Abstract
Intracranial hypertension is a common phenomenon in patients with aneurysmal subarachnoid hemorrhage (aSAH). Elevated intracranial pressure (ICP) plays an important role in early brain injuries and is associated with unfavorable outcomes. Despite advances in the management of aSAH, there is no consensus about the mechanisms involved in ICP increases after aSAH. Recently, a growing body of evidence suggests that oxidative stress (OS) may play a crucial role in physio-pathological changes following aSAH, which may also contribute to increased ICP. Herein, we discuss a potential relation between increased ICP and OS, and resultantly propose antioxidant mechanisms as a potential therapeutic strategy for the treatment of ICP elevation following aSAH.
1. Introduction
Aneurysmal subarachnoid hemorrhage (aSAH) is a devastating neurological disease associated with unfavorable outcome and significant mortality as high as 45% of the cases, especially in young adults [,,]. Unfortunately, recent developments in the diagnostics as well as the treatment of modalities have failed to arrive to an outstanding improvement in the functional outcomes of patients with aSAH [,].
Intracranial hypertension is very common in patients with aSAH, particularly in those with high-grade hemorrhages, in whom high intracranial pressure can lead to a vicious cycle that can be life-threatening if not treated in time []. However, the pathophysiological mechanism of intracranial hypertension after aSAH is complex and has not been fully clarified yet. Currently, there is no consensus guideline for the management of intracranial hypertension after aSAH.
In recent years, a growing body of evidence has suggested that oxidative stress may play a crucial role in pathological changes following aSAH [,,]. Early brain injuries (EBIs), including the disruption of the blood–brain barrier (BBB), cerebral edema, and impaired cerebrovascular autoregulation, are thought to be responsible for elevated intracranial pressure after aSAH [,,,]. Since these pathological changes are significantly associated with oxidative stress, antioxidants may be potential candidates to treat intracranial hypertension after aSAH. Therefore, in this work, we review the mechanisms of OS and intracranial hypertension and their relationships, and propose antioxidants as potential therapeutic strategies for increased ICP after aSAH.
2. Characteristics of ICP in aSAH
The rupture of an intracranial aneurysm causes blood spread into the subarachnoid space, resulting with a sharp increase in the ICP, which can even transcend the average arterial blood pressure (MABP). In fact, there are several factors contributing to the elevated ICP after aSAH, including the presence of extravasated blood in the subarachnoid space, intracerebral hematoma formation, hydrocephalus, cerebral edema, and venous drainage disorders. The typical pattern of ICP changes after aSAH includes a subsequent decrease in the ICP value to a steady state within a few minutes, which nevertheless remains significantly high above the baseline. This phenomenon of declining ICP is explained by the Monro–Kellie hypothesis: the absorption of cerebrospinal fluid into the veins, the transference from the skull to the spinal canal, and the displacement of some of the venous and arterial blood from the cranial cavity []. When these compensations are insufficient, aSAH can lead to elevated ICP and even brain herniation.
2.1. Peak of ICP
The initial elevation of ICP is considered as a protective mechanism that prevents aneurysm from rebleeding []. On the other hand, elevated ICP leads to decreased cerebral blood flow (CBF) and subsequent cerebral ischemia, leading to vascular-derived and cytotoxic cerebral edema, which in turn leads to further elevation of ICP [,]. Therefore, a vicious cycle that constantly causes severe damage to the brain is triggered.
The main reason for the rapid rise in ICP after aSAH is due to the mass effect caused by blood extravasation into the subarachnoid space and the decreased buffering capacity of the intracranial space. The alterations in the dynamics of the cerebrospinal fluid system as well as the presence of blood cells and proteins in the subarachnoid space leading to disturbed cerebrospinal fluid (CSF) circulation and increased outflow resistance significantly contribute to the ICP elevation following aSAH []. Similarly, cerebral congestion caused by the vasoparalysis of distal cerebral arterioles [], acute cerebral edema, and venous drainage disorders can be counted among other pathophysiological mechanisms, inducing an initial sharp increase in ICP. Moreover, the increased ICP has been shown to activate the sympathetic nervous system, which can trigger inflammation and cause an imbalance between the vasodilative and contractile factors [,,,]. Interestingly, an imbalance of brain tissue electrolytes occurs right after aSAH and plays a critical role in disease progression [,], but its relationship with elevated ICP remains unclear.
2.2. Steady State
The elevated ICP value almost always falls down over a few minutes reaching a steady state, the level of which is much lower than the peak level but significantly higher than the baseline level [,,]. It is important to note that, although the ICP values at this stage are well below the peak, the occurrence of intracranial hypertension at this stage is common and associated with poor outcomes [,].
At this stage, the blood breaks into the subarachnoid space, which could block the arachnoid granules and narrow the CSF passages such as the midbrain aqueduct and foramen of Monroe. The subsequent hydrocephalus contributes to the increase in ICP levels and are reported in more than half of the patients with aSAH []. In addition, it has been found that cerebral edema is the main reason for the increase in ICP levels at this stage, both in animal studies and clinical practice [,]. The cerebral edema can be observed in gray and white matter, as well as the deep cerebral nuclei and sometimes in the parasagittal watershed areas [], while the oxidative stress-induced disruption of the BBB and brain ischemia have been proposed as key mechanisms for the development of cerebral edema after aSAH.
3. ICP Monitoring in aSAH
Although a strong association between increased ICP and aSAH has been reported in both experimental and clinical studies, currently, there is a lack of specific recommendations regarding the indications for ICP monitoring in patients with aSAH. ICP monitoring can provide more timely and intensive treatment for patients with aSAH when it comes to preventing secondary brain injury. In an international multicenter observational study enrolled 521 patients with aSAH, Chiara Robba et al. found that ICP monitoring might be associated with lower 6-month mortality, particularly in more severe cases []. Of note, ICP monitoring was probably carried out in selected patients with highly severe aSAH in the reported studies. Nevertheless, future studies are warranted in order to compose new protocols for the indications of ICP monitoring in aSAH.
4. ICP and Outcome
Elevated ICP, conventionally and somewhat arbitrarily defined as ICP above 20 mmHg, is a common phenomenon after aSAH and may contribute to clinical deterioration. Clinical studies have revealed higher mortality rates and poor neurological outcome in patients with elevated ICP after aSAH [,]. The association between elevated ICP after aSAH and poor prognosis has been mainly conducted in retrospective studies, and the interpretation of the results is controversial due to the lack of large prospective studies. Interestingly, the results of a prospective observational study, including a total of 116 aSAH patients by Zoerle et al. on the relationship between elevated ICP and clinical outcome, revealed that although intracranial hypertension was associated with increased mortality, it was not independently related to unfavorable outcomes [,]. In another prospective study, Magni et al. have employed pressure–time dose (PTDICP) to quantify the burden and the time above four predefined thresholds (15, 20, 25, and 30 mmHg), and the high levels of PTDICP with thresholds set at 20, 25, and 30 mmHg were found to be associated with higher mortality at discharge, while moderate PTDICP 30 was related with a poor 6-month outcome []. Similarly, a recent retrospective multicenter study by Giogia Carra et al. demonstrated that the ICP pressure–time burden (duration and intensity of episodes of intracranial hypertension) was independently associated with 12-month outcome []. In this study, the researcher employed an ICP “dose” instead of a particular ICP cutoff and found ICP pressure–time burden to be an independent predictor of functional outcomes.
5. Oxidative Stress in aSAH
Oxidative stress refers to the imbalance between the production of reactive oxygen species (ROS) and defending antioxidant systems. Several sources for the excessive generation of oxidative stress after aSAH have been introduced so far, such as hemoglobin degradation, disrupted mitochondrial respiration, intracellular peroxidases pathways, and disrupted antioxidant systems [].
