
Structure–Activity Relationships and Transcriptomic Analysis of Hypoxia-Inducible Factor Prolyl Hydroxylase Inhibitors

 , , , , , ,
, , , , , ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
3. Results and Discussion
3.1. Comparison of HIF PHD Inhibitors in HIF1 ODD-luc Reporter Assay
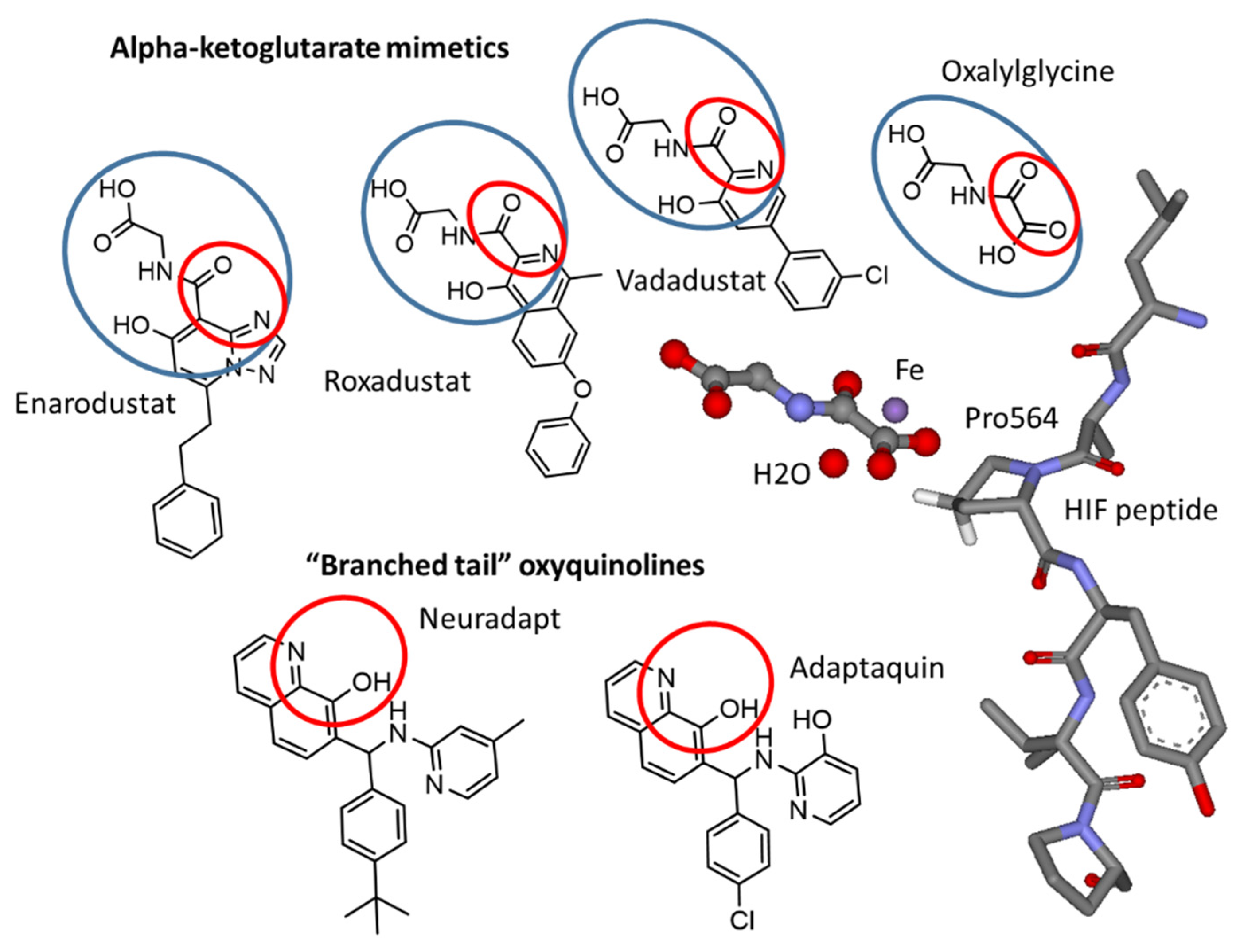
3.2. Refining Structure-Activity Relationships for “Branched-Tail” Oxyquinoline Inhibitors
3.3. “Branched-Tail” Oxyquinoline Inhibitors in Oxygen Glucose Deprivation (OGD) Model
3.4. Comparative Transcriptomic Analysis of HIF Prolyl Hydroxylase Inhibitors
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jaakkola, P.; Mole, D.R.; Tian, Y.-M.; Wilson, M.I.; Gielbert, J.; Gaskell, S.J.; von Kriegsheim, A.; Hebestreit, H.F.; Mukherji, M.; Schofield, C.J.; et al. Targeting of HIF-α to the von Hippel-Lindau Ubiquitylation Complex by O2-Regulated Prolyl Hydroxylation. Science 2001, 292, 468–472. [Google Scholar] [CrossRef] [PubMed]
- Hirota, K. HIF-α Prolyl Hydroxylase Inhibitors and Their Implications for Biomedicine: A Comprehensive Review. Biomedicines 2021, 9, 468. [Google Scholar] [CrossRef] [PubMed]
- Qie, S.; Jiao, N.; Duan, K.; Li, J.; Liu, Y.; Liu, G. The efficacy and safety of roxadustat treatment for anemia in patients with kidney disease: A meta-analysis and systematic review. Int. Urol. Nephrol. 2021, 53, 985–997. [Google Scholar] [CrossRef]
- Zhang, S.; Guo, J.; Xie, S.; Chen, J.; Yu, S.; Yu, Y. Efficacy and safety of hypoxia-inducible factor prolyl hydroxylase inhibitor (HIF-PHI) on anemia in non-dialysis-dependent chronic kidney disease (NDD-CKD): A systematic review and meta-analysis. Int. Urol. Nephrol. 2021, 53, 1139–1147. [Google Scholar] [CrossRef]
- Markham, A. Enarodustat: First Approval. Drugs 2021, 81, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Lun, J.; Zhang, H.; Zhu, L.; Zhang, G.; Fang, J. The non-canonical functions of HIF prolyl hydroxylases and their dual roles in cancer. Int. J. Biochem. Cell Biol. 2021, 135, 105982. [Google Scholar] [CrossRef] [PubMed]
- Klaus, S.J.; Molineaux, C.J.; Neff, T.B.; Guenzler-Pukall, V.; Lansetmo Parobok, I.; Seeley, T.W.; Stephenson, R.C. Use of HIF Alpha Stabilizers for Enhancing Erythropoiesis. WO2004108121, 16 December.
- Smirnova, N.A.; Rakhman, I.; Moroz, N.; Basso, M.; Payappilly, J.; Kazakov, S.; Hernandez-Guzman, F.; Gaisina, I.N.; Kozikowski, A.P.; Ratan, R.R.; et al. Utilization of an in vivo reporter for high throughput identification of branched small molecule regulators of hypoxic adaptation. Chem. Biol. 2010, 17, 380–391. [Google Scholar] [CrossRef] [Green Version]
- Pobbati, A.V.; Mejuch, T.; Chakraborty, S.; Karatas, H.; Bharath, S.R.; Guéret, S.M.; Goy, P.-A.; Hahne, G.; Pahl, A.; Sievers, S.; et al. Identification of Quinolinols as Activators of TEAD-Dependent Transcription. ACS Chem. Biol. 2019, 14, 2909–2921. [Google Scholar] [CrossRef]
