
Thiocyanate Reduces Motor Impairment in the hMPO-A53T PD Mouse Model While Reducing MPO-Oxidation of Alpha Synuclein in Enlarged LYVE1/AQP4 Positive Periventricular Glymphatic Vessels

Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Antibodies
2.3. Immunohistochemistry (IHC)
2.4. Quantitative Immunohistochemical Analysis
2.5. Animals
2.6. Transgenic Mice
2.7. Behavior Tests
2.7.1. Wire Hanging Test
2.7.2. RotaRod Test
2.7.3. Balance Beam
2.7.4. Statistical Analysis
3. Results
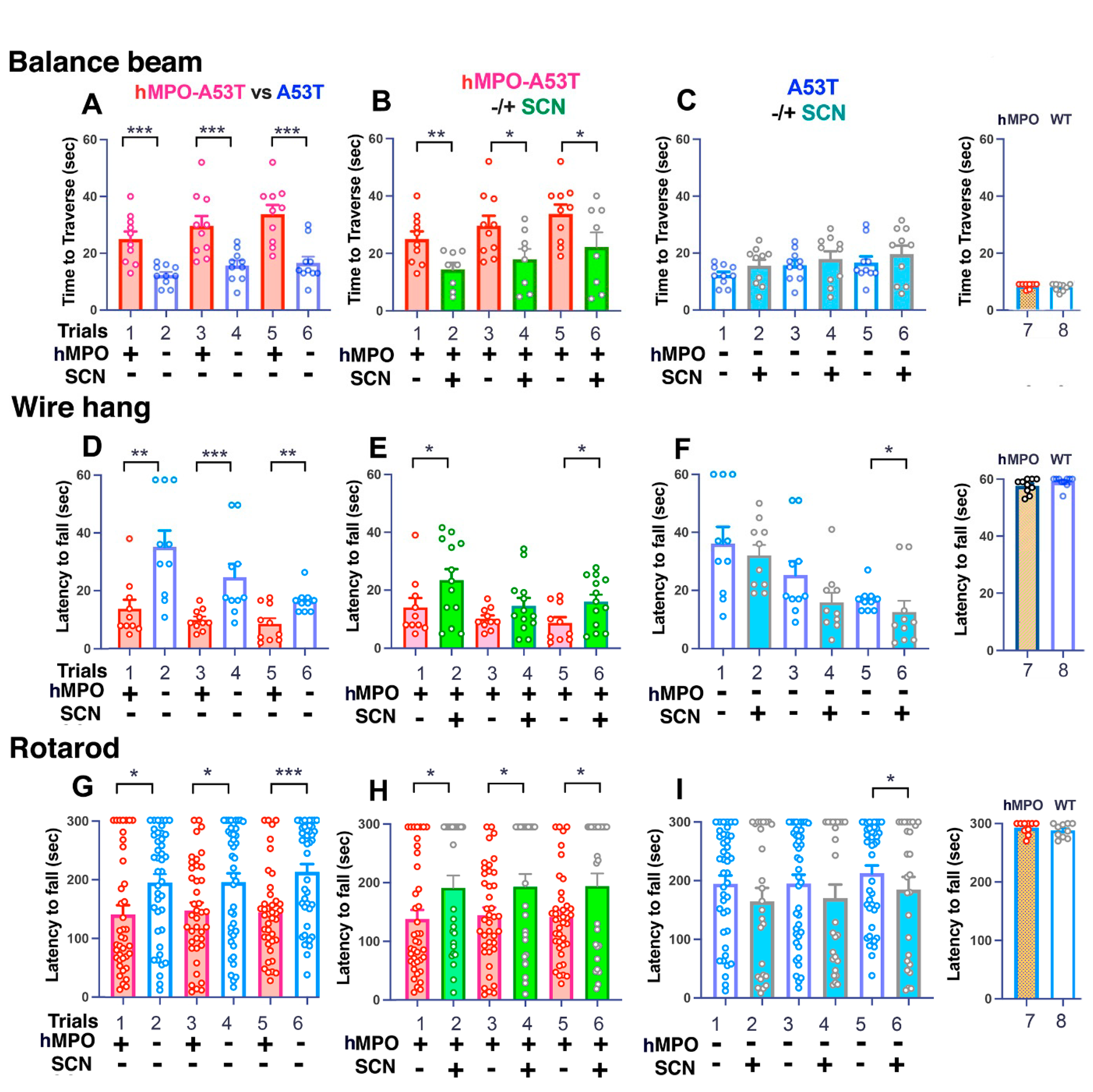
3.1. Motor Behavior
3.1.1. Balance Beam
3.1.2. Wire Hang
3.1.3. Rotarod
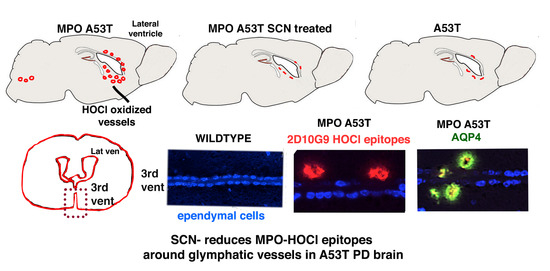
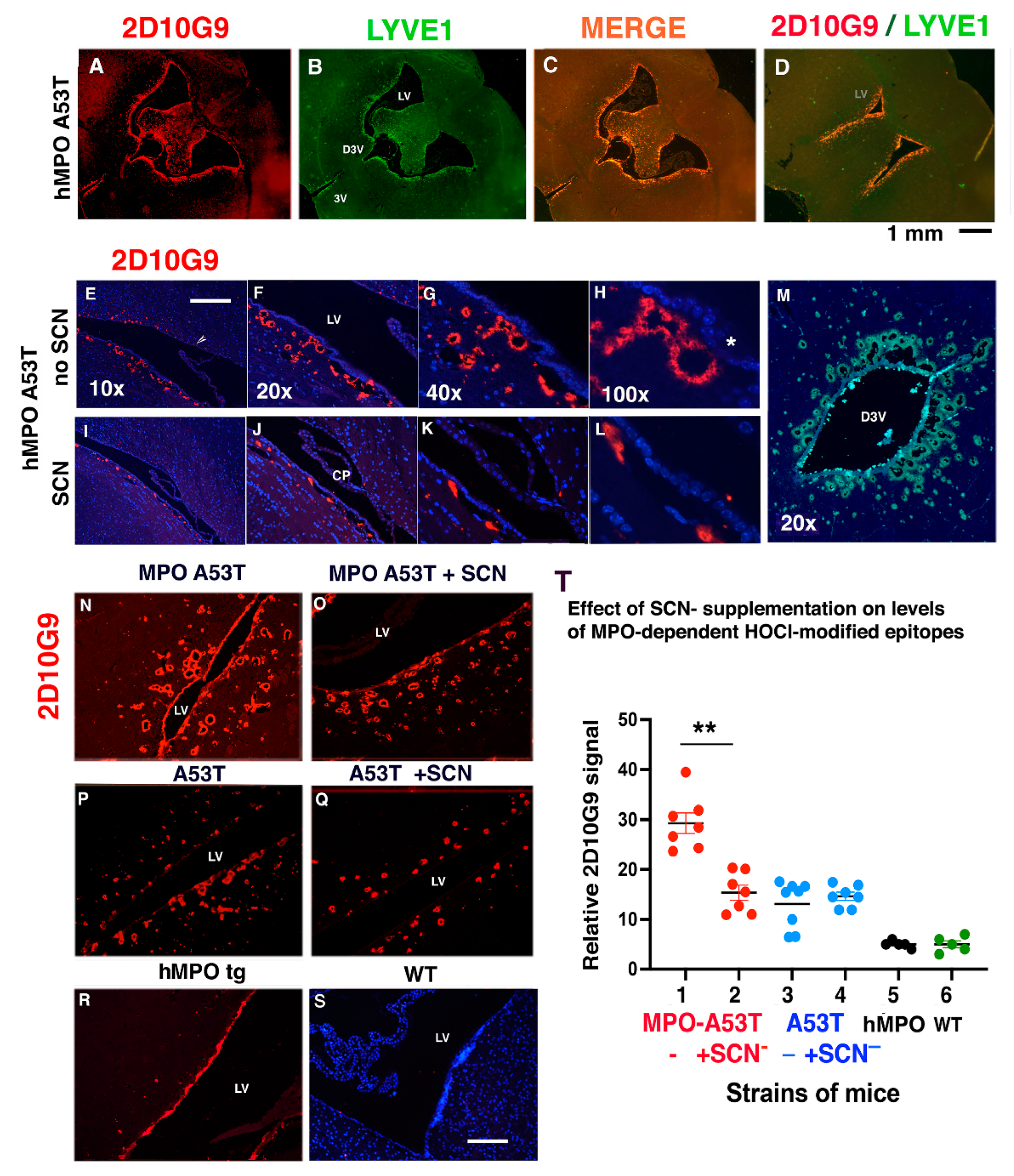
3.2. MPO, MPO-Generated HOCl Epitopes and LYVE1 Colocalize in Vessels around the Lateral Ventricles in the hMPO-A53T Mouse
3.3. SCN− Treatment Reduces the Size and Number of 2D10G9 MPO Modified HOCl Epitopes in the hMPO-A53T Mice but Not in A53T Lacking the hMPO Transgene
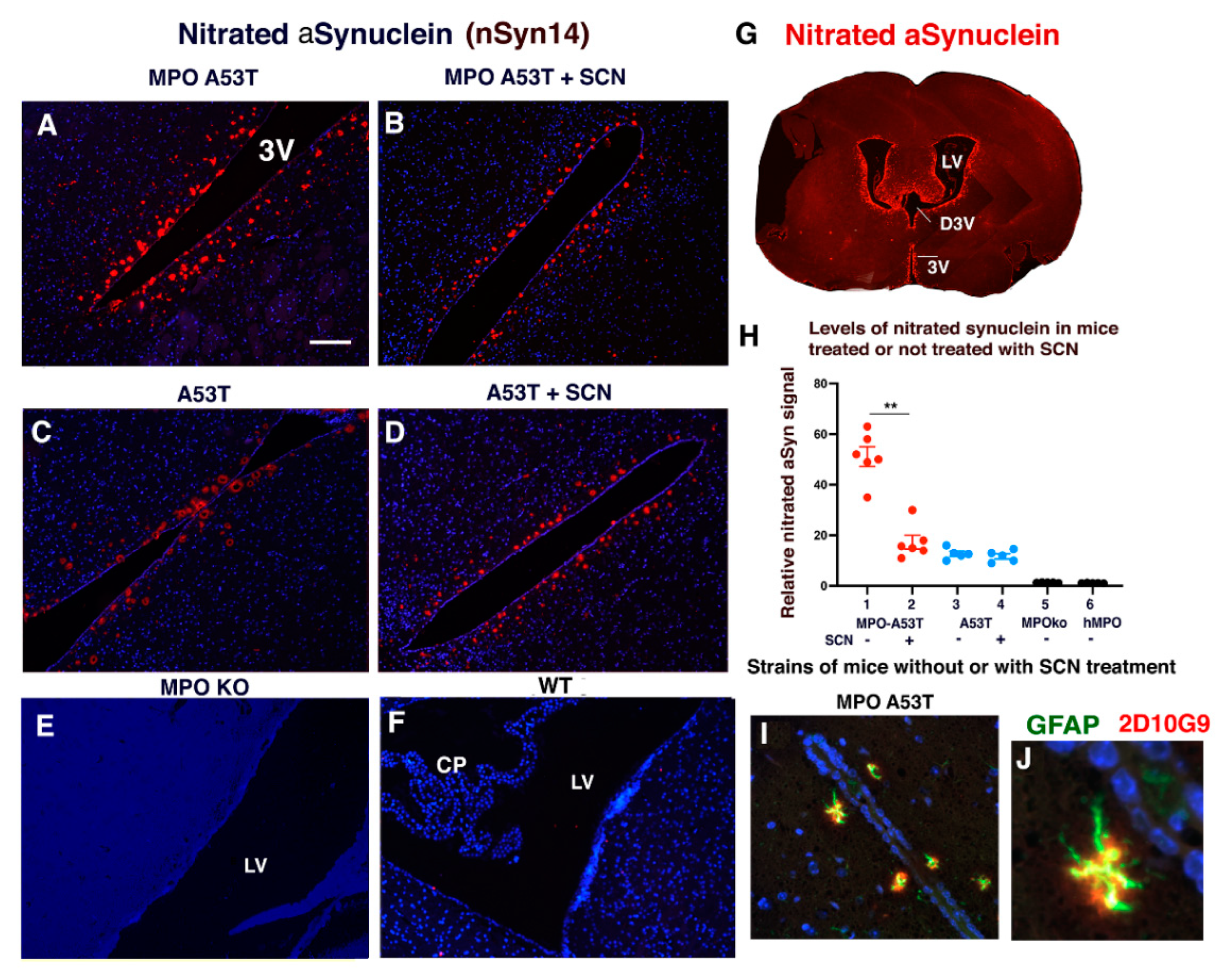
3.4. SCN− Treatment of hMPO-A53T Mice Reduces Levels of Nitrated αSyn in Glymphatic Vessels Surrounding the Ventricles
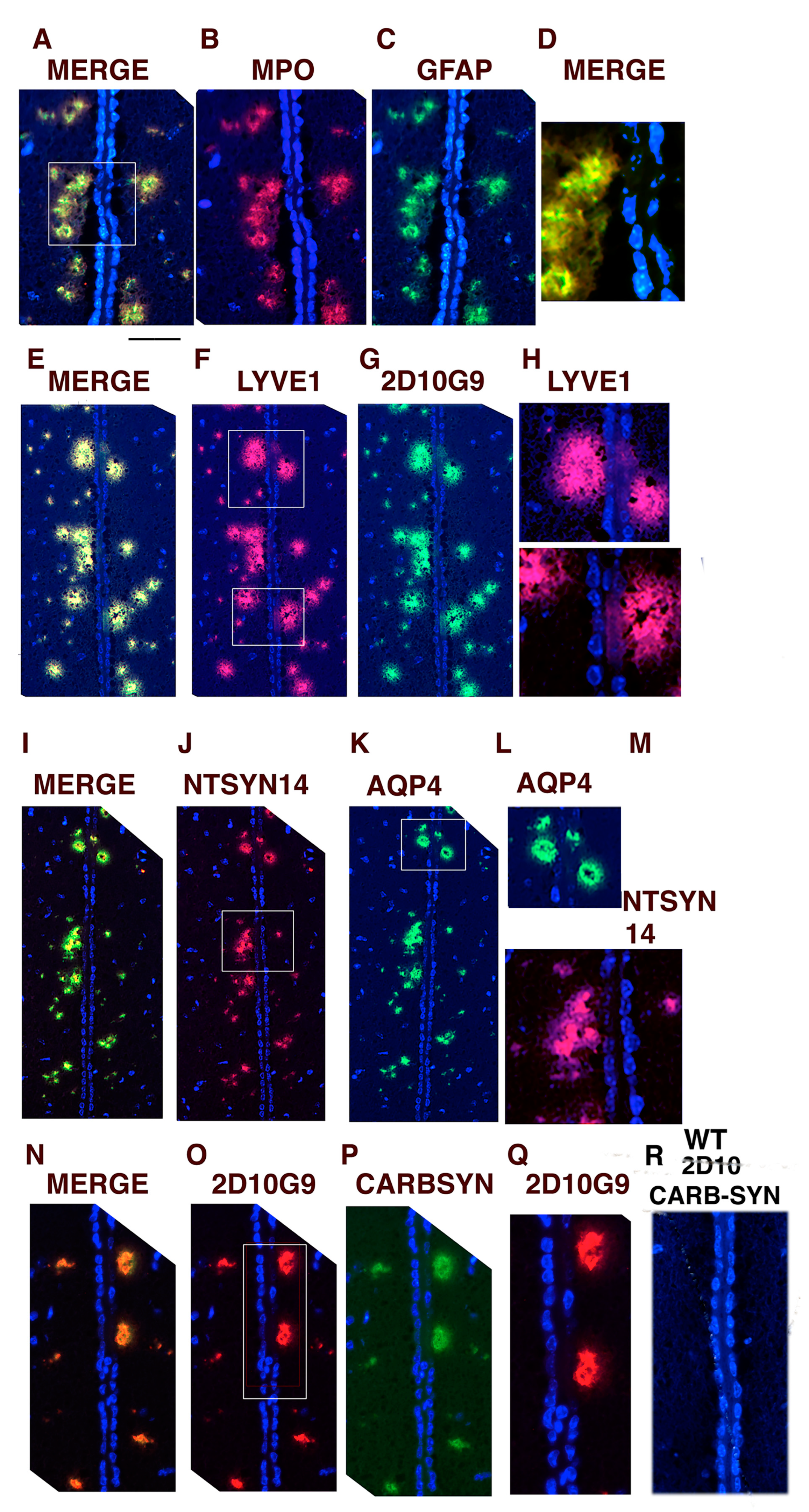
3.5. Glymphatic Vessels Colocalizing for MPO, LYVE1, AQP4, GFAP, 2D10G9, ntSyn14, and Carbamylated αSyn Can Be Toxic to Ependymal Cells Lining the Third Ventricle in hMPO-A53T Mice
3.6. AQP4 and HOCl-Epitopes in Periventricular Vessels Suggest MPO Contributes to Glymphatic Impairment
4. Discussion
4.1. Deleterious Versus Protective Effects of MPO
4.2. Impairment of the Glymphatic Waste Clearance System in hMPO-A53T Mice
4.3. The Possible Role of MPO in Glymphatics in PD
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| αSyn | Alpha synuclein |
| AD | Alzheimer’s disease |
| AQP4 | aquaporin 4 |
| Br− | bromide |
| Cl− | chloride |
| CSF | cerebral spinal fluid |
| DAPI | 4′, 6-diamidino-2-phenylindole |
| GFAP | glial fibrillary acidic protein |
| hMPO | human MPO |
| HOBr | hypobromous acid |
| HOCl | hypochlorous acid |
| HOI | hypoiodous acid |
| HOSCN | hypothiocyanous acid |
| I− | iodide |
| IHC | immunohistochemistry |
| LV | lateral ventricle |
| LYVE1 | lymphatic vessel endothelial hyaluronan receptor 1 |
| mAb | monoclonal antibody |
| mMPO | murine MPO |
| MPO | myeloperoxidase |
| NaSCN | sodium thiocyanate |
| pAb | polyclonal antibody |
| PBS | phosphate-buffered saline |
| PBST | PBS + 0.05% Tween 20 |
| PD | Parkinson’s disease |
| ROS | reactive oxygen species |
| SCN− | thiocyanate |
| Thy1 | human thymus cell antigen 1 |
References
- Goetz, C.G.; McGhiey, A. The movement disorder society and movement disorders: A modern history. Mov. Disord. 2011, 26, 939–946. [Google Scholar] [CrossRef] [PubMed]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Prim. 2017, 3, 17013. [Google Scholar] [CrossRef]
- Chavarria, C.; Souza, J.M. Oxidation and nitration of alpha-synuclein and their implications in neurodegenerative diseases. Arch. Biochem. Biophys. 2013, 533, 25–32. [Google Scholar] [CrossRef] [PubMed]
- He, Y.X.; Yu, Z.W.; Chen, S.D. Alpha-synuclein nitration and its implications in Parkinson’s disease. ACS Chem. Neurosci. 2018, 10, 777–782. [Google Scholar] [CrossRef] [PubMed]
- Perry, T.L.; Yong, V.W. Idiopathic Parkinson’s disease, progressive supranuclear palsy and glutathione metabolism in the substantia nigra of patients. Neurosci. Lett. 1986, 67, 269–274. Available online: https://www.ncbi.nlm.nih.gov/pubmed/3737015 (accessed on 21 October 2022). [CrossRef]
