
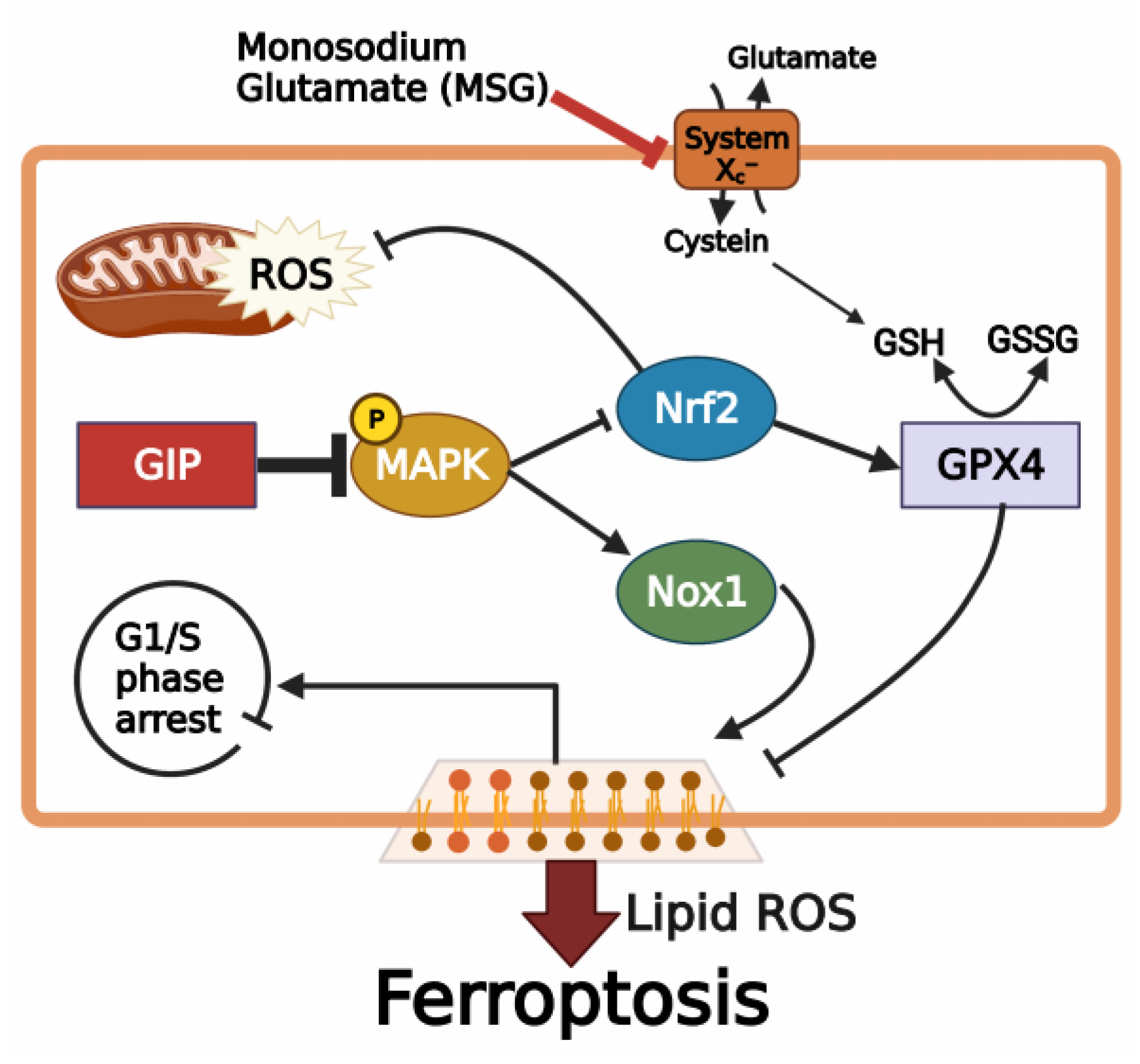
Protective Effect of GIP against Monosodium Glutamate-Induced Ferroptosis in Mouse Hippocampal HT-22 Cells through the MAPK Signaling Pathway

 , , , , ,
, , , , ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Reagents
2.2. Establishment of GIP-Overexpressing Cell Lines
2.3. Cell Counting Kit-8 (CCK-8) Assay
2.4. RNA Isolation and Quantitative Reverse Transcription Polymerase Chain Reaction (qRT-PCR) Analysis
2.5. Western Blotting
2.6. Flow Cytometric Analysis
2.7. Measurement of MDA
2.8. Immunocytochemistry
2.9. Statistical Analysis
3. Results
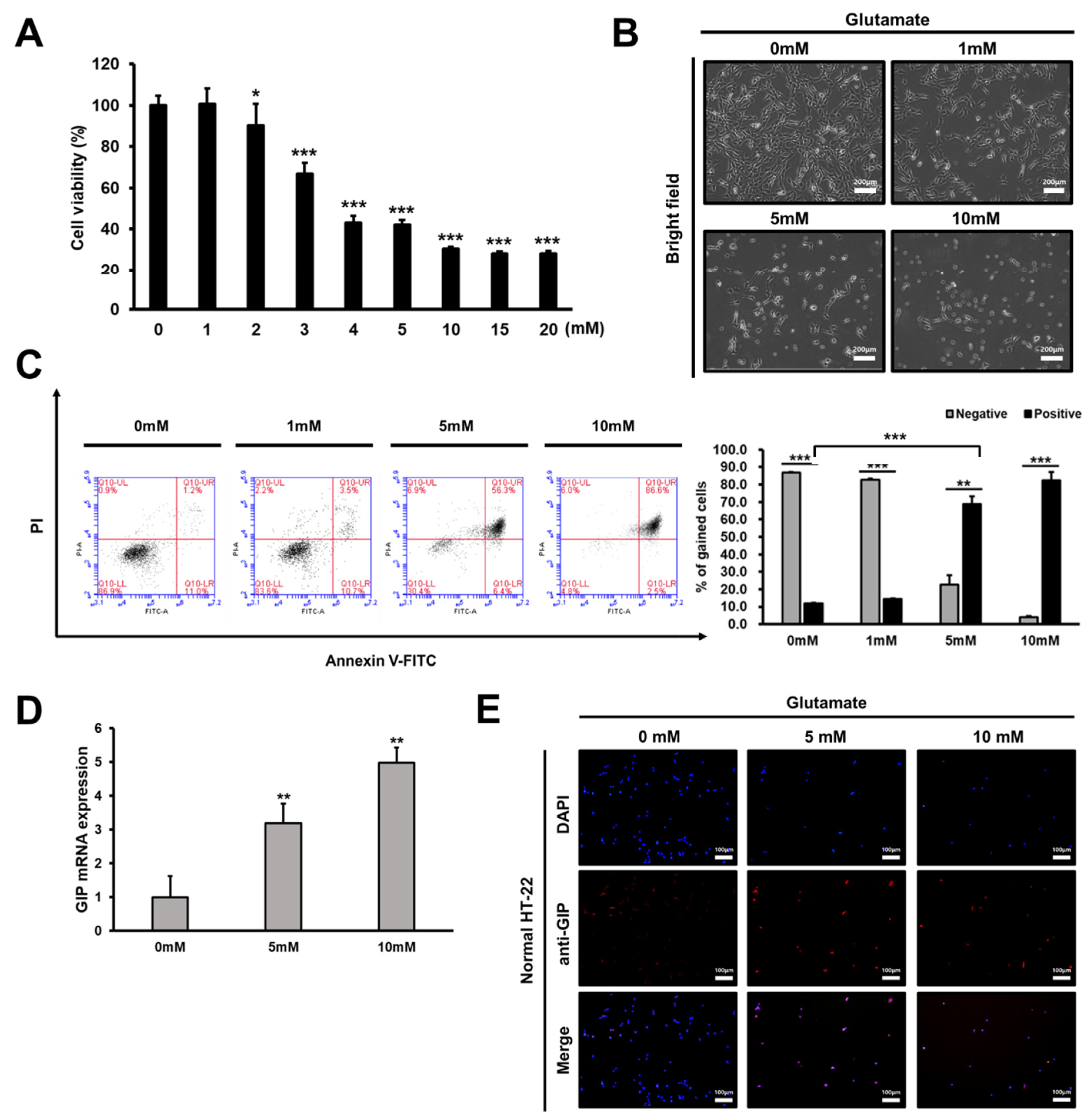
3.1. Glutamate Treatment Reduces the Viability of HT-22 Cells in a Dose-Dependent Manner
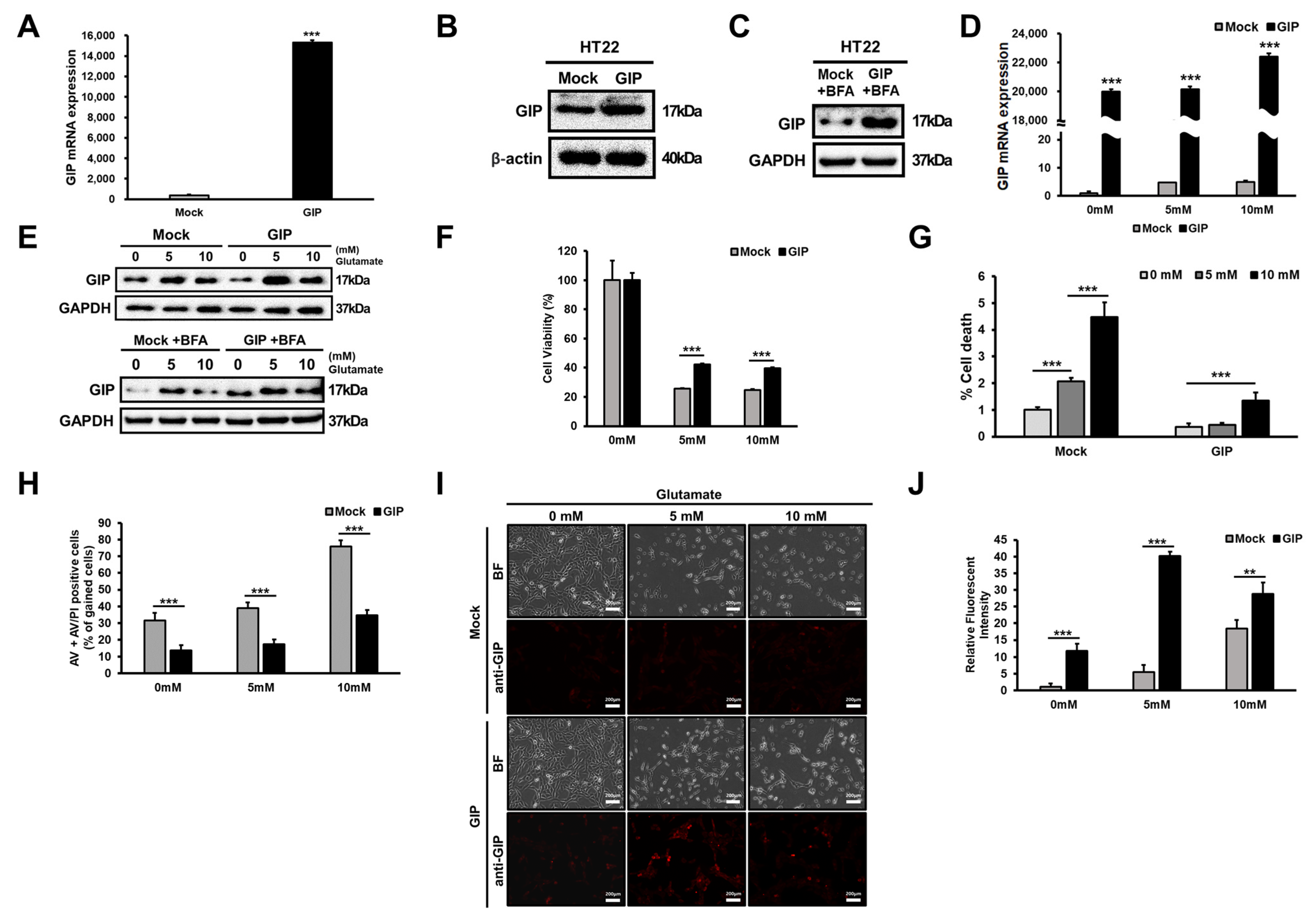
3.2. Association between GIP Overexpression and Glutamate-Induced Neuronal Cell Death
3.3. Glutamate-Induced Neuronal Cell Death Is Distinct from Apoptosis
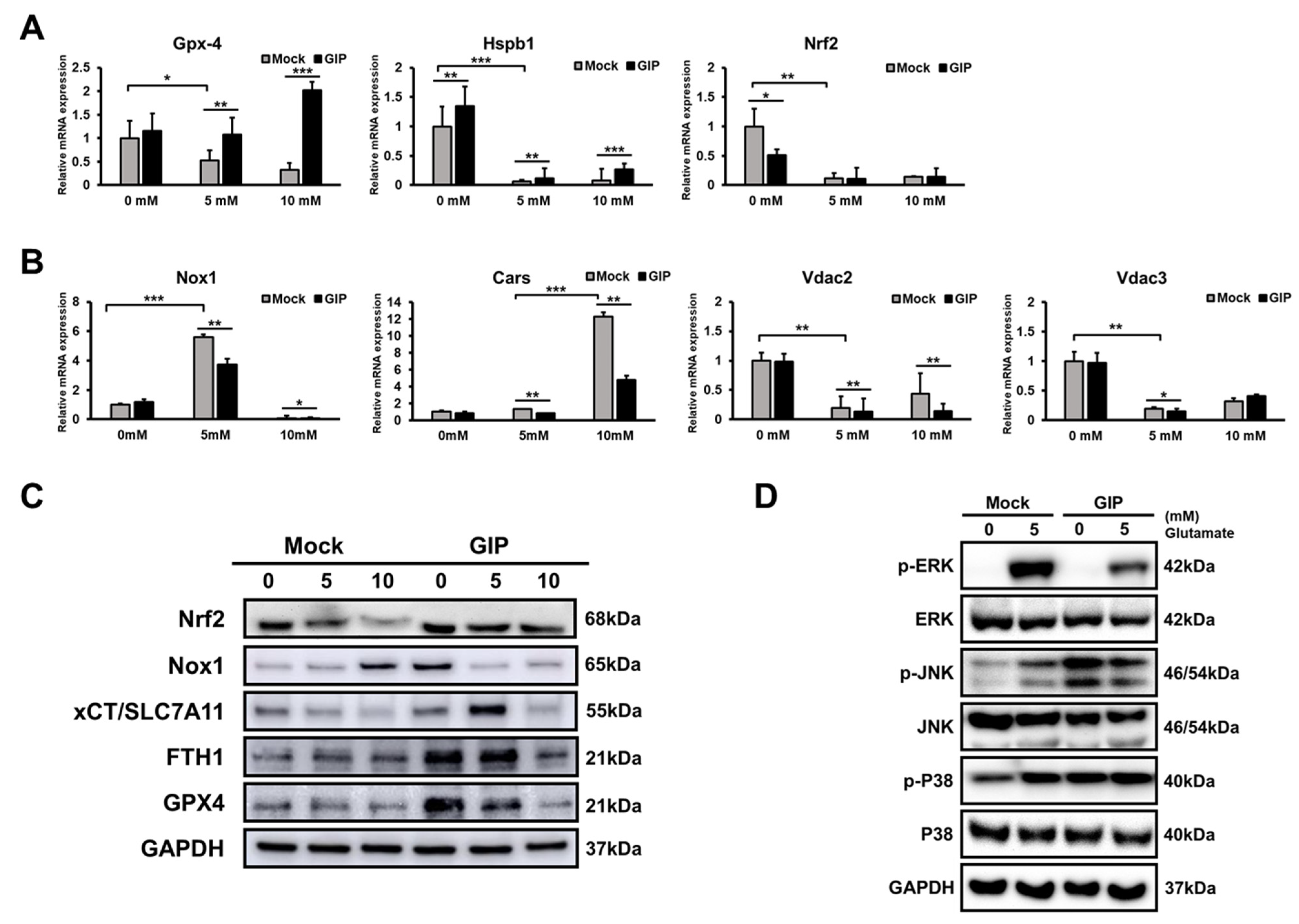
3.4. GIP Reduces Glutamate-Induced Neuronal Oxidative Stress in HT-22 Cells
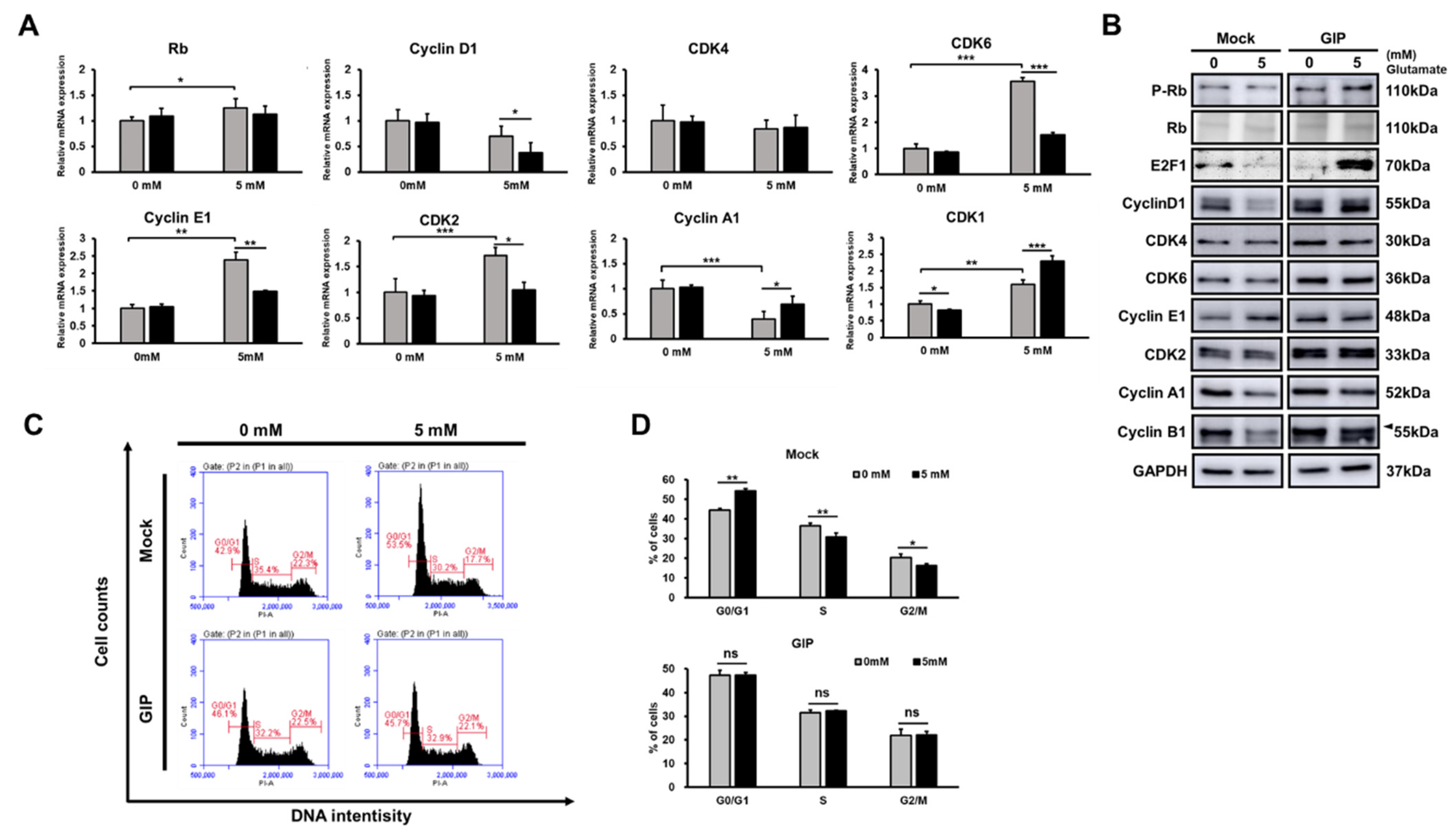
3.5. GIP Makes the Cell Cycle Run Stably under Glutamate-Induced Stress Conditions
3.6. GIP Ameliorates Glutamate-Induced Ferroptosis through the Suppression of MAPK Activation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Beyreuther, K.; Biesalski, H.K.; Fernstrom, J.D.; Grimm, P.; Hammes, W.P.; Heinemann, U.; Kempski, O.; Stehle, P.; Steinhart, H.; Walker, R. Consensus meeting: Monosodium glutamate-an update. Eur. J. Clin. Nutr. 2007, 61, 304–313. [Google Scholar] [CrossRef] [PubMed]
- Obayashi, Y.; Nagamura, Y. Does monosodium glutamate really cause headache? A systematic review of human studies. J. Headache Pain 2016, 17, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, K.; Zhao, L.; Daviglus, M.L.; Dyer, A.R.; Van Horn, L.; Garside, D.; Zhu, L.; Guo, D.; Wu, Y.; Zhou, B.; et al. Association of monosodium glutamate intake with overweight in Chinese adults: The INTERMAP Study. Obes. Silver Spring 2008, 16, 1875–1880. [Google Scholar] [CrossRef] [PubMed]
- Kostandy, B.B. The role of glutamate in neuronal ischemic injury: The role of spark in fire. Neurol. Sci. 2012, 33, 223–237. [Google Scholar] [CrossRef]
- Fuchs, Y.; Steller, H. Programmed cell death in animal development and disease. Cell 2011, 147, 742–758. [Google Scholar] [CrossRef] [Green Version]
- Lemasters, J.J.; Nieminen, A.L.; Qian, T.; Trost, L.C.; Elmore, S.P.; Nishimura, Y.; Crowe, R.A.; Cascio, W.E.; Bradham, C.A.; Brenner, D.A.; et al. The mitochondrial permeability transition in cell death: A common mechanism in necrosis, apoptosis and autophagy. Biochim. Biophys. Acta BBA Bioenerg. 1998, 1366, 177–196. [Google Scholar] [CrossRef] [Green Version]
- Tang, D.; Kang, R.; Berghe, T.V.; Vandenabeele, P.; Kroemer, G. The molecular machinery of regulated cell death. Cell Res. 2019, 29, 347–364. [Google Scholar] [CrossRef] [Green Version]
