
Hydroxamic Acid as a Potent Metal-Binding Group for Inhibiting Tyrosinase


Abstract
:1. Introduction
2. Materials and Methods
3. Results
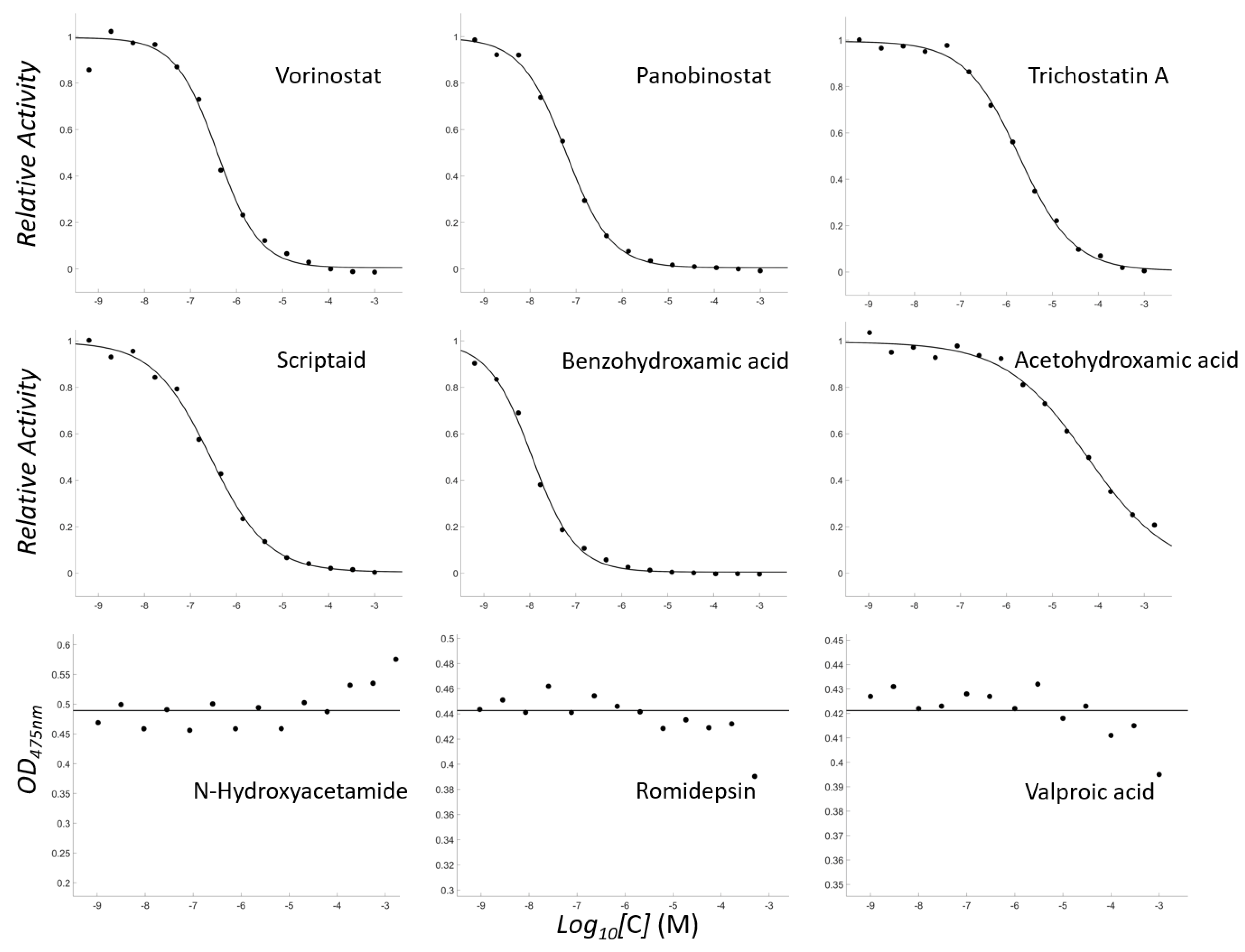
3.1. Vorinostat and Hydroxamate-Containing HDAC Inhibitors Are Inhibitory for Tyrosinase
3.2. Biophysical Experiments Confirmed the Direct Interaction between Hydroxamate-Containing Molecules and Tyrosinase
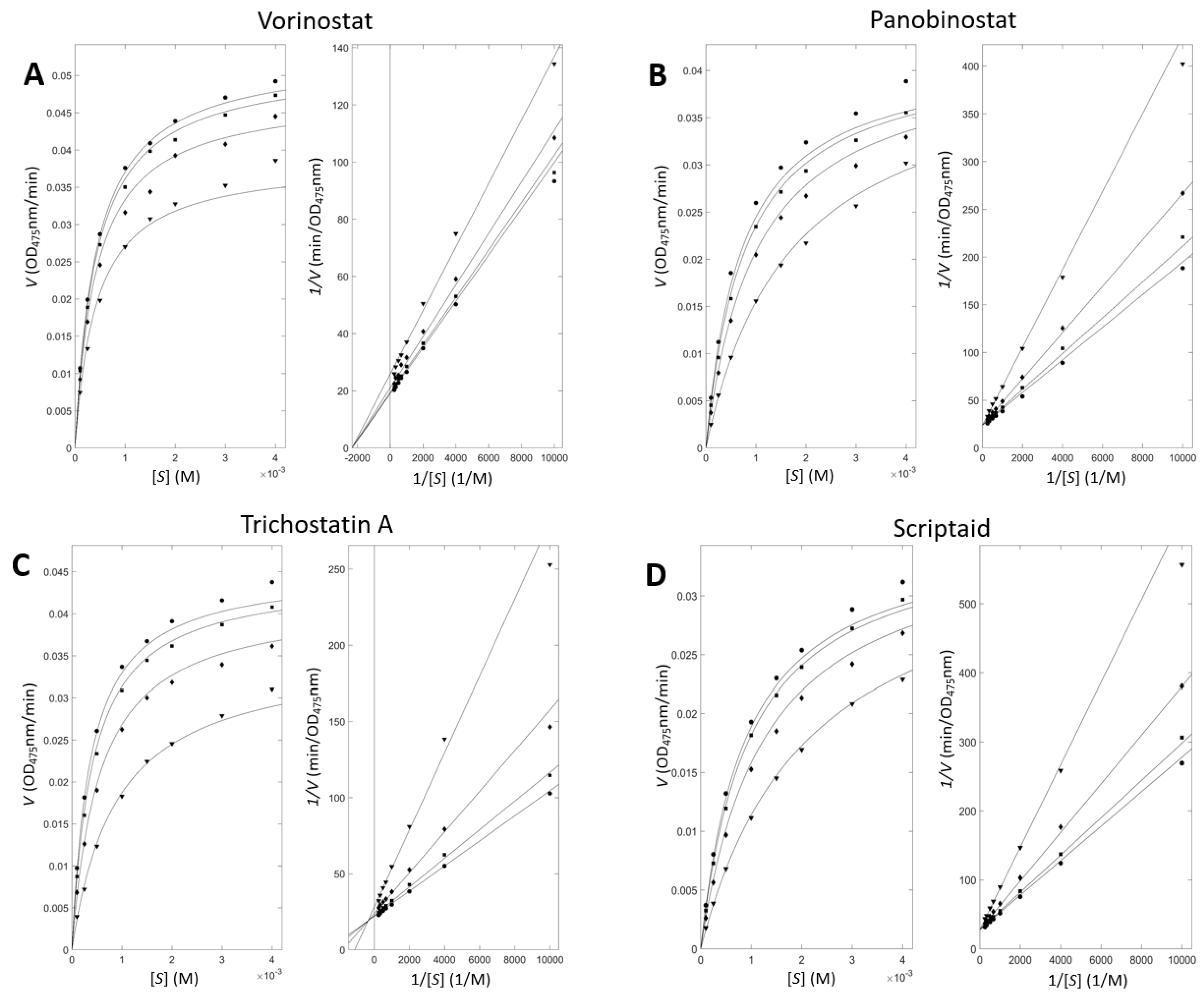
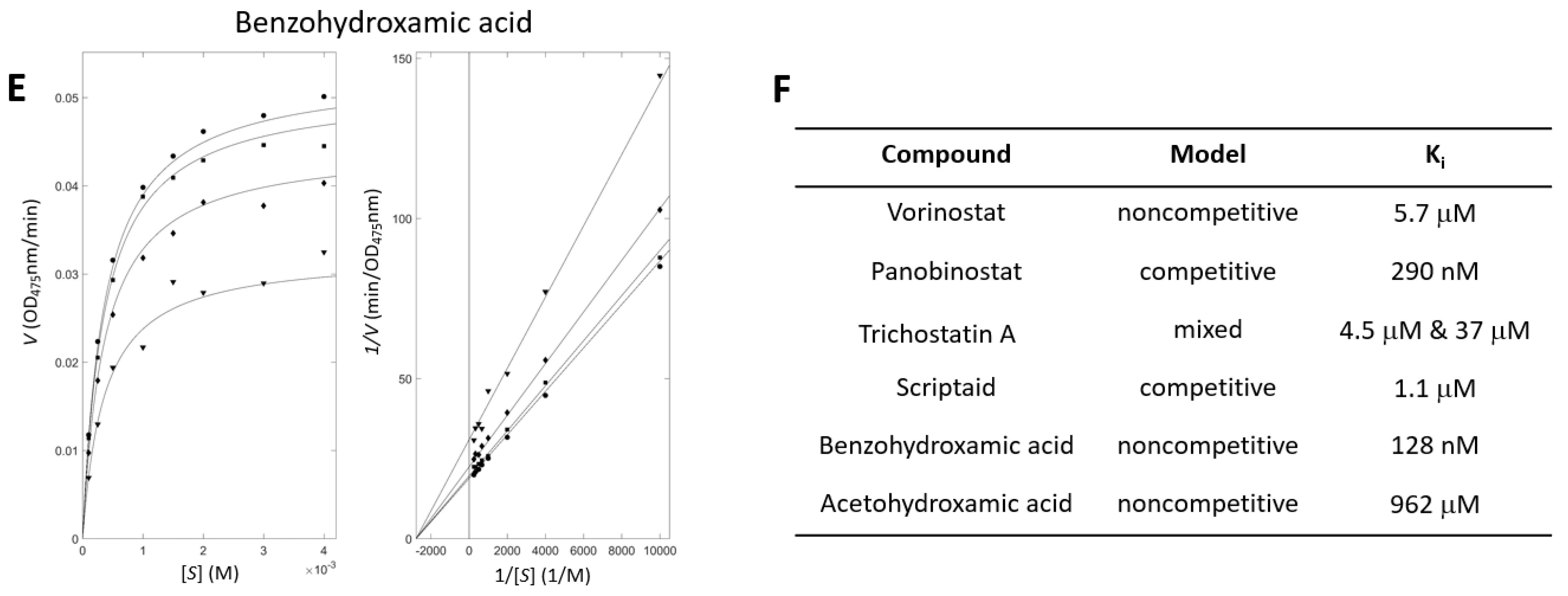
3.3. Enzyme Inhibitory Kinetic Experiments Characterize the Modes of the Inhibition
3.4. Hydroxamate-Containing Molecules Were Inhibitory for Mammalian Tyrosinase as Well
3.5. Cheminformatics and Docking Studies Suggested the Binding Modes of Hydroxamate-Containing Inhibitors
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Mostert, A.B. Melanin, the What, the Why and the How: An Introductory Review for Materials Scientists Interested in Flexible and Versatile Polymers. Polymers 2021, 13, 1670. [Google Scholar] [CrossRef]
- Solano, F. Melanins: Skin Pigments and Much More—Types, Structural Models, Biological Functions, and Formation Routes. New J. Sci. 2014, 2014, 1–28. [Google Scholar] [CrossRef] [Green Version]
- McNamara, M.E.; Rossi, V.; Slater, T.S.; Rogers, C.S.; Ducrest, A.L.; Dubey, S.; Roulin, A. Decoding the Evolution of Melanin in Vertebrates. Trends Ecol. Evol. 2021, 36, 430–443. [Google Scholar] [CrossRef]
- Chang, T.S. An updated review of tyrosinase inhibitors. Int. J. Mol. Sci. 2009, 10, 2440–2475. [Google Scholar] [CrossRef] [Green Version]
- Loizzo, M.R.; Tundis, R.; Menichini, F. Natural and Synthetic Tyrosinase Inhibitors as Antibrowning Agents: An Update. Compr. Rev. Food Sci. Food Saf. 2012, 11, 378–398. [Google Scholar] [CrossRef]
- Khan, M.T. Novel tyrosinase inhibitors from natural resources—Their computational studies. Curr. Med. Chem. 2012, 19, 2262–2272. [Google Scholar] [CrossRef]
- Kim, Y.J.; Uyama, H. Tyrosinase inhibitors from natural and synthetic sources: Structure, inhibition mechanism and perspective for the future. Cell. Mol. Life Sci. 2005, 62, 1707–1723. [Google Scholar] [CrossRef]