- Jenner, P.; Dexter, D.T.; Sian, J.; Schapira, A.H.; Marsden, C.D. Oxidative stress as a cause of nigral cell death in Parkinson’s disease and incidental Lewy body disease. The Royal Kings and Queens Parkinson’s Disease Research Group. Ann. Neurol. 1992, 32, S82–S87. Available online: https://www.ncbi.nlm.nih.gov/pubmed/1510385 (accessed on 1 June 2021). [CrossRef] [PubMed]
- Yoritaka, A.; Hattori, N.; Uchida, K.; Tanaka, M.; Stadtman, E.R.; Mizuno, Y. Immunohistochemical detection of 4-hydroxynonenal protein adducts in Parkinson disease. Proc. Natl. Acad. Sci. USA 1996, 93, 2696–2701. Available online: https://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=8610103 (accessed on 21 October 2022). [CrossRef] [PubMed]
- Duda, J.E.; Giasson, B.I.; Mabon, M.E.; Lee, V.M.; Trojanowski, J.Q. Novel antibodies to synuclein show abundant striatal pathology in Lewy body diseases. Ann. Neurol. 2002, 52, 205–210. [Google Scholar] [CrossRef]
- Nunomura, A.; Moreira, P.I.; Lee, H.G.; Zhu, X.; Castellani, R.J.; Smith, M.A.; Perry, G. Neuronal death and survival under oxidative stress in Alzheimer and Parkinson diseases. CNS Neurol. Disord. Drug Targets 2007, 6, 411–423. Available online: https://www.ncbi.nlm.nih.gov/pubmed/18220780 (accessed on 1 May 2019). [CrossRef]
- Percario, S.; Barbosa, A.d.; Varela, E.L.; Gomes, A.R.Q.; Ferreira, M.E.S.; Moreira, T.d.A.; Dolabela, M.F. Oxidative Stress in Parkinson’s Disease: Potential Benefits of Antioxidant Supplementation. Oxid. Med. Cell Longev. 2020, 2020, 2360872. [Google Scholar] [CrossRef]
- Malle, E.; Buch, T.; Grone, H.J. Myeloperoxidase in kidney disease. Kidney Int. 2003, 64, 1956–1967. [Google Scholar] [CrossRef] [PubMed]
- Arnhold, J.; Malle, E. Halogenation Activity of Mammalian Heme Peroxidases. Antioxidants 2022, 11, 890. [Google Scholar] [CrossRef]
- Klebanoff, S.J.; Kettle, A.J.; Rosen, H.; Winterbourn, C.C.; Nauseef, W.M. Myeloperoxidase: A front-line defender against phagocytosed microorganisms. J. Leukoc Biol. 2013, 93, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Winterbourn, C.C.; Kettle, A.J.; Hampton, M.B. Reactive Oxygen Species and Neutrophil Function. Ann. Rev. Biochem. 2016, 85, 765–792. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, C.L.; Davies, M.J. Role of myeloperoxidase and oxidant formation in the extracellular environment in inflammation-induced tissue damage. Free Radic. Biol. Med. 2021, 172, 633–651. [Google Scholar] [CrossRef]
- Davies, M.J. Myeloperoxidase: Mechanisms, reactions and inhibition as a therapeutic strategy in inflammatory diseases. Pharmacol. Ther. 2021, 218, 107685. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Chuang, C.Y.; Malle, E.; Gamon, L.F.; Hawkins, C.L.; Davies, M.J. Influence of plasma halide, pseudohalide and nitrite ions on myeloperoxidase-mediated protein and extracellular matrix damage. Free Radic. Biol. Med. 2022, 188, 162–174. [Google Scholar] [CrossRef]
- Siraki, A.G. The many roles of myeloperoxidase: From inflammation and immunity to biomarkers, drug metabolism and drug discovery. Redox. Biol. 2021, 46, 102109. [Google Scholar] [CrossRef]
- Davies, M.J.; Hawkins, C.L. The Role of Myeloperoxidase in Biomolecule Modification, Chronic Inflammation, and Disease. Antioxid. Redox. Signal 2020, 32, 957–981. [Google Scholar] [CrossRef]
- Hoy, A.; Tregouet, D.; Leininger-Muller, B.; Poirier, O.; Maurice, M.; Sass, C.; Siest, G.; Tiret, L.; Visvikis, S. Serum myeloperoxidase concentration in a healthy population: Biological variations, familial resemblance and new genetic polymorphisms. Eur. J. Hum. Genet 2001, 9, 780–786. [Google Scholar] [CrossRef]