- Picon, C.; Jayaraman, A.; James, R.; Beck, C.; Gallego, P.; Witte, M.E.; van Horssen, J.; Mazarakis, N.D.; Reynolds, R. Neuron-specific activation of necroptosis signaling in multiple sclerosis cortical grey matter. Acta Neuropathol. 2021, 141, 585–604. [Google Scholar] [CrossRef]
- Fatokun, A.A.; Dawson, V.L.; Dawson, T.M. Parthanatos: Mitochondrial-linked mechanisms and therapeutic opportunities. Br. J. Pharmacol. 2014, 171, 2000–2016. [Google Scholar] [CrossRef] [Green Version]
- Koehler, R.C.; Dawson, V.L.; Dawson, T.M. Targeting Parthanatos in Ischemic Stroke. Front. Neurol. 2021, 12, 662034. [Google Scholar] [CrossRef]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascon, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef]
- Mou, Y.; Wang, J.; Wu, J.; He, D.; Zhang, C.; Duan, C.; Li, B. Ferroptosis, a new form of cell death: Opportunities and challenges in cancer. J. Hematol. Oncol. 2019, 12, 34. [Google Scholar] [CrossRef] [Green Version]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [Green Version]
- Caudle, W.M.; Zhang, J. Glutamate, excitotoxicity, and programmed cell death in Parkinson disease. Exp. Neurol. 2009, 220, 230–233. [Google Scholar] [CrossRef]
- Coyle, J.T.; Puttfarcken, P. Oxidative stress, glutamate, and neurodegenerative disorders. Science 1993, 262, 689–695. [Google Scholar] [CrossRef]
- Kritis, A.A.; Stamoula, E.G.; Paniskaki, K.A.; Vavilis, T.D. Researching glutamate-induced cytotoxicity in different cell lines: A comparative/collective analysis/study. Front. Cell Neurosci. 2015, 9, 91. [Google Scholar] [CrossRef] [Green Version]
- Arundine, M.; Tymianski, M. Molecular mechanisms of glutamate-dependent neurodegeneration in ischemia and traumatic brain injury. Cell Mol. Life Sci. 2004, 61, 657–668. [Google Scholar] [CrossRef]
- Choi, D.W. Glutamate neurotoxicity and diseases of the nervous system. Neuron 1988, 1, 623–634. [Google Scholar] [CrossRef]
- Kuang, F.; Liu, J.; Tang, D.; Kang, R. Oxidative damage and antioxidant defense in ferroptosis. Front. Cell Dev. Biol 2020, 8, 586578. [Google Scholar] [CrossRef]
- Tan, S.; Wood, M.; Maher, P. Oxidative stress induces a form of programmed cell death with characteristics of both apoptosis and necrosis in neuronal cells. J. Neurochem. 1998, 71, 95–105. [Google Scholar] [CrossRef]
- Fukui, M.; Song, J.H.; Choi, J.; Choi, H.J.; Zhu, B.T. Mechanism of glutamate-induced neurotoxicity in HT22 mouse hippocampal cells. Eur J. Pharmacol. 2009, 617, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.F.; Zou, T.; Tuo, Q.Z.; Xu, S.; Li, H.; Belaidi, A.A.; Lei, P. Ferroptosis: Mechanisms and links with diseases. Signal. Transduct. Target. Ther. 2021, 6, 49. [Google Scholar] [CrossRef] [PubMed]
- Su, L.J.; Zhang, J.H.; Gomez, H.; Murugan, R.; Hong, X.; Xu, D.; Jiang, F.; Peng, Z.Y. Reactive oxygen species-induced lipid peroxidation in apoptosis, autophagy, and ferroptosis. Oxid. Med. Cell Longev. 2019, 2019, 5080843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Son, Y.; Kim, S.; Chung, H.T.; Pae, H.O. Reactive oxygen species in the activation of MAP kinases. Methods Enzymol. 2013, 528, 27–48. [Google Scholar] [PubMed]
