
Peroxisomal Stress Response and Inter-Organelle Communication in Cellular Homeostasis and Aging


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Peroxisome Biogenesis and Peroxisomal Import Machinery
3. Key Metabolic Functions of Peroxisome
4. Cellular Responses to Peroxisomal Dysfunction
4.1. Transcriptional Changes upon Peroxisomal Dysfunction
4.2. Impaired ROS Homeostasis
4.3. Dysregulated Lipid Metabolism
4.4. Mitochondrial Dysfunction
4.5. Endoplasmic Reticulum (ER) Stress
4.6. Apoptosis and Ferroptosis
4.7. Autophagy and Pexophagy
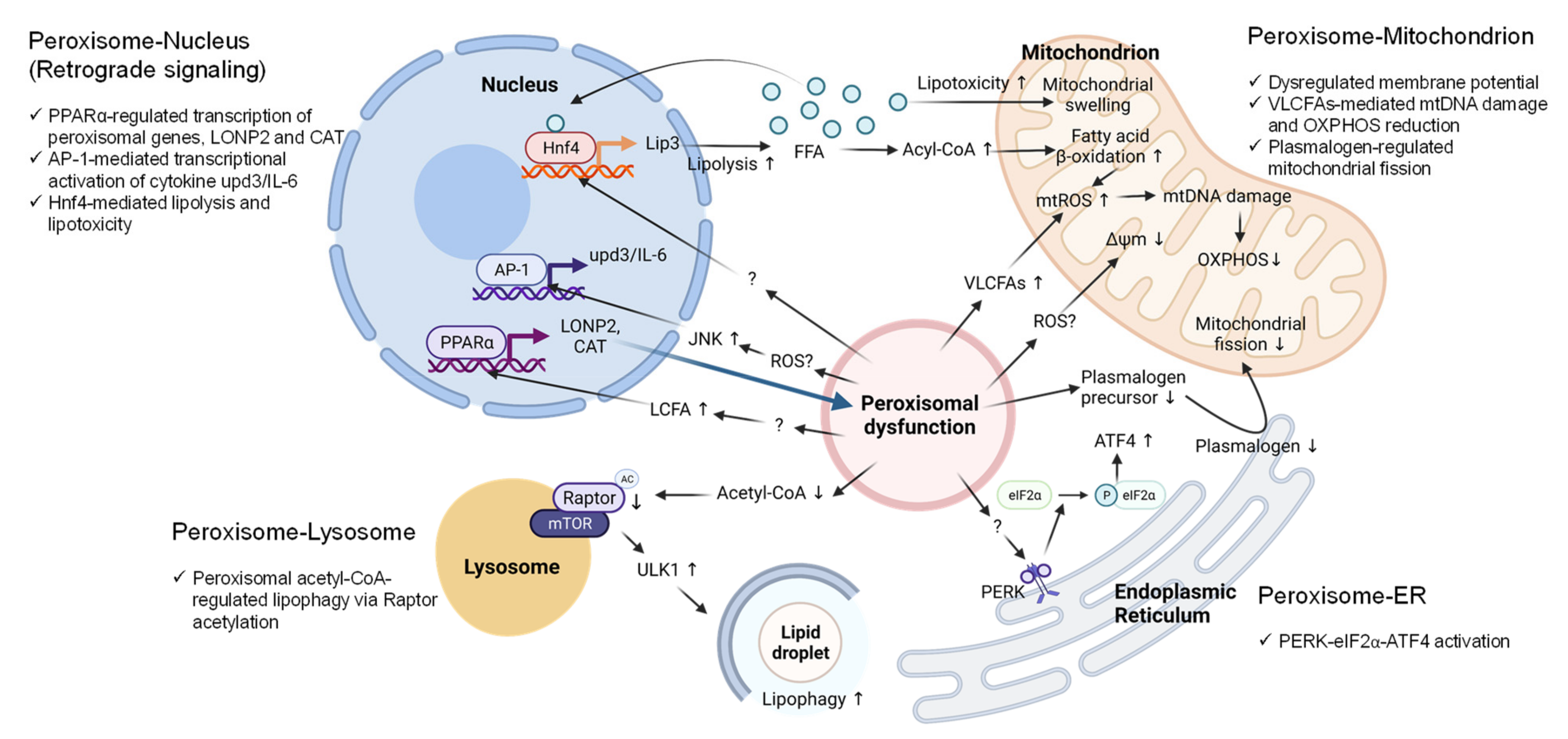
5. Peroxisome-Organelle Communication
5.1. Peroxisome-Nucleus Crosstalk
5.2. Peroxisome-Mitochondrion Crosstalk
5.3. Peroxisome-ER Crosstalk
5.4. Peroxisome-Lysosome Crosstalk
6. Peroxisomal Dysfunction in Aging and Aging-Related Diseases
7. Concluding Remark
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Klouwer, F.C.; Berendse, K.; Ferdinandusse, S.; Wanders, R.J.; Engelen, M. Zellweger spectrum disorders: Clinical overview and management approach. Orphanet J. Rare. Dis. 2015, 10, 151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waterham, H.R.; Ferdinandusse, S.; Wanders, R.J. Human disorders of peroxisome metabolism and biogenesis. Biochim. Biophys. Acta 2016, 1863, 922–933. [Google Scholar] [CrossRef]
- He, A.; Dean, J.M.; Lodhi, I.J. Peroxisomes as cellular adaptors to metabolic and environmental stress. Trends Cell Biol. 2021, 31, 656–670. [Google Scholar] [CrossRef] [PubMed]
- Wanders, R.J. Metabolic functions of peroxisomes in health and disease. Biochimie 2014, 98, 36–44. [Google Scholar] [CrossRef]
- Schrader, M.; Kamoshita, M.; Islinger, M. Organelle interplay-peroxisome interactions in health and disease. J. Inherit. Metab. Dis. 2020, 43, 71–89. [Google Scholar] [CrossRef]
- Lin, T.K.; Lin, K.J.; Lin, K.L.; Liou, C.W.; Chen, S.D.; Chuang, Y.C.; Wang, P.W.; Chuang, J.H.; Wang, T.J. When Friendship Turns Sour: Effective Communication Between Mitochondria and Intracellular Organelles in Parkinson’s Disease. Front. Cell Dev. Biol. 2020, 8, 607392. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Strunk, B.S.; Weisman, L.S. Close encounters of the lysosome-peroxisome kind. Cell 2015, 161, 197–198. [Google Scholar] [CrossRef] [Green Version]
- Huang, K.; Chen, W.; Zhu, F.; Li, P.; Li, P.W.-L.; Kapahi, P.; Bai, H. RiboTag translatomic profiling of Drosophila oenocytes under aging and induced oxidative stress. BMC Genom. 2019, 20, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, K.; Kim, J.; Vo, P.; Miao, T.; Bai, H. Peroxisome import stress impairs ribosome biogenesis and induces integrative stress response through eIF2α phosphorylation. bioRxiv 2021. [Google Scholar] [CrossRef]
- Rackles, E.; Witting, M.; Forné, I.; Zhang, X.; Zacherl, J.; Schrott, S.; Fischer, C.; Ewbank, J.J.; Osman, C.; Imhof, A.; et al. Reduced peroxisomal import triggers peroxisomal retrograde signaling. Cell Rep. 2021, 34, 108653. [Google Scholar] [CrossRef] [PubMed]
- Legakis, J.E.; Koepke, J.I.; Jedeszko, C.; Barlaskar, F.; Terlecky, L.J.; Edwards, H.J.; Walton, P.A.; Terlecky, S.R. Peroxisome senescence in human fibroblasts. Mol. Biol. Cell 2002, 13, 4243–4255. [Google Scholar] [CrossRef]
- Giordano, C.R.; Terlecky, S.R. Peroxisomes, cell senescence, and rates of aging. Biochim. Biophys. Acta Mol. Basis Dis. 2012, 1822, 1358–1362. [Google Scholar] [CrossRef] [Green Version]
- Narayan, V.; Ly, T.; Pourkarimi, E.; Murillo, A.B.; Gartner, A.; Lamond, A.; Kenyon, C. Deep Proteome Analysis Identifies Age-Related Processes in C. elegans. Cell Syst. 2016, 3, 144–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, K.; Miao, T.; Chang, K.; Kim, J.; Kang, P.; Jiang, Q.; Simmonds, A.J.; Di Cara, F.; Bai, H. Impaired peroxisomal import in Drosophila oenocytes causes cardiac dysfunction by inducing upd3 as a peroxikine. Nat. Commun. 2020, 11, 2943. [Google Scholar] [CrossRef]
- Islinger, M.; Cardoso, M.; Schrader, M. Be different—The diversity of peroxisomes in the animal kingdom. Biochim. et Biophys. Acta 2010, 1803, 881–897. [Google Scholar] [CrossRef] [Green Version]
- Huybrechts, S.J.; Van Veldhoven, P.P.; Brees, C.; Mannaerts, G.P.; Los, G.V.; Fransen, M. Peroxisome Dynamics in Cultured Mammalian Cells. Traffic 2009, 10, 1722–1733. [Google Scholar] [CrossRef]
- Lodhi, I.J.; Semenkovich, C.F. Peroxisomes: A Nexus for Lipid Metabolism and Cellular Signaling. Cell Metab. 2014, 19, 380–392. [Google Scholar] [CrossRef] [Green Version]
- Sugiura, A.; Mattie, S.; Prudent, J.; McBride, H.M. Newly born peroxisomes are a hybrid of mitochondrial and ER-derived pre-peroxisomes. Nature 2017, 542, 251–254. [Google Scholar] [CrossRef] [PubMed]
- Van der Zand, A.; Gent, J.; Braakman, I.; Tabak, H.F. Biochemically distinct vesicles from the endoplasmic reticulum fuse to form peroxisomes. Cell 2012, 149, 397–409. [Google Scholar] [CrossRef] [Green Version]
- Matsuzaki, T.; Fujiki, Y. The peroxisomal membrane protein import receptor Pex3p is directly transported to peroxisomes by a novel Pex19p- and Pex16p-dependent pathway. J. Cell Biol. 2008, 183, 1275–1286. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Morrell, J.C.; Jones, J.M.; Gould, S.J. PEX3 functions as a PEX19 docking factor in the import of class I peroxisomal membrane proteins. J. Cell Biol. 2004, 164, 863–875. [Google Scholar] [CrossRef] [Green Version]
- Lazarow, P.B.; Fujiki, Y. Biogenesis of Peroxisomes. Annu. Rev. Cell Biol. 1985, 1, 489–530. [Google Scholar] [CrossRef] [PubMed]
- Francisco, T.; Rodrigues, T.; Dias, A.F.; Barros-Barbosa, A.; Bicho, D.; Azevedo, J.E. Protein transport into peroxisomes: Knowns and unknowns. BioEssays 2017, 39, 39. [Google Scholar] [CrossRef] [PubMed]
- Platta, H.; Erdmann, R. The peroxisomal protein import machinery. FEBS Lett. 2007, 581, 2811–2819. [Google Scholar] [CrossRef] [PubMed]
- Gould, S.J.; Keller, G.-A.; Hosken, N.; Wilkinson, J.; Subramani, S. A conserved tripeptide sorts proteins to peroxisomes. J. Cell Biol. 1989, 108, 1657–1664. [Google Scholar] [CrossRef] [Green Version]
- Lazarow, P.B. Chapter 3.1.7. The import receptor Pex7p and the PTS2 targeting sequence. Biochim. Biophys. Acta Bioenerg. 2006, 1763, 1599–1604. [Google Scholar] [CrossRef] [Green Version]