- Pecoits-Filho, R.; Stenvinkel, P.; Marchlewska, A.; Heimburger, O.; Bárány, P.; Hoff, C.M.; Holmes, C.J.; Suliman, M.; Lindholm, B.; Schalling, M.; et al. A functional variant of the myeloperoxidase gene is associated with cardiovascular disease in end-stage renal disease patients. Kidney Int. Suppl 2003, 63, S172–S176. [Google Scholar] [CrossRef] [PubMed]
- Makela, R.; Karhunen, P.J.; Kunnas, T.A.; Ilveskoski, E.; Kajander, O.A.; Mikkelsson, J.; Perola, M.; Penttila, A.; Lehtimaki, T. Myeloperoxidase gene variation as a determinant of atherosclerosis progression in the abdominal and thoracic aorta: An autopsy study. Lab. Invest. 2003, 83, 919–925. Available online: https://www.ncbi.nlm.nih.gov/pubmed/12861032 (accessed on 20 February 2010). [CrossRef] [PubMed][Green Version]
- Makela, R.; Laaksonen, R.; Janatuinen, T.; Vesalainen, R.; Nuutila, P.; Jaakkola, O.; Knuuti, J.; Lehtimaki, T. Myeloperoxidase gene variation and coronary flow reserve in young healthy men. J. Biomed. Sci. 2004, 11, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Asselbergs, F.W.; Reynolds, W.F.; Cohen-Tervaert, J.W.; Jessurun, G.A.; Tio, R.A. Myeloperoxidase polymorphism related to cardiovascular events in coronary artery disease. Am. J. Med. 2004, 116, 429–430. [Google Scholar] [CrossRef] [PubMed]
- Rudolph, V.; Rudolph, T.K.; Kubala, L.; Clauberg, N.; Maas, R.; Pekarova, M.; Klinke, A.; Lau, D.; Szöcs, K.; Meinertz, T.; et al. A myeloperoxidase promoter polymorphism is independently associated with mortality in patients with impaired left ventricular function. Free Radic. Biol. Med. 2009, 47, 1584–1590. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, W.F.; Rhees, J.; Maciejewski, D.; Paladino, T.; Sieburg, H.; Maki, R.A.; Masliah, E. Myeloperoxidase polymorphism is associated with gender specific risk for Alzheimer’s disease. Exp. Neurol. 1999, 155, 31–41. [Google Scholar] [CrossRef]
- Maki, R.A.; Tyurin, V.A.; Lyon, R.C.; Hamilton, R.L.; DeKosky, S.T.; Kagan, V.E.; Reynolds, W.F. Aberrant expression of myeloperoxidase in astrocytes promotes phospholipid oxidation and memory deficits in a mouse model of Alzheimer disease. J. Biol. Chem. 2009, 284, 3158–3169. [Google Scholar] [CrossRef]
- Green, P.S.; Mendez, A.J.; Jacob, J.S.; Crowley, J.R.; Growdon, W.; Hyman, B.T.; Heinecke, J.W. Neuronal expression of myeloperoxidase is increased in Alzheimer’s disease. J. Neurochem. 2004, 90, 724–733. [Google Scholar] [CrossRef]
- Choi, D.-K.; Pennathur, S.; Perier, C.; Tieu, K.; Teismann, P.; Wu, D.-C.; Jackson-Lewis, V.; Vila, M.; Vonsattel, J.-P.; Heinecke, J.W.; et al. Ablation of the inflammatory enzyme myeloperoxidase mitigates features of Parkinson’s disease in mice. J. Neurosci. 2005, 25, 6594–6600. [Google Scholar] [CrossRef]
- Gellhaar, S.; Sunnemark, D.; Eriksson, H.; Olson, L.; Galter, D. Myeloperoxidase-immunoreactive cells are significantly increased in brain areas affected by neurodegeneration in Parkinson’s and Alzheimer’s disease. Cell Tissue Res. 2017, 369, 445–454. [Google Scholar] [CrossRef]
- Maki, R.A.; Holzer, M.; Motamedchaboki, K.; Malle, E.; Masliah, E.; Marsche, G.; Reynolds, W.F. Human myeloperoxidase (hMPO) is expressed in neurons in the substantia nigra in Parkinson’s disease and in the hMPO-alpha-synuclein-A53T mouse model, correlating with increased nitration and aggregation of alpha-synuclein and exacerbation of motor impairment. Free Radic. Biol. Med. 2019, 141, 115–140. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, W.F.; Maki, R.A. The role of myeloperoxidase in neurodegenerative disease. In Mammalian Heme Peroxidases: Diverse Roles in Health and Disease, 1st ed.; Hawkins, C., Nauseef, W.M., Eds.; CRC Press: Boca Raton, FL, USA, 2021; Chapter 15; pp. 249–273. [Google Scholar]