- Chang, W.T.; Bow, Y.D.; Fu, P.J.; Li, C.Y.; Wu, C.Y.; Chang, Y.H.; Teng, Y.N.; Li, R.N.; Lu, M.C.; Liu, Y.C.; et al. A Marine Terpenoid, Heteronemin, Induces Both the Apoptosis and Ferroptosis of Hepatocellular Carcinoma Cells and Involves the ROS and MAPK Pathways. Oxid. Med. Cell Longev. 2021, 2021, 7689045. [Google Scholar] [CrossRef] [PubMed]
- Hunter, K.; Holscher, C. Drugs developed to treat diabetes, liraglutide and lixisenatide, cross the blood brain barrier and enhance neurogenesis. BMC Neurosci. 2012, 13, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.Q.; Holscher, C. GIP has neuroprotective effects in Alzheimer and Parkinson’s disease models. Peptides 2020, 125, 170184. [Google Scholar] [CrossRef]
- Nyberg, J.; Anderson, M.F.; Meister, B.; Alborn, A.M.; Strom, A.K.; Brederlau, A.; Illerskog, A.C.; Nilsson, O.; Kieffer, T.J.; Hietala, M.A.; et al. Glucose-dependent insulinotropic polypeptide is expressed in adult hippocampus and induces progenitor cell proliferation. J. Neurosci. 2005, 25, 1816–1825. [Google Scholar] [CrossRef] [Green Version]
- Ding, K.H.; Zhong, Q.; Xie, D.; Chen, H.X.; Della-Fera, M.A.; Bollag, R.J.; Bollag, W.B.; Gujral, R.; Kang, B.; Sridhar, S.; et al. Effects of glucose-dependent insulinotropic peptide on behavior. Peptides 2006, 27, 2750–2755. [Google Scholar] [CrossRef]
- Gault, V.A.; Holscher, C. Protease-resistant glucose-dependent insulinotropic polypeptide agonists facilitate hippocampal LTP and reverse the impairment of LTP induced by beta-amyloid. J. Neurophysiol. 2008, 99, 1590–1595. [Google Scholar] [CrossRef] [Green Version]
- Spielman, L.J.; Gibson, D.L.; Klegeris, A. Incretin hormones regulate microglia oxidative stress, survival and expression of trophic factors. Eur. J. Cell Biol. 2017, 96, 240–253. [Google Scholar] [CrossRef]
- Meldrum, B.S. Glutamate as a neurotransmitter in the brain: Review of physiology and pathology. J. Nutr. 2000, 130, 1007S–1015S. [Google Scholar] [CrossRef] [Green Version]
- Maino, B.; Ciotti, M.T.; Calissano, P.; Cavallaro, S. Transcriptional analysis of apoptotic cerebellar granule neurons following rescue by gastric inhibitory polypeptide. Int. J. Mol. Sci. 2014, 15, 5596–5622. [Google Scholar] [CrossRef] [Green Version]
- Cao, H.; Zuo, C.; Huang, Y.; Zhu, L.; Zhao, J.; Yang, Y.; Jiang, Y.; Wang, F. Hippocampal proteomic analysis reveals activation of necroptosis and ferroptosis in a mouse model of chronic unpredictable mild stress-induced depression. Behav. Brain Res. 2021, 407, 113261. [Google Scholar] [CrossRef]
- Gabe, M.B.N.; Sparre-Ulrich, A.H.; Pedersen, M.F.; Gasbjerg, L.S.; Inoue, A.; Brauner-Osborne, H.; Hartmann, B.; Rosenkilde, M.M. Human GIP(3-30)NH2 inhibits G protein-dependent as well as G protein-independent signaling and is selective for the GIP receptor with high-affinity binding to primate but not rodent GIP receptors. Biochem. Pharmacol. 2018, 150, 97–107. [Google Scholar] [CrossRef]
