
The Role of Heme Oxygenase-1 as an Immunomodulator in Kidney Disease

{kind=link}
{kind=link}
Abstract
:1. Introduction
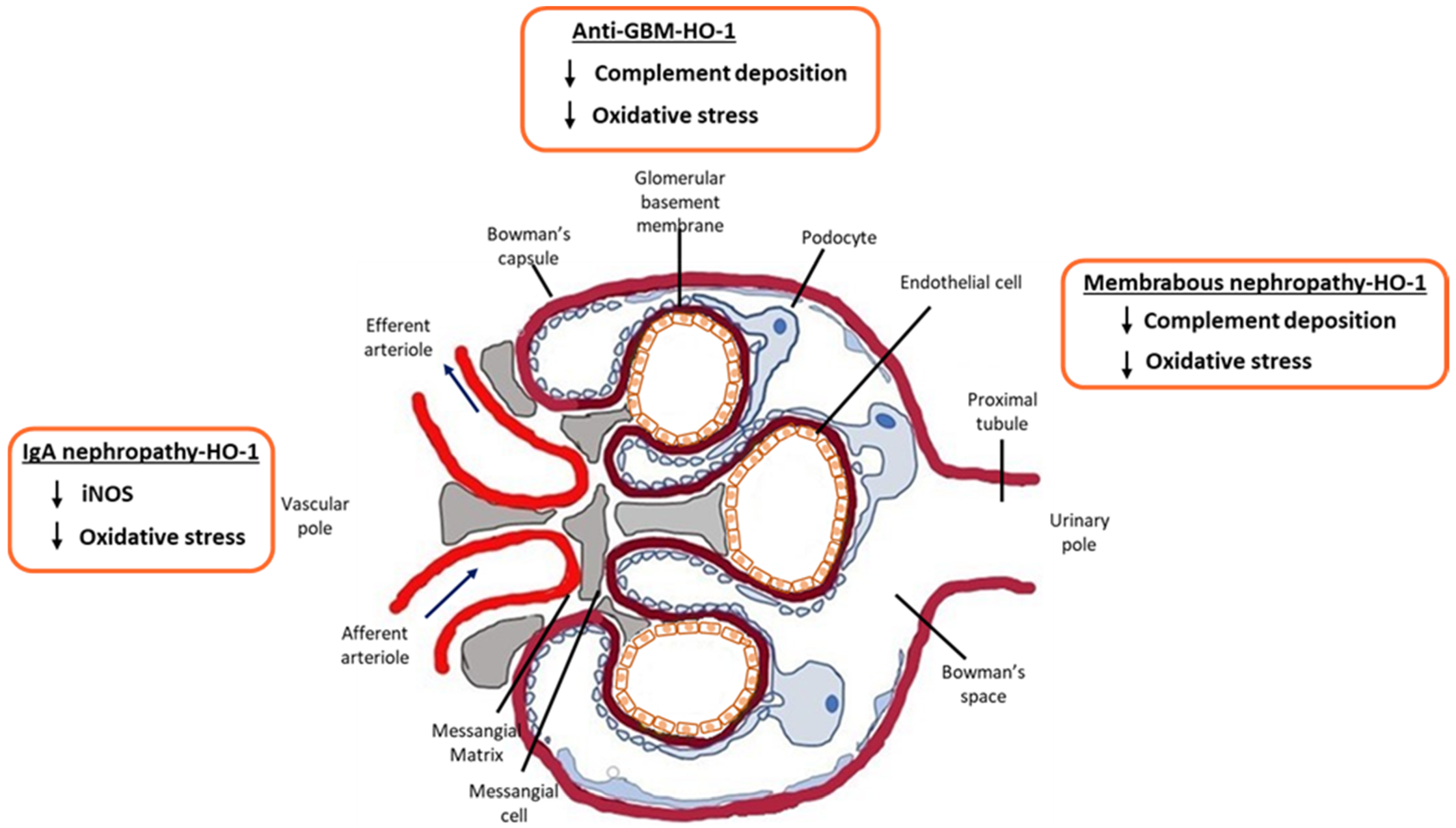
2. HO-1 and IgA Nephropathy
3. HO-1 and Membranous Nephropathy (MN)
4. HO-1 and Anti-GBM Disease
5. HO-1 and Lupus Nephritis
6. HO-1 and Acute Kidney Injury (AKI)
7. HO-1 in Renal Ischemia/Reperfusion Injury (IRI)
8. HO-1 Polymorphisms and Kidney Disease
9. Clinical Applications
10. Conclusions
Funding
Conflicts of Interest
References
- Barratt, J.; Feehally, J. IgA nephropathy. J. Am. Soc. Nephrol. JASN 2005, 16, 2088–2097. [Google Scholar] [CrossRef] [Green Version]
- Saha, M.K.; Julian, B.A.; Novak, J.; Rizk, D.V. Secondary IgA nephropathy. Kidney Int. 2018, 94, 674–681. [Google Scholar] [CrossRef]
- Penfold, R.S.; Prendecki, M.; McAdoo, S.; Tam, F.W. Primary IgA nephropathy: Current challenges and future prospects. Int. J. Nephrol. Renov. Dis. 2018, 11, 137–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, H. Biomarkers for IgA nephropathy on the basis of multi-hit pathogenesis. Clin. Exp. Nephrol. 2019, 23, 26–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dotz, V.; Visconti, A.; Lomax-Browne, H.J.; Clerc, F.; Hipgrave Ederveen, A.L.; Medjeral-Thomas, N.R.; Cook, H.T.; Pickering, M.C.; Wuhrer, M.; Falchi, M. O- and N-Glycosylation of Serum Immunoglobulin A is Associated with IgA Nephropathy and Glomerular Function. J. Am. Soc. Nephrol. JASN 2021, 32, 2455–2465. [Google Scholar] [CrossRef]
- Yau, T.; Korbet, S.M.; Schwartz, M.M.; Cimbaluk, D.J. The Oxford classification of IgA nephropathy: A retrospective analysis. Am. J. Nephrol. 2011, 34, 435–444. [Google Scholar] [CrossRef]
- Descamps-Latscha, B.; Witko-Sarsat, V.; Nguyen-Khoa, T.; Nguyen, A.T.; Gausson, V.; Mothu, N.; Cardoso, C.; Noel, L.H.; Guerin, A.P.; London, G.M.; et al. Early prediction of IgA nephropathy progression: Proteinuria and AOPP are strong prognostic markers. Kidney Int. 2004, 66, 1606–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, T.; Inoue, T.; Sugaya, T.; Kawagoe, Y.; Suzuki, T.; Ueda, Y.; Koide, H.; Node, K. Beneficial effects of olmesartan and temocapril on urinary liver-type fatty acid-binding protein levels in normotensive patients with immunoglobin A nephropathy. Am. J. Hypertens. 2007, 20, 1195–1201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Courtney, A.E.; McNamee, P.T.; Heggarty, S.; Middleton, D.; Maxwell, A.P. Association of functional haem oxygenase-1 gene promoter polymorphism with polycystic kidney disease and IgA nephropathy. Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transpl. Assoc.-Eur. Ren. Assoc. 2008, 23, 608–611. [Google Scholar] [CrossRef] [Green Version]
- Chin, H.J.; Cho, H.J.; Lee, T.W.; Na, K.Y.; Yoon, H.J.; Chae, D.W.; Kim, S.; Jeon, U.S.; Do, J.Y.; Park, J.W.; et al. The heme oxygenase-1 genotype is a risk factor to renal impairment of IgA nephropathy at diagnosis, which is a strong predictor of mortality. J. Korean Med. Sci. 2009, 24, S30–S37. [Google Scholar] [CrossRef]