5.1. Hemoglobin Degradation
Extracellular hemoglobin and its metabolites (hemoglobin–heme–iron axis) are the main sources of ROS during the pathophysiological process after aSAH. After hemolysis, tetrameric hemoglobin is released from red blood cells and it degrades gradually, producing toxic intermediates. In the ferrous (Fe2+) and trivalent (Fe3+) states, heme can react with hydrogen peroxide to generate hydroxyl radicals through the Fenton reaction, which can damage lipid membranes, leading to the production of lipid ROS, cell dysfunction, and even ferroptosis [,,].
5.2. Disrupted Mitochondrial Respiration
During normal mitochondrial respiration, electron transfer is accompanied by electron leakage from the transport chain and subsequent reaction with O2 to produce superoxides. This free radical is usually scavenged by the catalyzing enzyme superoxide dismutase (SOD). Three distinct types of SOD isozymes have been identified in mammals: cytosolic copper zinc SOD (SOD1), mitochondrial manganese SOD (SOD2), and extracellular SOD (SOD3) []. SOD2, the main mitochondrial antioxidant factor and ROS scavenger, is localized to the mitochondrial matrix and has been found to be involved in mitochondrial O2− to H2O2 conversion []. During an ischemic phase (e.g., the ischemic phase after aSAH), the mitochondria become a source of excess free radical production, and the antioxidant enzyme is unable to scavenge this free radical []. Therefore, mitochondrial dysfunction due to ischemic injury following aSAH can lead to the leakage of superoxide anions and the excessive production of ROS. Studies on mitochondria activities after aSAH have consistently found disrupted mitochondrial respiration favoring the production of ROS. Marzatico and Baena et al. have reported increased levels of state 4 mitochondrial respiration and decreased respiratory control ratios following aSAH in association with increased ROS production [,]. Moreover, the overproduction of ROS due to mitochondrial dysfunction has been shown to be a key mechanism for cognitive dysfunction and poor prognosis [,].
5.3. Intracellular Peroxidases Pathways
In addition to the hemoglobin and mitochondria, a number of other enzymatic pathways in association with the production of free radicals have been investigated so far. Among these, several pro-oxidant enzymatic pathways have been thought to be associated with the overproduction of free radicals, including NADPH oxidase (NOX), myeloperoxidase (MPO), and nitric oxide synthase (NOS) [,,,,].
The NOX family is a well-known and important source of ROS which is widely expressed in the central nervous system (CNS) cells except the oligodendrocytes [,]. The NOX catalyzes the transfer of two electrons through the biofilm to produce superoxide anion O2− by using intracellular NADPH as an electron donor and extracellular molecular oxygen as a receptor. Then, O2− is progressively metabolized to H2O2 and∙OH. The two major subtypes in the brain, NOX2 and NOX4, have been shown to significantly increase in the neurons as well as astrocytes around the hematomas of patients with aSAH []. Furthermore, a significant association has been shown between the NOX and delayed cerebrovascular spasm in experimental animal models, suggesting NOX as a potential risk factor for delayed ischemic neurologic deficit (DIND) after aSAH [].
Myeloxidase (MPO) is a heme-containing peroxidase mainly found in the primary azurophilic granules of neutrophils and also in the primary lysosomes of monocytes in small amounts. After aSAH, the neutrophils are recruited in the subarachnoid space by the chemokines and produce large amounts of hypochlorous acid through MPO which in turn causes damage to the lipids, proteins, and DNA. MPO-mediated oxidative injury in CNS has been shown to cause cognitive impairment and neurodegeneration [,,]. Consistent with these findings, clinical studies have revealed a positive correlation between the serum levels of MPO and the occurrence of DCI in patients with aSAH, and this has also been confirmed by experimental animal models [].
The nitric oxide synthase (NOS) family consists of three subtypes: endothelial NOS (eNOS), neuronal NOS (nNOS), and inductive NOS (iNOS). The eNOS and nNOS are expressed constitutively, whereas the expression of iNOS requires a stimulation by the cytokines or other inflammatory products []. As the resident innate immune cells of the CNS, the microglia are initially activated following aSAH []. The microglia can then transform into the M1 phenotype and express the iNOS depending on the transcription factors, including hypoxia-inducible factor-1 and NF-κB. Subsequently, the iNOS increases the levels of NO, leading to free-radical-mediated neuronal damage [,].
5.4. Disrupted Antioxidant Systems
Nuclear factor erythroid-derived 2-related factor 2 (Nrf2), a major regulator of cellular antioxidant response, is widely expressed in the central nervous system and significantly upregulated in neurons, astrocytes, microglia, endothelin cells, and smooth muscle cells after SAH [,]. Nrf2 is a well-known redox-sensitive transcription factor; it is always located in the cytoplasm and is degraded by Kelch-Like Epichlorohydrin-Associated Protein 1 (Keap1). In response to the acute stress of SAH, Nrf2 is upregulated, released from Keap1 and transferred to nucleus, then bound to the antioxidant response element (ARE), which rapidly regulates the transcription of many detoxifying and antioxidant enzymes [,]. These include the catalase, SOD, glutathione reductase, and so on. Meanwhile, the activation of Keap1-Nrf2-ARE can also promote the degradation of erythrocytes and their degradation products through the upregulation of haptoglobin (Hp), hemopexin, HO-1, and ferritin [,,,]. However, these enzyme systems are disrupted and heavily consumed after aSAH, significantly reducing the antioxidant capacity of the brain tissue. In experimental animal models, aSAH has been shown to lead to a decrease in the activities of Zn and Cu-SOD, and studies of human aSAH have revealed a significant increase in the proportion of SOD/GSH-Px activity. In addition, endogenous antioxidant molecules such as glutathione, ascorbic acid, and tocopherols could be depleted after aSAH, which can be another cause of oxidative stress [,]. Moreover, the overexpression of Nrf2 and c-Jun could upregulate the ARE-mediated expression of gamma-glutamylcysteine synthetase (gamma-GCS), a scavenger of ROS []. However, studies on aSAH showed the deleterious effect of the JNK/c-Jun pathway due to pro-inflammatory effects [,].
Mitophagy also plays an important role in the response to the OS after aSAH []. In response to mitochondrial stress after SAH, mitophagy could promote the clearance of irreversibly damaged mitochondria to encourage the survival of other mitochondria and thus the neurons. However, due to the severe injury stress, mitophagy is insufficient after SAH and neurons can inevitably undergo apoptosis [] (Figure 1).
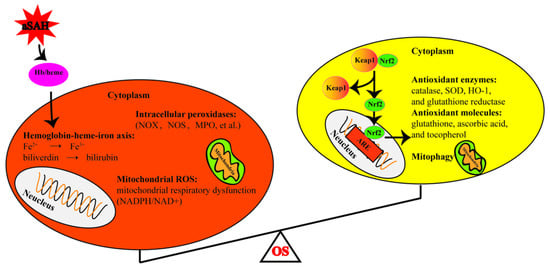
Figure 1.
Schematic diagram of sources of oxidative stress and antioxidant system after subarachnoid hemorrhage. The left circle represents the production of the oxidants, while the right represents the antioxidants. OS: oxidative stress, aSAH: aneurysmal subarachnoid hemorrhage, Hb: hemoglobin, NADPH: nicotinamide adenine dinucleotide phosphate, NOX: NADPH oxidase, MPO: myeloperoxidase, NOS: nitric oxide synthase, SOD: superoxide dismutase. HO-1: heme oxygenase 1, Keap1: Kelch-Like Epichlorohydrin-Associated Protein 1, ARE: antioxidant responsive element, Nrf2: nuclear erythroid-related factor 2.
6. Oxidative Stress and Increased ICP
Intracranial hypertension is a common complication of aSAH, which is also a critical determinator of the prognosis of patients with aSAH. Currently, there is evidence pointing to OS as a major contributor to increased ICP after aSAH. Importantly, impaired BBB is considered one of the most important causes of elevated ICP after aSAH. Specifically, OS can lead to the disruption of the BBB in several ways, including the apoptosis of the endothelial cells [,], damage to the tight-junction proteins [], the activation of matrix metalloproteinases [,], and the elevated levels of aquaporin-4 [] in patients with aSAH. Moreover, augmented levels of superoxide anions in the CSF after aSAH have been reported to display an association with cerebrovascular spasm, which may further lead to cerebral ischemia and elevated ICP [,]. OS-induced vasoconstriction is associated with NO depletion, the suppression of voltage-dependent K+ channels, and the upregulation of R-type Ca2+ channels in the cerebral arteries [,,,]. Moreover, OS-induced damage to the lipids, proteins, and DNA can lead to cytotoxic edema and the subsequent elevation of ICP as well (Figure 2).