- Zhang, Y.; Lu, X.; Bhavnani, B.R. Equine estrogens differentially inhibit DNA fragmentation induced by glutamate in neuronal cells by modulation of regulatory proteins involved in programmed cell death. BMC Neurosci. 2003, 4, 32. [Google Scholar] [CrossRef] [Green Version]
- van Leyen, K.; Siddiq, A.; Ratan, R.R.; Lo, E.H. Proteasome inhibition protects HT22 neuronal cells from oxidative glutamate toxicity. J. Neurochem. 2005, 92, 824–830. [Google Scholar] [CrossRef]
- Lee, J.S.; Kim, W.Y.; Jeon, Y.J.; Lee, S.K.; Son, C.G. Aquilariae Lignum extract attenuates glutamate-induced neuroexcitotoxicity in HT22 hippocampal cells. Biomed. Pharmacother 2018, 106, 1031–1038. [Google Scholar] [CrossRef]
- Soldani, C.; Scovassi, A.I. Poly(ADP-ribose) polymerase-1 cleavage during apoptosis: An update. Apoptosis 2002, 7, 321–328. [Google Scholar] [CrossRef]
- Andrabi, S.A.; Kang, H.C.; Haince, J.F.; Lee, Y.I.; Zhang, J.; Chi, Z.; West, A.B.; Koehler, R.C.; Poirier, G.G.; Dawson, T.M.; et al. Iduna protects the brain from glutamate excitotoxicity and stroke by interfering with poly(ADP-ribose) polymer-induced cell death. Nat. Med. 2011, 17, 692–699. [Google Scholar] [CrossRef]
- Eliasson, M.J.; Sampei, K.; Mandir, A.S.; Hurn, P.D.; Traystman, R.J.; Bao, J.; Pieper, A.; Wang, Z.Q.; Dawson, T.M.; Snyder, S.H.; et al. Poly(ADP-ribose) polymerase gene disruption renders mice resistant to cerebral ischemia. Nat. Med. 1997, 3, 1089–1095. [Google Scholar] [CrossRef]
- Abdalkader, M.; Lampinen, R.; Kanninen, K.M.; Malm, T.M.; Liddell, J.R. Targeting Nrf2 to Suppress Ferroptosis and Mitochondrial Dysfunction in Neurodegeneration. Front. Neurosci. 2018, 12, 466. [Google Scholar] [CrossRef] [Green Version]
- Jezek, J.; Cooper, K.F.; Strich, R. Reactive oxygen species and mitochondrial dynamics: The Yin and Yang of mitochondrial dysfunction and cancer progression. Antioxidants 2018, 7, 13. [Google Scholar] [CrossRef]
- Yang, W.S.; Stockwell, B.R. Ferroptosis: Death by lipid peroxidation. Trends Cell Biol. 2016, 26, 165–176. [Google Scholar] [CrossRef] [Green Version]
- Adams, P.D.; Kaelin, W.G., Jr. Transcriptional control by E2F. Semin. Cancer Biol. 1995, 6, 99–108. [Google Scholar] [CrossRef]
- Rodenak-Kladniew, B.; Castro, A.; Starkel, P.; De Saeger, C.; Garcia de Bravo, M.; Crespo, R. Linalool induces cell cycle arrest and apoptosis in HepG2 cells through oxidative stress generation and modulation of Ras/MAPK and Akt/mTOR pathways. Life Sci. 2018, 199, 48–59. [Google Scholar] [CrossRef] [Green Version]
- Jiang, T.; Cheng, H.; Su, J.; Wang, X.; Wang, Q.; Chu, J.; Li, Q. Gastrodin protects against glutamate-induced ferroptosis in HT-22 cells through Nrf2/HO-1 signaling pathway. Toxicol. In Vitro 2020, 62, 104715. [Google Scholar] [CrossRef]
