholar] [CrossRef]
- Jin, G.F.; Hurst, J.S.; Godley, B.F. Hydrogen peroxide stimulates apoptosis in cultured human retinal pigment epithelial cells. Curr. Eye Res. 2001, 22, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Hanus, J.; Zhang, H.; Wang, Z.; Liu, Q.; Zhou, Q.; Wang, S. Induction of necrotic cell death by oxidative stress in retinal pigment epithelial cells. Cell Death Dis. 2013, 4, e965. [Google Scholar] [CrossRef] [Green Version]
- Brown, E.E.; DeWeerd, A.J.; Ildefonso, C.J.; Lewin, A.S.; Ash, J.D. Mitochondrial oxidative stress in the retinal pigment epithelium (RPE) led to metabolic dysfunction in both the RPE and retinal photoreceptors. Redox Biol. 2019, 24, 101201. [Google Scholar] [CrossRef] [PubMed]
- Piippo, N.; Korhonen, E.; Hytti, M.; Kinnunen, K.; Kaarniranta, K.; Kauppinen, A. Oxidative Stress is the Principal Contributor to Inflammasome Activation in Retinal Pigment Epithelium Cells with Defunct Proteasomes and Autophagy. Cell. Physiol. Biochem. 2018, 49, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Yao, Y.; Zhu, X.; Zhang, K.; Zhou, F.; Zhu, L. Amyloid beta induces NLRP3 inflammasome activation in retinal pigment epithelial cells via NADPH oxidase- and mitochondria-dependent ROS production. J. Biochem. Mol. Toxicol. 2017, 31. [Google Scholar] [CrossRef] [PubMed]
- Blevins, H.M.; Xu, Y.; Biby, S.; Zhang, S. The NLRP3 Inflammasome Pathway: A Review of Mechanisms and Inhibitors for the Treatment of Inflammatory Diseases. Front. Aging Neurosci. 2022, 14, 879021. [Google Scholar] [CrossRef]
- Ruan, Y.; Jiang, S.; Gericke, A. Age-Related Macular Degeneration: Role of Oxidative Stress and Blood Vessels. Int. J. Mol. Sci 2021, 22, 1296. [Google Scholar] [CrossRef]
- Kim, Y.W.; Byzova, T.V. Oxidative stress in angiogenesis and vascular disease. Blood 2014, 123, 625–631. [Google Scholar] [CrossRef]
- Monaghan-Benson, E.; Hartmann, J.; Vendrov, A.E.; Budd, S.; Byfield, G.; Parker, A.; Ahmad, F.; Huang, W.; Runge, M.; Burridge, K.; et al. The role of vascular endothelial growth factor-induced activation of NADPH oxidase in choroidal endothelial cells and choroidal neovascularization. Am. J. Pathol. 2010, 177, 2091–2102. [Google Scholar] [CrossRef] [PubMed]
- Collado, M.; Blasco, M.A.; Serrano, M. Cellular senescence in cancer and aging. Cell 2007, 130, 223–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Micco, R.; Krizhanovsky, V.; Baker, D.; d’Adda di Fagagna, F. Cellular senescence in ageing: From mechanisms to therapeutic opportunities. Nat. Rev. Mol. Cell Biol. 2021, 22, 75–95. [Google Scholar] [CrossRef] [PubMed]
- Childs, B.G.; Durik, M.; Baker, D.J.; van Deursen, J.M. Cellular senescence in aging and age-related disease: From mechanisms to therapy. Nat. Med. 2015, 21, 1424–1435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumari, R.; Jat, P. Mechanisms of Cellular Senescence: Cell Cycle Arrest and Senescence Associated Secretory Phenotype. Front. Cell Dev. Biol. 2021, 9, 645593. [Google Scholar] [CrossRef]
- Terman, A.; Kurz, T.; Navratil, M.; Arriaga, E.A.; Brunk, U.T. Mitochondrial turnover and aging of long-lived postmitotic cells: The mitochondrial-lysosomal axis theory of aging. Antioxid. Redox Signal. 2010, 12, 503–535. [Google Scholar] [CrossRef] [Green Version]
- Moreno-Garcia, A.; Kun, A.; Calero, O.; Medina, M.; Calero, M. An Overview of the Role of Lipofuscin in Age-Related Neurodegeneration. Front. Neurosci. 2018, 12, 464. [Google Scholar] [CrossRef] [Green Version]
- Rizou, S.V.; Evangelou, K.; Myrianthopoulos, V.; Mourouzis, I.; Havaki, S.; Athanasiou, A.; Vasileiou, P.V.S.; Margetis, A.; Kotsinas, A.; Kastrinakis, N.G.; et al. A Novel Quantitative Method for the Detection of Lipofuscin, the Main By-Product of Cellular Senescence, in Fluids. Methods Mol. Biol. 2019, 1896, 119–138. [Google Scholar] [CrossRef]
- Georgakopoulou, E.A.; Tsimaratou, K.; Evangelou, K.; Fernandez Marcos, P.J.; Zoumpourlis, V.; Trougakos, I.P.; Kletsas, D.; Bartek, J.; Serrano, M.; Gorgoulis, V.G. Specific lipofuscin staining as a novel biomarker to detect replicative and stress-induced senescence. A method applicable in cryo-preserved and archival tissues. Aging (Albany NY) 2013, 5, 37–50. [Google Scholar] [CrossRef] [Green Version]