- Swinkels, B.W.; Gould, S.J.; Bodnar, A.G.; Rachubinski, R.A.; Subramani, S. A novel, cleavable peroxisomal targeting signal at the amino-terminus of the rat 3-ketoacyl-CoA thiolase. EMBO J. 1991, 10, 3255–3262. [Google Scholar] [CrossRef]
- Meinecke, M.; Cizmowski, C.; Schliebs, W.; Krüger, V.; Beck, S.; Wagner, R.H.; Erdmann, R. The peroxisomal importomer constitutes a large and highly dynamic pore. Nat. Cell Biol. 2010, 12, 273–277. [Google Scholar] [CrossRef]
- Rucktäschel, R.; Girzalsky, W.; Erdmann, R. Protein import machineries of peroxisomes. Biochim. Biophys. Acta Biomembr. 2011, 1808, 892–900. [Google Scholar] [CrossRef]
- Platta, H.W.; El Magraoui, F.; Baumer, B.E.; Schlee, D.; Girzalsky, W.; Erdmann, R. Pex2 and Pex12 Function as Protein-Ubiquitin Ligases in Peroxisomal Protein Import. Mol. Cell. Biol. 2009, 29, 5505–5516. [Google Scholar] [CrossRef] [Green Version]
- Platta, H.W.; Brinkmeier, R.; Reidick, C.; Galiani, S.; Clausen, M.P.; Eggeling, C. Regulation of peroxisomal matrix protein import by ubiquitination. Biochim. Biophys. Acta Bioenerg. 2016, 1863, 838–849. [Google Scholar] [CrossRef] [PubMed]
- Thoms, S.; Erdmann, R. Peroxisomal matrix protein receptor ubiquitination and recycling. Biochim. Biophys. Acta Bioenerg. 2006, 1763, 1620–1628. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, A.F.; Pinto, M.P.; Grou, C.P.; Alencastre, I.S.; Fransen, M.; Sa-Miranda, C.; Azevedo, J.E. Ubiquitination of Mammalian Pex5p, the Peroxisomal Import Receptor. J. Biol. Chem. 2007, 282, 31267–31272. [Google Scholar] [CrossRef] [Green Version]
- Grou, C.P.; Carvalho, A.F.; Pinto, M.P.; Alencastre, I.S.; Rodrigues, T.A.; Freitas, M.O.; Francisco, T.; Sá-Miranda, C.; Azevedo, J.E. The peroxisomal protein import machinery—A case report of transient ubiquitination with a new flavor. Cell. Mol. Life Sci. 2008, 66, 254–262. [Google Scholar] [CrossRef]
- Matsumoto, N.; Tamura, S.; Fujiki, Y. The pathogenic peroxin Pex26p recruits the Pex1p–Pex6p AAA ATPase complexes to peroxisomes. Nat. Cell Biol. 2003, 5, 454–460. [Google Scholar] [CrossRef]
- Blok, N.B.; Tan, D.; Wang, R.Y.; Penczek, P.A.; Baker, D.; DiMaio, F.; Rapoport, T.A.; Walz, T. Unique double-ring structure of the peroxisomal Pex1/Pex6 ATPase complex revealed by cryo-electron microscopy. Proc. Natl. Acad. Sci. USA 2015, 112, E4017–E4025. [Google Scholar] [CrossRef] [Green Version]
- Ciniawsky, S.; Grimm, I.; Saffian, D.; Girzalsky, W.; Erdmann, R.; Wendler, P. Molecular snapshots of the Pex1/6 AAA+ complex in action. Nat. Commun. 2015, 6, 7331. [Google Scholar] [CrossRef] [Green Version]
- Tan, D.; Blok, N.B.; Rapoport, T.A.; Walz, T. Structures of the double-ring AAA ATPase Pex1-Pex6 involved in peroxisome biogenesis. FEBS J. 2015, 283, 986–992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gardner, B.M.; Chowdhury, S.; Lander, G.C.; Martin, A. The Pex1/Pex6 complex is a heterohexameric AAA+ motor with alternating and highly coordinated subunits. J. Mol. Biol. 2015, 427, 1375–1388. [Google Scholar] [CrossRef] [Green Version]
- Debelyy, M.; Platta, H.; Saffian, D.; Hensel, A.; Thoms, S.; Meyer, H.E.; Warscheid, B.; Girzalsky, W.; Erdmann, R. Ubp15p, a Ubiquitin Hydrolase Associated with the Peroxisomal Export Machinery. J. Biol. Chem. 2011, 286, 28223–28234. [Google Scholar] [CrossRef] [Green Version]
- Grou, C.P.; Francisco, T.; Rodrigues, T.A.; Freitas, M.O.; Pinto, M.P.; Carvalho, A.F.; Domingues, P.; Wood, S.A.; Rodríguez-Borges, J.E.; Sá-Miranda, C.; et al. Identification of Ubiquitin-specific Protease 9X (USP9X) as a Deubiquitinase Acting on Ubiquitin-Peroxin 5 (PEX5) Thioester Conjugate. J. Biol. Chem. 2012, 287, 12815–12827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrader, M.; Reuber, B.E.; Morrell, J.C.; Jimenez-Sanchez, G.; Obie, C.; Stroh, T.A.; Valle, D.; Schroer, T.A.; Gould, S.J. Expression of PEX11beta mediates peroxisome proliferation in the absence of extracellular stimuli. J. Biol. Chem. 1998, 273, 29607–29614. [Google Scholar] [CrossRef] [Green Version]
- Schrader, M.; Fahimi, H.D. Growth and Division of Peroxisomes. Adv. Appl. Microbiol. 2006, 255, 237–290. [Google Scholar]
- Fagarasanu, A.; Fagarasanu, M.; Rachubinski, R.A. Maintaining peroxisome populations: A story of division and inheritance. Annu. Rev. Cell Dev. Biol. 2007, 23, 321–344. [Google Scholar] [CrossRef] [PubMed]
- Thoms, S.; Erdmann, R. Dynamin-related proteins and Pex11 proteins in peroxisome division and proliferation. FEBS J. 2005, 272, 5169–5181. [Google Scholar] [CrossRef]
- Okumoto, K.; Tamura, S.; Honsho, M.; Fujiki, Y. Peroxisome: Metabolic Functions and Biogenesis. Adv. Exp. Med. Biol. 2020, 1299, 3–17. [Google Scholar]
- Ebberink, M.S.; Koster, J.; Visser, G.; van Spronsen, F.; Stolte-Dijkstra, I.; Smit, G.P.; Fock, J.M.; Kemp, S.; Wanders, R.J.; Waterham, H.R. A novel defect of peroxisome division due to a homozygous non-sense mutation in the PEX11β gene. J. Med. Genet. 2012, 49, 307–313. [Google Scholar] [CrossRef] [PubMed]
- Itoyama, A.; Honsho, M.; Abe, Y.; Moser, A.; Yoshida, Y.; Fujiki, Y. Docosahexaenoic acid mediates peroxisomal elongation, a prerequisite for peroxisome division. J. Cell Sci. 2012, 125, 589–602. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, S.; Tanaka, A.; Fujiki, Y. Fis1, DLP1, and Pex11p coordinately regulate peroxisome morphogenesis. Exp. Cell Res. 2007, 313, 1675–1686. [Google Scholar] [CrossRef]
- Opaliński, Ł.; Kiel, J.A.; Williams, C.; Veenhuis, M.; Van Der Klei, I.J. Membrane curvature during peroxisome fission requires Pex11. EMBO J. 2011, 30, 5–16. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, Y.; Niwa, H.; Honsho, M.; Itoyama, A.; Fujiki, Y. Pex11mediates peroxisomal proliferation by promoting deformation of the lipid membrane. Biol. Open 2015, 4, 710–721. [Google Scholar] [CrossRef] [Green Version]
- Su, J.; Thomas, A.S.; Grabietz, T.; Landgraf, C.; Volkmer, R.; Marrink, S.J.; Williams, C.; Melo, M.N. The N-terminal amphipathic helix of Pex11p self-interacts to induce membrane remodelling during peroxisome fission. Biochim. Biophys. Acta Biomembr. 2018, 1860, 1292–1300. [Google Scholar] [CrossRef]
- Itoyama, A.; Michiyuki, S.; Honsho, M.; Yamamoto, T.; Moser, A.; Yoshida, Y.; Fujiki, Y. Mff functions with Pex11pβ and DLP1 in peroxisomal fission. Biol. Open 2013, 2, 998–1006. [Google Scholar] [CrossRef] [Green Version]