- Merle, N.S.; Grunenwald, A.; Rajaratnam, H.; Gnemmi, V.; Frimat, M.; Figueres, M.L.; Knockaert, S.; Bouzekri, S.; Charue, D.; Noe, R.; et al. Intravascular hemolysis activates complement via cell-free heme and heme-loaded microvesicles. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [Green Version]
- Frimat, M.; Tabarin, F.; Dimitrov, J.D.; Poitou, C.; Halbwachs-Mecarelli, L.; Fremeaux-Bacchi, V.; Roumenina, L.T. Complement activation by heme as a secondary hit for atypical hemolytic uremic syndrome. Blood 2013, 122, 282–292. [Google Scholar] [CrossRef]
- Li, Y.; Ma, K.; Han, Z.; Chi, M.; Sai, X.; Zhu, P.; Ding, Z.; Song, L.; Liu, C. Immunomodulatory Effects of Heme Oxygenase-1 in Kidney Disease. Front. Med. 2021, 8, 708453. [Google Scholar] [CrossRef]
- Gozzelino, R.; Jeney, V.; Soares, M.P. Mechanisms of cell protection by heme oxygenase-1. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 323–354. [Google Scholar] [CrossRef] [Green Version]
- Shepard, M.; Dhulipala, P.; Kabaria, S.; Abraham, N.G.; Lianos, E.A. Heme oxygenase-1 localization in the rat nephron. Nephron 2002, 92, 660–664. [Google Scholar] [CrossRef]
- Detsika, M.G.; Lianos, E.A. Regulation of Complement Activation by Heme Oxygenase-1 (HO-1) in Kidney Injury. Antioxidants 2021, 10, 60. [Google Scholar] [CrossRef] [PubMed]
- Trimarchi, H.; Barratt, J.; Cattran, D.C.; Cook, H.T.; Coppo, R.; Haas, M.; Liu, Z.H.; Roberts, I.S.; Yuzawa, Y.; Zhang, H.; et al. Oxford Classification of IgA nephropathy 2016: An update from the IgA Nephropathy Classification Working Group. Kidney Int. 2017, 91, 1014–1021. [Google Scholar] [CrossRef] [Green Version]
- Rovin, B.H.; Adler, S.G.; Barratt, J.; Bridoux, F.; Burdge, K.A.; Chan, T.M.; Cook, H.T.; Fervenza, F.C.; Gibson, K.L.; Glassock, R.J.; et al. Executive summary of the KDIGO 2021 Guideline for the Management of Glomerular Diseases. Kidney Int. 2021, 100, 753–779. [Google Scholar] [CrossRef]
- Barratt, J.; Floege, J. SGLT-2 inhibition in IgA nephropathy: The new standard of care? Kidney Int. 2021, 100, 24–26. [Google Scholar] [CrossRef] [PubMed]
- Kidney Disease: Improving Global Outcomes Glomerular Diseases Work Group. KDIGO 2021 Clinical Practice Guideline for the Management of Glomerular Diseases. Kidney Int. 2021, 100, S1–S276. [Google Scholar] [CrossRef] [PubMed]
- Keri, K.C.; Blumenthal, S.; Kulkarni, V.; Beck, L.; Chongkrairatanakul, T. Primary membranous nephropathy: Comprehensive review and historical perspective. Postgrad. Med. J. 2019, 95, 23–31. [Google Scholar] [CrossRef]
- Heymann, W.; Hackel, D.B.; Harwood, S.; Wilson, S.G.; Hunter, J.L. Production of nephrotic syndrome in rats by Freund’s adjuvants and rat kidney suspensions. Proc. Soc. Exp. Biol. Medicine. Soc. Exp. Biol. Med. 1959, 100, 660–664. [Google Scholar] [CrossRef]
- Farquhar, M.G.; Saito, A.; Kerjaschki, D.; Orlando, R.A. The Heymann nephritis antigenic complex: Megalin (gp330) and RAP. J. Am. Soc. Nephrol. JASN 1995, 6, 35–47. [Google Scholar] [CrossRef]