- Salmonowicz, H.; Passos, J.F. Detecting senescence: A new method for an old pigment. Aging Cell 2017, 16, 432–434. [Google Scholar] [CrossRef]
- Goulielmaki, E.; Ioannidou, A.; Tsekrekou, M.; Stratigi, K.; Poutakidou, I.K.; Gkirtzimanaki, K.; Aivaliotis, M.; Evangelou, K.; Topalis, P.; Altmuller, J.; et al. Tissue-infiltrating macrophages mediate an exosome-based metabolic reprogramming upon DNA damage. Nat. Commun. 2020, 11, 42. [Google Scholar] [CrossRef] [Green Version]
- Konig, J.; Ott, C.; Hugo, M.; Jung, T.; Bulteau, A.L.; Grune, T.; Hohn, A. Mitochondrial contribution to lipofuscin formation. Redox Biol. 2017, 11, 673–681. [Google Scholar] [CrossRef] [PubMed]
- Vida, C.; de Toda, I.M.; Cruces, J.; Garrido, A.; Gonzalez-Sanchez, M.; De la Fuente, M. Role of macrophages in age-related oxidative stress and lipofuscin accumulation in mice. Redox Biol. 2017, 12, 423–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- von Kobbe, C. Cellular senescence: A view throughout organismal life. Cell. Mol. Life Sci. 2018, 75, 3553–3567. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J. The biology of replicative senescence. Eur. J. Cancer 1997, 33, 703–709. [Google Scholar] [CrossRef]
- Beck, J.; Horikawa, I.; Harris, C. Cellular Senescence: Mechanisms, Morphology, and Mouse Models. Vet. Pathol. 2020, 57, 747–757. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Espin, D.; Canamero, M.; Maraver, A.; Gomez-Lopez, G.; Contreras, J.; Murillo-Cuesta, S.; Rodriguez-Baeza, A.; Varela-Nieto, I.; Ruberte, J.; Collado, M.; et al. Programmed cell senescence during mammalian embryonic development. Cell 2013, 155, 1104–1118. [Google Scholar] [CrossRef] [Green Version]
- Macip, S.; Igarashi, M.; Fang, L.; Chen, A.; Pan, Z.Q.; Lee, S.W.; Aaronson, S.A. Inhibition of p21-mediated ROS accumulation can rescue p21-induced senescence. EMBO J. 2002, 21, 2180–2188. [Google Scholar] [CrossRef]
- Chen, J.; Goligorsky, M.S. Premature senescence of endothelial cells: Methusaleh’s dilemma. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H1729–H1739. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.S.; Lee, B.Y.; Hwang, E.S. Dinstinct ROS and biochemical profiles in cells undergoing DNA damage-induced senescence and apoptosis. Mech. Ageing Dev. 2005, 126, 580–590. [Google Scholar] [CrossRef]
- McConnell, B.B.; Gregory, F.J.; Stott, F.J.; Hara, E.; Peters, G. Induced expression of p16(INK4a) inhibits both CDK4- and CDK2-associated kinase activity by reassortment of cyclin-CDK-inhibitor complexes. Mol. Cell. Biol. 1999, 19, 1981–1989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, W.; Zender, L.; Miething, C.; Dickins, R.A.; Hernando, E.; Krizhanovsky, V.; Cordon-Cardo, C.; Lowe, S.W. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007, 445, 656–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Sharpless, N.E. Tumor suppressor mechanisms in immune aging. Curr. Opin. Immunol. 2009, 21, 431–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quesnel, B. [Inhibitors of cyclins/CDK of the 9p21 chromosomal region and malignant hemopathies]. Bull. Cancer 1998, 85, 747–754. [Google Scholar] [PubMed]
- Romagosa, C.; Simonetti, S.; Lopez-Vicente, L.; Mazo, A.; Lleonart, M.E.; Castellvi, J.; Ramon y Cajal, S. p16(Ink4a) overexpression in cancer: A tumor suppressor gene associated with senescence and high-grade tumors. Oncogene 2011, 30, 2087–2097. [Google Scholar] [CrossRef] [Green Version]
- Zindy, F.; Quelle, D.E.; Roussel, M.F.; Sherr, C.J. Expression of the p16INK4a tumor suppressor versus other INK4 family members during mouse development and aging. Oncogene 1997, 15, 203–211. [Google Scholar] [CrossRef] [Green Version]
- Krishnamurthy, J.; Torrice, C.; Ramsey, M.R.; Kovalev, G.I.; Al-Regaiey, K.; Su, L.; Sharpless, N.E. Ink4a/Arf expression is a biomarker of aging. J. Clin. Invest. 2004, 114, 1299–1307. [Google Scholar] [CrossRef]
- Baker, D.J.; Childs, B.G.; Durik, M.; Wijers, M.E.; Sieben, C.J.; Zhong, J.; Saltness, R.A.; Jeganathan, K.B.; Verzosa, G.C.; Pezeshki, A.; et al. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature 2016, 530, 184–189. [Google Scholar] [CrossRef] [Green Version]