- Bonekamp, N.A.; Völkl, A.; Fahimi, H.D.; Schrader, M. Reactive oxygen species and peroxisomes: Struggling for balance. BioFactors 2009, 35, 346–355. [Google Scholar] [CrossRef]
- Islinger, M.; Li, K.W.; Seitz, J.; Völkl, A.; Lüers, G.H. Hitchhiking of Cu/Zn Superoxide Dismutase to Peroxisomes–Evidence for a Natural Piggyback Import Mechanism in Mammals. Traffic 2009, 10, 1711–1721. [Google Scholar] [CrossRef] [PubMed]
- del Río, L.A.; Corpas, F.J.; Sandalio, L.M.; Palma, J.M.; Gómez, M.; Barroso, J.B. Reactive oxygen species, antioxidant systems and nitric oxide in peroxisomes. J. Exp. Bot. 2002, 53, 1255–1272. [Google Scholar] [CrossRef] [PubMed]
- Wanders, R.J.; Waterham, H.R. Biochemistry of Mammalian Peroxisomes Revisited. Annu. Rev. Biochem. 2006, 75, 295–332. [Google Scholar] [CrossRef]
- Cipolla, C.M.; Lodhi, I.J. Peroxisomal Dysfunction in Age-Related Diseases. Trends Endocrinol. Metab. 2017, 28, 297–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wanders, R.J.A.; Waterham, H.R.; Ferdinandusse, S. Metabolic Interplay between Peroxisomes and Other Subcellular Organelles Including Mitochondria and the Endoplasmic Reticulum. Front. Cell Dev. Biol. 2016, 3, 83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagan, N.; Zoeller, R.A. Plasmalogens: Biosynthesis and functions. Prog. Lipid Res. 2001, 40, 199–229. [Google Scholar] [CrossRef]
- Dean, J.M.; Lodhi, I.J. Structural and functional roles of ether lipids. Protein Cell 2018, 9, 196–206. [Google Scholar] [CrossRef] [PubMed]
- Honsho, M.; Tanaka, M.; Zoeller, R.A.; Fujiki, Y. Distinct Functions of Acyl/Alkyl Dihydroxyacetonephosphate Reductase in Peroxisomes and Endoplasmic Reticulum. Front. Cell Dev. Biol. 2020, 8, 855. [Google Scholar] [CrossRef] [PubMed]
- Gallego-García, A.; Monera-Girona, A.J.; Pajares-Martínez, E.; Bastida-Martínez, E.; Pérez-Castaño, R.; Iniesta, A.A.; Fontes, M.; Padmanabhan, S.; Elías-Arnanz, M. A bacterial light response reveals an orphan desaturase for human plasmalogen synthesis. Science 2019, 366, 128–132. [Google Scholar] [CrossRef]
- Werner, E.R.; Keller, M.A.; Sailer, S.; Lackner, K.; Koch, J.; Hermann, M.; Coassin, S.; Golderer, G.; Werner-Felmayer, G.; Zo-eller, R.A.; et al. The TMEM189 gene encodes plasmanylethanolamine desaturase which introduces the characteristic vinyl ether double bond into plasmalogens. Proc. Natl. Acad. Sci. USA 2020, 117, 7792–7798. [Google Scholar] [CrossRef] [Green Version]
- De Duve, C.; Baudhuin, P. Peroxisomes (microbodies and related particles). Physiol. Rev. 1966, 46, 323–357. [Google Scholar] [CrossRef]
- Boveris, A.; Oshino, N.; Chance, B. The cellular production of hydrogen peroxide. Biochem. J. 1972, 128, 617–630. [Google Scholar] [CrossRef]
- Fransen, M.; Nordgren, M.; Wang, B.; Apanasets, O. Role of peroxisomes in ROS/RNS-metabolism: Implications for human disease. Biochim. et Biophys. Acta Mol. Basis Dis. 2011, 1822, 1363–1373. [Google Scholar] [CrossRef] [Green Version]
- Peeters, A.; Fraisl, P.; Berg, S.V.D.; van Themaat, E.V.L.; Van Kampen, A.; Rider, M.H.; Takemori, H.; van Dijk, K.W.; Van Veldhoven, P.P.; Carmeliet, P.; et al. Carbohydrate Metabolism Is Perturbed in Peroxisome-deficient Hepatocytes Due to Mitochondrial Dysfunction, AMP-activated Protein Kinase (AMPK) Activation, and Peroxisome Proliferator-activated Receptor γ Coactivator 1α (PGC-1α) Suppression. J. Biol. Chem. 2011, 286, 42162–42179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baboota, R.K.; Shinde, A.B.; Lemaire, K.; Fransen, M.; Vinckier, S.; Van Veldhoven, P.P.; Schuit, F.; Baes, M. Functional peroxisomes are required for beta-cell integrity in mice. Mol. Metab. 2019, 22, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Mosser, J.; Douar, A.-M.; Sarde, C.-O.; Kioschis, P.; Feil, R.; Moser, H.; Poustka, A.-M.; Mandel, J.-L.; Aubourg, P. Putative X-linked adrenoleukodystrophy gene shares unexpected homology with ABC transporters. Nature 1993, 361, 726–730. [Google Scholar] [CrossRef] [PubMed]
- Schluter, A.; Espinosa, L.; Fourcade, S.; Galino, J.; López, E.; Ilieva, E.; Morató, L.; Asheuer, M.; Cook, T.; McLaren, A.; et al. Functional genomic analysis unravels a metabolic-inflammatory interplay in adrenoleukodystrophy. Hum. Mol. Genet. 2011, 21, 1062–1077. [Google Scholar] [CrossRef] [Green Version]
- Heit, C.; Marshall, S.; Singh, S.; Yu, X.; Charkoftaki, G.; Zhao, H.; Orlicky, D.J.; Fritz, K.; Thompson, D.C.; Vasiliou, V. Catalase deletion promotes prediabetic phenotype in mice. Free Radic. Biol. Med. 2017, 103, 48–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peeters, A.; Shinde, A.B.; Dirkx, R.; Smet, J.; De Bock, K.; Espeel, M.; Vanhorebeek, I.; Vanlander, A.; Van Coster, R.; Carmeliet, P.; et al. Mitochondria in peroxisome-deficient hepatocytes exhibit impaired respiration, depleted DNA, and PGC-1α independent proliferation. Biochim. Biophys. Acta Bioenerg. 2015, 1853, 285–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrer, I.; Aubourg, P.; Pujol, A. General Aspects and Neuropathology of X-Linked Adrenoleukodystrophy. Brain Pathol. 2009, 20, 817–830. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.-F.; Lawler, A.M.; Watkins, P.A.; Powers, J.M.; Moser, A.B.; Moser, H.W.; Smith, K.D. A mouse model for X-linked adrenoleukodystrophy. Proc. Natl. Acad. Sci. USA 1997, 94, 9366–9371. [Google Scholar] [CrossRef] [Green Version]
- Piao, L.; Dorotea, D.; Jiang, S.; Koh, E.H.; Oh, G.T.; Ha, H. Impaired Peroxisomal Fitness in Obese Mice, a Vicious Cycle Exacerbating Adipocyte Dysfunction via Oxidative Stress. Antioxid. Redox Signal. 2019, 31, 1339–1351. [Google Scholar] [CrossRef] [PubMed]
- Oruqaj, G.; Karnati, S.; Vijayan, V.; Kotarkonda, L.K.; Boateng, E.; Zhang, W.; Ruppert, C.; Günther, A.; Shi, W.; Baumgart-Vogt, E. Compromised peroxisomes in idiopathic pulmonary fibrosis, a vicious cycle inducing a higher fibrotic response via TGF-β signaling. Proc. Natl. Acad. Sci. USA 2015, 112, E2048–E2057. [Google Scholar] [CrossRef] [Green Version]
- Ahlemeyer, B.; Gottwald, M.; Baumgart-Vogt, E. Deletion of a single allele of the Pex11β gene is sufficient to cause oxidative stress, delayed differentiation and neuronal death in mouse brain. Dis. Model. Mech. 2012, 5, 125–140. [Google Scholar] [CrossRef] [Green Version]