- Castellani, L.W.; Chang, J.J.; Wang, X.; Lusis, A.J.; Reynolds, W.F. Transgenic mice express human MPO -463G/A alleles at atherosclerotic lesions, developing hyperlipidemia and obesity in -463G males. J. Lipid. Res. 2006, 47, 1366–1377. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.P.; Piedrafita, F.J.; Reynolds, W.F. Peroxisome proliferator-activated receptor gamma ligands regulate myeloperoxidase expression in macrophages by an estrogen-dependent mechanism involving the -463GA promoter polymorphism. J. Biol. Chem. 2004, 279, 8300–8315. [Google Scholar] [CrossRef]
- Piedrafita, F.J.; Molander, R.B.; Vansant, G.; Orlova, E.A.; Pfahl, M.; Reynolds, W.F. An Alu element in the myeloperoxidase promoter contains a composite SP1-thyroid hormone-retinoic acid response element. J. Biol. Chem. 1996, 271, 14412–14420. Available online: https://www.ncbi.nlm.nih.gov/pubmed/8662930 (accessed on 7 May 2000). [CrossRef] [PubMed]
- Reynolds, W.F.; Kumar, A.; Piedrafita, F.J. The human myeloperoxidase gene is regulated by LXR and PPARalpha ligands. Biochem. Biophys. Res. Commun. 2006, 349, 846–854. [Google Scholar] [CrossRef] [PubMed]
- Vansant, G.; Reynolds, W.F. The consensus sequence of a major Alu subfamily contains a functional retinoic acid response element. Proc. Natl. Acad. Sci. USA 1995, 92, 8229–8233. Available online: https://www.ncbi.nlm.nih.gov/pubmed/7667273 (accessed on 13 June 2015). [CrossRef]
- Reynolds, W.F.; Hiltunen, M.; Pirskanen, M.; Mannermaa, A.; Helisalmi, S.; Lehtovirta, M.; Alafuzoff, I.; Soininen, H. MPO and APOEepsilon4 polymorphisms interact to increase risk for AD in Finnish males. Neurology 2000, 55, 1284–1290. Available online: https://www.ncbi.nlm.nih.gov/pubmed/11087769 (accessed on 21 October 2022). [CrossRef]
- Crawford, F.C.; Freeman, M.J.; Schinka, J.A.; Morris, M.D.; Abdullah, L.I.; Richards, D.; Sevush, S.; Duara, R.; Mullan, M.J. Association between Alzheimer’s disease and a functional polymorphism in the Myeloperoxidase gene. Exp. Neurol. 2001, 167, 456–459. [Google Scholar] [CrossRef]
- Leininger-Muller, B.; Hoy, A.; Herbeth, B.; Pfister, M.; Serot, J.M.; Stavljenic-Rukavina, M.; Massana, L.; Passmore, P.; Siest, G.; Visvikis, S. Myeloperoxidase G-463A polymorphism and Alzheimer’s disease in the ApoEurope study. Neurosci. Lett. 2003, 349, 95–98. Available online: https://www.ncbi.nlm.nih.gov/pubmed/12946561 (accessed on 12 March 2009). [CrossRef]
- Zappia, M.; Manna, I.; Serra, P.; Cittadella, R.; Andreoli, V.; La Russa, A.; Annesi, F.; Spadafora, P.; Romeo, N.; Nicoletti, G.; et al. Increased risk for Alzheimer disease with the interaction of MPO and A2M polymorphisms. Arch. Neurol. 2004, 61, 341–344. [Google Scholar] [CrossRef]
- Pope, S.K.; Kritchevsky, S.B.; Ambrosone, C.; Yaffe, K.; Tylavsky, F.; Simonsick, E.M.; Rosano, C.; Stewart, S.; Harris, T. Myeloperoxidase polymorphism and cognitive decline in older adults in the Health, Aging, and Body Composition Study. Am. J. Epidemiol. 2006, 163, 1084–1090. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Schabath, M.B.; Spitz, M.R.; Hong, W.K.; Delclos, G.L.; Reynolds, W.F.; Gunn, G.B.; Whitehead, L.W.; Wu, X. A myeloperoxidase polymorphism associated with reduced risk of lung cancer. Lung Cancer 2002, 37, 35–40. Available online: https://www.ncbi.nlm.nih.gov/pubmed/12057865 (accessed on 21 October 2022). [CrossRef] [PubMed]