- Jung, Y.S.; Qian, Y.; Chen, X. Examination of the expanding pathways for the regulation of p21 expression and activity. Cell Signal. 2010, 22, 1003–1012. [Google Scholar] [CrossRef] [Green Version]
- Al Bitar, S.; Gali-Muhtasib, H. The Role of the Cyclin Dependent Kinase Inhibitor p21(cip1/waf1) in Targeting Cancer: Molecular Mechanisms and Novel Therapeutics. Cancers 2019, 11, 1475. [Google Scholar] [CrossRef]
- Yosef, R.; Pilpel, N.; Papismadov, N.; Gal, H.; Ovadya, Y.; Vadai, E.; Miller, S.; Porat, Z.; Ben-Dor, S.; Krizhanovsky, V. p21 maintains senescent cell viability under persistent DNA damage response by restraining JNK and caspase signaling. EMBO J. 2017, 36, 2280–2295. [Google Scholar] [CrossRef] [PubMed]
- Karimian, A.; Ahmadi, Y.; Yousefi, B. Multiple functions of p21 in cell cycle, apoptosis and transcriptional regulation after DNA damage. DNA Repair (Amst) 2016, 42, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.Y.; Han, J.A.; Im, J.S.; Morrone, A.; Johung, K.; Goodwin, E.C.; Kleijer, W.J.; DiMaio, D.; Hwang, E.S. Senescence-associated beta-galactosidase is lysosomal beta-galactosidase. Aging Cell 2006, 5, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Katsuumi, G.; Shimizu, I.; Yoshida, Y.; Minamino, T. Vascular Senescence in Cardiovascular and Metabolic Diseases. Front. Cardiovasc. Med. 2018, 5, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ksiazek, K.; Piwocka, K.; Brzezinska, A.; Sikora, E.; Zabel, M.; Breborowicz, A.; Jorres, A.; Witowski, J. Early loss of proliferative potential of human peritoneal mesothelial cells in culture: The role of p16INK4a-mediated premature senescence. J. Appl. Physiol. 2006, 100, 988–995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patil, P.; Falabella, M.; Saeed, A.; Lee, D.; Kaufman, B.; Shiva, S.; Croix, C.S.; Van Houten, B.; Niedernhofer, L.J.; Robbins, P.D.; et al. Oxidative stress-induced senescence markedly increases disc cell bioenergetics. Mech. Ageing Dev. 2019, 180, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Ok, C.Y.; Park, S.; Jang, H.O.; Takata, T.; Lee, O.H.; Bae, M.K.; Bae, S.K. FK866 Protects Human Dental Pulp Cells against Oxidative Stress-Induced Cellular Senescence. Antioxidants 2021, 10, 271. [Google Scholar] [CrossRef]
- Faget, D.V.; Ren, Q.; Stewart, S.A. Unmasking senescence: Context-dependent effects of SASP in cancer. Nat. Rev. Cancer 2019, 19, 439–453. [Google Scholar] [CrossRef]
- Han, X.; Zhang, T.; Zhang, X.; Zhang, R.; Lao, K.; Mi, Y.; Gou, X. AMPK alleviates oxidative stressinduced premature senescence via inhibition of NF-kappaB/STAT3 axis-mediated positive feedback loop. Mech. Ageing Dev. 2020, 191, 111347. [Google Scholar] [CrossRef]
- Salminen, A.; Kauppinen, A.; Kaarniranta, K. Emerging role of NF-kappaB signaling in the induction of senescence-associated secretory phenotype (SASP). Cell Signal. 2012, 24, 835–845. [Google Scholar] [CrossRef]
- Chien, Y.; Scuoppo, C.; Wang, X.; Fang, X.; Balgley, B.; Bolden, J.E.; Premsrirut, P.; Luo, W.; Chicas, A.; Lee, C.S.; et al. Control of the senescence-associated secretory phenotype by NF-kappaB promotes senescence and enhances chemosensitivity. Genes Dev. 2011, 25, 2125–2136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rovillain, E.; Mansfield, L.; Caetano, C.; Alvarez-Fernandez, M.; Caballero, O.L.; Medema, R.H.; Hummerich, H.; Jat, P.S. Activation of nuclear factor-kappa B signalling promotes cellular senescence. Oncogene 2011, 30, 2356–2366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Davies, K.J.A.; Forman, H.J. Oxidative stress response and Nrf2 signaling in aging. Free Radic Biol. Med. 2015, 88, 314–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suh, J.H.; Shenvi, S.V.; Dixon, B.M.; Liu, H.; Jaiswal, A.K.; Liu, R.M.; Hagen, T.M. Decline in transcriptional activity of Nrf2 causes age-related loss of glutathione synthesis, which is reversible with lipoic acid. Proc. Natl. Acad. Sci. USA 2004, 101, 3381–3386. [Google Scholar] [CrossRef] [Green Version]