- Walton, P.A.; Pizzitelli, M. Effects of peroxisomal catalase inhibition on mitochondrial function. Front. Physiol. 2012, 3, 108. [Google Scholar] [CrossRef] [Green Version]
- Ivashchenko, O.; Van Veldhoven, P.P.; Brees, C.; Ho, Y.-S.; Terlecky, S.R.; Fransen, M. Intraperoxisomal redox balance in mammalian cells: Oxidative stress and interorganellar cross-talk. Mol. Biol. Cell 2011, 22, 1440–1451. [Google Scholar] [CrossRef]
- Hwang, I.; Uddin, J.; Pak, E.S.; Kang, H.; Jin, E.-J.; Jo, S.; Kang, D.; Lee, H.; Ha, H. The impaired redox balance in peroxisomes of catalase knockout mice accelerates nonalcoholic fatty liver disease through endoplasmic reticulum stress. Free. Radic. Biol. Med. 2020, 148, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Moser, A.B.; Kreiter, N.; Bezman, L.; Lu, S.-E.; Raymond, G.V.; Naidu, S.; Moser, H.W. Plasma very long chain fatty acids in 3,000 peroxisome disease patients and 29,000 controls. Ann. Neurol. 1999, 45, 100–110. [Google Scholar] [CrossRef]
- Dasouki, M. Chapter 11-Peroxisomal disorders: Clinical and biochemical laboratory aspects. Biomark. Inborn Errors Metab. 2017, 2, 80087–80096. [Google Scholar]
- Stradomska, T.J.; Syczewska, M.; Jamroz, E.; Pleskaczyńska, A.; Kruczek, P.; Ciara, E.; Tylki-Szymanska, A. Serum very long-chain fatty acids (VLCFA) levels as predictive biomarkers of diseases severity and probability of survival in peroxisomal disorders. PLoS ONE 2020, 15, e0238796. [Google Scholar] [CrossRef]
- Aubourg, P.; Adamsbaum, C.; Lavallard-Rousseau, M.C.; Rocchiccioli, F.; Cartier, N.; Jambaque, I.; Jakobezak, C.; Lemaitre, A.; Boureau, F.; Wolf, C.; et al. A two-year trial of oleic and erucic acids (“Lorenzo’s oil”) as treatment for adrenomyeloneuropathy. N. Engl. J. Med. 1993, 329, 745–752. [Google Scholar] [CrossRef]
- van Geel, B.M.; Assies, J.; Haverkort, E.B.; Koelman, J.H.; Verbeeten, B.W.; Wanders, R.J.; Barth, P.G. Progression of abnormalities in adrenomyeloneuropathy and neurologically asymptomatic X-linked adrenoleukodystrophy despite treatment with “Lorenzo’s oil”. J. Neurol. Neurosurg. Psychiatry 1999, 67, 290–299. [Google Scholar] [CrossRef] [PubMed]
- Baes, M.; Van Veldhoven, P.P. Generalised and conditional inactivation of Pex genes in mice. Biochim. Biophys. Acta 2006, 1763, 1785–1793. [Google Scholar] [CrossRef] [Green Version]
- Faust, P.L.; Hatten, M.E. Targeted Deletion of the PEX2 Peroxisome Assembly Gene in Mice Provides a Model for Zellweger Syndrome, a Human Neuronal Migration Disorder. J. Cell Biol. 1997, 139, 1293–1305. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, M.; Bjorkman, J.; Nguyen, T.; Sharp, P.; Finnie, J.; Paterson, C.; Tonks, I.; Paton, B.C.; Kay, G.F.; Crane, D.I. Pex13 Inactivation in the Mouse Disrupts Peroxisome Biogenesis and Leads to a Zellweger Syndrome Phenotype. Mol. Cell. Biol. 2003, 23, 5947–5957. [Google Scholar] [CrossRef] [Green Version]
- Baes, M.; Gressens, P.; Baumgart-Vogt, E.; Carmeliet, P.; Casteels, M.; Fransen, M.; Evrard, P.; Fahimi, D.; Declercq, P.E.; Collen, D.; et al. A mouse model for Zellweger syndrome. Nat. Genet. 1997, 17, 49–57. [Google Scholar] [CrossRef]
- Janssen, A.; Baes, M.; Gressens, P.; Mannaerts, G.P.; Declercq, P.; Van Veldhoven, P.P. Docosahexaenoic Acid Deficit Is Not a Major Pathogenic Factor in Peroxisome-Deficient Mice. Lab. Investig. 2000, 80, 31–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brites, P.; Ferreira, A.S.; da Silva, T.F.; Sousa, V.F.; Malheiro, A.R.; Duran, M.; Waterham, H.R.; Baes, M.; Wanders, R.J.A. Alkyl-Glycerol Rescues Plasmalogen Levels and Pathology of Ether-Phospholipid Deficient Mice. PLoS ONE 2011, 6, e28539. [Google Scholar] [CrossRef] [Green Version]
- Hofer, D.; Pessentheiner, A.; Pelzmann, H.J.; Schlager, S.; Madreiter-Sokolowski, C.T.; Kolb, D.; Eichmann, T.O.; Rechberger, G.; Bilban, M.; Graier, W.; et al. Critical role of the peroxisomal protein PEX16 in white adipocyte development and lipid homeostasis. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 358–368. [Google Scholar] [CrossRef]
- Sellin, J.; Wingen, C.; Gosejacob, D.; Senyilmaz, D.; Hänschke, L.; Büttner, S.; Meyer, K.; Bano, D.; Nicotera, P.; Teleman, A.; et al. Dietary rescue of lipotoxicity-induced mitochondrial damage in Peroxin19 mutants. PLoS Biol. 2018, 16, e2004893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovacs, W.J.; Shackelford, J.E.; Tape, K.N.; Richards, M.J.; Faust, P.L.; Fliesler, S.J.; Krisans, S.K. Disturbed Cholesterol Homeostasis in a Peroxisome-Deficient PEX2 Knockout Mouse Model. Mol. Cell. Biol. 2004, 24, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ridgway, N.; McLeod, R. Biochemistry of Lipids, Lipoproteins and Membranes; Elsevier Science: Amsterdam, The Netherlands, 2008. [Google Scholar]
- Krisans, S.K.; Ericsson, J.; Edwards, P.A.; Keller, G.A. Farnesyl-diphosphate synthase is localized in peroxisomes. J. Biol. Chem. 1994, 269, 14165–14169. [Google Scholar] [CrossRef]
- Kovacs, W.J.; Tape, K.N.; Shackelford, J.E.; Wikander, T.M.; Richards, M.J.; Fliesler, S.J.; Krisans, S.K.; Faust, P.L. Peroxisome Deficiency Causes a Complex Phenotype because of Hepatic SREBP/Insig Dysregulation Associated with Endoplasmic Reticulum Stress. J. Biol. Chem. 2009, 284, 7232–7245. [Google Scholar] [CrossRef] [Green Version]
- Hogenboom, S.; Romeijn, G.J.; Houten, S.M.; Baes, M.; Wanders, R.J.A.; Waterham, H.R. Absence of functional peroxisomes does not lead to deficiency of enzymes involved in cholesterol biosynthesis. J. Lipid Res. 2002, 43, 90–98. [Google Scholar] [CrossRef]
- Hogenboom, S.; Wanders, R.J.; Waterham, H.R. Cholesterol biosynthesis is not defective in peroxisome biogenesis defective fibroblasts. Mol. Genet. Metab. 2003, 80, 290–295. [Google Scholar] [CrossRef]
- Hogenboom, S.; Tuyp, J.J.M.; Espeel, M.; Koster, J.; Wanders, R.J.A.; Waterham, H.R. Mevalonate kinase is a cytosolic enzyme in humans. J. Cell Sci. 2004, 117, 631–639. [Google Scholar] [CrossRef] [Green Version]
- Hogenboom, S.; Tuyp, J.J.M.; Espeel, M.; Koster, J.; Wanders, R.J.A.; Waterham, H.R. Phosphomevalonate kinase is a cytosolic protein in humans. J. Lipid Res. 2004, 45, 697–705. [Google Scholar] [CrossRef] [Green Version]