- Seo, S.Y.; Sharma, V.K.; Sharma, N. Mushroom tyrosinase: Recent prospects. J. Agric. Food Chem. 2003, 51, 2837–2853. [Google Scholar] [CrossRef]
- Ubeid, A.A.; Do, S.; Nye, C.; Hantash, B.M. Potent low toxicity inhibition of human melanogenesis by novel indole-containing octapeptides. Biochim. Biophys. Acta 2012, 1820, 1481–1489. [Google Scholar] [CrossRef]
- Solano, F.; Briganti, S.; Picardo, M.; Ghanem, G. Hypopigmenting agents: An updated review on biological, chemical and clinical aspects. Pigment. Cell Res. / Spons. By Eur. Soc. Pigment. Cell Res. Int. Pigment. Cell Soc. 2006, 19, 550–571. [Google Scholar] [CrossRef]
- Halaouli, S.; Asther, M.; Sigoillot, J.C.; Hamdi, M.; Lomascolo, A. Fungal tyrosinases: New prospects in molecular characteristics, bioengineering and biotechnological applications. J. Appl. Microbiol. 2006, 100, 219–232. [Google Scholar] [CrossRef]
- Pillaiyar, T.; Manickam, M.; Namasivayam, V. Skin whitening agents: Medicinal chemistry perspective of tyrosinase inhibitors. J. Enzym. Inhib. Med. Chem. 2017, 32, 403–425. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.; Choi, K.E.; Park, S.J.; Kim, S.Y.; Jee, J.G. Ensemble-Based Virtual Screening Led to the Discovery of New Classes of Potent Tyrosinase Inhibitors. J. Chem. Inf. Modeling 2016, 56, 354–367. [Google Scholar] [CrossRef]
- Waldron, K.J.; Robinson, N.J. How do bacterial cells ensure that metalloproteins get the correct metal? Nat. Rev. Microbiol. 2009, 7, 25–35. [Google Scholar] [CrossRef]
- Waldron, K.J.; Rutherford, J.C.; Ford, D.; Robinson, N.J. Metalloproteins and metal sensing. Nature 2009, 460, 823–830. [Google Scholar] [CrossRef]
- Yang, Y.; Hu, X.Q.; Li, Q.S.; Zhang, X.X.; Ruan, B.F.; Xu, J.; Liao, C. Metalloprotein Inhibitors for the Treatment of Human Diseases. Curr. Top. Med. Chem. 2016, 16, 384–396. [Google Scholar] [CrossRef]
- Johnstone, T.C.; Nolan, E.M. Beyond iron: Non-classical biological functions of bacterial siderophores. Dalton Trans. 2015, 44, 6320–6339. [Google Scholar] [CrossRef] [Green Version]