- Savyuk, M.; Krivonosov, M.; Mishchenko, T.; Gazaryan, I.; Ivanchenko, M.; Khristichenko, A.; Poloznikov, A.; Hushpulian, D.; Nikulin, S.; Tonevitsky, E.; et al. Neuroprotective Effect of HIF Prolyl Hydroxylase Inhibition in an In Vitro Hypoxia Model. Antioxidants 2020, 9, 662. [Google Scholar] [CrossRef]
- Gaisina, I.N.; Lee, S.H.; Kaidery, N.A.; Ben Aissa, M.; Ahuja, M.; Smirnova, N.N.; Wakade, S.; Gaisin, A.; Bourassa, M.W.; Ratan, R.R.; et al. Activation of Nrf2 and Hypoxic Adaptive Response Contribute to Neuroprotection Elicited by Phenylhydroxamic Acid Selective HDAC6 Inhibitors. ACS Chem. Neurosci. 2018, 9, 894–900. [Google Scholar] [CrossRef]
- Hushpulian, D.M.; Nikulin, S.V.; Chubar, T.A.; Khristichenko, A.Y.; Poloznikov, A.A.; Gazaryan, I.G. Fast Responding Genes to HIF Prolyl Hydroxylase Inhibitors. Moscow Univ. Chem. Bull. 2021, 76, 114–121. [Google Scholar] [CrossRef]
- Maltseva, D.; Raygorodskaya, M.; Knyazev, E.; Zgoda, V.; Tikhonova, O.; Zaidi, S.; Nikulin, S.; Baranova, A.; Turchinovich, A.; Rodin, S.; et al. Knockdown of the α5 laminin chain affects differentiation of colorectal cancer cells and their sensitivity to chemotherapy. Biochimie 2020, 174, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Kudriaeva, A.; Galatenko, V.; Maltseva, D.; Khaustova, N.; Kuzina, E.; Tonevitsky, A.; Gabibov, A.; Belogurov, A. The Transcriptome of Type I Murine Astrocytes under Interferon-Gamma Exposure and Remyelination Stimulus. Molecules 2017, 22, 808. [Google Scholar] [CrossRef]
- Thirstrup, K.; Christensen, S.; Møller, H.A.; Ritzén, A.; Bergström, A.-L.; Sager, T.N.; Jensen, H.S. Endogenous 2-oxoglutarate levels impact potencies of competitive HIF prolyl hydroxylase inhibitors. Pharmacol. Res. 2011, 64, 268–273. [Google Scholar] [CrossRef]
- Zhdanov, A.V.; Okkelman, I.A.; Collins, F.W.J.; Melgar, S.; Papkovsky, D.B. A novel effect of DMOG on cell metabolism: Direct inhibition of mitochondrial function precedes HIF target gene expression. Biochim. Biophys. Acta 2015, 1847, 1254–1266. [Google Scholar] [CrossRef] [Green Version]
- Roger, S.D.; Coyne, D.W. HIF-Prolyl Hydroxylase Inhibitors: Confirmed Efficacy with Uncertain Safety. Am. J. Nephrol. 2021, 52, 894–898. [Google Scholar] [CrossRef]
- Ratan, R.R. The Chemical Biology of Ferroptosis in the Central Nervous System. Cell Chem. Biol. 2020. [Google Scholar] [CrossRef]
- Carmo dos Santos, N.A.; Natali, M.; Badetti, E.; Wurst, K.; Licini, G.; Zonta, C. Cobalt, nickel, and iron complexes of 8-hydroxyquinoline-di(2-picolyl)amine for light-driven hydrogen evolution. Dalt. Trans. 2017, 46, 16455–16464. [Google Scholar] [CrossRef]