- Chu, B.-B.; Liao, Y.-C.; Qi, W.; Xie, C.; Du, X.; Wang, J.; Yang, H.; Miao, H.-H.; Li, B.-L.; Song, B.-L. Cholesterol Transport through Lysosome-Peroxisome Membrane Contacts. Cell 2015, 161, 291–306. [Google Scholar] [CrossRef] [Green Version]
- Fransen, M.; Lismont, C.; Walton, P. The Peroxisome-Mitochondria Connection: How and Why? Int. J. Mol. Sci. 2017, 18, 1126. [Google Scholar] [CrossRef]
- Baes, M.; Van Veldhoven, P.P. Hepatic dysfunction in peroxisomal disorders. Biochim. Biophys. Acta Bioenerg. 2016, 1863, 956–970. [Google Scholar] [CrossRef] [PubMed]
- Dirkx, R.; Vanhorebeek, I.; Martens, K.; Schad, A.; Grabenbauer, M.; Fahimi, D.; Declercq, P.; Van Veldhoven, P.P.; Baes, M. Absence of peroxisomes in mouse hepatocytes causes mitochondrial and ER abnormalities. Hepatology 2005, 41, 868–878. [Google Scholar] [CrossRef]
- Keane, M.H.; Overmars, H.; Wikander, T.M.; Ferdinandusse, S.; Duran, M.; Wanders, R.J.A.; Faust, P.L. Bile acid treatment alters hepatic disease and bile acid transport in peroxisome-deficientPEX2Zellweger mice. Hepatology 2007, 45, 982–997. [Google Scholar] [CrossRef] [PubMed]
- López-Erauskin, J.; Galino, J.; Ruiz, M.; Cuezva, J.; Fabregat, I.; Cacabelos, D.; Boada, J.; Martínez, J.; Ferrer, I.; Pamplona, R.; et al. Impaired mitochondrial oxidative phosphorylation in the peroxisomal disease X-linked adrenoleukodystrophy. Hum. Mol. Genet. 2013, 22, 3296–3305. [Google Scholar] [CrossRef] [Green Version]
- Bülow, M.H.; Wingen, C.; Senyilmaz, D.; Gosejacob, D.; Sociale, M.; Bauer, R.; Schulze, H.; Sandhoff, K.; Teleman, A.A.; Hoch, M.; et al. Unbalanced lipolysis results in lipotoxicity and mitochondrial damage in peroxisome-deficient Pex19 mutants. Mol. Biol. Cell 2018, 29, 396–407. [Google Scholar] [CrossRef]
- Park, H.; He, A.; Tan, M.; Johnson, J.; Dean, J.M.; Pietka, T.A.; Chen, Y.; Zhang, X.; Hsu, F.-F.; Razani, B.; et al. Peroxisome-derived lipids regulate adipose thermogenesis by mediating cold-induced mitochondrial fission. J. Clin. Investig. 2019, 129, 694–711. [Google Scholar] [CrossRef] [Green Version]
- Mathis, R.K.; Watkins, J.B.; Leeuwen, P.S.-V.; Lott, I.T. Liver in the cerebro-hepato-renal syndrome: Defective bile acid synthesis and abnormal mitochondria. Gastroenterology 1980, 79, 1311–1317. [Google Scholar] [CrossRef]
- Trijbels, J.M.F.; A Berden, J.; Monnens, L.A.H.; Willems, J.L.; Janssen, A.J.M.; Schutgens, R.B.H.; Essen, M.V.D.B.-V. Biochemical Studies in the Liver and Muscle of Patients with Zellweger Syndrome. Pediatr. Res. 1983, 17, 514–517. [Google Scholar] [CrossRef]
- Goldfischer, S.; Moore, C.L.; Johnson, A.B.; Spiro, A.J.; Valsamis, M.P.; Wisniewski, H.K.; Ritch, R.H.; Norton, W.T.; Rapin, I.; Gartner, L.M. Peroxisomal and Mitochondrial Defects in the Cerebro-Hepato-Renal Syndrome. Science 1973, 182, 62–64. [Google Scholar] [CrossRef]
- Hughes, J.L.; Poulos, A.; Robertson, E.; Chow, C.W.; Sheffield, L.J.; Christodoulou, J.; Carter, R.F. Pathology of hepatic peroxisomes and mitochondria in patients with peroxisomal disorders. Virchows Arch. A 1990, 416, 255–264. [Google Scholar] [CrossRef]
- Yoshida, H. ER stress and diseases. FEBS J. 2007, 274, 630–658. [Google Scholar] [CrossRef]
- Harding, H.; Zhang, Y.; Bertolotti, A.; Zeng, H.; Ron, D. Perk Is Essential for Translational Regulation and Cell Survival during the Unfolded Protein Response. Mol. Cell 2000, 5, 897–904. [Google Scholar] [CrossRef]
- Harding, H.; Novoa, I.; Zhang, Y.; Zeng, H.; Wek, R.; Schapira, M.; Ron, D. Regulated Translation Initiation Controls Stress-Induced Gene Expression in Mammalian Cells. Mol. Cell 2000, 6, 1099–1108. [Google Scholar] [CrossRef]
- Baird, T.; Wek, R.C. Eukaryotic Initiation Factor 2 Phosphorylation and Translational Control in Metabolism. Adv. Nutr. Int. Rev. J. 2012, 3, 307–321. [Google Scholar] [CrossRef] [Green Version]
- Cox, J.S.; Shamu, C.E.; Walter, P. Transcriptional induction of genes encoding endoplasmic reticulum resident proteins requires a transmembrane protein kinase. Cell 1993, 73, 1197–1206. [Google Scholar] [CrossRef]
- Haze, K.; Yoshida, H.; Yanagi, H.; Yura, T.; Mori, K. Mammalian Transcription Factor ATF6 Is Synthesized as a Transmembrane Protein and Activated by Proteolysis in Response to Endoplasmic Reticulum Stress. Mol. Biol. Cell 1999, 10, 3787–3799. [Google Scholar] [CrossRef] [Green Version]
- Kovacs, W.J.; Charles, K.N.; Walter, K.M.; Shackelford, J.E.; Wikander, T.M.; Richards, M.J.; Fliesler, S.J.; Krisans, S.K.; Faust, P.L. Peroxisome deficiency-induced ER stress and SREBP-2 pathway activation in the liver of newborn PEX2 knock-out mice. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2012, 1821, 895–907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, M.; Sun, X.; Wang, W.; Lian, Z.; Wu, P.; Han, S.; Chen, H.; Zhang, P. Disruption of peroxisome function leads to metabolic stress, mTOR inhibition, and lethality in liver cancer cells. Cancer Lett. 2018, 421, 82–93. [Google Scholar] [CrossRef]
- Faust, P.L.; Kovacs, W.J. Cholesterol biosynthesis and ER stress in peroxisome deficiency. Biochimie 2014, 98, 75–85. [Google Scholar] [CrossRef]
- Krysko, O.; Hulshagen, L.; Janssen, A.; Schütz, G.; Klein, R.; De Bruycker, M.; Espeel, M.; Gressens, P.; Baes, M. Neocortical and cerebellar developmental abnormalities in conditions of selective elimination of peroxisomes from brain or from liver. J. Neurosci. Res. 2007, 85, 58–72. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Song, J.; Ahn, C.; Kang, Y.; Chun, C.-H.; Jin, E.-J. Peroxisomal dysfunction is associated with up-regulation of apoptotic cell death via miR-223 induction in knee osteoarthritis patients with type 2 diabetes mellitus. Bone 2014, 64, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Okazaki, T.; Aoyama, S.; Yokota, M.; Koike, M.; Okada, Y.; Fujiki, Y.; Gotoh, Y. Peroxisomes control mitochondrial dynamics and the mitochondrion-dependent apoptosis pathway. J. Cell Sci. 2019, 132, jcs224766. [Google Scholar] [CrossRef] [Green Version]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [Green Version]
- Zou, Y.; Henry, W.S.; Ricq, E.L.; Graham, E.T.; Phadnis, V.V.; Maretich, P.; Paradkar, S.; Boehnke, N.; Deik, A.A.; Reinhardt, F.; et al. Plasticity of ether lipids promotes ferroptosis susceptibility and evasion. Nature 2020, 585, 603–608. [Google Scholar] [CrossRef] [PubMed]