- Das, M.; Das, B.; Samanta, A. Antioxidant and anticancer activity of synthesized 4-amino-5-((aryl substituted)-4H-1,2,4-triazole-3-yl)thio-linked hydroxamic acid derivatives. J. Pharm. Pharmacol. 2019, 71, 1400–1411. [Google Scholar] [CrossRef]
- Dwibedi, V.; Kalia, S.; Saxena, S. Isolation and enhancement of resveratrol production in Xylaria psidii by exploring the phenomenon of epigenetics: Using DNA methyltransferases and histone deacetylase as epigenetic modifiers. Mol. Biol. Rep. 2019, 46, 4123–4137. [Google Scholar] [CrossRef]
- Liu, Y.H.; Lin, S.Y.; Lee, C.C.; Hou, W.C. Antioxidant and nitric oxide production inhibitory activities of galacturonyl hydroxamic acid. Food Chem. 2008, 109, 159–166. [Google Scholar] [CrossRef]
- Chaudhary, D.; Chong, F.; Neupane, T.; Choi, J.; Jee, J.G. New Inhibitors of Laccase and Tyrosinase by Examination of Cross-Inhibition between Copper-Containing Enzymes. Int. J. Mol. Sci. 2021, 22, 13661. [Google Scholar] [CrossRef]
- Choi, J.; Lee, Y.M.; Jee, J.G. Thiopurine Drugs Repositioned as Tyrosinase Inhibitors. Int. J. Mol. Sci. 2018, 19, 77. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.; Park, S.J.; Jee, J.G. Analogues of ethionamide, a drug used for multidrug-resistant tuberculosis, exhibit potent inhibition of tyrosinase. Eur. J. Med. Chem. 2015, 106, 157–166. [Google Scholar] [CrossRef]
- Choi, J.; Jee, J.G. Repositioning of Thiourea-Containing Drugs as Tyrosinase Inhibitors. Int. J. Mol. Sci. 2015, 16, 28534–28548. [Google Scholar] [CrossRef] [Green Version]
- Yung-Chi, C.; Prusoff, W.H. Relationship between the inhibition constant (KI) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. [Google Scholar] [CrossRef]
- Dias, A.A.; Pinto, P.A.; Fraga, I.; Bezerra, R.M. Diagnosis of enzyme inhibition using Excel Solver: A combined dry and wet laboratory exercise. J. Chem. Educ. 2014, 91, 1017–1021. [Google Scholar] [CrossRef]
- Mendez, D.; Gaulton, A.; Bento, A.P.; Chambers, J.; de Veij, M.; Felix, E.; Magarinos, M.P.; Mosquera, J.F.; Mutowo, P.; Nowotka, M.; et al. ChEMBL: Towards direct deposition of bioassay data. Nucleic Acids Res. 2019, 47, D930–D940. [Google Scholar] [CrossRef]