- Cybulsky, A.V.; Quigg, R.J.; Salant, D.J. Experimental membranous nephropathy redux. Am. J. Physiol. Ren. Physiol. 2005, 289, F660–F671. [Google Scholar] [CrossRef]
- Ma, H.; Sandor, D.G.; Beck, L.H., Jr. The role of complement in membranous nephropathy. Semin. Nephrol. 2013, 33, 531–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miwa, T.; Song, W.C. Membrane complement regulatory proteins: Insight from animal studies and relevance to human diseases. Int. Immunopharmacol. 2001, 1, 445–459. [Google Scholar] [CrossRef] [PubMed]
- Bao, L.; Spiller, O.B.; St John, P.L.; Haas, M.; Hack, B.K.; Ren, G.; Cunningham, P.N.; Doshi, M.; Abrahamson, D.R.; Morgan, B.P.; et al. Decay-accelerating factor expression in the rat kidney is restricted to the apical surface of podocytes. Kidney Int. 2002, 62, 2010–2021. [Google Scholar] [CrossRef] [Green Version]
- Detsika, M.G.; Goudevenou, K.; Geurts, A.M.; Gakiopoulou, H.; Grapsa, E.; Lianos, E.A. Generation of a novel decay accelerating factor (DAF) knock-out rat model using clustered regularly-interspaced short palindromic repeats, (CRISPR)/associated protein 9 (Cas9), genome editing. Transgenic Res. 2021, 30, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.C.; Lu, K.C.; Lin, G.J.; Hsieh, H.Y.; Chu, P.; Lin, S.H.; Sytwu, H.K. Melatonin enhances endogenous heme oxygenase-1 and represses immune responses to ameliorate experimental murine membranous nephropathy. J. Pineal Res. 2012, 52, 460–469. [Google Scholar] [CrossRef]
- Yu, P.; Zhang, X.; Liu, N.; Tang, L.; Peng, C.; Chen, X. Pyroptosis: Mechanisms and diseases. Signal Transduct. Target. Ther. 2021, 6, 128. [Google Scholar] [CrossRef]
- Wang, H.; Lv, D.; Jiang, S.; Hou, Q.; Zhang, L.; Li, S.; Zhu, X.; Xu, X.; Wen, J.; Zeng, C.; et al. Complement induces podocyte pyroptosis in membranous nephropathy by mediating mitochondrial dysfunction. Cell Death Dis. 2022, 13, 281. [Google Scholar] [CrossRef]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef]
- Fu, L.; Zhang, D.X.; Zhang, L.M.; Song, Y.C.; Liu, F.H.; Li, Y.; Wang, X.P.; Zheng, W.C.; Wang, X.D.; Gui, C.X.; et al. Exogenous carbon monoxide protects against mitochondrial DNAinduced hippocampal pyroptosis in a model of hemorrhagic shock and resuscitation. Int. J. Mol. Med. 2020, 45, 1176–1186. [Google Scholar] [CrossRef] [Green Version]
- Ryter, S.W. Heme Oxgenase-1, a Cardinal Modulator of Regulated Cell Death and Inflammation. Cells 2021, 10, 515. [Google Scholar] [CrossRef]
- Nakatani, Y.; Inagi, R. Epigenetic Regulation Through SIRT1 in Podocytes. Curr. Hypertens. Rev. 2016, 12, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.J.; Wu, W.J.; Zhou, Q.; Jie, J.P.; Chen, X.; Wang, F.; Gong, X.H. Advanced glycation end-products induce oxidative stress through the Sirt1/Nrf2 axis by interacting with the receptor of AGEs under diabetic conditions. J. Cell. Biochem. 2018. [Google Scholar] [CrossRef] [PubMed]