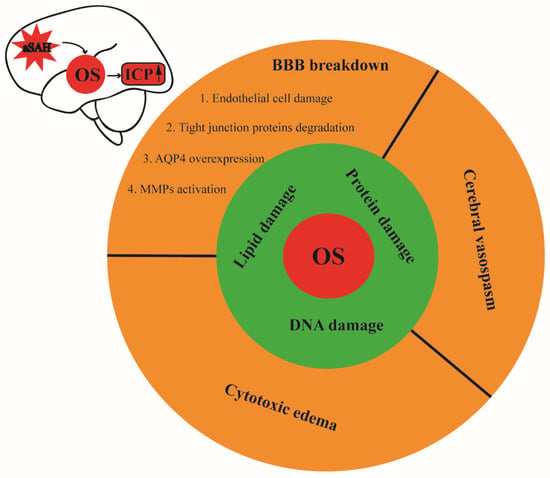
Figure 2.
Schematic illustration of oxidative stress and its contribution to intracranial hypertension after aSAH. Oxidative damage to the lipids, proteins, and DNA can disrupt the BBB, vasoconstriction, and cytotoxic edema after SAH, leading to subsequent elevation of the intracranial pressure. OS: oxidative stress, ICP: intracranial pressure, aSAH: aneurysmal subarachnoid hemorrhage, BBB: blood–brain barrier.
7. Treatment
Intracranial hypertension is common among patients with aSAH. However, there are no specific treatment guidelines, and many current treatment recommendations are based on traumatic brain injury (TBI) guidelines. These recommendations include standard medical therapy (temperature control, head elevation, controlled hyperventilation with PaCO2 between 30 and 35 mmHg, osmotherapy, and CSF drainage), the surgical removal of space-occupying intracranial hematoma, and decompressive craniectomy [,,,]. Although patients potentially benefit from standard medical therapy, intracranial hypertension constitutes a major challenge when it is refractory []. We comparatively reviewed the two main treatment strategies (CSF draining and anti-OS therapy), and propose the best possible method in the management of increased ICP following aSAH.
Guidelines for the management of aSAH by AHA/ASA and the European Stroke Organization both recommend the use of CSF drainage to treat SAH-associated hydrocephalus [,]. The placement of an external ventricular drainage (EVD), continuous lumbar drainage, and lumbar puncture are the three commonly used strategies for CSF drainage in clinical practice. CSF drainage not only reduces intracranial pressure by alleviating hydrocephalus, but also allows the removal of accumulated intraventricular blood, thereby diminishing secondary damage to the brain tissue [,,]. Consistently, decreasing elevated ICP via CSF drainage has been shown to correlate with improved cerebral microcirculation in patients with aSAH []. However, there is no consensus on the daily volume of required drainage, which is generally kept below 200 mL, adjusted according to the sign and symptoms of hydrocephalus or the ICP treatment threshold [,,]. It should be noted that CSF over drainage and compensatory brain hyperemia can result in the malabsorption of CSF due to increased intracranial venous pressure and impaired CSF outflow. Therefore, impaired CSF circulation may lead to the aggravation of clinical symptoms and even coma, which can be reversed by immediately quitting the drainage []. The simultaneous monitoring of ICP and lumbar pressure was previously used as a treatment strategy to avoid lumbar over drainage by identifying progressive pressure gradients in advance []. With the development of technologies in new external drainage systems, it is now possible to regulate flow while monitoring ICP []. The system can detect the changes in the brain compliant by monitoring the ICP, thereby allowing the regulation of the CSF drainage based on the ICP levels. This avoids large fluctuations in the ICP and effectively prevents from excessive or insufficient drainage of the CSF. In fact, optimal CSF drainage should eliminate the blood degradations from the CSF circulation routes, and balance the formation and absorption of the CSF by ensuring proper brain compliance and cerebral blood supply.
During the last decade, the results of scientific research have shown that the genetic and pharmacological inhibition of OS alleviates multiple components of EBI, including elevated ICP. Hemoglobin is the main source of ROS after aSAH and has been shown as a potential therapeutic target. The physical clearance of blood either by surgically removing the hematoma or via CSF drainage has been proven to reduce the incidence of cerebral vasospasm, which might reduce intracranial hypertension [,]. Nrf2 is a transcription factor widely expressed in the CNS, and plays an important role in attenuating oxidative insults by regulating the expression of the genes involved in antioxidative response. Nrf2 has been shown to attenuate the disruption of the BBB, cerebral edema, and apoptosis through the Keap1-Nrf2-ARE pathway. Specifically, a variety of Nrf2 system activators, including the andrographolide, oleanolic acid, paeoniflorin, salvianolic acid A and B, aloperine, mangiferin, dimethylfumarate, astaxanthin, and L-cysteine, have been shown to significantly attenuate EBI after aSAH [,,,,,,,,]. Similarly, docosahexaenoic acid, SS31 (a cell-permeable novel mitochondria-targeted peptide), Mdivi-1 (a selective Drp1 inhibitor), Mfn1-βIIPKC, fucoxanthin, bakuchiol, hydrogen, and metformin have been shown to have antioxidant stress and reduced cerebral edema after aSAH, and may be associated with its reduction in mitochondrial ROS [,,,,,,,,]. The pharmacologic agents that potentially attenuate aSAH-induced OS and subsequent disruptions in the BBB and related pathways are summarized in Table 1 [,,]. Since preclinical trials of several antioxidants mentioned above have shown them to protect the BBB and reduce brain edema, therapeutic strategies against ROS/OS may prevent ICP elevation after aSAH.

Table 1.
The results of experimental studies on antioxidant systems and intracranial hypertension following aSAH.
8. Conclusions and Future Directions
The mechanism of intracranial hypertension caused by aSAH is multifaceted and contributes significantly to the prognosis of the patient. Oxidative stress is an important pathophysiological mechanism that causes intracranial hypertension, rendering oxidative stress as a potential target for the treatment of intracranial hypertension. The complex network among oxidative stress, intracranial hypertension after aSAH, and the outcome is still unclear and requires further accurate identification. Thus, future research on the contribution of different pathways of oxidative stress to raised ICP after aSAH is of great importance. Preclinical trials of several antioxidants have been shown to protect the BBB, reduce brain edema, and improve prognosis in aSAH animal models. Thus, we propose antioxidant mechanisms as a potential therapeutic strategy for the treatment of ICP elevation after aSAH. However, future large randomized controlled trials are warranted to acquire a better understanding on the relationship among oxidative stress, raised ICP, and the clinical outcomes in patient with aSAH.