- Gaulton, A.; Hersey, A.; Nowotka, M.; Bento, A.P.; Chambers, J.; Mendez, D.; Mutowo, P.; Atkinson, F.; Bellis, L.J.; Cibrian-Uhalte, E.; et al. The ChEMBL database in 2017. Nucleic Acids Res. 2017, 45, D945–D954. [Google Scholar] [CrossRef]
- Liu, Z.; Li, Y.; Han, L.; Li, J.; Liu, J.; Zhao, Z.; Nie, W.; Liu, Y.; Wang, R. PDB-wide collection of binding data: Current status of the PDBbind database. Bioinformatics 2015, 31, 405–412. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Mysinger, M.M.; Shoichet, B.K. Rapid context-dependent ligand desolvation in molecular docking. J. Chem. Inf. Modeling 2010, 50, 1561–1573. [Google Scholar] [CrossRef]
- Irwin, J.J.; Shoichet, B.K.; Mysinger, M.M.; Huang, N.; Colizzi, F.; Wassam, P.; Cao, Y. Automated docking screens: A feasibility study. J. Med. Chem. 2009, 52, 5712–5720. [Google Scholar] [CrossRef] [Green Version]
- McGann, M. FRED and HYBRID docking performance on standardized datasets. J. Comput. Aided Mol. Des. 2012, 26, 897–906. [Google Scholar] [CrossRef]
- McGann, M. FRED pose prediction and virtual screening accuracy. J. Chem. Inf. Modeling 2011, 51, 578–596. [Google Scholar] [CrossRef]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Lin, Y.S.; Wu, W.C.; Lin, S.Y.; Hou, W.C. Glycine hydroxamate inhibits tyrosinase activity and melanin contents through downregulating cAMP/PKA signaling pathways. Amino Acids 2015, 47, 617–625. [Google Scholar] [CrossRef]
- Kwak, S.Y.; Yang, J.K.; Choi, H.R.; Park, K.C.; Kim, Y.B.; Lee, Y.S. Synthesis and dual biological effects of hydroxycinnamoyl phenylalanyl/prolyl hydroxamic acid derivatives as tyrosinase inhibitor and antioxidant. Bioorganic Med. Chem. Lett. 2013, 23, 1136–1142. [Google Scholar] [CrossRef]
- Kwak, S.Y.; Lee, S.; Choi, H.R.; Park, K.C.; Lee, Y.S. Dual effects of caffeoyl-amino acidyl-hydroxamic acid as an antioxidant and depigmenting agent. Bioorganic Med. Chem. Lett. 2011, 21, 5155–5158. [Google Scholar] [CrossRef]
- Day, J.A.; Cohen, S.M. Investigating the selectivity of metalloenzyme inhibitors. J. Med. Chem. 2013, 56, 7997–8007. [Google Scholar] [CrossRef] [Green Version]