- Pomatto, L.C.D.; Dill, T.; Carboneau, B.; Levan, S.; Kato, J.; Mercken, E.M.; Pearson, K.J.; Bernier, M.; de Cabo, R. Deletion of Nrf2 shortens lifespan in C57BL6/J male mice but does not alter the health and survival benefits of caloric restriction. Free Radic. Biol. Med. 2020, 152, 650–658. [Google Scholar] [CrossRef]
- Gorbunova, V.; Rezazadeh, S.; Seluanov, A. Dangerous Entrapment for NRF2. Cell 2016, 165, 1312–1313. [Google Scholar] [CrossRef] [Green Version]
- Kapeta, S.; Chondrogianni, N.; Gonos, E.S. Nuclear erythroid factor 2-mediated proteasome activation delays senescence in human fibroblasts. J. Biol. Chem. 2010, 285, 8171–8184. [Google Scholar] [CrossRef] [Green Version]
- Wati, S.M.; Matsumaru, D.; Motohashi, H. NRF2 pathway activation by KEAP1 inhibition attenuates the manifestation of aging phenotypes in salivary glands. Redox Biol. 2020, 36, 101603. [Google Scholar] [CrossRef]
- Fulop, G.A.; Kiss, T.; Tarantini, S.; Balasubramanian, P.; Yabluchanskiy, A.; Farkas, E.; Bari, F.; Ungvari, Z.; Csiszar, A. Nrf2 deficiency in aged mice exacerbates cellular senescence promoting cerebrovascular inflammation. Geroscience 2018, 40, 513–521. [Google Scholar] [CrossRef]
- Chen, W.; Sun, Z.; Wang, X.J.; Jiang, T.; Huang, Z.; Fang, D.; Zhang, D.D. Direct interaction between Nrf2 and p21(Cip1/WAF1) upregulates the Nrf2-mediated antioxidant response. Mol. Cell 2009, 34, 663–673. [Google Scholar] [CrossRef]
- Nakano-Kobayashi, A.; Fukumoto, A.; Morizane, A.; Nguyen, D.T.; Le, T.M.; Hashida, K.; Hosoya, T.; Takahashi, R.; Takahashi, J.; Hori, O.; et al. Therapeutics potentiating microglial p21-Nrf2 axis can rescue neurodegeneration caused by neuroinflammation. Sci. Adv. 2020, 6, eabc1428. [Google Scholar] [CrossRef] [PubMed]
- Matsumaru, D.; Motohashi, H. The KEAP1-NRF2 System in Healthy Aging and Longevity. Antioxidants 2021, 10, 1929. [Google Scholar] [CrossRef] [PubMed]
- Powell, J.D.; Pollizzi, K.N.; Heikamp, E.B.; Horton, M.R. Regulation of immune responses by mTOR. Annu. Rev. Immunol. 2012, 30, 39–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weichhart, T. mTOR as Regulator of Lifespan, Aging, and Cellular Senescence: A Mini-Review. Gerontology 2018, 64, 127–134. [Google Scholar] [CrossRef]
- Ehninger, D.; Neff, F.; Xie, K. Longevity, aging and rapamycin. Cell. Mol. Life Sci. 2014, 71, 4325–4346. [Google Scholar] [CrossRef] [Green Version]
- Xue, Q.L.; Yang, H.; Li, H.F.; Abadir, P.M.; Burks, T.N.; Koch, L.G.; Britton, S.L.; Carlson, J.; Chen, L.; Walston, J.D.; et al. Rapamycin increases grip strength and attenuates age-related decline in maximal running distance in old low capacity runner rats. Aging (Albany NY) 2016, 8, 769–776. [Google Scholar] [CrossRef] [Green Version]
- Laberge, R.M.; Sun, Y.; Orjalo, A.V.; Patil, C.K.; Freund, A.; Zhou, L.; Curran, S.C.; Davalos, A.R.; Wilson-Edell, K.A.; Liu, S.; et al. MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat. Cell Biol. 2015, 17, 1049–1061. [Google Scholar] [CrossRef]
- Mannick, J.B.; Del Giudice, G.; Lattanzi, M.; Valiante, N.M.; Praestgaard, J.; Huang, B.; Lonetto, M.A.; Maecker, H.T.; Kovarik, J.; Carson, S.; et al. mTOR inhibition improves immune function in the elderly. Sci. Transl. Med. 2014, 6, 268ra179. [Google Scholar] [CrossRef]
- Wang, R.; Yu, Z.; Sunchu, B.; Shoaf, J.; Dang, I.; Zhao, S.; Caples, K.; Bradley, L.; Beaver, L.M.; Ho, E.; et al. Rapamycin inhibits the secretory phenotype of senescent cells by a Nrf2-independent mechanism. Aging Cell 2017, 16, 564–574. [Google Scholar] [CrossRef]
- Wagner, K.D.; Wagner, N. The Senescence Markers p16INK4A, p14ARF/p19ARF, and p21 in Organ Development and Homeostasis. Cells 2022, 11, 1966. [Google Scholar] [CrossRef]
- Takeda, S.; Sasagawa, S.; Oyama, T.; Searleman, A.C.; Westergard, T.D.; Cheng, E.H.; Hsieh, J.J. Taspase1-dependent TFIIA cleavage coordinates head morphogenesis by limiting Cdkn2a locus transcription. J. Clin. Invest. 2015, 125, 1203–1214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheong, C.; Sung, Y.H.; Lee, J.; Choi, Y.S.; Song, J.; Kee, C.; Lee, H.W. Role of INK4a locus in normal eye development and cataract genesis. Mech. Ageing Dev. 2006, 127, 633–638. [Google Scholar] [CrossRef] [PubMed]