- Yang, J.-P.; Wang, W.-B.; Yang, X.-X.; Yang, L.; Ren, L.; Zhou, F.-X.; Hu, L.; He, W.; Li, B.-Y.; Zhu, Y.; et al. The MPO-463G>A polymorphism and lung cancer risk: A meta-analysis based on 22 case-control studies. PLoS ONE 2013, 8, e65778. [Google Scholar] [CrossRef]
- Castillo-Tong, D.C.; Pils, D.; Heinze, G.; Braicu, I.; Sehouli, J.; Reinthaller, A.; Schuster, E.; Wolf, A.; Watrowski, R.; Maki, R.A.; et al. Association of myeloperoxidase with ovarian cancer. Tumour Biol. 2014, 35, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Davies, M.J.; Hawkins, C.L. Role of thiocyanate in the modulation of myeloperoxidase-derived oxidant induced damage to macrophages. Redox Biol. 2020, 36, 101666. [Google Scholar] [CrossRef]
- Vanichkitrungruang, S.; Chuang, C.Y.; Hawkins, C.L.; Hammer, A.; Hoefler, G.; Malle, E.; Davies, M.J. Oxidation of human plasma fibronectin by inflammatory oxidants perturbs endothelial cell function. Free Radic. Biol. Med. 2019, 136, 118–134. [Google Scholar] [CrossRef]
- Flouda, K.; Gammelgaard, B.; Davies, M.J.; Hawkins, C.L. Modulation of hypochlorous acid (HOCl) induced damage to vascular smooth muscle cells by thiocyanate and selenium analogues. Redox Biol. 2021, 41, 101873. [Google Scholar] [CrossRef]
- Morgan, P.E.; Laura, R.; Maki, R.A.; Reynolds, W.F.; Davies, M.J. Thiocyanate supplementation decreases atherosclerotic plaque in mice expressing human myeloperoxidase. Free Radic. Res. 2015, 49, 743–749. [Google Scholar] [CrossRef]
- Hablitz, L.M.; Nedergaard, M. The glymphatic system. Curr. Biol. 2021, 31, R1371–R1375. [Google Scholar] [CrossRef]
- Rasmussen, M.K.; Mestre, H.; Nedergaard, M. Fluid transport in the brain. Physiol. Rev. 2022, 102, 1025–1151. [Google Scholar] [CrossRef]
- Laura, R.P.; Dong, D.; Reynolds, W.F.; Maki, R.A. T47D Cells Expressing Myeloperoxidase Are Able to Process, Traffic and Store the Mature Protein in Lysosomes: Studies in T47D Cells Reveal a Role for Cys319 in MPO Biosynthesis that Precedes Its Known Role in Inter-Molecular Disulfide Bond Formation. PLoS ONE 2016, 11, e0149391. [Google Scholar] [CrossRef] [PubMed]
- Baba, M.; Nakajo, S.; Tu, P.H.; Tomita, T.; Nakaya, K.; Lee, V.M.; Trojanowski, J.Q.; Iwatsubo, T. Aggregation of alpha-synuclein in Lewy bodies of sporadic Parkinson’s disease and dementia with Lewy bodies. Am. J. Pathol. 1998, 152, 879–884. Available online: https://www.ncbi.nlm.nih.gov/pubmed/9546347 (accessed on 21 October 2022). [PubMed]
- Giasson, B.I.; Duda, J.E.; Murray, I.V.; Chen, Q.; Souza, J.M.; Hurtig, H.I.; Ischiropoulos, H.; Trojanowski, J.Q.; Lee, V.M. Oxidative damage linked to neurodegeneration by selective alpha-synuclein nitration in synucleinopathy lesions. Science 2000, 290, 985–989. Available online: https://www.ncbi.nlm.nih.gov/pubmed/11062131 (accessed on 21 October 2022). [CrossRef] [PubMed]
- Malle, E.; Woenckhaus, C.; Waeg, G.; Esterbauer, H.; Grone, E.F.; Grone, H.J. Immunological evidence for hypochlorite-modified proteins in human kidney. Am. J. Pathol. 1997, 150, 603–615. Available online: https://www.ncbi.nlm.nih.gov/pubmed/9033274 (accessed on 23 August 2005). [PubMed]
- Malle, E.; Hazell, L.; Stocker, R.; Sattler, W.; Esterbauer, H.; Waeg, G. Immunologic detection and measurement of hypochlorite-modified LDL with specific monoclonal antibodies. Arter. Thromb. Vasc. Biol. 1995, 15, 982–989. Available online: https://www.ncbi.nlm.nih.gov/pubmed/7541296 (accessed on 21 October 2022). [CrossRef]