Author Contributions
Writing—original draft preparation, G.H.; writing—review and editing, P.E. and J.M.; funding acquisition, J.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Natural Science Foundation of China, grant number 81901336.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Johnston, S.C.; Selvin, S.; Gress, D.R. The Burden, Trends, and Demographics of Mortality from Subarachnoid Hemorrhage. Neurology 1998, 50, 1413–1418. [Google Scholar] [CrossRef]
- le Roux, A.A.; Wallace, M.C. Outcome and Cost of Aneurysmal Subarachnoid Hemorrhage. Neurosurg. Clin. N. Am. 2010, 21, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Sehba, F.A.; Hou, J.; Pluta, R.M.; Zhang, J.H. The Importance of Early Brain Injury after Subarachnoid Hemorrhage. Prog. Neurobiol. 2012, 97, 14–37. [Google Scholar] [CrossRef] [PubMed]
- Ingelmo Ingelmo, I.; Fabregas Julia, N.; Rama-Maceiras, P.; Hernandez-Palazon, J.; Rubio Romero, R.; Carmona Aurioles, J.; Grupo Ad Hoc de la Seccion de Neurociencia de la Sociedad Espanola de Anestesiologia; Reanimación y Terapéutica del Dolor. Subarachnoid Hemorrhage: Epidemiology, Social Impact and A Multidisciplinary Approach. Rev. Esp. Anestesiol. Reanim. 2010, 57 (Suppl. 2), S4–S15. [Google Scholar] [PubMed]
- van Gijn, J.; Rinkel, G.J. Subarachnoid Haemorrhage: Diagnosis, Causes and Management. Brain 2001, 124, 249–278. [Google Scholar] [CrossRef] [PubMed]
- Heuer, G.G.; Smith, M.J.; Elliott, J.P.; Winn, H.R.; LeRoux, P.D. Relationship between Intracranial Pressure and Other Clinical Variables in Patients with Aneurysmal Subarachnoid Hemorrhage. J. Neurosurg. 2004, 101, 408–416. [Google Scholar] [CrossRef]
- Wu, F.; Liu, Z.; Li, G.; Zhou, L.; Huang, K.; Wu, Z.; Zhan, R.; Shen, J. Inflammation and Oxidative Stress: Potential Targets for Improving Prognosis after Subarachnoid Hemorrhage. Front. Cell. Neurosci. 2021, 15, 739506. [Google Scholar] [CrossRef]
- Yang, Y.; Chen, S.; Zhang, J.M. The Updated Role of Oxidative Stress in Subarachnoid Hemorrhage. Curr. Drug Deliv. 2017, 14, 832–842. [Google Scholar] [CrossRef]
- Lin, F.; Li, R.; Tu, W.J.; Chen, Y.; Wang, K.; Chen, X.; Zhao, J. An Update on Antioxidative Stress Therapy Research for Early Brain Injury after Subarachnoid Hemorrhage. Front. Aging Neurosci. 2021, 13, 772036. [Google Scholar] [CrossRef]
- Warren, M.C.; Bump, E.A.; Medeiros, D.; Braunhut, S.J. Oxidative Stress-Induced Apoptosis of Endothelial Cells. Free Radic. Biol. Med. 2000, 29, 537–547. [Google Scholar] [CrossRef]
- Qing, W.G.; Dong, Y.Q.; Ping, T.Q.; Lai, L.G.; Fang, L.D.; Min, H.W.; Xia, L.; Heng, P.Y. Brain Edema after Intracerebral Hemorrhage in Rats: The Role of Iron Overload and Aquaporin 4. J. Neurosurg. 2009, 110, 462–468. [Google Scholar] [CrossRef]
- Fumoto, T.; Naraoka, M.; Katagai, T.; Li, Y.; Shimamura, N.; Ohkuma, H. The Role of Oxidative Stress in Microvascular Disturbances after Experimental Subarachnoid Hemorrhage. Transl. Stroke Res. 2019, 10, 684–694. [Google Scholar] [CrossRef] [PubMed]
- Schenck, H.; Netti, E.; Teernstra, O.; De Ridder, I.; Dings, J.; Niemela, M.; Temel, Y.; Hoogland, G.; Haeren, R. The Role of the Glycocalyx in the Pathophysiology of Subarachnoid Hemorrhage-Induced Delayed Cerebral Ischemia. Front. Cell. Dev. Biol. 2021, 9, 731641. [Google Scholar] [CrossRef]
- Mokri, B. The Monro-Kellie Hypothesis: Applications in CSF Volume Depletion. Neurology 2001, 56, 1746–1748. [Google Scholar] [CrossRef] [PubMed]
- Nornes, H. The Role of Intracranial Pressure in The Arrest of Hemorrhage in Patients with Ruptured Intracranial Aneurysm. J. Neurosurg. 1973, 39, 226–234. [Google Scholar] [CrossRef] [PubMed]
- Keep, R.F.; Andjelkovic, A.V.; Stamatovic, S.M.; Shakui, P.; Ennis, S.R. Ischemia-Induced Endothelial Cell Dysfunction. Acta Neurochir. Suppl. 2005, 95, 399–402. [Google Scholar] [CrossRef]
- Sabri, M.; Lass, E.; Macdonald, R.L. Early Brain Injury: A Common Mechanism in Subarachnoid Hemorrhage and Global Cerebral Ischemia. Stroke Res. Treat. 2013, 2013, 394036. [Google Scholar] [CrossRef]
- Brinker, T.; Seifert, V.; Stolke, D. Acute Changes in The Dynamics of The Cerebrospinal Fluid System during Experimental Subarachnoid Hemorrhage. Neurosurgery 1990, 27, 369–372. [Google Scholar] [CrossRef]
- Grote, E.; Hassler, W. The Critical First Minutes after Subarachnoid Hemorrhage. Neurosurgery 1988, 22, 654–661. [Google Scholar] [CrossRef]
- Furuichi, S.; Endo, S.; Haji, A.; Takeda, R.; Nisijima, M.; Takaku, A. Related Changes in Sympathetic Activity, Cerebral Blood Flow and Intracranial Pressure, and Effect of an Alpha-blocker in Experimental Subarachnoid Haemorrhage. Acta Neurochir. 1999, 141, 415–423, discussion 414–423. [Google Scholar] [CrossRef]
- Westermaier, T.; Jauss, A.; Eriskat, J.; Kunze, E.; Roosen, K. Acute Vasoconstriction: Decrease and Recovery of Cerebral Blood Flow after Various Intensities of Experimental Subarachnoid Hemorrhage in Rats. J. Neurosurg. 2009, 110, 996–1002. [Google Scholar] [CrossRef] [PubMed]
- Graetz, D.; Nagel, A.; Schlenk, F.; Sakowitz, O.; Vajkoczy, P.; Sarrafzadeh, A. High ICP as Trigger of Proinflammatory IL-6 Cytokine Activation in Aneurysmal Subarachnoid Hemorrhage. Neurol. Res. 2010, 32, 728–735. [Google Scholar] [CrossRef]
- Makino, K.; Osuka, K.; Watanabe, Y.; Usuda, N.; Hara, M.; Aoyama, M.; Takayasu, M.; Wakabayashi, T. Increased ICP Promotes CaMKII-Mediated Phosphorylation of Neuronal NOS at Ser(8)(4)(7) in The Hippocampus Immediately after Subarachnoid Hemorrhage. Brain Res. 2015, 1616, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Trojanowski, T. Early Effects of Experimental Arterial Subarachnoid Haemorrhage on The Cerebral Circulation. Part I: Experimental Subarachnoid Haemorrhage in Cat and Its Pathophysiological Effects. Methods of Regional Cerebral Blood Flow Measurement and Evaluation of Microcirculation. Acta Neurochir. 1984, 72, 79–94. [Google Scholar] [CrossRef] [PubMed]