- Li, J.; Cao, F.; Yin, H.L.; Huang, Z.J.; Lin, Z.T.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, present and future. Cell Death Dis. 2020, 11, 88. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Lu, J.; Hao, X.; Li, H.; Zhang, G.; Liu, X.; Li, X.; Zhao, C.; Kuang, W.; Chen, D.; et al. FTH1 Inhibits Ferroptosis Through Ferritinophagy in the 6-OHDA Model of Parkinson’s Disease. Neurotherapeutics 2020, 17, 1796–1812. [Google Scholar] [CrossRef]
- Xie, Y.; Hou, W.; Song, X.; Yu, Y.; Huang, J.; Sun, X.; Kang, R.; Tang, D. Ferroptosis: Process and function. Cell Death Differ. 2016, 23, 369–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.Y.; Jung, W.J.; Kang, J.S.; Kim, C.M.; Park, G.; Choi, Y.W. Neuroprotective effects of alpha-iso-cubebene against glutamate-induced damage in the HT22 hippocampal neuronal cell line. Int. J. Mol. Med. 2015, 35, 525–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, J.; Liu, C.X.; Song, R.; Li, Q.L. Ferrostatin-1 protects HT-22 cells from oxidative toxicity. Neural. Regen. Res. 2020, 15, 528–536. [Google Scholar] [PubMed]
- Zhao, Z.Y.; Luan, P.; Huang, S.X.; Xiao, S.H.; Zhao, J.; Zhang, B.; Gu, B.B.; Pi, R.B.; Liu, J. Edaravone protects HT22 neurons from H2O2-induced apoptosis by inhibiting the MAPK signaling pathway. CNS Neurosci. Ther. 2013, 19, 163–169. [Google Scholar] [CrossRef]
- Song, J.H.; Kang, K.S.; Choi, Y.K. Protective effect of casuarinin against glutamate-induced apoptosis in HT22 cells through inhibition of oxidative stress-mediated MAPK phosphorylation. Bioorg. Med. Chem. Lett. 2017, 27, 5109–5113. [Google Scholar] [CrossRef]
- Trumper, A.; Trumper, K.; Horsch, D. Mechanisms of mitogenic and anti-apoptotic signaling by glucose-dependent insulinotropic polypeptide in beta(INS-1)-cells. J. Endocrinol. 2002, 174, 233–246. [Google Scholar] [CrossRef] [Green Version]
- Ji, C.; Xue, G.F.; Li, G.; Li, D.; Holscher, C. Neuroprotective effects of glucose-dependent insulinotropic polypeptide in Alzheimer’s disease. Rev. Neurosci. 2016, 27, 61–70. [Google Scholar] [CrossRef]
- Creutzfeldt, W.; Ebert, R.; Willms, B.; Frerichs, H.; Brown, J.C. Gastric inhibitory polypeptide (GIP) and insulin in obesity: Increased response to stimulation and defective feedback control of serum levels. Diabetologia 1978, 14, 15–24. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Yamada, Y.; Tsukiyama, K.; Miyawaki, K.; Hosokawa, M.; Nagashima, K.; Toyoda, K.; Naitoh, R.; Mizunoya, W.; Fushiki, T.; et al. Gastric inhibitory polypeptide modulates adiposity and fat oxidation under diminished insulin action. Biochem. Biophys. Res. Commun. 2005, 335, 937–942. [Google Scholar] [CrossRef] [Green Version]
- Zhong, Q.; Itokawa, T.; Sridhar, S.; Ding, K.H.; Xie, D.; Kang, B.; Bollag, W.B.; Bollag, R.J.; Hamrick, M.; Insogna, K.; et al. Effects of glucose-dependent insulinotropic peptide on osteoclast function. Am. J. Physiol. Endocrinol. Metab. 2007, 292, E543–E548. [Google Scholar] [CrossRef]