- Diao, C.; Chen, Z.; Qiu, T.; Liu, H.; Yang, Y.; Liu, X.; Wu, J.; Wang, L. Inhibition of PRMT5 Attenuates Oxidative Stress-Induced Pyroptosis via Activation of the Nrf2/HO-1 Signal Pathway in a Mouse Model of Renal Ischemia-Reperfusion Injury. Oxidative Med. Cell. Longev. 2019, 2019, 2345658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.; Ronai, Z.A. PRMT5 function and targeting in cancer. Cell Stress 2020, 4, 199–215. [Google Scholar] [CrossRef]
- Fernandez-Juarez, G.; Rojas-Rivera, J.; Logt, A.V.; Justino, J.; Sevillano, A.; Caravaca-Fontan, F.; Avila, A.; Rabasco, C.; Cabello, V.; Varela, A.; et al. The STARMEN trial indicates that alternating treatment with corticosteroids and cyclophosphamide is superior to sequential treatment with tacrolimus and rituximab in primary membranous nephropathy. Kidney Int. 2021, 99, 986–998. [Google Scholar] [CrossRef]
- Kant, S.; Kronbichler, A.; Sharma, P.; Geetha, D. Advances in Understanding of Pathogenesis and Treatment of Immune-Mediated Kidney Disease: A Review. Am. J. Kidney Dis. Off. J. Natl. Kidney Found. 2022, 79, 582–600. [Google Scholar] [CrossRef] [PubMed]
- Segelmark, M.; Hellmark, T. Anti-glomerular basement membrane disease: An update on subgroups, pathogenesis and therapies. Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transpl. Assoc.-Eur. Ren. Assoc. 2019, 34, 1826–1832. [Google Scholar] [CrossRef] [PubMed]
- Steblay, R.W. Glomerulonephritis induced in sheep by injections of heterologous glomerular basement membrane and Freund’s complete adjuvant. J. Exp. Med. 1962, 116, 253–272. [Google Scholar] [CrossRef] [PubMed]
- Ryan, J.J.; Reynolds, J.; Norgan, V.A.; Pusey, C.D. Expression and characterization of recombinant rat alpha 3(IV)NC1 and its use in induction of experimental autoimmune glomerulonephritis. Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transpl. Assoc.-Eur. Ren. Assoc. 2001, 16, 253–261. [Google Scholar] [CrossRef] [Green Version]
- Otten, M.A.; Groeneveld, T.W.; Flierman, R.; Rastaldi, M.P.; Trouw, L.A.; Faber-Krol, M.C.; Visser, A.; Essers, M.C.; Claassens, J.; Verbeek, J.S.; et al. Both complement and IgG fc receptors are required for development of attenuated antiglomerular basement membrane nephritis in mice. J. Immunol. 2009, 183, 3980–3988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sogabe, H.; Nangaku, M.; Ishibashi, Y.; Wada, T.; Fujita, T.; Sun, X.; Miwa, T.; Madaio, M.P.; Song, W.C. Increased susceptibility of decay-accelerating factor deficient mice to anti-glomerular basement membrane glomerulonephritis. J. Immunol. 2001, 167, 2791–2797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cattell, V.; Largen, P.; de Heer, E.; Cook, T. Glomeruli synthesize nitrite in active Heymann nephritis; the source is infiltrating macrophages. Kidney Int. 1991, 40, 847–851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Datta, P.K.; Koukouritaki, S.B.; Hopp, K.A.; Lianos, E.A. Heme oxygenase-1 induction attenuates inducible nitric oxide synthase expression and proteinuria in glomerulonephritis. J. Am. Soc. Nephrol. JASN 1999, 10, 2540–2550. [Google Scholar] [CrossRef]