- Sterling, T.; Irwin, J.J. ZINC 15--Ligand Discovery for Everyone. J. Chem. Inf. Modeling 2015, 55, 2324–2337. [Google Scholar] [CrossRef] [PubMed]
- Furumai, R.; Matsuyama, A.; Kobashi, N.; Lee, K.H.; Nishiyama, N.; Nakajima, I.; Tanaka, A.; Komatsu, Y.; Nishino, N.; Yoshida, M.; et al. FK228 (depsipeptide) as a natural prodrug that inhibits class I histone deacetylases. Cancer Res. 2002, 62, 4916–4921. [Google Scholar] [PubMed]
- Gottlicher, M.; Minucci, S.; Zhu, P.; Kramer, O.H.; Schimpf, A.; Giavara, S.; Sleeman, J.P.; Lo Coco, F.; Nervi, C.; Pelicci, P.G.; et al. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J. 2001, 20, 6969–6978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, D.; Torosyan, H.; Elnatan, D.; McLaughlin, C.K.; Logie, J.; Shoichet, M.S.; Agard, D.A.; Shoichet, B.K. Internal Structure and Preferential Protein Binding of Colloidal Aggregates. ACS Chem. Biol. 2017, 12, 282–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLaughlin, C.K.; Duan, D.; Ganesh, A.N.; Torosyan, H.; Shoichet, B.K.; Shoichet, M.S. Stable Colloidal Drug Aggregates Catch and Release Active Enzymes. ACS Chem. Biol. 2016, 11, 992–1000. [Google Scholar] [CrossRef] [Green Version]
- Owen, S.C.; Doak, A.K.; Wassam, P.; Shoichet, M.S.; Shoichet, B.K. Colloidal aggregation affects the efficacy of anticancer drugs in cell culture. ACS Chem. Biol. 2012, 7, 1429–1435. [Google Scholar] [CrossRef]
- Coan, K.E.; Shoichet, B.K. Stoichiometry and physical chemistry of promiscuous aggregate-based inhibitors. J. Am. Chem. Soc. 2008, 130, 9606–9612. [Google Scholar] [CrossRef] [Green Version]
- Tunyasuvunakool, K.; Adler, J.; Wu, Z.; Green, T.; Zielinski, M.; Zidek, A.; Bridgland, A.; Cowie, A.; Meyer, C.; Laydon, A.; et al. Highly accurate protein structure prediction for the human proteome. Nature 2021, 596, 590–596. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Zhang, Y.; Skolnick, J. TM-align: A protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 2005, 33, 2302–2309. [Google Scholar] [CrossRef]