- Karuppagounder, S.S.; Alim, I.; Khim, S.J.; Bourassa, M.W.; Sleiman, S.F.; John, R.; Thinnes, C.C.; Yeh, T.-L.; Demetriades, M.; Neitemeier, S.; et al. Therapeutic targeting of oxygen-sensing prolyl hydroxylases abrogates ATF4-dependent neuronal death and improves outcomes after brain hemorrhage in several rodent models. Sci. Transl. Med. 2016, 8, 328ra29. [Google Scholar] [CrossRef] [Green Version]
- Neitemeier, S.; Dolga, A.M.; Honrath, B.; Karuppagounder, S.S.; Alim, I.; Ratan, R.R.; Culmsee, C. Inhibition of HIF-prolyl-4-hydroxylases prevents mitochondrial impairment and cell death in a model of neuronal oxytosis. Cell Death Dis. 2016, 7, e2214. [Google Scholar] [CrossRef] [Green Version]
- David, B.T.; Curtin, J.J.; Brown, J.L.; Coutts, D.J.C.; Boles, N.C.; Hill, C.E. Treatment with hypoxia-mimetics protects cultured rat Schwann cells against oxidative stress-induced cell death. Glia 2021, 69, 2215–2234. [Google Scholar] [CrossRef]
- Ogle, M.E.; Gu, X.; Espinera, A.R.; Wei, L. Inhibition of prolyl hydroxylases by dimethyloxaloylglycine after stroke reduces ischemic brain injury and requires hypoxia inducible factor-1α. Neurobiol. Dis. 2012, 45, 733–742. [Google Scholar] [CrossRef] [Green Version]
- Reischl, S.; Li, L.; Walkinshaw, G.; Flippin, L.A.; Marti, H.H.; Kunze, R. Inhibition of HIF prolyl-4-hydroxylases by FG-4497 Reduces Brain Tissue Injury and Edema Formation during Ischemic Stroke. PLoS ONE 2014, 9, e84767. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.L.; Ogunshola, O.O.; Yeoh, K.K.; Jani, A.; Papadakis, M.; Nagel, S.; Schofield, C.J.; Buchan, A.M. HIF prolyl hydroxylase inhibition prior to transient focal cerebral ischaemia is neuroprotective in mice. J. Neurochem. 2014, 131, 177–189. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Wilson, J.W.; Schofield, C.J.; Chen, R. Author Correction: Hypoxia-inducible factor (HIF) prolyl hydroxylase inhibitors induce autophagy and have a protective effect in an in-vitro ischaemia model. Sci. Rep. 2020, 10, 6041. [Google Scholar] [CrossRef] [Green Version]
- Feng, H.; Hu, L.; Zhu, H.; Tao, L.; Wu, L.; Zhao, Q.; Gao, Y.; Gong, Q.; Mao, F.; Li, X.; et al. Repurposing antimycotic ciclopirox olamine as a promising anti-ischemic stroke agent. Acta Pharm. Sin. B 2020, 10, 434–446. [Google Scholar] [CrossRef]
- Rodriguez, J.; Herrero, A.; Li, S.; Rauch, N.; Quintanilla, A.; Wynne, K.; Krstic, A.; Acosta, J.C.; Taylor, C.; Schlisio, S.; et al. PHD3 Regulates p53 Protein Stability by Hydroxylating Proline 359. Cell Rep. 2018, 24, 1316–1329. [Google Scholar] [CrossRef] [Green Version]
- Fitzpatrick, S.F.; Fabian, Z.; Schaible, B.; Lenihan, C.R.; Schwarzl, T.; Rodriguez, J.; Zheng, X.; Li, Z.; Tambuwala, M.M.; Higgins, D.G.; et al. Prolyl hydroxylase-1 regulates hepatocyte apoptosis in an NF-κB-dependent manner. Biochem. Biophys. Res. Commun. 2016, 474, 579–586. [Google Scholar] [CrossRef] [Green Version]