- Datta, P.K.; Lianos, E.A. Nitric oxide induces heme oxygenase-1 gene expression in mesangial cells. Kidney Int. 1999, 55, 1734–1739. [Google Scholar] [CrossRef] [Green Version]
- Liang, M.; Croatt, A.J.; Nath, K.A. Mechanisms underlying induction of heme oxygenase-1 by nitric oxide in renal tubular epithelial cells. Am. J. Physiol. Ren. Physiol. 2000, 279, F728–F735. [Google Scholar] [CrossRef]
- Datta, P.K.; Gross, E.J.; Lianos, E.A. Interactions between inducible nitric oxide synthase and heme oxygenase-1 in glomerulonephritis. Kidney Int. 2002, 61, 847–850. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Agesta, M.; Rabasco, C.; Soler, M.J.; Shabaka, A.; Canllavi, E.; Fernandez, S.J.; Cazorla, J.M.; Lopez-Rubio, E.; Romera, A.; Barroso, S.; et al. Anti-glomerular Basement Membrane Glomerulonephritis: A Study in Real Life. Front. Med. 2022, 9, 889185. [Google Scholar] [CrossRef]
- Zucchi, D.; Elefante, E.; Schiliro, D.; Signorini, V.; Trentin, F.; Bortoluzzi, A.; Tani, C. One year in review 2022: Systemic lupus erythematosus. Clin. Exp. Rheumatol. 2022, 40, 4–14. [Google Scholar] [CrossRef]
- Ocampo-Piraquive, V.; Nieto-Aristizabal, I.; Canas, C.A.; Tobon, G.J. Mortality in systemic lupus erythematosus: Causes, predictors and interventions. Expert Rev. Clin. Immunol. 2018, 14, 1043–1053. [Google Scholar] [CrossRef]
- Frangou, E.; Georgakis, S.; Bertsias, G. Update on the cellular and molecular aspects of lupus nephritis. Clin. Immunol. 2020, 216, 108445. [Google Scholar] [CrossRef]
- Maria, N.I.; Davidson, A. Renal Macrophages and Dendritic Cells in SLE Nephritis. Curr. Rheumatol. Rep. 2017, 19, 81. [Google Scholar] [CrossRef] [PubMed]
- Kishimoto, D.; Kirino, Y.; Tamura, M.; Takeno, M.; Kunishita, Y.; Takase-Minegishi, K.; Nakano, H.; Kato, I.; Nagahama, K.; Yoshimi, R.; et al. Dysregulated heme oxygenase-1(low) M2-like macrophages augment lupus nephritis via Bach1 induced by type I interferons. Arthritis Res. Ther. 2018, 20, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuitino, L.; Obreque, J.; Gajardo-Meneses, P.; Villarroel, A.; Crisostomo, N.; San Francisco, I.F.; Valenzuela, R.A.; Mendez, G.P.; Llanos, C. Heme-Oxygenase-1 Is Decreased in Circulating Monocytes and Is Associated With Impaired Phagocytosis and ROS Production in Lupus Nephritis. Front. Immunol. 2019, 10, 2868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lech, M.; Anders, H.J. The pathogenesis of lupus nephritis. J. Am. Soc. Nephrol. JASN 2013, 24, 1357–1366. [Google Scholar] [CrossRef] [Green Version]
- Funes, S.C.; Manrique de Lara, A.; Altamirano-Lagos, M.J.; Mackern-Oberti, J.P.; Escobar-Vera, J.; Kalergis, A.M. Immune checkpoints and the regulation of tolerogenicity in dendritic cells: Implications for autoimmunity and immunotherapy. Autoimmun. Rev. 2019, 18, 359–368. [Google Scholar] [CrossRef]