- Roulier, B.; Peres, B.; Haudecoeur, R. Advances in the Design of Genuine Human Tyrosinase Inhibitors for Targeting Melanogenesis and Related Pigmentations. J. Med. Chem. 2020, 63, 13428–13443. [Google Scholar] [CrossRef] [PubMed]
- Gerdemann, C.; Eicken, C.; Krebs, B. The crystal structure of catechol oxidase: New insight into the function of type-3 copper proteins. Acc. Chem. Res. 2002, 35, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Eicken, C.; Krebs, B.; Sacchettini, J.C. Catechol oxidase—Structure and activity. Curr. Opin. Struct. Biol. 1999, 9, 677–683. [Google Scholar] [CrossRef] [Green Version]
- Ramsden, C.A.; Riley, P.A. Tyrosinase: The four oxidation states of the active site and their relevance to enzymatic activation, oxidation and inactivation. Bioorganic Med. Chem. 2014, 22, 2388–2395. [Google Scholar] [CrossRef] [PubMed]
- Ballatore, C.; Huryn, D.M.; Smith, A.B., III. Carboxylic acid (bio)isosteres in drug design. ChemMedChem 2013, 8, 385–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, F.F.; Olson, D.E.; Gale, J.P.; Kaya, T.; Weiwer, M.; Aidoud, N.; Thomas, M.; Davoine, E.L.; Lemercier, B.C.; Zhang, Y.L.; et al. Potent and selective inhibition of histone deacetylase 6 (HDAC6) does not require a surface-binding motif. J. Med. Chem. 2013, 56, 1772–1776. [Google Scholar] [CrossRef]
- Di Fiore, A.; Maresca, A.; Supuran, C.T.; De Simone, G. Hydroxamate represents a versatile zinc binding group for the development of new carbonic anhydrase inhibitors. Chem. Commun. 2012, 48, 8838–8840. [Google Scholar] [CrossRef] [Green Version]
- Malachowski, W.P.; Winters, M.; DuHadaway, J.B.; Lewis-Ballester, A.; Badir, S.; Wai, J.; Rahman, M.; Sheikh, E.; LaLonde, J.M.; Yeh, S.R.; et al. O-alkylhydroxylamines as rationally-designed mechanism-based inhibitors of indoleamine 2,3-dioxygenase-1. Eur. J. Med. Chem. 2016, 108, 564–576. [Google Scholar] [CrossRef] [Green Version]
- Summers, J.B.; Mazdiyasni, H.; Holms, J.H.; Ratajczyk, J.D.; Dyer, R.D.; Carter, G.W. Hydroxamic acid inhibitors of 5-lipoxygenase. J. Med. Chem. 1987, 30, 574–580. [Google Scholar] [CrossRef]
- Bourque, J.R.; Burley, R.K.; Bearne, S.L. Intermediate analogue inhibitors of mandelate racemase: N-Hydroxyformanilide and cupferron. Bioorganic Med. Chem. Lett. 2007, 17, 105–108. [Google Scholar] [CrossRef]