- Germain, K.; Kim, P.K. Pexophagy: A Model for Selective Autophagy. Int. J. Mol. Sci. 2020, 21, 578. [Google Scholar] [CrossRef] [Green Version]
- Zientara-Rytter, K.; Subramani, S. Autophagic degradation of peroxisomes in mammals. Biochem. Soc. Trans. 2016, 44, 431–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yokota, S.; Haraguchi, C.M.; Oda, T. Induction of peroxisomal Lon protease in rat liver after di-(2-ethylhexyl)phthalate treatment. Histochem. Cell Biol. 2007, 129, 73–83. [Google Scholar] [CrossRef]
- Yokota, S.; Fahimi, H.D. Degradation of excess peroxisomes in mammalian liver cells by autophagy and other mechanisms. Histochem. Cell Biol. 2009, 131, 455–458. [Google Scholar] [CrossRef] [PubMed]
- Van Leyen, K.; Duvoisin, R.M.; Engelhardt, H.; Wiedmann, M. A function for lipoxygenase in programmed organelle degradation. Nature 1998, 395, 392–395. [Google Scholar] [CrossRef]
- Komatsu, M.; Waguri, S.; Ueno, T.; Iwata, J.; Murata, S.; Tanida, I.; Ezaki, J.; Mizushima, N.; Ohsumi, Y.; Uchiyama, Y.; et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J. Cell Biol. 2005, 169, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, W. Mechanisms and Functions of Pexophagy in Mammalian Cells. Cells 2021, 10, 1094. [Google Scholar] [CrossRef]
- Steinberg, S.J.; Dodt, G.; Raymond, G.V.; Braverman, N.E.; Moser, A.B.; Moser, H.W. Peroxisome biogenesis disorders. Biochim. Biophys. Acta Mol. Cell Res. 2006, 1763, 1733–1748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Law, K.B.; Bronte-Tinkew, D.; Di Pietro, E.; Snowden, A.; Jones, R.O.; Moser, A.; Brumell, J.H.; Braverman, N.; Kim, P.K. The peroxisomal AAA ATPase complex prevents pexophagy and development of peroxisome biogenesis disorders. Autophagy 2017, 13, 868–884. [Google Scholar] [CrossRef] [PubMed]
- Klouwer, F.C.C.; Falkenberg, K.D.; Ofman, R.; Koster, J.; van Gent, D.; Ferdinandusse, S.; Wanders, R.J.A.; Waterham, H.R. Autophagy Inhibitors Do Not Restore Peroxisomal Functions in Cells with the Most Common Peroxisome Biogenesis Defect. Front. Cell Dev. Biol. 2021, 9, 661298. [Google Scholar] [CrossRef] [PubMed]
- He, A.; Chen, X.; Tan, M.; Chen, Y.; Lu, D.; Zhang, X.; Dean, J.M.; Razani, B.; Lodhi, I.J. Acetyl-CoA Derived from Hepatic Peroxisomal β-Oxidation Inhibits Autophagy and Promotes Steatosis via mTORC1 Activation. Mol. Cell 2020, 79, 30–42. [Google Scholar] [CrossRef]
- He, A.; Dean, J.M.; Lu, D.; Chen, Y.; Lodhi, I.J. Hepatic peroxisomal β-oxidation suppresses lipophagy via RPTOR acetylation and MTOR activation. Autophagy 2020, 16, 1727–1728. [Google Scholar] [CrossRef] [PubMed]
- Zechner, R.; Madeo, F.; Kratky, D. Cytosolic lipolysis and lipophagy: Two sides of the same coin. Nat. Rev. Mol. Cell Biol. 2017, 18, 671–684. [Google Scholar] [CrossRef]
- Li, W.; He, P.; Huang, Y.; Li, Y.-F.; Lu, J.; Li, M.; Kurihara, H.; Luo, Z.; Meng, T.; Onishi, M.; et al. Selective autophagy of intracellular organelles: Recent research advances. Theranostics 2021, 11, 222–256. [Google Scholar] [CrossRef] [PubMed]
- Schrader, M.; Grille, S.; Fahimi, H.D.; Islinger, M. Peroxisome Interactions and Cross-Talk with Other Subcellular Compartments in Animal Cells. Subcell. Biochem. 2013, 69, 1–22. [Google Scholar] [PubMed]
- Rakhshandehroo, M.; Knoch, B.; Müller, M.; Kersten, S. Peroxisome Proliferator-Activated Receptor Alpha Target Genes. PPAR Res. 2010, 2010, 393–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rakhshandehroo, M.; Hooiveld, G.; Müller, M.; Kersten, S. Comparative analysis of gene regulation by the transcription factor PPARalpha between mouse and human. PLoS ONE 2009, 4, e6796. [Google Scholar] [CrossRef] [Green Version]
- Palanker, L.; Tennessen, J.M.; Lam, G.; Thummel, C.S. Drosophila HNF4 regulates lipid mobilization and beta-oxidation. Cell Metab. 2009, 9, 228–239. [Google Scholar] [CrossRef] [Green Version]
- E Barry, W.; Thummel, C.S. The Drosophila HNF4 nuclear receptor promotes glucose-stimulated insulin secretion and mitochondrial function in adults. eLife 2016, 5, e11183. [Google Scholar] [CrossRef]
- Poirier, Y.; Antonenkov, V.D.; Glumoff, T.; Hiltunen, J.K. Peroxisomal beta-oxidation-a metabolic pathway with multiple functions. Biochim. Et Biophys. Acta Mol. Cell Res. 2006, 1763, 1413–1426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shai, N.; Yifrach, E.; Van Roermund, C.W.T.; Cohen, N.; Bibi, C.; Ijlst, L.; Cavellini, L.; Meurisse, J.; Schuster, R.; Zada, L.; et al. Systematic mapping of contact sites reveals tethers and a function for the peroxisome-mitochondria contact. Nat. Commun. 2018, 9, 1761. [Google Scholar] [CrossRef] [PubMed]
- Usaj, M.M.; Brložnik, M.; Kaferle, P.; Žitnik, M.; Wolinski, H.; Leitner, F.; Kohlwein, S.D.; Zupan, B.; Petrovič, U. Genome-Wide Localization Study of Yeast Pex11 Identifies Peroxisome–Mitochondria Interactions through the ERMES Complex. J. Mol. Biol. 2015, 427, 2072–2087. [Google Scholar] [CrossRef]
- McGuinness, M.C.; Lu, J.-F.; Zhang, H.-P.; Dong, G.-X.; Heinzer, A.K.; Watkins, P.A.; Powers, J.; Smith, K.D. Role of ALDP (ABCD1) and Mitochondria in X-Linked Adrenoleukodystrophy. Mol. Cell. Biol. 2003, 23, 744–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, J.; Li, X.; Issop, L.; Culty, M. ACBD2/ECI2-Mediated Peroxisome-Mitochondria Interactions in Leydig Cell Steroid Biosynthesis. Mol. Endocrinol. 2016, 30, 763–782. [Google Scholar] [CrossRef] [Green Version]
- Neuspiel, M.; Schauss, A.C.; Braschi, E.; Zunino, R.; Rippstein, P.; Rachubinski, R.A.; Andrade-Navarro, M.A.; McBride, H.M. Cargo-Selected Transport from the Mitochondria to Peroxisomes Is Mediated by Vesicular Carriers. Curr. Biol. 2008, 18, 102–108. [Google Scholar] [CrossRef] [Green Version]
- Forss-Petter, S.; Werner, H.; Berger, J.; Lassmann, H.; Molzer, B.; Schwab, M.H.; Bernheimer, H.; Zimmermann, F.; Nave, K.A. Targeted inactivation of the X-linked adrenoleukodystrophy gene in mice. J. Neurosci. Res. 1997, 50, 829–843. [Google Scholar] [CrossRef]