- Holscher, C. The incretin hormones glucagonlike peptide 1 and glucose-dependent insulinotropic polypeptide are neuroprotective in mouse models of Alzheimer’s disease. Alzheimers Dement. 2014, 10, S47–S54. [Google Scholar] [CrossRef]
- Kuhn-Wache, K.; Manhart, S.; Hoffmann, T.; Hinke, S.A.; Gelling, R.; Pederson, R.A.; McIntosh, C.H.; Demuth, H.U. Analogs of glucose-dependent insulinotropic polypeptide with increased dipeptidyl peptidase IV resistance. Adv. Exp. Med. Biol. 2000, 477, 187–195. [Google Scholar]
- Lamont, B.J.; Drucker, D.J. Differential antidiabetic efficacy of incretin agonists versus DPP-4 inhibition in high fat fed mice. Diabetes 2008, 57, 190–198. [Google Scholar] [CrossRef] [Green Version]
- Rossowski, W.J.; Zacharia, S.; Mungan, Z.; Ozmen, V.; Ertan, A.; Baylor, L.M.; Jiang, N.Y.; Coy, D.H. Reduced gastric acid inhibitory effect of a pGIP(1-30)NH2 fragment with potent pancreatic amylase inhibitory activity. Regul. Pept. 1992, 39, 9–17. [Google Scholar] [CrossRef]
- Harada, N.; Yamada, Y.; Tsukiyama, K.; Yamada, C.; Nakamura, Y.; Mukai, E.; Hamasaki, A.; Liu, X.; Toyoda, K.; Seino, Y.; et al. A novel GIP receptor splice variant influences GIP sensitivity of pancreatic beta-cells in obese mice. Am. J. Physiol. Endocrinol. Metab. 2008, 294, E61–E68. [Google Scholar] [CrossRef]
- Xie, D.; Zhong, Q.; Ding, K.H.; Cheng, H.; Williams, S.; Correa, D.; Bollag, W.B.; Bollag, R.J.; Insogna, K.; Troiano, N.; et al. Glucose-dependent insulinotropic peptide-overexpressing transgenic mice have increased bone mass. Bone 2007, 40, 1352–1360. [Google Scholar] [CrossRef]
- Kim, S.J.; Nian, C.; Karunakaran, S.; Clee, S.M.; Isales, C.M.; McIntosh, C.H. GIP-overexpressing mice demonstrate reduced diet-induced obesity and steatosis, and improved glucose homeostasis. PLoS ONE 2012, 7, e40156. [Google Scholar] [CrossRef]
- Faivre, E.; Gault, V.A.; Thorens, B.; Holscher, C. Glucose-dependent insulinotropic polypeptide receptor knockout mice are impaired in learning, synaptic plasticity, and neurogenesis. J. Neurophysiol. 2011, 105, 1574–1580. [Google Scholar] [CrossRef] [Green Version]
- Lennox, R.; Moffett, R.C.; Porter, D.W.; Irwin, N.; Gault, V.A.; Flatt, P.R. Effects of glucose-dependent insulinotropic polypeptide receptor knockout and a high-fat diet on cognitive function and hippocampal gene expression in mice. Mol. Med. Rep. 2015, 12, 1544–1548. [Google Scholar] [CrossRef] [Green Version]
- Moon, J.L.; Kim, S.Y.; Shin, S.W.; Park, J.W. Regulation of brefeldin A-induced ER stress and apoptosis by mitochondrial NADP(+)-dependent isocitrate dehydrogenase. Biochem. Biophys. Res. Commun. 2012, 417, 760–764. [Google Scholar] [CrossRef]
- Kang, J.H.; Kim, M.H.; Lee, H.J.; Huh, J.W.; Lee, H.S.; Lee, D.S. Peroxiredoxin 4 attenuates glutamate-induced neuronal cell death through inhibition of endoplasmic reticulum stress. Free Radic. Res. 2020, 54, 207–220. [Google Scholar] [CrossRef]