- Caner, B.; Hou, J.; Altay, O.; Fujii, M.; Zhang, J.H. Transition of Research Focus from Vasospasm to Early Brain Injury after Subarachnoid Hemorrhage. J. Neurochem. 2012, 123 (Suppl. 2), 12–21. [Google Scholar] [CrossRef]
- Shimizu, T.; Hishikawa, T.; Nishihiro, S.; Shinji, Y.; Takasugi, Y.; Haruma, J.; Hiramatsu, M.; Kawase, H.; Sato, S.; Mizoue, R.; et al. NADH Fluorescence Imaging and The Histological Impact of Cortical Spreading Depolarization during the Acute Phase of Subarachnoid Hemorrhage in Rats. J. Neurosurg. 2018, 128, 137–143. [Google Scholar] [CrossRef]
- Lu, J.; Chen, F.; Cai, B.; Chen, F.; Kang, D. A Rabbit Model of Aneurysmal Subarachnoid Hemorrhage by Ear Central Artery-Suprasellar Cistern Shunt. J. Clin. Neurosci. 2017, 44, 300–305. [Google Scholar] [CrossRef]
- Friedrich, V.; Bederson, J.B.; Sehba, F.A. Gender Influences the Initial Impact of Subarachnoid Hemorrhage: An Experimental Investigation. PLoS ONE 2013, 8, e80101. [Google Scholar] [CrossRef]
- Lv, Y.; Wang, D.; Lei, J.; Tan, G. Clinical Observation of the Time Course of Raised Intracranial Pressure after Subarachnoid Hemorrhage. Neurol. Sci. 2015, 36, 1203–1210. [Google Scholar] [CrossRef]
- Macdonald, R.L.; Schweizer, T.A. Spontaneous Subarachnoid Haemorrhage. Lancet 2017, 389, 655–666. [Google Scholar] [CrossRef]
- Altay, O.; Suzuki, H.; Hasegawa, Y.; Caner, B.; Krafft, P.R.; Fujii, M.; Tang, J.; Zhang, J.H. Isoflurane Attenuates Blood-Brain Barrier Disruption in Ipsilateral Hemisphere after Subarachnoid Hemorrhage in Mice. Stroke 2012, 43, 2513–2516. [Google Scholar] [CrossRef]
- Ahn, S.H.; Savarraj, J.P.; Pervez, M.; Jones, W.; Park, J.; Jeon, S.B.; Kwon, S.U.; Chang, T.R.; Lee, K.; Kim, D.H.; et al. The Subarachnoid Hemorrhage Early Brain Edema Score Predicts Delayed Cerebral Ischemia and Clinical Outcomes. Neurosurgery 2018, 83, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Kamiya, K.; Kuyama, H.; Symon, L. An Experimental Study of the Acute Stage of subarachnoid hemorrhage. J. Neurosurg. 1983, 59, 917–924. [Google Scholar] [CrossRef]
- Robba, C.; Graziano, F.; Rebora, P.; Elli, F.; Giussani, C.; Oddo, M.; Meyfroidt, G.; Helbok, R.; Taccone, F.S.; Prisco, L.; et al. Intracranial Pressure Monitoring in Patients with Acute Brain Injury in the Intensive Care Unit (SYNAPSE-ICU): An International, Prospective Observational Cohort Study. Lancet Neurol. 2021, 20, 548–558. [Google Scholar] [CrossRef] [PubMed]
- Zoerle, T.; Lombardo, A.; Colombo, A.; Longhi, L.; Zanier, E.R.; Rampini, P.; Stocchetti, N. Intracranial Pressure after Subarachnoid Hemorrhage. Crit. Care Med. 2015, 43, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Cossu, G.; Messerer, M.; Stocchetti, N.; Levivier, M.; Daniel, R.T.; Oddo, M. Intracranial Pressure and Outcome in Critically Ill Patients with Aneurysmal Subarachnoid Hemorrhage: A Systematic Review. Minerva Anestesiol. 2016, 82, 684–696. [Google Scholar]
- Magni, F.; Pozzi, M.; Rota, M.; Vargiolu, A.; Citerio, G. High-Resolution Intracranial Pressure Burden and Outcome in Subarachnoid Hemorrhage. Stroke 2015, 46, 2464–2469. [Google Scholar] [CrossRef]
- Carra, G.; Elli, F.; Ianosi, B.; Flechet, M.; Huber, L.; Rass, V.; Depreitere, B.; Guiza, F.; Meyfroidt, G.; Citerio, G.; et al. Association of Dose of Intracranial Hypertension with Outcome in Subarachnoid Hemorrhage. Neurocrit Care. 2021, 34, 722–730. [Google Scholar] [CrossRef]
- Ayer, R.E.; Zhang, J.H. Oxidative Stress in Subarachnoid Haemorrhage: Significance in Acute Brain Injury and Vasospasm. Acta Neurochir. Suppl. 2008, 104, 33–41. [Google Scholar] [CrossRef]
- Sehba, F.A.; Pluta, R.M.; Zhang, J.H. Metamorphosis of Subarachnoid Hemorrhage Research: From Delayed Vasospasm to Early Brain Injury. Mol. Neurobiol. 2011, 43, 27–40. [Google Scholar] [CrossRef]
- Blackburn, S.L.; Kumar, P.T.; McBride, D.; Zeineddine, H.A.; Leclerc, J.; Choi, H.A.; Dash, P.K.; Grotta, J.; Aronowski, J.; Cardenas, J.C.; et al. Unique Contribution of Haptoglobin and Haptoglobin Genotype in Aneurysmal Subarachnoid Hemorrhage. Front. Physiol. 2018, 9, 592. [Google Scholar] [CrossRef] [PubMed]
- Che, M.; Wang, R.; Li, X.; Wang, H.Y.; Zheng, X.F.S. Expanding Roles of Superoxide Dismutases in Cell Regulation and Cancer. Drug Discov Today. 2016, 21, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.H.; Kim, E.M.; Song, J.Y.; Park, J.K.; Um, H.D. Mitochondrial Superoxide dismutase 2 Mediates Gamma-Irradiation-Induced Cancer Cell Invasion. Exp. Mol. Med. 2019, 51, 1–10. [Google Scholar] [CrossRef]
- Papay, F.A.; Levine, H.L.; Schiavone, W.A. Facial Fuzz and Funny Findings. Facial Hair Causing Otalgia and Oropharyngeal pain. Cleve Clin J Med. 1989, 56, 273–276. [Google Scholar] [CrossRef]
- Marzatico, F.; Gaetani, P.; Silvani, V.; Lombardi, D.; Sinforiani, E.; Rodriguez y Baena, R. Experimental Isobaric Subarachnoid Hemorrhage: Regional Mitochondrial Function during the Acute and Late Phase. Surg. Neurol. 1990, 34, 294–300. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez y Baena, R.; Gaetani, P.; Silvani, V.; Spanu, G.; Marzatico, F. Effect of Nimodipine on Mitochondrial Respiration in Different Rat Brain Areas after Subarachnoid Haemorrhage. Acta Neurochir. Suppl. 1988, 43, 177–181. [Google Scholar] [CrossRef]
- Moro, M.A.; Almeida, A.; Bolanos, J.P.; Lizasoain, I. Mitochondrial Respiratory Chain and Free Radical Generation in stroke. Free Radic. Biol. Med. 2005, 39, 1291–1304. [Google Scholar] [CrossRef] [PubMed]
- Cobley, J.N. Mechanisms of Mitochondrial ROS Production in Assisted Reproduction: The Known, the Unknown, and the Intriguing. Antioxidants 2020, 9, 933. [Google Scholar] [CrossRef]
- Ma, M.W.; Wang, J.; Zhang, Q.; Wang, R.; Dhandapani, K.M.; Vadlamudi, R.K.; Brann, D.W. NADPH Oxidase in Brain Injury and Neurodegenerative Disorders. Mol. Neurodegener. 2017, 12, 7. [Google Scholar] [CrossRef]
- Cantu-Medellin, N.; Kelley, E.E. Xanthine Oxidoreductase-Catalyzed Reactive Species Generation: A Process in Critical Need of Reevaluation. Redox Biol. 2013, 1, 353–358. [Google Scholar] [CrossRef]