- Pujol, A.; Hindelang, C.; Callizot, N.; Bartsch, U.; Schachner, M.; Mandel, J.L. Late onset neurological phenotype of the X-ALD gene inactivation in mice: A mouse model for adrenomyeloneuropathy. Hum. Mol. Genet. 2002, 11, 499–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oezen, I.; Rossmanith, W.; Forss-Petter, S.; Kemp, S.; Voigtländer, T.; Moser-Thier, K.; Wanders, R.J.; Bittner, R.E.; Berger, J. Accumulation of very long-chain fatty acids does not affect mitochondrial function in adrenoleukodystrophy protein deficiency. Hum. Mol. Genet. 2005, 14, 1127–1137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novikoff, P.M.; Novikoff, A.B. Peroxisomes in absorptive cells of mammalian small intestine. J. Cell Biol. 1972, 53, 532–560. [Google Scholar] [CrossRef] [Green Version]
- Zaar, K.; Völkl, A.; Fahimi, H. Association of isolated bovine kidney cortex peroxisomes with endoplasmic reticulum. Biochim. Biophys. Acta Biomembr. 1987, 897, 135–142. [Google Scholar] [CrossRef]
- Gronemeyer, T.; Wiese, S.; Ofman, R.; Bunse, C.; Pawlas, M.; Hayen, H.; Eisenacher, M.; Stephan, C.; Meyer, H.E.; Waterham, H.R.; et al. The proteome of human liver peroxisomes: Identification of five new peroxisomal constituents by a label-free quantitative proteomics survey. PLoS ONE 2013, 8, e57395. [Google Scholar] [CrossRef]
- Costello, J.; Castro, I.G.; Hacker, C.; Schrader, T.A.; Metz, J.; Zeuschner, D.; Azadi, A.S.; Godinho, L.F.; Costina, V.; Findeisen, P.; et al. ACBD5 and VAPB mediate membrane associations between peroxisomes and the ER. J. Cell Biol. 2017, 216, 331–342. [Google Scholar] [CrossRef] [Green Version]
- Hua, R.; Cheng, D.; Coyaud, É.; Freeman, S.; Di Pietro, E.; Wang, Y.; Vissa, A.; Yip, C.M.; Fairn, G.D.; Braverman, N.; et al. VAPs and ACBD5 tether peroxisomes to the ER for peroxisome maintenance and lipid homeostasis. J. Cell Biol. 2017, 216, 367–377. [Google Scholar] [CrossRef]
- Wanders, R.J.A.; Vaz, F.M.; Waterham, H.R.; Ferdinandusse, S. Fatty Acid Oxidation in Peroxisomes: Enzymology, Metabolic Crosstalk with Other Organelles and Peroxisomal Disorders. Adv. Exp. Med. Biol. 2020, 1299, 55–70. [Google Scholar]
- Ferdinandusse, S.; Falkenberg, K.; Koster, J.; Mooyer, P.A.; Jones, R.; Van Roermund, C.W.T.; A Pizzino, A.; Schrader, M.; Wanders, R.J.A.; Vanderver, A.; et al. ACBD5 deficiency causes a defect in peroxisomal very long-chain fatty acid metabolism. J. Med Genet. 2017, 54, 330–337. [Google Scholar] [CrossRef] [Green Version]
- Herzog, K.; Pras-Raves, M.L.; Ferdinandusse, S.; Vervaart, M.A.T.; Luyf, A.C.M.; van Kampen, A.H.C.; Wanders, R.J.A.; Waterham, H.R.; Vaz, F.M. Functional characterisation of peroxisomal β-oxidation disorders in fibroblasts using lipidomics. J. Inherit. Metab. Dis. 2018, 41, 479–487. [Google Scholar] [CrossRef] [Green Version]
- Torres, S.E.; Gallagher, C.M.; Plate, L.; Gupta, M.; Liem, C.R.; Guo, X.; Tian, R.; Stroud, R.M.; Kampmann, M.; Weissman, J.S.; et al. Ceapins block the unfolded protein response sensor ATF6α by inducing a neomorphic inter-organelle tether. eLife 2019, 8, e46595. [Google Scholar] [CrossRef]
- Gallagher, C.M.; Walter, P. Ceapins inhibit ATF6α signaling by selectively preventing transport of ATF6α to the Golgi apparatus during ER stress. eLife 2016, 5, e11880. [Google Scholar] [CrossRef]
- Gallagher, C.M.; Garri, C.; Cain, E.L.; Ang, K.K.-H.; Wilson, C.G.; Chen, S.; Hearn, B.; Jaishankar, P.; Aranda-Diaz, A.; Arkin, M.R.; et al. Ceapins are a new class of unfolded protein response inhibitors, selectively targeting the ATF6α branch. eLife 2016, 5, e11878. [Google Scholar] [CrossRef] [PubMed]
- Kleinecke, S.; Richert, S.; De Hoz, L.; Brügger, B.; Kungl, T.; Asadollahi, E.; Quintes, S.; Blanz, J.; McGonigal, R.; Naseri, K.; et al. Peroxisomal dysfunctions cause lysosomal storage and axonal Kv1 channel redistribution in peripheral neuropathy. eLife 2017, 6, e23332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sebastiani, P.; Federico, A.; Morris, M.; Gurinovich, A.; Tanaka, T.; Chandler, K.B.; Andersen, S.L.; Denis, G.; Costello, C.E.; Ferrucci, L.; et al. Protein signatures of centenarians and their offspring suggest centenarians age slower than other humans. Aging Cell 2021, 20, e13290. [Google Scholar] [CrossRef]
- Titorenko, V.I.; Terlecky, S.R. Peroxisome Metabolism and Cellular Aging. Traffic 2010, 12, 252–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lizard, G.; Rouaud, O.; Demarquoy, J.; Cherkaoui-Malki, M.; Iuliano, L. Potential roles of peroxisomes in Alzheimer’s disease and in dementia of the Alzheimer’s type. J. Alzheimer Dis. 2012, 29, 241–254. [Google Scholar] [CrossRef]
- Jo, D.S.; Park, N.Y.; Cho, D.-H. Peroxisome quality control and dysregulated lipid metabolism in neurodegenerative diseases. Exp. Mol. Med. 2020, 52, 1486–1495. [Google Scholar] [CrossRef]
- Pharaoh, G.; Sataranatarajan, K.; Street, K.; Hill, S.; Gregston, J.; Ahn, B.; Kinter, C.; Kinter, M.; Van Remmen, H. Metabolic and Stress Response Changes Precede Disease Onset in the Spinal Cord of Mutant SOD1 ALS Mice. Front. Neurosci. 2019, 13, 487. [Google Scholar] [CrossRef] [PubMed]
- Qin, F.; Lennon-Edwards, S.; Lancel, S.; Biolo, A.; Siwik, D.A.; Pimentel, D.R.; Dorn, G.W.; Kang, Y.J.; Colucci, W.S. Cardiac-specific overexpression of catalase identifies hydrogen peroxide-dependent and -independent phases of myocardial remodeling and prevents the progression to overt heart failure in G(alpha)q-overexpressing transgenic mice. Circ. Heart Fail. 2010, 3, 306–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, I.; Lee, J.; Huh, J.Y.; Park, J.; Lee, H.B.; Ho, Y.-S.; Ha, H. Catalase Deficiency Accelerates Diabetic Renal Injury Through Peroxisomal Dysfunction. Diabetes 2012, 61, 728–738. [Google Scholar] [CrossRef] [Green Version]
- Francque, S.; Szabo, G.; Abdelmalek, M.F.; Byrne, C.D.; Cusi, K.; Dufour, J.-F.; Roden, M.; Sacks, F.; Tacke, F. Nonalcoholic steatohepatitis: The role of peroxisome proliferator-activated receptors. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 24–39. [Google Scholar] [CrossRef]
- Marcus, D.L.; Thomas, C.; Rodriguez, C.; Simberkoff, K.; Tsai, J.S.; Strafaci, J.A.; Freedman, M.L. Increased peroxidation and reduced antioxidant enzyme activity in Alzheimer’s disease. Exp. Neurol. 1998, 150, 40–44. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, F.G.; Pahan, K.; Khan, M.; Barbosa, E.; Singh, I. Abnormality in catalase import into peroxisomes leads to severe neurological disorder. Proc. Natl. Acad. Sci. USA 1998, 95, 2961–2966. [Google Scholar] [CrossRef] [Green Version]