- McKeller, R.N.; Fowler, J.L.; Cunningham, J.J.; Warner, N.; Smeyne, R.J.; Zindy, F.; Skapek, S.X. The Arf tumor suppressor gene promotes hyaloid vascular regression during mouse eye development. Proc. Natl. Acad. Sci. USA 2002, 99, 3848–3853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, R.L.; Thornton, J.D.; Martin, A.C.; Rehg, J.E.; Bertwistle, D.; Zindy, F.; Skapek, S.X. Arf-dependent regulation of Pdgf signaling in perivascular cells in the developing mouse eye. EMBO J. 2005, 24, 2803–2814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Mera-Rodriguez, J.A.; Alvarez-Hernan, G.; Ganan, Y.; Martin-Partido, G.; Rodriguez-Leon, J.; Francisco-Morcillo, J. Senescence-associated beta-galactosidase activity in the developing avian retina. Dev. Dyn. 2019, 248, 850–865. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Luppo, M.; Catita, J.; Ramos, D.; Navarro, M.; Carretero, A.; Mendes-Jorge, L.; Munoz-Canoves, P.; Rodriguez-Baeza, A.; Nacher, V.; Ruberte, J. Cellular Senescence Is Associated With Human Retinal Microaneurysm Formation During Aging. Invest. Ophthalmol. Vis. Sci. 2017, 58, 2832–2842. [Google Scholar] [CrossRef] [Green Version]
- Blasiak, J. Senescence in the pathogenesis of age-related macular degeneration. Cell. Mol. Life Sci. 2020, 77, 789–805. [Google Scholar] [CrossRef]
- Vuong, L.; Conley, S.M.; Al-Ubaidi, M.R. Expression and role of p53 in the retina. Invest. Ophthalmol. Vis. Sci. 2012, 53, 1362–1371. [Google Scholar] [CrossRef]
- Sreekumar, P.G.; Reddy, S.T.; Hinton, D.R.; Kannan, R. Mechanisms of RPE senescence and potential role of alphaB crystallin peptide as a senolytic agent in experimental AMD. Exp. Eye Res. 2022, 215, 108918. [Google Scholar] [CrossRef]
- Mishima, K.; Handa, J.T.; Aotaki-Keen, A.; Lutty, G.A.; Morse, L.S.; Hjelmeland, L.M. Senescence-associated beta-galactosidase histochemistry for the primate eye. Invest. Ophthalmol. Vis. Sci. 1999, 40, 1590–1593. [Google Scholar]
- Shimizu, H.; Yamada, K.; Suzumura, A.; Kataoka, K.; Takayama, K.; Sugimoto, M.; Terasaki, H.; Kaneko, H. Caveolin-1 Promotes Cellular Senescence in Exchange for Blocking Subretinal Fibrosis in Age-Related Macular Degeneration. Invest. Ophthalmol. Vis. Sci. 2020, 61, 21. [Google Scholar] [CrossRef] [PubMed]
- Ach, T.; Tolstik, E.; Messinger, J.D.; Zarubina, A.V.; Heintzmann, R.; Curcio, C.A. Lipofuscin redistribution and loss accompanied by cytoskeletal stress in retinal pigment epithelium of eyes with age-related macular degeneration. Invest. Ophthalmol. Vis. Sci. 2015, 56, 3242–3252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marazita, M.C.; Dugour, A.; Marquioni-Ramella, M.D.; Figueroa, J.M.; Suburo, A.M. Oxidative stress-induced premature senescence dysregulates VEGF and CFH expression in retinal pigment epithelial cells: Implications for Age-related Macular Degeneration. Redox Biol. 2016, 7, 78–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, A.L.; Fuchshofer, R.; Kook, D.; Kampik, A.; Bloemendal, H.; Welge-Lussen, U. Subtoxic oxidative stress induces senescence in retinal pigment epithelial cells via TGF-beta release. Invest. Ophthalmol. Vis. Sci. 2009, 50, 926–935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, D.; Wu, J.; Spee, C.; Ryan, S.J.; Hinton, D.R. BMP4 mediates oxidative stress-induced retinal pigment epithelial cell senescence and is overexpressed in age-related macular degeneration. J. Biol. Chem. 2009, 284, 9529–9539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.; Forrester, J.V.; Xu, H. Synthesis of complement factor H by retinal pigment epithelial cells is down-regulated by oxidized photoreceptor outer segments. Exp. Eye Res. 2007, 84, 635–645. [Google Scholar] [CrossRef]
- Westlund, B.S.; Cai, B.; Zhou, J.; Sparrow, J.R. Involvement of c-Abl, p53 and the MAP kinase JNK in the cell death program initiated in A2E-laden ARPE-19 cells by exposure to blue light. Apoptosis 2009, 14, 31–41. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Wang, J.; Cai, J.; Sternberg, P. Altered mTOR signaling in senescent retinal pigment epithelium. Invest. Ophthalmol. Vis. Sci. 2010, 51, 5314–5319. [Google Scholar] [CrossRef] [Green Version]