- Hou, L.; Zhang, L.; Hong, J.S.; Zhang, D.; Zhao, J.; Wang, Q. Nicotinamide Adenine Dinucleotide Phosphate Oxidase and Neurodegenerative Diseases: Mechanisms and Therapy. Antioxid. Redox Signal. 2020, 33, 374–393. [Google Scholar] [CrossRef] [PubMed]
- Chu, Y.; Wilson, K.; Gu, H.; Wegman-Points, L.; Dooley, S.A.; Pierce, G.L.; Cheng, G.; Pena Silva, R.A.; Heistad, D.D.; Hasan, D. Myeloperoxidase is Increased in Human Cerebral Aneurysms and Increases Formation and Rupture of Cerebral Aneurysms in Mice. Stroke 2015, 46, 1651–1656. [Google Scholar] [CrossRef] [PubMed]
- Cooney, S.J.; Bermudez-Sabogal, S.L.; Byrnes, K.R. Cellular and Temporal Expression of NADPH Oxidase (NOX) Isotypes after Brain Injury. J. Neuroinflammation 2013, 10, 155. [Google Scholar] [CrossRef] [PubMed]
- Sorce, S.; Krause, K.H. NOX Enzymes in the Central Nervous System: From Signaling to Disease. Antioxid. Redox Signal. 2009, 11, 2481–2504. [Google Scholar] [CrossRef]
- Zhang, L.; Li, Z.; Feng, D.; Shen, H.; Tian, X.; Li, H.; Wang, Z.; Chen, G. Involvement of Nox2 and Nox4 NADPH Oxidases in Early Brain Injury after Subarachnoid Hemorrhage. Free Radic. Res. 2017, 51, 316–328. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.E.; Suh, Y.S.; Lee, M.S.; Kim, K.Y.; Lee, J.H.; Lee, H.S.; Hong, K.W.; Kim, C.D. Vascular NAD(P)H Oxidase Triggers Delayed Cerebral Vasospasm after Subarachnoid Hemorrhage in Rats. Stroke 2002, 33, 2687–2691. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.K.; Pennathur, S.; Perier, C.; Tieu, K.; Teismann, P.; Wu, D.C.; Jackson-Lewis, V.; Vila, M.; Vonsattel, J.P.; Heinecke, J.W.; et al. Ablation of The Inflammatory Enzyme Myeloperoxidase Mitigates Features of Parkinson’s Disease in Mice. J. Neurosci. 2005, 25, 6594–6600. [Google Scholar] [CrossRef] [PubMed]
- Green, P.S.; Mendez, A.J.; Jacob, J.S.; Crowley, J.R.; Growdon, W.; Hyman, B.T.; Heinecke, J.W. Neuronal Expression of Myeloperoxidase is Increased in Alzheimer’s Disease. J. Neurochem. 2004, 90, 724–733. [Google Scholar] [CrossRef]
- Nagra, R.M.; Becher, B.; Tourtellotte, W.W.; Antel, J.P.; Gold, D.; Paladino, T.; Smith, R.A.; Nelson, J.R.; Reynolds, W.F. Immunohistochemical and Genetic Evidence of Myeloperoxidase Involvement in Multiple Sclerosis. J. Neuroimmunol. 1997, 78, 97–107. [Google Scholar] [CrossRef]
- Chen, Z.Q.; Mou, R.T.; Feng, D.X.; Wang, Z.; Chen, G. The Role of Nitric Oxide in stroke. Med. Gas Res. 2017, 7, 194–203. [Google Scholar] [CrossRef]
- Khey, K.M.W.; Huard, A.; Mahmoud, S.H. Inflammatory Pathways Following Subarachnoid Hemorrhage. Cell. Mol. Neurobiol. 2020, 40, 675–693. [Google Scholar] [CrossRef] [PubMed]
- Lenz, I.J.; Plesnila, N.; Terpolilli, N.A. Role of Endothelial Nitric Oxide Synthase for Early Brain Injury after Subarachnoid Hemorrhage in Mice. J. Cereb. Blood Flow Metab. 2021, 41, 1669–1681. [Google Scholar] [CrossRef] [PubMed]
- Sehba, F.A.; Bederson, J.B. Nitric Oxide in Early Brain Injury after Subarachnoid Hemorrhage. Acta Neurochir. Suppl. 2011, 110, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Chen, G.; Zhu, W.W.; Zhou, D. Activation of Nuclear Factor-Erythroid 2-Related Factor 2 (Nrf2) in the Basilar Artery after Subarachnoid Hemorrhage in Rats. Ann. Clin. Lab. Sci. 2010, 40, 233–239. [Google Scholar] [PubMed]
- Zolnourian, A.; Galea, I.; Bulters, D. Neuroprotective Role of the Nrf2 Pathway in Subarachnoid Haemorrhage and Its Therapeutic Potential. Oxidative Med. Cell. Longev. 2019, 2019, 6218239. [Google Scholar] [CrossRef]
- Antognelli, C.; Trapani, E.; Delle Monache, S.; Perrelli, A.; Daga, M.; Pizzimenti, S.; Barrera, G.; Cassoni, P.; Angelucci, A.; Trabalzini, L.; et al. KRIT1 Loss-of-Function Induces A Chronic Nrf2-Mediated Adaptive Homeostasis That Sensitizes Cells to Oxidative Stress: Implication for Cerebral Cavernous Malformation disease. Free Radic. Biol. Med. 2018, 115, 202–218. [Google Scholar] [CrossRef]
- Harada, N.; Kanayama, M.; Maruyama, A.; Yoshida, A.; Tazumi, K.; Hosoya, T.; Mimura, J.; Toki, T.; Maher, J.M.; Yamamoto, M.; et al. Nrf2 Regulates Ferroportin 1-Mediated Iron Efflux and Counteracts Lipopolysaccharide-Induced Ferroportin 1 mRNA Suppression in Macrophages. Arch. Biochem. Biophys. 2011, 508, 101–109. [Google Scholar] [CrossRef]
- Chen, M.; Regan, R.F. Time Course of Increased Heme Oxygenase Activity and Expression after Experimental Intracerebral Hemorrhage: Correlation with Oxidative Injury. J. Neurochem. 2007, 103, 2015–2021. [Google Scholar] [CrossRef]
- Morris, C.M.; Candy, J.M.; Edwardson, J.A.; Bloxham, C.A.; Smith, A. Evidence For the Localization of Haemopexin Immunoreactivity in Neurones in the Human Brain. Neurosci. Lett. 1993, 149, 141–144. [Google Scholar] [CrossRef]
- Zhao, X.; Song, S.; Sun, G.; Strong, R.; Zhang, J.; Grotta, J.C.; Aronowski, J. Neuroprotective Role of Haptoglobin after Intracerebral Hemorrhage. J. Neurosci. 2009, 29, 15819–15827. [Google Scholar] [CrossRef]
- Jeyapaul, J.; Jaiswal, A.K. Nrf2 and c-Jun Regulation of Antioxidant Response Element (ARE)-Mediated Expression and Induction of Gamma-Glutamylcysteine Synthetase Heavy Subunit Gene. Biochem. Pharmacol. 2000, 59, 1433–1439. [Google Scholar] [CrossRef] [PubMed]
- Ling, G.Q.; Li, X.F.; Lei, X.H.; Wang, Z.Y.; Ma, D.Y.; Wang, Y.N.; Ye, W. c-Jun N-terminal Kinase Inhibition Attenuates Early Brain Injury Induced Neuronal Apoptosis via Decreasing p53 Phosphorylation and Mitochondrial Apoptotic Pathway Activation in Subarachnoid Hemorrhage Rats. Mol. Med. Rep. 2019, 19, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Yatsushige, H.; Ostrowski, R.P.; Tsubokawa, T.; Colohan, A.; Zhang, J.H. Role of c-Jun N-Terminal Kinase in Early Brain Injury after Subarachnoid Hemorrhage. J. Neurosci. Res. 2007, 85, 1436–1448. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.L.; Chiang, S.; Kalinowski, D.S.; Bae, D.H.; Sahni, S.; Richardson, D.R. The Role of the Antioxidant Response in Mitochondrial Dysfunction in Degenerative Diseases: Cross-Talk between Antioxidant Defense, Autophagy, and Apoptosis. Oxidative Med. Cell. Longev. 2019, 2019, 6392763. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Wu, P.; Budbazar, E.; Zhu, Q.; Sun, C.; Mo, J.; Peng, J.; Gospodarev, V.; Tang, J.; Shi, H.; et al. Mitophagy Reduces Oxidative Stress Via Keap1 (Kelch-Like Epichlorohydrin-Associated Protein 1)/Nrf2 (Nuclear Factor-E2-Related Factor 2)/PHB2 (Prohibitin 2) Pathway after Subarachnoid Hemorrhage in Rats. Stroke 2019, 50, 978–988. [Google Scholar] [CrossRef]