- Ha, Y.M.; Park, J.Y.; Park, Y.J.; Park, D.; Choi, Y.J.; Kim, J.M.; Lee, E.K.; Han, Y.K.; Kim, J.A.; Lee, J.Y.; et al. Synthesis and biological activity of hydroxy substituted phenyl-benzo[d]thiazole analogues for antityrosinase activity in B16 cells. Bioorganic Med. Chem. Lett. 2011, 21, 2445–2449. [Google Scholar] [CrossRef] [PubMed]
- Ullah, S.; Kang, D.; Lee, S.; Ikram, M.; Park, C.; Park, Y.; Yoon, S.; Chun, P.; Moon, H.R. Synthesis of cinnamic amide derivatives and their anti-melanogenic effect in alpha-MSH-stimulated B16F10 melanoma cells. Eur. J. Med. Chem. 2019, 161, 78–92. [Google Scholar] [CrossRef] [PubMed]
- Mann, T.; Gerwat, W.; Batzer, J.; Eggers, K.; Scherner, C.; Wenck, H.; Stab, F.; Hearing, V.J.; Rohm, K.H.; Kolbe, L. Inhibition of Human Tyrosinase Requires Molecular Motifs Distinctively Different from Mushroom Tyrosinase. J. Investig. Dermatol. 2018, 138, 1601–1608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haudecoeur, R.; Carotti, M.; Gouron, A.; Maresca, M.; Buitrago, E.; Hardre, R.; Bergantino, E.; Jamet, H.; Belle, C.; Reglier, M.; et al. 2-Hydroxypyridine-N-oxide-Embedded Aurones as Potent Human Tyrosinase Inhibitors. ACS Med. Chem. Lett. 2017, 8, 55–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buitrago, E.; Hardre, R.; Haudecoeur, R.; Jamet, H.; Belle, C.; Boumendjel, A.; Bubacco, L.; Reglier, M. Are Human Tyrosinase and Related Proteins Suitable Targets for Melanoma Therapy? Curr. Top. Med. Chem. 2016, 16, 3033–3047. [Google Scholar] [CrossRef]
- Vargas, A.J.; Sittadjody, S.; Thangasamy, T.; Mendoza, E.E.; Limesand, K.H.; Burd, R. Exploiting tyrosinase expression and activity in melanocytic tumors: Quercetin and the central role of p53. Integr. Cancer Ther. 2011, 10, 328–340. [Google Scholar] [CrossRef]
- Yeon, M.; Kim, Y.; Jung, H.S.; Jeoung, D. Histone Deacetylase Inhibitors to Overcome Resistance to Targeted and Immuno Therapy in Metastatic Melanoma. Front. Cell Dev. Biol. 2020, 8, 486. [Google Scholar] [CrossRef]