- Masliah, E.; Rockenstein, E.; Veinbergs, I.; Mallory, M.; Hashimoto, M.; Takeda, A.; Sagara, Y.; Sisk, A.; Mucke, L. Dopaminergic loss and inclusion body formation in alpha-synuclein mice: Implications for neurodegenerative disorders. Science 2000, 287, 1265–1269. Available online: https://www.ncbi.nlm.nih.gov/pubmed/10678833 (accessed on 7 February 2011). [CrossRef]
- Games, D.; Seubert, P.; Rockenstein, E.; Patrick, C.; Trejo, M.; Ubhi, K.; Ettle, B.; Ghassemiam, M.; Barbour, R.; Schenk, D.; et al. Axonopathy in an alpha-synuclein transgenic model of Lewy body disease is associated with extensive accumulation of C-terminal-truncated alpha-synuclein. Am. J. Pathol. 2013, 182, 940–953. [Google Scholar] [CrossRef]
- Kumar, A.P.; Ryan, C.; Cordy, V.; Reynolds, W.F. Inducible nitric oxide synthase expression is inhibited by myeloperoxidase. Nitric Oxide 2005, 13, 42–53. [Google Scholar] [CrossRef]
- Chandra, S.; Gallardo, G.; Fernandez-Chacon, R.; Schluter, O.M.; Sudhof, T.C. Alpha-synuclein cooperates with CSPalpha in preventing neurodegeneration. Cell 2005, 123, 383–396. [Google Scholar] [CrossRef]
- Martin, L.J.; Semenkow, S.; Hanaford, A.; Wong, M. Mitochondrial permeability transition pore regulates Parkinson’s disease development in mutant alpha-synuclein transgenic mice. Neurobiol. Aging 2014, 35, 1132–1152. [Google Scholar] [CrossRef]
- Rothman, S.M.; Griffioen, K.J.; Vranis, N.; Ladenheim, B.; Cong, W.N.; Cadet, J.L.; Haran, J.; Martin, B.; Mattson, M. Neuronal expression of familial Parkinson’s disease A53T alpha-synuclein causes early motor impairment, reduced anxiety and potential sleep disturbances in mice. J. Park. Dis. 2013, 3, 215–229. [Google Scholar] [CrossRef]
- van Dalen, C.J.; Whitehouse, M.W.; Winterbourn, C.C.; Kettle, A.J. Thiocyanate and chloride as competing substrates for myeloperoxidase. Biochem. J. 1997, 327 (Pt 2), 487–492. Available online: https://www.ncbi.nlm.nih.gov/pubmed/9359420 (accessed on 20 February 2006). [CrossRef]
- Delanghe, S.; Delanghe, J.R.; Speeckaert, R.; Van Biesen, W.; Speeckaert, M.M. Mechanisms and consequences of carbamoylation. Nat. Rev. Nephrol. 2017, 13, 580–593. [Google Scholar] [CrossRef]
- Wang, Z.; Nicholls, S.; Rodriguez, E.R.; Kummu, O.; Hörkkö, S.; Barnard, J.W.; Reynolds, W.F.; Topol, E.; A DiDonato, J.; Hazen, S.L. Protein carbamylation links inflammation, smoking, uremia and atherogenesis. Nat. Med. 2007, 13, 1176–1184. [Google Scholar] [CrossRef]
- Holzer, M.; Gauster, M.; Pfeifer, T.; Wadsack, C.; Fauler, G.; Stiegler, P.; Koefeler, H.; Beubler, E.; Schuligoi, R.; Heinemann, A.; et al. Protein carbamylation renders high-density lipoprotein dysfunctional. Antioxid. Redox. Signal 2011, 14, 2337–2346. [Google Scholar] [CrossRef]
- Marsche, G.; Stadler, J.T.; Kargl, J.; Holzer, M. Understanding Myeloperoxidase-Induced Damage to HDL Structure and Function in the Vessel Wall: Implications for HDL-Based Therapies. Antioxidants 2022, 11, 556. [Google Scholar] [CrossRef]
- Nagahara, N.; Okazaki, T.; Nishino, T. Cytosolic mercaptopyruvate sulfurtransferase is evolutionarily related to mitochondrial rhodanese. Striking similarity in active site amino acid sequence and the increase in the mercaptopyruvate sulfurtransferase activity of rhodanese by site-directed mutagenesis. J. Biol. Chem. 1995, 270, 16230–16235. Available online: https://www.ncbi.nlm.nih.gov/pubmed/7608189 (accessed on 4 November 2015).