- Saleem, U.; Sabir, S.; Niazi, S.G.; Naeem, M.; Ahmad, B. Role of Oxidative Stress and Antioxidant Defense Biomarkers in Neurodegenerative Diseases. Crit. Rev. Eukaryot. Gene Expr. 2020, 30, 311–322. [Google Scholar] [CrossRef]
- Lancelot, E.; Beal, M.F. Glutamate toxicity in chronic neurodegenerative disease. Prog. Brain Res. 1998, 116, 331–347. [Google Scholar]
- Park, E.; Bell, J.D.; Baker, A.J. Traumatic brain injury: Can the consequences be stopped? CMAJ 2008, 178, 1163–1170. [Google Scholar] [CrossRef] [Green Version]
- Belov Kirdajova, D.; Kriska, J.; Tureckova, J.; Anderova, M. Ischemia-Triggered Glutamate Excitotoxicity From the Perspective of Glial Cells. Front. Cell Neurosci. 2020, 14, 51. [Google Scholar] [CrossRef] [Green Version]
- Faivre, E.; Hamilton, A.; Holscher, C. Effects of acute and chronic administration of GIP analogues on cognition, synaptic plasticity and neurogenesis in mice. Eur. J. Pharmacol. 2012, 674, 294–306. [Google Scholar] [CrossRef]
- Latunde-Dada, G.O. Ferroptosis: Role of lipid peroxidation, iron and ferritinophagy. Biochim. Et Biophys. Acta BBA-Bioenerg. Gen. Subj. 2017, 1861, 1893–1900. [Google Scholar] [CrossRef] [Green Version]
- Park, S.Y.; Jin, M.L.; Kim, Y.H.; Kim, C.M.; Lee, S.J.; Park, G. Involvement of heme oxygenase-1 in neuroprotection by sanguinarine against glutamate-triggered apoptosis in HT22 neuronal cells. Environ. Toxicol. Pharmacol. 2014, 38, 701–710. [Google Scholar] [CrossRef]
- New, D.C.; Wong, Y.H. Molecular mechanisms mediating the G protein-coupled receptor regulation of cell cycle progression. J. Mol. Signal. 2007, 2, 2. [Google Scholar] [CrossRef] [Green Version]
- Son, Y.; Cheong, Y.K.; Kim, N.H.; Chung, H.T.; Kang, D.G.; Pae, H.O. Mitogen-Activated Protein Kinases and Reactive Oxygen Species: How Can ROS Activate MAPK Pathways? J. Signal. Transduct. 2011, 2011, 792639. [Google Scholar] [CrossRef] [PubMed]
- Atkins, C.M.; Selcher, J.C.; Petraitis, J.J.; Trzaskos, J.M.; Sweatt, J.D. The MAPK cascade is required for mammalian associative learning. Nat. Neurosci. 1998, 1, 602–609. [Google Scholar] [CrossRef] [PubMed]
- Cavanaugh, J.E. Role of extracellular signal regulated kinase 5 in neuronal survival. Eur. J. Biochem. 2004, 271, 2056–2059. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Chen, L.; Zhao, Q.; Du, X.; Bi, M.; Li, Y.; Jiao, Q.; Jiang, H. Ferroptosis was more initial in cell death caused by iron overload and its underlying mechanism in Parkinson’s disease. Free Radic. Biol. Med. 2020, 152, 227–234. [Google Scholar] [CrossRef]
- Widenmaier, S.B.; Ao, Z.; Kim, S.J.; Warnock, G.; McIntosh, C.H. Suppression of p38 MAPK and JNK via Akt-mediated inhibition of apoptosis signal-regulating kinase 1 constitutes a core component of the beta-cell pro-survival effects of glucose-dependent insulinotropic polypeptide. J. Biol. Chem. 2009, 284, 30372–30382. [Google Scholar] [CrossRef] [Green Version]