- Felszeghy, S.; Viiri, J.; Paterno, J.J.; Hyttinen, J.M.T.; Koskela, A.; Chen, M.; Leinonen, H.; Tanila, H.; Kivinen, N.; Koistinen, A.; et al. Loss of NRF-2 and PGC-1alpha genes leads to retinal pigment epithelium damage resembling dry age-related macular degeneration. Redox Biol. 2019, 20, 1–12. [Google Scholar] [CrossRef]
- Wang, S.; Wang, X.; Cheng, Y.; Ouyang, W.; Sang, X.; Liu, J.; Su, Y.; Liu, Y.; Li, C.; Yang, L.; et al. Autophagy Dysfunction, Cellular Senescence, and Abnormal Immune-Inflammatory Responses in AMD: From Mechanisms to Therapeutic Potential. Oxid. Med. Cell. Longev. 2019, 2019, 3632169. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Zheng, Y.; Wang, C.; Liu, Y. Glutathione depletion induces ferroptosis, autophagy, and premature cell senescence in retinal pigment epithelial cells. Cell Death Dis. 2018, 9, 753. [Google Scholar] [CrossRef] [PubMed]
- Shao, Z.; Wang, B.; Shi, Y.; Xie, C.; Huang, C.; Chen, B.; Zhang, H.; Zeng, G.; Liang, H.; Wu, Y.; et al. Senolytic agent Quercetin ameliorates intervertebral disc degeneration via the Nrf2/NF-kappaB axis. Osteoarthr. Cartil. 2021, 29, 413–422. [Google Scholar] [CrossRef] [PubMed]
- Chae, S.Y.; Park, S.Y.; Park, G. Lutein protects human retinal pigment epithelial cells from oxidative stressinduced cellular senescence. Mol. Med. Rep. 2018, 18, 5182–5190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, T.J.; Schneider, R.J.; Miller, J.A.; Martin, B.P.; Al-Ubaidi, M.R.; Agarwal, N.; Dethloff, L.A.; Philbert, M.A. Photoreceptor cell apoptosis induced by the 2-nitroimidazole radiosensitizer, CI-1010, is mediated by p53-linked activation of caspase-3. Neurotoxicology 2006, 27, 44–59. [Google Scholar] [CrossRef] [PubMed]
- Sagiv, A.; Krizhanovsky, V. Immunosurveillance of senescent cells: The bright side of the senescence program. Biogerontology 2013, 14, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Song, P.; An, J.; Zou, M.H. Immune Clearance of Senescent Cells to Combat Ageing and Chronic Diseases. Cells 2020, 9, 671. [Google Scholar] [CrossRef] [Green Version]
- Kelly, J.; Ali Khan, A.; Yin, J.; Ferguson, T.A.; Apte, R.S. Senescence regulates macrophage activation and angiogenic fate at sites of tissue injury in mice. J. Clin. Invest. 2007, 117, 3421–3426. [Google Scholar] [CrossRef]
- Ambati, J.; Atkinson, J.P.; Gelfand, B.D. Immunology of age-related macular degeneration. Nat. Rev. Immunol. 2013, 13, 438–451. [Google Scholar] [CrossRef] [Green Version]
- Ma, W.; Cojocaru, R.; Gotoh, N.; Gieser, L.; Villasmil, R.; Cogliati, T.; Swaroop, A.; Wong, W.T. Gene expression changes in aging retinal microglia: Relationship to microglial support functions and regulation of activation. Neurobiol. Aging 2013, 34, 2310–2321. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Geng, J.; Gao, J.; Zhao, H.; Li, J.; Shi, Y.; Yang, B.; Xiao, C.; Linghu, Y.; Sun, X.; et al. Macrophage achieves self-protection against oxidative stress-induced ageing through the Mst-Nrf2 axis. Nat. Commun. 2019, 10, 755. [Google Scholar] [CrossRef]
- Sadhu, S.; Decker, C.; Sansbury, B.E.; Marinello, M.; Seyfried, A.; Howard, J.; Mori, M.; Hosseini, Z.; Arunachalam, T.; Finn, A.V.; et al. Radiation-Induced Macrophage Senescence Impairs Resolution Programs and Drives Cardiovascular Inflammation. J. Immunol. 2021, 207, 1812–1823. [Google Scholar] [CrossRef] [PubMed]
- Doyle, S.L.; Campbell, M.; Ozaki, E.; Salomon, R.G.; Mori, A.; Kenna, P.F.; Farrar, G.J.; Kiang, A.S.; Humphries, M.M.; Lavelle, E.C.; et al. NLRP3 has a protective role in age-related macular degeneration through the induction of IL-18 by drusen components. Nat. Med. 2012, 18, 791–798. [Google Scholar] [CrossRef] [PubMed]
- Singh Kushwaha, S.; Patro, N.; Kumar Patro, I. A Sequential Study of Age-Related Lipofuscin Accumulation in Hippocampus and Striate Cortex of Rats. Ann. Neurosci. 2018, 25, 223–233. [Google Scholar] [CrossRef]
- Xu, H.; Chen, M.; Manivannan, A.; Lois, N.; Forrester, J.V. Age-dependent accumulation of lipofuscin in perivascular and subretinal microglia in experimental mice. Aging Cell 2008, 7, 58–68. [Google Scholar] [CrossRef] [PubMed]