- Funes, S.C.; Rios, M.; Gomez-Santander, F.; Fernandez-Fierro, A.; Altamirano-Lagos, M.J.; Rivera-Perez, D.; Pulgar-Sepulveda, R.; Jara, E.L.; Rebolledo-Zelada, D.; Villarroel, A.; et al. Tolerogenic dendritic cell transfer ameliorates systemic lupus erythematosus in mice. Immunology 2019, 158, 322–339. [Google Scholar] [CrossRef]
- Takeda, Y.; Takeno, M.; Iwasaki, M.; Kobayashi, H.; Kirino, Y.; Ueda, A.; Nagahama, K.; Aoki, I.; Ishigatsubo, Y. Chemical induction of HO-1 suppresses lupus nephritis by reducing local iNOS expression and synthesis of anti-dsDNA antibody. Clin. Exp. Immunol. 2004, 138, 237–244. [Google Scholar] [CrossRef]
- Mackern-Oberti, J.P.; Llanos, C.; Carreno, L.J.; Riquelme, S.A.; Jacobelli, S.H.; Anegon, I.; Kalergis, A.M. Carbon monoxide exposure improves immune function in lupus-prone mice. Immunology 2013, 140, 123–132. [Google Scholar] [CrossRef]
- Mackern-Oberti, J.P.; Obreque, J.; Mendez, G.P.; Llanos, C.; Kalergis, A.M. Carbon monoxide inhibits T cell activation in target organs during systemic lupus erythematosus. Clin. Exp. Immunol. 2015, 182, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanly, J.G.; O’Keeffe, A.G.; Su, L.; Urowitz, M.B.; Romero-Diaz, J.; Gordon, C.; Bae, S.C.; Bernatsky, S.; Clarke, A.E.; Wallace, D.J.; et al. The frequency and outcome of lupus nephritis: Results from an international inception cohort study. Rheumatology 2016, 55, 252–262. [Google Scholar] [CrossRef] [Green Version]
- Tektonidou, M.G.; Dasgupta, A.; Ward, M.M. Risk of End-Stage Renal Disease in Patients With Lupus Nephritis, 1971-2015: A Systematic Review and Bayesian Meta-Analysis. Arthritis Rheumatol. 2016, 68, 1432–1441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yap, D.Y.; Tang, C.S.; Ma, M.K.; Lam, M.F.; Chan, T.M. Survival analysis and causes of mortality in patients with lupus nephritis. Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transpl. Assoc.-Eur. Ren. Assoc. 2012, 27, 3248–3254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mok, C.C.; Kwok, R.C.; Yip, P.S. Effect of renal disease on the standardized mortality ratio and life expectancy of patients with systemic lupus erythematosus. Arthritis Rheum. 2013, 65, 2154–2160. [Google Scholar] [CrossRef]
- Chen, Y.E.; Korbet, S.M.; Katz, R.S.; Schwartz, M.M.; Lewis, E.J.; Collaborative Study Group. Value of a complete or partial remission in severe lupus nephritis. Clin. J. Am. Soc. Nephrol. CJASN 2008, 3, 46–53. [Google Scholar] [CrossRef] [Green Version]
- Bajema, I.M.; Wilhelmus, S.; Alpers, C.E.; Bruijn, J.A.; Colvin, R.B.; Cook, H.T.; D’Agati, V.D.; Ferrario, F.; Haas, M.; Jennette, J.C.; et al. Revision of the International Society of Nephrology/Renal Pathology Society classification for lupus nephritis: Clarification of definitions, and modified National Institutes of Health activity and chronicity indices. Kidney Int. 2018, 93, 789–796. [Google Scholar] [CrossRef]