- Köditz, J.; Nesper, J.; Wottawa, M.; Stiehl, D.P.; Camenisch, G.; Franke, C.; Myllyharju, J.; Wenger, R.H.; Katschinski, D.M. Oxygen-dependent ATF-4 stability is mediated by the PHD3 oxygen sensor. Blood 2007, 110, 3610–3617. [Google Scholar] [CrossRef] [Green Version]
- Wei, G.Z.; Saraswat Ohri, S.; Khattar, N.K.; Listerman, A.W.; Doyle, C.H.; Andres, K.R.; Karuppagounder, S.S.; Ratan, R.R.; Whittemore, S.R.; Hetman, M. Hypoxia-inducible factor prolyl hydroxylase domain (PHD) inhibition after contusive spinal cord injury does not improve locomotor recovery. PLoS ONE 2021, 16, e0249591. [Google Scholar] [CrossRef]
- Aimé, P.; Karuppagounder, S.S.; Rao, A.; Chen, Y.; Burke, R.E.; Ratan, R.R.; Greene, L.A. The drug adaptaquin blocks ATF4/CHOP-dependent pro-death Trib3 induction and protects in cellular and mouse models of Parkinson’s disease. Neurobiol. Dis. 2020, 136, 104725. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Li, T.; Wang, Y.; Xu, Y.; Zhang, S.; Culmsee, C.; Wang, X.; Zhu, C. Sex differences in neonatal mouse brain injury after hypoxia-ischemia and adaptaquin treatment. J. Neurochem. 2019, 150, 759–775. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poloznikov, A.A.; Nikulin, S.V.; Hushpulian, D.M.; Khristichenko, A.Y.; Osipyants, A.I.; Asachenko, A.F.; Shurupova, O.V.; Savin, S.S.; Lee, S.H.; Gaisina, I.N.; et al. Structure–Activity Relationships and Transcriptomic Analysis of Hypoxia-Inducible Factor Prolyl Hydroxylase Inhibitors. Antioxidants 2022, 11, 220. https://doi.org/10.3390/antiox11020220
Poloznikov AA, Nikulin SV, Hushpulian DM, Khristichenko AY, Osipyants AI, Asachenko AF, Shurupova OV, Savin SS, Lee SH, Gaisina IN, et al. Structure–Activity Relationships and Transcriptomic Analysis of Hypoxia-Inducible Factor Prolyl Hydroxylase Inhibitors. Antioxidants. 2022; 11(2):220. https://doi.org/10.3390/antiox11020220
Chicago/Turabian StylePoloznikov, Andrey A., Sergey V. Nikulin, Dmitry M. Hushpulian, Anna Yu. Khristichenko, Andrey I. Osipyants, Andrey F. Asachenko, Olga V. Shurupova, Svyatoslav S. Savin, Sue H. Lee, Irina N. Gaisina, and et al. 2022. "Structure–Activity Relationships and Transcriptomic Analysis of Hypoxia-Inducible Factor Prolyl Hydroxylase Inhibitors" Antioxidants 11, no. 2: 220. https://doi.org/10.3390/antiox11020220
APA StylePoloznikov, A. A., Nikulin, S. V., Hushpulian, D. M., Khristichenko, A. Y., Osipyants, A. I., Asachenko, A. F., Shurupova, O. V., Savin, S. S., Lee, S. H., Gaisina, I. N., Thatcher, G. R. J., Narciso, A., Chang, E. P., Kazakov, S. V., Krucher, N., Tishkov, V. I., Thomas, B., & Gazaryan, I. G. (2022). Structure–Activity Relationships and Transcriptomic Analysis of Hypoxia-Inducible Factor Prolyl Hydroxylase Inhibitors. Antioxidants, 11(2), 220. https://doi.org/10.3390/antiox11020220