- Moschos, M.M.; Dettoraki, M.; Androudi, S.; Kalogeropoulos, D.; Lavaris, A.; Garmpis, N.; Damaskos, C.; Garmpi, A.; Tsatsos, M. The Role of Histone Deacetylase Inhibitors in Uveal Melanoma: Current Evidence. Anticancer Res. 2018, 38, 3817–3824. [Google Scholar] [CrossRef]
- Croce, M.; Ferrini, S.; Pfeffer, U.; Gangemi, R. Targeted Therapy of Uveal Melanoma: Recent Failures and New Perspectives. Cancers 2019, 11, 846. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | ZINC ID | MW | cLogP | Ki |
|---|---|---|---|---|
| Vorinostat | ZINC1543873 | 264 | 2.47 | 257 ± 26 nM |
| Panobinostat | ZINC22010649 | 349 | 3.33 | 40 ± 5 nM |
| Trichostatin A | ZINC100014731 | 302 | 2.58 | 1.0 ± 0.1 μM |
| Scriptaid | ZINC3873638 | 326 | 2.50 | 180 ± 21 nM |
| Benzohydroxamic acid | ZINC4701351 | 137 | 1.38 | 7 ± 1 nM |
| Acetohydroxamic acid | ZINC4658603 | 75 | −0.49 | 38 ± 8 μM |
| N-Hydroxyacetamide | ZINC4692578 | 75 | −1.54 | >1 mM |
| Romidepsin | ZINC100371951 | 542 | 1.99 | >500 μM |
| Valproic acid | ZINC3008621 | 144 | 2.29 | >500 μM |
| Suberohydroxamic acid | ZINC3873635 | 204 | 0.34 | 1.0 ± 0.2 μM |
| Butyrylhydroxamic acid | ZINC4809144 | 103 | 0.29 | 32 ± 5 μM |
| PTU * | ZINC3875720 | 152 | 1.34 | 62 ± 3 nM |
| Tropolone * | ZINC392003 | 122 | 0.75 | 43 ± 3 nM |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, J.; Neupane, T.; Baral, R.; Jee, J.-G. Hydroxamic Acid as a Potent Metal-Binding Group for Inhibiting Tyrosinase. Antioxidants 2022, 11, 280. https://doi.org/10.3390/antiox11020280
Choi J, Neupane T, Baral R, Jee J-G. Hydroxamic Acid as a Potent Metal-Binding Group for Inhibiting Tyrosinase. Antioxidants. 2022; 11(2):280. https://doi.org/10.3390/antiox11020280
Chicago/Turabian StyleChoi, Joonhyeok, Trilok Neupane, Rishiram Baral, and Jun-Goo Jee. 2022. "Hydroxamic Acid as a Potent Metal-Binding Group for Inhibiting Tyrosinase" Antioxidants 11, no. 2: 280. https://doi.org/10.3390/antiox11020280
APA StyleChoi, J., Neupane, T., Baral, R., & Jee, J.-G. (2022). Hydroxamic Acid as a Potent Metal-Binding Group for Inhibiting Tyrosinase. Antioxidants, 11(2), 280. https://doi.org/10.3390/antiox11020280