- Link, T.E.; Murakami, K.; Beem-Miller, M.; Tranmer, B.I.; Wellman, G.C. Oxyhemoglobin-Induced Expression of R-type Ca2+ Channels in Cerebral Arteries. Stroke 2008, 39, 2122–2128. [Google Scholar] [CrossRef]
- Ishiguro, M.; Morielli, A.D.; Zvarova, K.; Tranmer, B.I.; Penar, P.L.; Wellman, G.C. Oxyhemoglobin-Induced Suppression of Voltage-Dependent K+ Channels in Cerebral Arteries by Enhanced Tyrosine Kinase Activity. Circ. Res. 2006, 99, 1252–1260. [Google Scholar] [CrossRef]
- Sabri, M.; Ai, J.; Knight, B.; Tariq, A.; Jeon, H.; Shang, X.; Marsden, P.A.; Loch Macdonald, R. Uncoupling of Endothelial Nitric Oxide Synthase after Experimental Subarachnoid Hemorrhage. J. Cereb. Blood Flow Metab. 2011, 31, 190–199. [Google Scholar] [CrossRef]
- Rangel-Castilla, L.; Gopinath, S.; Robertson, C.S. Management of Intracranial Hypertension. Neurol. Clin. 2008, 26, 521–541. [Google Scholar] [CrossRef]
- Mak, C.H.; Lu, Y.Y.; Wong, G.K. Review and Recommendations on Management of Refractory Raised Intracranial Pressure in Aneurysmal Subarachnoid Hemorrhage. Vasc. Health Risk Manag. 2013, 9, 353–359. [Google Scholar] [CrossRef][Green Version]
- Carney, N.; Totten, A.M.; O’Reilly, C.; Ullman, J.S.; Hawryluk, G.W.; Bell, M.J.; Bratton, S.L.; Chesnut, R.; Harris, O.A.; Kissoon, N.; et al. Guidelines for the Management of Severe Traumatic Brain Injury, Fourth Edition. Neurosurgery 2017, 80, 6–15. [Google Scholar] [CrossRef] [PubMed]
- Alotaibi, N.M.; Wang, J.Z.; Pasarikovski, C.R.; Guha, D.; Al-Mufti, F.; Mamdani, M.; Saposnik, G.; Schweizer, T.A.; Macdonald, R.L. Management of Raised Intracranial Pressure in Aneurysmal Subarachnoid Hemorrhage: Time for a consensus? Neurosurg. Focus 2017, 43, E13. [Google Scholar] [CrossRef] [PubMed]
- Steiner, T.; Juvela, S.; Unterberg, A.; Jung, C.; Forsting, M.; Rinkel, G.; European Stroke, O. European Stroke Organization Guidelines for the Management of Intracranial Aneurysms and Subarachnoid Haemorrhage. Cerebrovasc Dis. 2013, 35, 93–112. [Google Scholar] [CrossRef] [PubMed]
- Connolly, E.S., Jr.; Rabinstein, A.A.; Carhuapoma, J.R.; Derdeyn, C.P.; Dion, J.; Higashida, R.T.; Hoh, B.L.; Kirkness, C.J.; Naidech, A.M.; Ogilvy, C.S.; et al. Guidelines for the Management of Aneurysmal Subarachnoid Hemorrhage: A Guideline for Healthcare Professionals from the American Heart Association/american Stroke Association. Stroke 2012, 43, 1711–1737. [Google Scholar] [CrossRef]
- Li, H.; Wang, W. Evaluation of the Effectiveness of Lumbar Punctures in Aneurysmal Subarachnoid Hemorrhage Patient with External Ventricular Drainage. World Neurosurg. 2021, 151, e1–e9. [Google Scholar] [CrossRef]
- Konovalov, A.; Shekhtman, O.; Pilipenko, Y.; Okishev, D.; Ershova, O.; Oshorov, A.; Abramyan, A.; Kurzakova, I.; Eliava, S. External Ventricular Drainage in Patients with Acute Aneurysmal Subarachnoid Hemorrhage after Microsurgical Clipping: Our 2006–2018 Experience and a Literature Review. Cureus 2021, 13, e12951. [Google Scholar] [CrossRef]
- Duan, F.; Wang, G.; Ma, X.; Zhao, Y.; Xu, X.; Dong, F. A Controlled Study of Continuous Lumbar Drainage of Fluid and Lumbar Puncture Drainage for Aneurysmal SAH after Intracranial Aneurysm Clipping. J. Healthcare Eng. 2021, 2021, 2827493. [Google Scholar] [CrossRef]
- Wang, A.Y.; Hsieh, P.C.; Chen, C.C.; Chin, S.C.; Wu, Y.M.; Chen, C.T.; Chang, C.H.; Wu, T.E. Effect of Intracranial Pressure Control on Improvement of Cerebral Perfusion after Acute Subarachnoid Hemorrhage: A Comparative Angiography Study Based on Temporal Changes of Intracranial Pressure and Systemic Pressure. World Neurosurg. 2018, 120, e290–e296. [Google Scholar] [CrossRef]
- van Lieshout, J.H.; Pumplun, I.; Fischer, I.; Kamp, M.A.; Cornelius, J.F.; Steiger, H.J.; Boogaarts, H.D.; Petridis, A.K.; Beseoglu, K. Volume of Cerebrospinal Fluid Drainage as a Predictor for Pretreatment Aneurysmal Rebleeding. J. Neurosurg. 2018, 128, 1778–1784. [Google Scholar] [CrossRef]
- Olson, D.M.; Zomorodi, M.; Britz, G.W.; Zomorodi, A.R.; Amato, A.; Graffagnino, C. Continuous Cerebral Spinal Fluid Drainage Associated with Complications in Patients Admitted with Subarachnoid Hemorrhage. J. Neurosurg. 2013, 119, 974–980. [Google Scholar] [CrossRef]
- Cagnazzo, F.; Chalard, K.; Lefevre, P.H.; Garnier, O.; Derraz, I.; Dargazanli, C.; Gascou, G.; Riquelme, C.; Bonafe, A.; Perrini, P.; et al. Optimal Intracranial Pressure in Patients with Aneurysmal Subarachnoid Hemorrhage Treated with Coiling and Requiring External Ventricular Drainage. Neurosurg. Rev. 2021, 44, 1191–1204. [Google Scholar] [CrossRef] [PubMed]
- Ozeki, T.; Kubota, A.; Murai, Y.; Morita, A. A Case of Suspected Low-Pressure Hydrocephalus Caused by Spinal Drainage Following Subarachnoid Hemorrhage. J. Nippon. Med. Sch. 2021, 89, 238–243. [Google Scholar] [CrossRef] [PubMed]
- Linsler, S.; Schmidtke, M.; Steudel, W.I.; Kiefer, M.; Oertel, J. Automated Intracranial Pressure-Controlled Cerebrospinal Fluid External Drainage with LiquoGuard. Acta Neurochir. 2013, 155, 1589–1594, discussion 1585–1594. [Google Scholar] [CrossRef]
- Al-Tamimi, Y.Z.; Bhargava, D.; Feltbower, R.G.; Hall, G.; Goddard, A.J.; Quinn, A.C.; Ross, S.A. Lumbar Drainage of Cerebrospinal Fluid after Aneurysmal Subarachnoid Hemorrhage: A Prospective, Randomized, Controlled trial (LUMAS). Stroke 2012, 43, 677–682. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Xin, D.Q.; Hu, Q.; Wang, L.X.; Qiu, J.; Yuan, H.T.; Chu, X.L.; Liu, D.X.; Li, G.; Wang, Z. Neuroprotective Mechanism of L-cysteine after Subarachnoid Hemorrhage. Neural Regen. Res. 2020, 15, 1920–1930. [Google Scholar] [CrossRef]