- Shu, W.; Baumann, B.H.; Song, Y.; Liu, Y.; Wu, X.; Dunaief, J.L. Ferrous but not ferric iron sulfate kills photoreceptors and induces photoreceptor-dependent RPE autofluorescence. Redox Biol. 2020, 34, 101469. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Bell, B.A.; Song, Y.; Kim, H.J.; Sterling, J.K.; Kim, B.J.; Poli, M.; Guo, M.; Zhang, K.; Rao, A.; et al. Intraocular iron injection induces oxidative stress followed by elements of geographic atrophy and sympathetic ophthalmia. Aging Cell 2021, 20, e13490. [Google Scholar] [CrossRef]
- Oubaha, M.; Miloudi, K.; Dejda, A.; Guber, V.; Mawambo, G.; Germain, M.A.; Bourdel, G.; Popovic, N.; Rezende, F.A.; Kaufman, R.J.; et al. Senescence-associated secretory phenotype contributes to pathological angiogenesis in retinopathy. Sci. Transl. Med. 2016, 8, 362ra144. [Google Scholar] [CrossRef]
- Crespo-Garcia, S.; Tsuruda, P.R.; Dejda, A.; Ryan, R.D.; Fournier, F.; Chaney, S.Y.; Pilon, F.; Dogan, T.; Cagnone, G.; Patel, P.; et al. Pathological angiogenesis in retinopathy engages cellular senescence and is amenable to therapeutic elimination via BCL-xL inhibition. Cell Metab. 2021, 33, 818–832.e817. [Google Scholar] [CrossRef]
- Binet, F.; Cagnone, G.; Crespo-Garcia, S.; Hata, M.; Neault, M.; Dejda, A.; Wilson, A.M.; Buscarlet, M.; Mawambo, G.T.; Howard, J.P.; et al. Neutrophil extracellular traps target senescent vasculature for tissue remodeling in retinopathy. Science 2020, 369, eaay5356. [Google Scholar] [CrossRef]
- Osman, W.; Low, S.K.; Takahashi, A.; Kubo, M.; Nakamura, Y. A genome-wide association study in the Japanese population confirms 9p21 and 14q23 as susceptibility loci for primary open angle glaucoma. Hum. Mol. Genet. 2012, 21, 2836–2842. [Google Scholar] [CrossRef]
- Liton, P.B.; Challa, P.; Stinnett, S.; Luna, C.; Epstein, D.L.; Gonzalez, P. Cellular senescence in the glaucomatous outflow pathway. Exp. Gerontol. 2005, 40, 745–748. [Google Scholar] [CrossRef] [PubMed]
- Skowronska-Krawczyk, D.; Zhao, L.; Zhu, J.; Weinreb, R.N.; Cao, G.; Luo, J.; Flagg, K.; Patel, S.; Wen, C.; Krupa, M.; et al. P16INK4a Upregulation Mediated by SIX6 Defines Retinal Ganglion Cell Pathogenesis in Glaucoma. Mol. Cell 2015, 59, 931–940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, D.; Xu, Y.; Shi, Y.; Ji, J.; Lu, X.; Chen, H.; Huang, R.; Lu, P.; Li, Y.; Cheng, L.; et al. Occurrence of Oxidative Stress and Premature Senescence in the Anterior Segment of Acute Primary Angle-Closure Eyes. Invest. Ophthalmol. Vis. Sci. 2022, 63, 34. [Google Scholar] [CrossRef] [PubMed]
- Jeon, O.H.; Kim, C.; Laberge, R.M.; Demaria, M.; Rathod, S.; Vasserot, A.P.; Chung, J.W.; Kim, D.H.; Poon, Y.; David, N.; et al. Local clearance of senescent cells attenuates the development of post-traumatic osteoarthritis and creates a pro-regenerative environment. Nat. Med. 2017, 23, 775–781. [Google Scholar] [CrossRef] [Green Version]
- Xu, M.; Pirtskhalava, T.; Farr, J.N.; Weigand, B.M.; Palmer, A.K.; Weivoda, M.M.; Inman, C.L.; Ogrodnik, M.B.; Hachfeld, C.M.; Fraser, D.G.; et al. Senolytics improve physical function and increase lifespan in old age. Nat. Med. 2018, 24, 1246–1256. [Google Scholar] [CrossRef]
- Chang, J.; Wang, Y.; Shao, L.; Laberge, R.M.; Demaria, M.; Campisi, J.; Janakiraman, K.; Sharpless, N.E.; Ding, S.; Feng, W.; et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat. Med. 2016, 22, 78–83. [Google Scholar] [CrossRef] [Green Version]
- Yosef, R.; Pilpel, N.; Tokarsky-Amiel, R.; Biran, A.; Ovadya, Y.; Cohen, S.; Vadai, E.; Dassa, L.; Shahar, E.; Condiotti, R.; et al. Directed elimination of senescent cells by in.nhibition of BCL-W and BCL-XL. Nat. Commun. 2016, 7, 11190. [Google Scholar] [CrossRef]
- Safety, Tolerability, and Efficacy Study of UBX1325 in Patients With Neovascular Age-Related Macular Degeneration (ENVISION). Available online: https://clinicaltrials.gov/ct2/show/NCT05275205?term=Safety%2C+Tolerability%2C+and+Efficacy+Study+of+UBX1325+in+Patients+With+Neovascular+Age-Related+Macular+Degeneration+%28ENVISION%29&draw=2&rank=1 (accessed on 1 October 2022).