- Lavine, J.A.; Attie, A.D. Gastrointestinal hormones and the regulation of beta-cell mass. Ann. N. Y. Acad. Sci. 2010, 1212, 41–58. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Forward Sequence | Reverse Sequence | Ala(bp) |
|---|---|---|---|
| GIP (NM_008119.2) | CTCTTTGCCCAAGAGCCTCA | ATCAGAAGGTCCCTCAGCACA | 92 |
| CDK1 (NM_007659.4) | CAGAACTGGCCACCAAGAAG | TTGTTAGGAGTGCCCAGAGC | 131 |
| CDK2 (NM_183417.3) | ATGGACGGAGCTTGTTATCG | CATCCTGGAAGAAAGGGTGA | 133 |
| CDK4 (NM_009870.4) | GGCCCTCAAGAGTGTGAGAG | CCTCCTTAACAAGGCCACCT | 131 |
| CDK6 (NM_009873.3) | AGAAGTCCTGCTCCAGTCCA | AAGAGGCTTTCT GCGAAACA | 131 |
| Cyclin D1 (NM_001379248.1) | TTGACTGCCGAGAAGTTGTG | CCACTTGAGCTTGTTCACCA | 136 |
| Cyclin E1 (NM_007633.2) | CCCTCTGACCATTGTGTCCT | ACCTGCTGTGGGTACTGAGG | 136 |
| Cyclin A1 (NM_001305221.1) | TCCACTTCCTGCTGGATTTC | CTGAACCAAAATCCGTTGCT | 133 |
| Nox1 (NM_172203.2) | CTCCAGCCTATCTCATCCTGAG | AGTGGCAATCACTCCAGTAAGGC | 166 |
| VDAC2 (NM_011695.2) | TCGGCAAAGCTGCCAGAGACAT | GTCTCCAAGGTCCCGCTAACTT | 195 |
| VDAC3 (NM_001198998.1) | GCCTTTGAAGGTTGGCTTGCTG | GAGCCTCCAAACTCAGTGCCAT | 190 |
| CARS (NM_013742.5) | GGGCTCTGCTGGAGAACATT | AGGGCATGACTGTTGACTCG | 178 |
| GPX4 (NM_008162.4) | CGCTCCATGCACGAATTCTC | GTGACGATGCACACGAAACC | 126 |
| HSPB1 (NM_013560.2) | GCTCACAGTGAAGACCAAGGAAG | TGAAGCACCGAGAGATGTAGCC | 137 |
| Nrf2 (NM_010902.4) | CAGCATAGAGCAGGACATGGAG | GAACAGCGGTAGTATCAGCCAG | 151 |
| Bax (NM_007527.3) | GGCGAATTGGAGATGAACTG | CAAAGTAGAAGAGGGCAACCAC | 201 |
| Bcl-2 (NM_009741.5) | TCGCCCTGTGGATGACTGA | CACTTGTGGCCCAGGTATG | 240 |
| Caspase-3 (NM_001284409.1) | ATGGGAGCAAGTCAGTGGAC | CGTACCAGAGCGAGATGACA | 177 |
| Caspase-9 (NM_015733.5) | GGCGGAGCTCATGATGTCTGTG | TTCCGGTGTGCCATCTCCATCA | 313 |
| PARP (NM_007415.3) | CTCTCCCAGAACAAGGACGAAG | CCGCTTTCACTTCCTCCATCTTC | 190 |
| GAPDH (NM_001289726.1) | TGAGGCCGGTGCTGAGTATGTCG | CCACAGTCTTCTGGGTGGCAGTG | 348 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ko, J.; Jang, S.; Kwon, W.; Kim, S.-Y.; Jang, S.; Kim, E.; Ji, Y.-R.; Park, S.; Kim, M.-O.; Choi, S.-K.; et al. Protective Effect of GIP against Monosodium Glutamate-Induced Ferroptosis in Mouse Hippocampal HT-22 Cells through the MAPK Signaling Pathway. Antioxidants 2022, 11, 189. https://doi.org/10.3390/antiox11020189
Ko J, Jang S, Kwon W, Kim S-Y, Jang S, Kim E, Ji Y-R, Park S, Kim M-O, Choi S-K, et al. Protective Effect of GIP against Monosodium Glutamate-Induced Ferroptosis in Mouse Hippocampal HT-22 Cells through the MAPK Signaling Pathway. Antioxidants. 2022; 11(2):189. https://doi.org/10.3390/antiox11020189
Chicago/Turabian StyleKo, Jiwon, Soyoung Jang, Wookbong Kwon, Si-Yong Kim, Soyeon Jang, Eungyung Kim, Young-Rae Ji, Sijun Park, Myoung-Ok Kim, Seong-Kyoon Choi, and et al. 2022. "Protective Effect of GIP against Monosodium Glutamate-Induced Ferroptosis in Mouse Hippocampal HT-22 Cells through the MAPK Signaling Pathway" Antioxidants 11, no. 2: 189. https://doi.org/10.3390/antiox11020189