- Wu, Q.; Zhang, X.S.; Wang, H.D.; Zhang, X.; Yu, Q.; Li, W.; Zhou, M.L.; Wang, X.L. Astaxanthin Activates Nuclear Factor Erythroid-related Factor 2 and the Antioxidant Responsive Element (Nrf2-ARE) Pathway in the Brain after Subarachnoid Hemorrhage in Rats and Attenuates Early Brain Injury. Mar. Drugs 2014, 12, 6125–6141. [Google Scholar] [CrossRef]
- Liu, Y.; Qiu, J.; Wang, Z.; You, W.; Wu, L.; Ji, C.; Chen, G. Dimethylfumarate Alleviates Early Brain Injury and Secondary Cognitive Deficits after Experimental Subarachnoid Hemorrhage via Activation of Keap1-Nrf2-ARE System. J. Neurosurg. 2015, 123, 915–923. [Google Scholar] [CrossRef]
- Wang, Z.; Guo, S.; Wang, J.; Shen, Y.; Zhang, J.; Wu, Q. Nrf2/HO-1 Mediates the Neuroprotective Effect of Mangiferin on Early Brain Injury after Subarachnoid Hemorrhage by Attenuating Mitochondria-Related Apoptosis and Neuroinflammation. Sci. Rep. 2017, 7, 11883. [Google Scholar] [CrossRef]
- Song, S.; Chen, Y.; Han, F.; Dong, M.; Xiang, X.; Sui, J.; Li, Y.; Yang, H.; Liu, J. Aloperine Activates the Nrf2-ARE Pathway When Ameliorating Early Brain Injury in A Subarachnoid Hemorrhage Model. Exp. Ther. Med. 2018, 15, 3847–3855. [Google Scholar] [CrossRef]
- Zhang, X.; Wu, Q.; Lu, Y.; Wan, J.; Dai, H.; Zhou, X.; Lv, S.; Chen, X.; Zhang, X.; Hang, C.; et al. Cerebroprotection by Salvianolic Acid B after Experimental Subarachnoid Hemorrhage Occurs via Nrf2- and SIRT1-Dependent Pathways. Free Radic. Biol. Med. 2018, 124, 504–516. [Google Scholar] [CrossRef]
- Wang, T.; Xu, L.; Gao, L.; Zhao, L.; Liu, X.H.; Chang, Y.Y.; Liu, Y.L. Paeoniflorin Attenuates Early Brain Injury through Reducing Oxidative Sress and Neuronal Apoptosis after Subarachnoid Hemorrhage in Rats. Metab. Brain Dis. 2020, 35, 959–970. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Wang, C.; Li, X.; Liang, G. Oleanolic Acid Reduces Oxidative Stress and Neuronal Apoptosis after Experimental Subarachnoid Hemorrhage by Regulating Nrf2/HO-1 Pathway. Drug Dev Res. 2022, 83, 680–687. [Google Scholar] [CrossRef] [PubMed]
- Gong, P.; Zhang, W.; Zou, C.; Han, S.; Tian, Q.; Wang, J.; He, P.; Guo, Y.; Li, M. Andrographolide Attenuates Blood-Brain Barrier Disruption, Neuronal Apoptosis, and Oxidative Stress Through Activation of Nrf2/HO-1 Signaling Pathway in Subarachnoid Hemorrhage. Neurotox Res. 2022, 40, 508–519. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Wang, Y.; Wang, Y.H.; Hang, C.H. The Mfn1-betaIIPKC Interaction Regulates Mitochondrial Dysfunction via Sirt3 Following Experimental Subarachnoid Hemorrhage. Transl. Stroke Res. 2022, 13, 845–857. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Li, Y.; Zhu, S.; Wang, C.; Dai, J.; Zhang, G.; Zheng, B.; Xu, S.; Wang, L.; Zhang, T.; et al. Mdivi-1 Alleviates Early Brain Injury after Experimental Subarachnoid Hemorrhage in Rats, Possibly via Inhibition of Drp1-Activated Mitochondrial Fission and Oxidative Stress. Neurochem. Res. 2017, 42, 1449–1458. [Google Scholar] [CrossRef] [PubMed]
- Shen, R.; Zhou, J.; Li, G.; Chen, W.; Zhong, W.; Chen, Z. SS31 Attenuates Oxidative Stress and Neuronal Apoptosis in Early Brain Injury following Subarachnoid Hemorrhage Possibly by the Mitochondrial Pathway. Neurosci. Lett. 2020, 717, 134654. [Google Scholar] [CrossRef]
- Zhang, T.; Wu, P.; Zhang, J.H.; Li, Y.; Xu, S.; Wang, C.; Wang, L.; Zhang, G.; Dai, J.; Zhu, S.; et al. Docosahexaenoic Acid Alleviates Oxidative Stress-Based Apoptosis via Improving Mitochondrial Dynamics in Early Brain Injury after Subarachnoid Hemorrhage. Cell. Mol. Neurobiol. 2018, 38, 1413–1423. [Google Scholar] [CrossRef]
- Fan, L.F.; He, P.Y.; Peng, Y.C.; Du, Q.H.; Ma, Y.J.; Jin, J.X.; Xu, H.Z.; Li, J.R.; Wang, Z.J.; Cao, S.L.; et al. Mdivi-1 Ameliorates Early Brain Injury after Subarachnoid Hemorrhage via the Suppression of Inflammation-Related Blood-Brain Barrier Disruption and Endoplasmic Reticulum Stress-Based Apoptosis. Free Radic. Biol. Med. 2017, 112, 336–349. [Google Scholar] [CrossRef]
- Zhang, X.S.; Lu, Y.; Tao, T.; Wang, H.; Liu, G.J.; Liu, X.Z.; Liu, C.; Xia, D.Y.; Hang, C.H.; Li, W. Fucoxanthin Mitigates Subarachnoid Hemorrhage-Induced Oxidative Damage via Sirtuin 1-Dependent Pathway. Mol. Neurobiol. 2020, 57, 5286–5298. [Google Scholar] [CrossRef]
- Liu, H.; Guo, W.; Guo, H.; Zhao, L.; Yue, L.; Li, X.; Feng, D.; Luo, J.; Wu, X.; Cui, W.; et al. Bakuchiol Attenuates Oxidative Stress and Neuron Damage by Regulating Trx1/TXNIP and the Phosphorylation of AMPK after Subarachnoid Hemorrhage in Mice. Front. Pharmacol. 2020, 11, 712. [Google Scholar] [CrossRef]
- Zhuang, K.; Zuo, Y.C.; Sherchan, P.; Wang, J.K.; Yan, X.X.; Liu, F. Hydrogen Inhalation Attenuates Oxidative Stress Related Endothelial Cells Injury after Subarachnoid Hemorrhage in Rats. Front. Neurosci. 2019, 13, 1441. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, T.; Li, Y.; Guo, Y.; Liu, B.; Tian, Y.; Wu, P.; Shi, H. Metformin Attenuates Early Brain Injury after Subarachnoid Hemorrhage in Rats via AMPK-Dependent Mitophagy. Exp. Neurol. 2022, 353, 114055. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Li, T.; Gao, L.; Zheng, J.; Yan, J.; Zhang, J.; Shao, A. Apelin-13/APJ System Attenuates Early Brain Injury via Suppression of Endoplasmic Reticulum Stress-Associated TXNIP/NLRP3 Inflammasome Activation and Oxidative Stress in A AMPK-Dependent Manner after Subarachnoid Hemorrhage in Rats. J. Neuroinflammation 2019, 16, 247. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Su, J.; Liu, X.; Zhao, Y.; Wang, C.; Li, X. Naringin Alleviates Early Brain Injury after Experimental Subarachnoid Hemorrhage by Reducing Oxidative Stress and Inhibiting Apoptosis. Brain Res. Bull. 2017, 133, 42–50. [Google Scholar] [CrossRef]
- Fu, P.; Hu, Q. 3,4-Dihydroxyphenylethanol Alleviates Early Brain Injury by Modulating Oxidative Stress and Akt and Nuclear Factor-KappaB Pathways in A Rat Model of Subarachnoid Hemorrhage. Exp. Ther. Med. 2016, 11, 1999–2004. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).