- Bolisetty, S.; Zarjou, A.; Agarwal, A. Heme Oxygenase 1 as a Therapeutic Target in Acute Kidney Injury. Am. J. Kidney Dis. Off. J. Natl. Kidney Found. 2017, 69, 531–545. [Google Scholar] [CrossRef]
- Hull, T.D.; Kamal, A.I.; Boddu, R.; Bolisetty, S.; Guo, L.; Tisher, C.C.; Rangarajan, S.; Chen, B.; Curtis, L.M.; George, J.F.; et al. Heme Oxygenase-1 Regulates Myeloid Cell Trafficking in AKI. J. Am. Soc. Nephrol. JASN 2015, 26, 2139–2151. [Google Scholar] [CrossRef] [Green Version]
- Ferenbach, D.A.; Ramdas, V.; Spencer, N.; Marson, L.; Anegon, I.; Hughes, J.; Kluth, D.C. Macrophages expressing heme oxygenase-1 improve renal function in ischemia/reperfusion injury. Mol. Ther. J. Am. Soc. Gene Ther. 2010, 18, 1706–1713. [Google Scholar] [CrossRef]
- Kwong, A.M.; Luke, P.P.W.; Bhattacharjee, R.N. Carbon monoxide mechanism of protection against renal ischemia and reperfusion injury. Biochem. Pharmacol. 2022, 202, 115156. [Google Scholar] [CrossRef] [PubMed]
- Song, R.; Mahidhara, R.S.; Zhou, Z.; Hoffman, R.A.; Seol, D.W.; Flavell, R.A.; Billiar, T.R.; Otterbein, L.E.; Choi, A.M. Carbon monoxide inhibits T lymphocyte proliferation via caspase-dependent pathway. J. Immunol. 2004, 172, 1220–1226. [Google Scholar] [CrossRef] [Green Version]
- Nath, K.A. Heme oxygenase-1 and acute kidney injury. Curr. Opin. Nephrol. Hypertens. 2014, 23, 17–24. [Google Scholar] [CrossRef] [Green Version]
- Eltzschig, H.K.; Eckle, T. Ischemia and reperfusion--from mechanism to translation. Nat. Med. 2011, 17, 1391–1401. [Google Scholar] [CrossRef] [Green Version]
- Salvadori, M.; Rosso, G.; Bertoni, E. Update on ischemia-reperfusion injury in kidney transplantation: Pathogenesis and treatment. World J. Transplant. 2015, 5, 52–67. [Google Scholar] [CrossRef]
- Ferenbach, D.A.; Kluth, D.C.; Hughes, J. Hemeoxygenase-1 and renal ischaemia-reperfusion injury. Nephron. Exp. Nephrol. 2010, 115, e33–e37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Y.; Li, H.; Yao, J.; Zhong, H.; Kuang, Y.; Li, X.; Bian, W. HO-1 knockdown upregulates the expression of VCAM-1 to induce neutrophil recruitment during renal ischemia-reperfusion injury. Int. J. Mol. Med. 2021, 48. [Google Scholar] [CrossRef] [PubMed]
- Ortega-Trejo, J.A.; Pérez-Villalva, R.; Sánchez-Navarro, A.; Marquina, B.; Rodríguez-Iturbe, B.; Bobadilla, N.A. Repeated Episodes of Ischemia/Reperfusion Induce Heme-Oxygenase-1 (HO-1) and Anti-Inflammatory Responses and Protects against Chronic Kidney Disease. Int. J. Mol. Sci. 2022, 23, 14573. [Google Scholar] [CrossRef]
- Rossi, M.; Thierry, A.; Delbauve, S.; Preyat, N.; Soares, M.P.; Roumeguere, T.; Leo, O.; Flamand, V.; Le Moine, A.; Hougardy, J.M. Specific expression of heme oxygenase-1 by myeloid cells modulates renal ischemia-reperfusion injury. Sci. Rep. 2017, 7, 197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baan, C.; Peeters, A.; Lemos, F.; Uitterlinden, A.; Doxiadis, I.; Claas, F.; Ijzermans, J.; Roodnat, J.; Weimar, W. Fundamental role for HO-1 in the self-protection of renal allografts. Am. J. Transplant. Off. J. Am. Soc. Transplant. Am. Soc. Transpl. Surg. 2004, 4, 811–818. [Google Scholar] [CrossRef]