- Delporte, C.; Boudjeltia, K.Z.; Furtmueller, P.G.; Maki, R.A.; Dieu, M.; Noyon, C.; Soudi, M.; Dufour, D.; Coremans, C.; Nuyens, V.; et al. Myeloperoxidase-catalyzed oxidation of cyanide to cyanate: A potential carbamylation route involved in the formation of atherosclerotic plaques? J. Biol. Chem. 2018, 293, 6374–6386. [Google Scholar] [CrossRef]
- Wagner, B.A.; Reszka, K.J.; McCormick, M.L.; Britigan, B.E.; Evig, C.B.; Burns, C. Role of thiocyanate, bromide and hypobromous acid in hydrogen peroxide-induced apoptosis. Free Radic. Res. 2004, 38, 167–175. Available online: https://www.ncbi.nlm.nih.gov/pubmed/15104210 (accessed on 21 October 2022). [CrossRef]
- Chandler, J.D.; Day, B.J. Biochemical mechanisms and therapeutic potential of pseudohalide thiocyanate in human health. Free Radic Res. 2015, 49, 695–710. [Google Scholar] [CrossRef]
- Skaff, O.; Pattison, D.I.; Davies, M.J. Hypothiocyanous acid reactivity with low-molecular-mass and protein thiols: Absolute rate constants and assessment of biological relevance. Biochem. J. 2009, 422, 111–117. [Google Scholar] [CrossRef]
- Talib, J.; Pattison, D.I.; Harmer, J.A.; Celermajer, D.S.; Davies, M.J. High plasma thiocyanate levels modulate protein damage induced by myeloperoxidase and perturb measurement of 3-chlorotyrosine. Free Radic. Biol. Med. 2012, 53, 20–29. [Google Scholar] [CrossRef]
- Guo, C.; Sileikaite, I.; Davies, M.J.; Hawkins, C.L. Myeloperoxidase Modulates Hydrogen Peroxide Mediated Cellular Damage in Murine Macrophages. Antioxidants 2020, 9, 1255. [Google Scholar] [CrossRef]
- Chandler, J.D.; Min, E.; Huang, J.; Nichols, D.; Day, B.J. Nebulized thiocyanate improves lung infection outcomes in mice. Br. J. Pharmacol. 2013, 169, 1166–1177. [Google Scholar] [CrossRef]
- Zietzer, A.; Niepmann, S.T.; Camara, B.; Lenart, M.A.; Jansen, F.; Becher, M.U.; Andrie, R.; Nickenig, G.; Tiyerili, V. Sodium thiocyanate treatment attenuates atherosclerotic plaque formation and improves endothelial regeneration in mice. PLoS ONE 2019, 14, e0214476. [Google Scholar] [CrossRef]
- Hall, L.; Guo, C.; Tandy, S.; Broadhouse, K.; Dona, A.C.; Malle, E.; Bartels, E.D.; Christoffersen, C.; Grieve, S.M.; Figtree, G.; et al. Oral pre-treatment with thiocyanate (SCN(-)) protects against myocardial ischaemia-reperfusion injury in rats. Sci. Rep. 2021, 11, 12712. [Google Scholar] [CrossRef]
- Love, D.T.; Guo, C.; Nikelshparg, E.I.; Brazhe, N.A.; Sosnovtseva, O.; Hawkins, C.L. The role of the myeloperoxidase-derived oxidant hypothiocyanous acid (HOSCN) in the induction of mitochondrial dysfunction in macrophages. Redox Biol. 2020, 36, 101602. [Google Scholar] [CrossRef]
- Rayner, B.S.; Love, D.T.; Hawkins, C.L. Comparative reactivity of myeloperoxidase-derived oxidants with mammalian cells. Free. Radic. Biol. Med. 2014, 71, 240–255. [Google Scholar] [CrossRef]
- Day, B.J. The science of licking your wounds: Function of oxidants in the innate immune system. Biochem. Pharmacol. 2019, 163, 451–457. [Google Scholar] [CrossRef]
- Iliff, J.; Simon, M. CrossTalk proposal: The glymphatic system supports convective exchange of cerebrospinal fluid and brain interstitial fluid that is mediated by perivascular aquaporin-4. J. Physiol. 2019, 597, 4417–4419. [Google Scholar] [CrossRef]
- Iliff, J.J.; Wang, M.; Liao, Y.; Plogg, B.A.; Peng, W.; Gundersen, G.A.; Benveniste, H.; Vates, G.E.; Deane, R.; Goldman, S.A.; et al. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid beta. Sci. Transl. Med. 2012, 4, 147ra111. [Google Scholar] [CrossRef] [PubMed]
- Mestre, H.; Hablitz, L.M.; Xavier, A.L.R.; Feng, W.; Zou, W.; Pu, T.; Monai, H.; Murlidharan, G.; Rivera, R.M.C.; Simon, M.J.; et al. Aquaporin-4-dependent glymphatic solute transport in the rodent brain. Elife 2018, 7, e40070. [Google Scholar] [CrossRef] [PubMed]
- Lewis, B. The relationships of the nerve-cell of the cortex to the llymphatic system of the brain. Proc. Roy. Soc. Lond. 2022, 26, 326–334. [Google Scholar]
- Obersteiner, H. Introduction to the Study of the Anatomy of the Central Nervous Organs in Helath and Disease; P. Blakiston, Son and Company: Philadelphia, PA, USA, 1890. [Google Scholar]
- Hablitz, L.M.; Nedergaard, M. The Glymphatic System: A Novel Component of Fundamental Neurobiology. J. Neurosci. 2021, 41, 7698–7711. [Google Scholar] [CrossRef]
- Louveau, A.; Smirnov, I.; Keyes, T.J.; Eccles, J.D.; Rouhani, S.J.; Peske, J.D.; Derecki, N.C.; Castle, D.; Mandell, J.W.; Lee, K.S.; et al. Structural and functional features of central nervous system lymphatic vessels. Nature 2015, 523, 337–341. [Google Scholar] [CrossRef]
- Mezey, E.; Szalayova, I.; Hogden, C.T.; Brady, A.; Dosa, A.; Sotonyi, P.; Palkovits, M. An immunohistochemical study of lymphatic elements in the human brain. Proc. Natl. Acad. Sci. USA 2021, 118, e2002574118. [Google Scholar] [CrossRef]
- Pizzo, M.E.; Wolak, D.J.; Kumar, N.N.; Brunette, E.; Brunnquell, C.L.; Hannocks, M.; Abbott, N.J.; Meyerand, M.E.; Sorokin, L.; Stanimirovic, D.B.; et al. Intrathecal antibody distribution in the rat brain: Surface diffusion, perivascular transport and osmotic enhancement of delivery. J. Physiol. 2018, 596, 445–475. [Google Scholar] [CrossRef]
- Asgari, M.; de Zelicourt, D.; Kurtcuoglu, V. Glymphatic solute transport does not require bulk flow. Sci. Rep. 2016, 6, 38635. [Google Scholar] [CrossRef]
- Smith, A.J.; Yao, X.; Dix, J.A.; Jin, B.J.; Verkman, A.S. Test of the ‘glymphatic’ hypothesis demonstrates diffusive and aquaporin-4-independent solute transport in rodent brain parenchyma. Elife 2017, 6, e27679. [Google Scholar] [CrossRef]
- Hladky, S.B.; Barrand, M.A. Mechanisms of fluid movement into, through and out of the brain: Evaluation of the evidence. Fluids Barriers CNS 2014, 11, 26. [Google Scholar] [CrossRef]