- Rocha, L.R.; Nguyen Huu, V.A.; Palomino La Torre, C.; Xu, Q.; Jabari, M.; Krawczyk, M.; Weinreb, R.N.; Skowronska-Krawczyk, D. Early removal of senescent cells protects retinal ganglion cells loss in experimental ocular hypertension. Aging Cell 2020, 19, e13089. [Google Scholar] [CrossRef] [Green Version]
- Cheng, S.C.; Huang, W.C.; JH, S.P.; Wu, Y.H.; Cheng, C.Y. Quercetin Inhibits the Production of IL-1beta-Induced Inflammatory Cytokines and Chemokines in ARPE-19 Cells via the MAPK and NF-kappaB Signaling Pathways. Int. J. Mol. Sci. 2019, 20, 2957. [Google Scholar] [CrossRef] [Green Version]
- Weng, S.; Mao, L.; Gong, Y.; Sun, T.; Gu, Q. Role of quercetin in protecting ARPE19 cells against H2O2induced injury via nuclear factor erythroid 2 like 2 pathway activation and endoplasmic reticulum stress inhibition. Mol. Med. Rep. 2017, 16, 3461–3468. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Q.; Liu, M.; He, Y.; Yang, B. Quercetin protect cigarette smoke extracts induced inflammation and apoptosis in RPE cells. Artif. Cells Nanomed. Biotechnol. 2019, 47, 2010–2015. [Google Scholar] [CrossRef] [PubMed]
- Ueda, S.; Nunn, B.M.; Chauhan, R.; McDonald, K.; Kaplan, H.J.; O’Toole, M.G.; Tamiya, S. Sustained dasatinib treatment prevents early fibrotic changes following ocular trauma. Graefes Arch. Clin. Exp. Ophthalmol. 2021, 259, 1103–1111. [Google Scholar] [CrossRef] [PubMed]
- Seo, S.; Suh, W. Antiangiogenic effect of dasatinib in murine models of oxygen-induced retinopathy and laser-induced choroidal neovascularization. Mol. Vis. 2017, 23, 823–831. [Google Scholar] [PubMed]
- Blitzer, A.L.; Ham, S.A.; Colby, K.A.; Skondra, D. Association of Metformin Use With Age-Related Macular Degeneration: A Case-Control Study. JAMA Ophthalmol. 2021, 139, 302–309. [Google Scholar] [CrossRef]
- Qu, S.; Zhang, C.; Liu, D.; Wu, J.; Tian, H.; Lu, L.; Xu, G.T.; Liu, F.; Zhang, J. Metformin Protects ARPE-19 Cells from Glyoxal-Induced Oxidative Stress. Oxid. Med. Cell. Longev. 2020, 2020, 1740943. [Google Scholar] [CrossRef]
- Zhao, X.; Liu, L.; Jiang, Y.; Silva, M.; Zhen, X.; Zheng, W. Protective Effect of Metformin against Hydrogen Peroxide-Induced Oxidative Damage in Human Retinal Pigment Epith.h.helial (RPE) Cells by Enhancing Autophagy through Activation of AMPK Pathway. Oxid. Med. Cell. Longev. 2020, 2020, 2524174. [Google Scholar] [CrossRef] [PubMed]
- Suzumura, A.; Terao, R.; Kaneko, H. Protective Effects and Molecular Signaling of n-3 Fatty Acids on Oxidative Stress and Inflammation in Retinal Diseases. Antioxidants 2020, 9, 920. [Google Scholar] [CrossRef]
- Dentchev, T.; Milam, A.H.; Lee, V.M.; Trojanowski, J.Q.; Dunaief, J.L. Amyloid-beta is found in drusen from some age-related macular degeneration retinas, but not in drusen from normal retinas. Mol. Vis. 2003, 9, 184–190. [Google Scholar]
- Guglielmotto, M.; Giliberto, L.; Tamagno, E.; Tabaton, M. Oxidative stress mediates the pathogenic effect of different Alzheimer’s disease risk factors. Front. Aging Neurosci. 2010, 2, 3. [Google Scholar] [CrossRef]
- Do, K.V.; Kautzmann, M.I.; Jun, B.; Gordon, W.C.; Nshimiyimana, R.; Yang, R.; Petasis, N.A.; Bazan, N.G. Elovanoids counteract oligomeric beta-amyloid-induced gene expression and protect photoreceptors. Proc. Natl. Acad. Sci. USA 2019, 116, 24317–24325. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Terao, R.; Ahmed, T.; Suzumura, A.; Terasaki, H. Oxidative Stress-Induced Cellular Senescence in Aging Retina and Age-Related Macular Degeneration. Antioxidants 2022, 11, 2189. https://doi.org/10.3390/antiox11112189
Terao R, Ahmed T, Suzumura A, Terasaki H. Oxidative Stress-Induced Cellular Senescence in Aging Retina and Age-Related Macular Degeneration. Antioxidants. 2022; 11(11):2189. https://doi.org/10.3390/antiox11112189
Chicago/Turabian StyleTerao, Ryo, Tazbir Ahmed, Ayana Suzumura, and Hiroko Terasaki. 2022. "Oxidative Stress-Induced Cellular Senescence in Aging Retina and Age-Related Macular Degeneration" Antioxidants 11, no. 11: 2189. https://doi.org/10.3390/antiox11112189
APA StyleTerao, R., Ahmed, T., Suzumura, A., & Terasaki, H. (2022). Oxidative Stress-Induced Cellular Senescence in Aging Retina and Age-Related Macular Degeneration. Antioxidants, 11(11), 2189. https://doi.org/10.3390/antiox11112189