- Funes, S.C.; Rios, M.; Fernandez-Fierro, A.; Covian, C.; Bueno, S.M.; Riedel, C.A.; Mackern-Oberti, J.P.; Kalergis, A.M. Naturally Derived Heme-Oxygenase 1 Inducers and Their Therapeutic Application to Immune-Mediated Diseases. Front. Immunol. 2020, 11, 1467. [Google Scholar] [CrossRef] [PubMed]
- Detsika, M.G.; Duann, P.; Atsaves, V.; Papalois, A.; Lianos, E.A. Heme Oxygenase 1 Up-Regulates Glomerular Decay Accelerating Factor Expression and Minimizes Complement Deposition and Injury. Am. J. Pathol. 2016, 186, 2833–2845. [Google Scholar] [CrossRef] [Green Version]
- Stec, D.E.; Hinds, T.D., Jr. Natural Product Heme Oxygenase Inducers as Treatment for Nonalcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2020, 21, 9493. [Google Scholar] [CrossRef] [PubMed]
- Nikolic, I.; Saksida, T.; Mangano, K.; Vujicic, M.; Stojanovic, I.; Nicoletti, F.; Stosic-Grujicic, S. Pharmacological application of carbon monoxide ameliorates islet-directed autoimmunity in mice via anti-inflammatory and anti-apoptotic effects. Diabetologia 2014, 57, 980–990. [Google Scholar] [CrossRef]
- Fagone, P.; Mangano, K.; Quattrocchi, C.; Motterlini, R.; Di Marco, R.; Magro, G.; Penacho, N.; Romao, C.C.; Nicoletti, F. Prevention of clinical and histological signs of proteolipid protein (PLP)-induced experimental allergic encephalomyelitis (EAE) in mice by the water-soluble carbon monoxide-releasing molecule (CORM)-A1. Clin. Exp. Immunol. 2011, 163, 368–374. [Google Scholar] [CrossRef]
- Chora, A.A.; Fontoura, P.; Cunha, A.; Pais, T.F.; Cardoso, S.; Ho, P.P.; Lee, L.Y.; Sobel, R.A.; Steinman, L.; Soares, M.P. Heme oxygenase-1 and carbon monoxide suppress autoimmune neuroinflammation. J. Clin. Investig. 2007, 117, 438–447. [Google Scholar] [CrossRef] [Green Version]
- Mangano, K.; Cavalli, E.; Mammana, S.; Basile, M.S.; Caltabiano, R.; Pesce, A.; Puleo, S.; Atanasov, A.G.; Magro, G.; Nicoletti, F.; et al. Involvement of the Nrf2/HO-1/CO axis and therapeutic intervention with the CO-releasing molecule CORM-A1, in a murine model of autoimmune hepatitis. J. Cell. Physiol. 2018, 233, 4156–4165. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Athanassiadou, V.; Plavoukou, S.; Grapsa, E.; Detsika, M.G. The Role of Heme Oxygenase-1 as an Immunomodulator in Kidney Disease. Antioxidants 2022, 11, 2454. https://doi.org/10.3390/antiox11122454
Athanassiadou V, Plavoukou S, Grapsa E, Detsika MG. The Role of Heme Oxygenase-1 as an Immunomodulator in Kidney Disease. Antioxidants. 2022; 11(12):2454. https://doi.org/10.3390/antiox11122454
Chicago/Turabian StyleAthanassiadou, Virginia, Stella Plavoukou, Eirini Grapsa, and Maria G. Detsika. 2022. "The Role of Heme Oxygenase-1 as an Immunomodulator in Kidney Disease" Antioxidants 11, no. 12: 2454. https://doi.org/10.3390/antiox11122454