- Proulx, S.T. Cerebrospinal fluid outflow: A review of the historical and contemporary evidence for arachnoid villi, perineural routes, and dural lymphatics. Cell Mol. Life Sci. 2021, 78, 2429–2457. [Google Scholar] [CrossRef] [PubMed]
- Rennels, M.L.; Gregory, T.F.; Blaumanis, O.R.; Fujimoto, K.; Grady, P.A. Evidence for a ‘paravascular’ fluid circulation in the mammalian central nervous system, provided by the rapid distribution of tracer protein throughout the brain from the subarachnoid space. Brain Res. 1985, 326, 47–63. [Google Scholar] [CrossRef] [PubMed]
- Balusu, S.; Van Wonterghem, E.; De Rycke, R.; Raemdonck, K.; Stremersch, S.; Gevaert, K.; Brkic, M.; Demeestere, D.; Vanhooren, V.; Hendrix, A.; et al. Identification of a novel mechanism of blood-brain communication during peripheral inflammation via choroid plexus-derived extracellular vesicles. EMBO Mol. Med. 2016, 8, 1162–1183. [Google Scholar] [CrossRef]
- Fernandez-Espejo, E.; de Fonseca, F.R.; Gavito, A.L.; Cordoba-Fernandez, A.; Chacon, J.; de Pablos, A.M. Myeloperoxidase and Advanced Oxidation Protein Products in the Cerebrospinal Fluid in Women and Men with Parkinson’s Disease. Antioxidants 2022, 11, 1088. [Google Scholar] [CrossRef] [PubMed]
- Teismann, P. Myeloperoxidase in the neurodegenerative process of Parkinson’s disease. Dtsch. Med. Wochenschr. 2014, 139, 99–102. [Google Scholar] [CrossRef] [PubMed]
- Yap, Y.W.; Whiteman, M.; Cheung, N.S. Chlorinative stress: An under appreciated mediator of neurodegeneration? Cell Signal 2007, 19, 219–228. [Google Scholar] [CrossRef]
- Ray, R.S.; Katyal, A. Myeloperoxidase: Bridging the gap in neurodegeneration. Neurosci. Biobehav. Rev. 2016, 68, 611–620. [Google Scholar] [CrossRef]
- Jeitner, T.M.; Kalogiannis, M.; Krasnikov, B.F.; Gomolin, I.; Peltier, M.R.; Moran, G.R. Linking Inflammation and Parkinson Disease: Hypochlorous Acid Generates Parkinsonian Poisons. Toxicol. Sci. 2016, 151, 388–402. [Google Scholar] [CrossRef]
- Chung, Y.C.; Kim, S.R.; Jin, B.K. Paroxetine prevents loss of nigrostriatal dopaminergic neurons by inhibiting brain inflammation and oxidative stress in an experimental model of Parkinson’s disease. J. Immunol. 2010, 185, 1230–1237. [Google Scholar] [CrossRef]
- Hirsch, E.C.; Hunot, S. Neuroinflammation in Parkinson’s disease: A target for neuroprotection? Lancet Neurol. 2009, 8, 382–397. [Google Scholar] [CrossRef]
- Soubhye, J.; Aldib, I.; Delporte, C.; Prevost, M.; Dufrasne, F.; Antwerpen, P.V. Myeloperoxidase as a Target for the Treatment of Inflammatory Syndromes: Mechanisms and Structure Activity Relationships of Inhibitors. Curr. Med. Chem. 2016, 23, 3975–4008. [Google Scholar] [CrossRef]
- Kargapolova, Y.; Geissen, S.; Zheng, R.; Baldus, S.; Winkels, H.; Adam, M. The Enzymatic and Non-Enzymatic Function of Myeloperoxidase (MPO) in Inflammatory Communication. Antioxidants 2021, 10, 562. [Google Scholar] [CrossRef] [PubMed]
- Jucaite, A.; Svenningsson, P.; Rinne, J.O.; Cselényi, Z.; Varnäs, K.; Johnström, P.; Amini, N.; Kirjavainen, A.K.; Helin, S.; Minkwitz, M.C.; et al. Effect of the myeloperoxidase inhibitor AZD3241 on microglia: A PET study in Parkinson’s disease. Brain 2015, 138 (Pt 9), 2687–2700. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Ray, A.; Miller, N.M.; Hartwig, D.; Pritchard, K.A.; Dittel, B.N. Inhibition of myeloperoxidase at the peak of experimental autoimmune encephalomyelitis restores blood-brain barrier integrity and ameliorates disease severity. J. Neurochem. 2016, 136, 826–836. [Google Scholar] [CrossRef] [PubMed]
- Stefanova, N.; Georgievska, B.; Eriksson, H.; Poewe, W.; Wenning, G.K. Myeloperoxidase inhibition ameliorates multiple system atrophy-like degeneration in a transgenic mouse model. Neurotox Res. 2012, 21, 393–404. [Google Scholar] [CrossRef]
- Kaindlstorfer, C.; Sommer, P.; Georgievska, B.; Mather, R.J.; Kugler, A.R.; Poewe, W.; Wenning, G.K.; Stefanova, N. Failure of Neuroprotection Despite Microglial Suppression by Delayed-Start Myeloperoxidase Inhibition in a Model of Advanced Multiple System Atrophy: Clinical Implications. Neurotox Res. 2015, 28, 185–194. [Google Scholar] [CrossRef]
- Peng, J.; Pan, J.; Mo, J.; Peng, Y. MPO/HOCl Facilitates Apoptosis and Ferroptosis in the SOD1(G93A) Motor Neuron of Amyotrophic Lateral Sclerosis. Oxid Med. Cell Longev. 2022, 2022, 8217663. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reynolds, W.F.; Malle, E.; Maki, R.A. Thiocyanate Reduces Motor Impairment in the hMPO-A53T PD Mouse Model While Reducing MPO-Oxidation of Alpha Synuclein in Enlarged LYVE1/AQP4 Positive Periventricular Glymphatic Vessels. Antioxidants 2022, 11, 2342. https://doi.org/10.3390/antiox11122342
Reynolds WF, Malle E, Maki RA. Thiocyanate Reduces Motor Impairment in the hMPO-A53T PD Mouse Model While Reducing MPO-Oxidation of Alpha Synuclein in Enlarged LYVE1/AQP4 Positive Periventricular Glymphatic Vessels. Antioxidants. 2022; 11(12):2342. https://doi.org/10.3390/antiox11122342
Chicago/Turabian StyleReynolds, Wanda F., Ernst Malle, and Richard A. Maki. 2022. "Thiocyanate Reduces Motor Impairment in the hMPO-A53T PD Mouse Model While Reducing MPO-Oxidation of Alpha Synuclein in Enlarged LYVE1/AQP4 Positive Periventricular Glymphatic Vessels" Antioxidants 11, no. 12: 2342. https://doi.org/10.3390/antiox11122342
APA StyleReynolds, W. F., Malle, E., & Maki, R. A. (2022). Thiocyanate Reduces Motor Impairment in the hMPO-A53T PD Mouse Model While Reducing MPO-Oxidation of Alpha Synuclein in Enlarged LYVE1/AQP4 Positive Periventricular Glymphatic Vessels. Antioxidants, 11(12), 2342. https://doi.org/10.3390/antiox11122342