- Goodenowe, D.B.; Cook, L.L.; Liu, J.; Lu, Y.; Jayasinghe, D.A.; Ahiahonu, P.W.; Heath, D.; Yamazaki, Y.; Flax, J.; Krenitsky, K.F.; et al. Peripheral ethanolamine plasmalogen deficiency: A logical causative factor in Alzheimer’s disease and dementia. J. Lipid Res. 2007, 48, 2485–2498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graham, W.V.; Bonito-Oliva, A.; Sakmar, T.P. Update on Alzheimer’s Disease Therapy and Prevention Strategies. Annu. Rev. Med. 2017, 68, 413–430. [Google Scholar] [CrossRef] [Green Version]
- Cimini, A.; Moreno, S.; D’Amelio, M.; Cristiano, L.; D’Angelo, B.; Falone, S.; Benedetti, E.; Carrara, P.; Fanelli, F.; Cecconi, F.; et al. Early biochemical and morphological modifications in the brain of a transgenic mouse model of Alzheimer’s disease: A role for peroxisomes. J. Alzheimer Dis. 2009, 18, 935–952. [Google Scholar] [CrossRef] [PubMed]
- Fanelli, F.; Sepe, S.; D’Amelio, M.; Bernardi, C.; Cristiano, L.; Cimini, A.; Cecconi, F.; Ceru’, M.P.; Moreno, S. Age-dependent roles of peroxisomes in the hippocampus of a transgenic mouse model of Alzheimer’s disease. Mol. Neurodegener. 2013, 8, 8. [Google Scholar] [CrossRef] [Green Version]
- Nunomura, A.; Tamaoki, T.; Motohashi, N.; Nakamura, M.; McKeel, D.W., Jr.; Tabaton, M.; Lee, H.-G.; Smith, M.A.; Perry, G.; Zhu, X. The Earliest Stage of Cognitive Impairment in Transition from Normal Aging to Alzheimer Disease Is Marked by Prominent RNA Oxidation in Vulnerable Neurons. J. Neuropathol. Exp. Neurol. 2012, 71, 233–241. [Google Scholar] [CrossRef]
- Kou, J.; Kovacs, G.G.; Höftberger, R.; Kulik, W.; Brodde, A.; Forss-Petter, S.; Hönigschnabl, S.; Gleiss, A.; Brügger, B.; Wanders, R.; et al. Peroxisomal alterations in Alzheimer’s disease. Acta Neuropathol. 2011, 122, 271–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Astarita, G.; Jung, K.M.; Berchtold, N.C.; Nguyen, V.Q.; Gillen, D.L.; Head, E.; Cotman, C.W.; Piomelli, D. Deficient liver biosynthesis of docosahexaenoic acid correlates with cognitive impairment in Alzheimer’s disease. PLoS ONE 2010, 5, e12538. [Google Scholar] [CrossRef] [Green Version]
- Shi, R.; Zhang, Y.; Shi, Y.; Shi, S.; Jiang, L. Inhibition of peroxisomal β-oxidation by thioridazine increases the amount of VLCFAs and Aβ generation in the rat brain. Neurosci. Lett. 2012, 528, 6–10. [Google Scholar] [CrossRef]
- Inestrosa, N.C.; Carvajal, F.J.; Zolezzi, J.M.; Tapia-Rojas, C.; Serrano, F.; Karmelic, D.; Toledo, E.M.; Toro, A.; Toro, J.; Santos, M.J. Peroxisome proliferators reduce spatial memory impairment, synaptic failure, and neurodegeneration in brains of a double transgenic mice model of Alzheimer’s disease. J. Alzheimer Dis. 2013, 33, 941–959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berger, J.; Dorninger, F.; Forss-Petter, S.; Kunze, M. Peroxisomes in brain development and function. Biochim. Biophys. Acta 2016, 1863, 934–955. [Google Scholar] [CrossRef] [Green Version]
- Dai, D.-F.; Chen, T.; Johnson, S.; Szeto, H.; Rabinovitch, P.S. Cardiac Aging: From Molecular Mechanisms to Significance in Human Health and Disease. Antioxid. Redox Signal. 2012, 16, 1492–1526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yazdanyar, A.; Newman, A.B. The Burden of Cardiovascular Disease in the Elderly: Morbidity, Mortality, and Costs. Clin. Geriatr. Med. 2009, 25, 563–577. [Google Scholar] [CrossRef] [Green Version]
- Rodgers, J.L.; Jones, J.; Bolleddu, S.I.; Vanthenapalli, S.; Rodgers, L.E.; Shah, K.; Karia, K.; Panguluri, S.K. Cardiovascular Risks Associated with Gender and Aging. J. Cardiovasc. Dev. Dis. 2019, 6, 19. [Google Scholar] [CrossRef] [Green Version]
- Mittler, R. Oxidative stress, antioxidants and stress tolerance. Trends Plant Sci. 2002, 7, 405–410. [Google Scholar] [CrossRef]
- Bäumer, A.T.; Flesch, M.; Wang, X.; Shen, Q.; Feuerstein, G.Z.; Böhm, M. Antioxidative Enzymes in Human Hearts with Idiopathic Dilated Cardiomyopathy. J. Mol. Cell. Cardiol. 2000, 32, 121–130. [Google Scholar] [CrossRef]
- Colasante, C.; Chen, J.; Ahlemeyer, B.; Baumgart-Vogt, E. Peroxisomes in cardiomyocytes and the peroxisome/peroxisome proliferator-activated receptor-loop. Thromb. Haemost. 2015, 113, 452–463. [Google Scholar] [CrossRef] [Green Version]
- Koh, J.T.; Choi, H.H.; Ahn, K.Y.; Kim, J.U.; Kim, J.H.; Chun, J.-Y.; Baik, Y.H.; Kim, K.K. Cardiac Characteristics of Transgenic Mice Overexpressing Refsum Disease Gene-Associated Protein within the Heart. Biochem. Biophys. Res. Commun. 2001, 286, 1107–1116. [Google Scholar] [CrossRef] [PubMed]
- Wanders, R.J.; Komen, J.C. Peroxisomes, Refsum’s disease and the alpha- and omega-oxidation of phytanic acid. Biochem. Soc. Trans. 2007, 35, 865–869. [Google Scholar] [CrossRef] [PubMed]
- Miard, S.; Picard, F. Obesity and aging have divergent genomic fingerprints. Int. J. Obes. 2008, 32, 1873–1874. [Google Scholar] [CrossRef] [Green Version]
- Barzilai, N.; Huffman, D.M.; Muzumdar, R.H.; Bartke, A. The Critical Role of Metabolic Pathways in Aging. Diabetes 2012, 61, 1315–1322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jura, M.; Kozak, L. Obesity and related consequences to ageing. AGE 2016, 38, 23. [Google Scholar] [CrossRef] [Green Version]
- Goth, L.; Eaton, J.W. Hereditary catalase deficiencies and increased risk of diabetes. Lancet 2000, 356, 1820–1821. [Google Scholar] [CrossRef]
- Milisav, I.; Poljsak, B.; Suput, D. Adaptive response, evidence of cross-resistance and its potential clinical use. Int. J. Mol. Sci. 2012, 13, 10771–10806. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.; Bai, H. Peroxisomal Stress Response and Inter-Organelle Communication in Cellular Homeostasis and Aging. Antioxidants 2022, 11, 192. https://doi.org/10.3390/antiox11020192
Kim J, Bai H. Peroxisomal Stress Response and Inter-Organelle Communication in Cellular Homeostasis and Aging. Antioxidants. 2022; 11(2):192. https://doi.org/10.3390/antiox11020192
Chicago/Turabian StyleKim, Jinoh, and Hua Bai. 2022. "Peroxisomal Stress Response and Inter-Organelle Communication in Cellular Homeostasis and Aging" Antioxidants 11, no. 2: 192. https://doi.org/10.3390/antiox11020192