
Oxidative Stress and Cellular Senescence Are Involved in the Aging Kidney

 , , , , , ,
, , , , , ,  , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Protein Studies
2.3. Histology and Immunohistochemistry
2.4. Gene-Expression Studies
2.5. Statistical Analysis
3. Results
3.1. Inflammation Precedes Tubular and Glomerular Lesions in Kidneys of Aged Mice
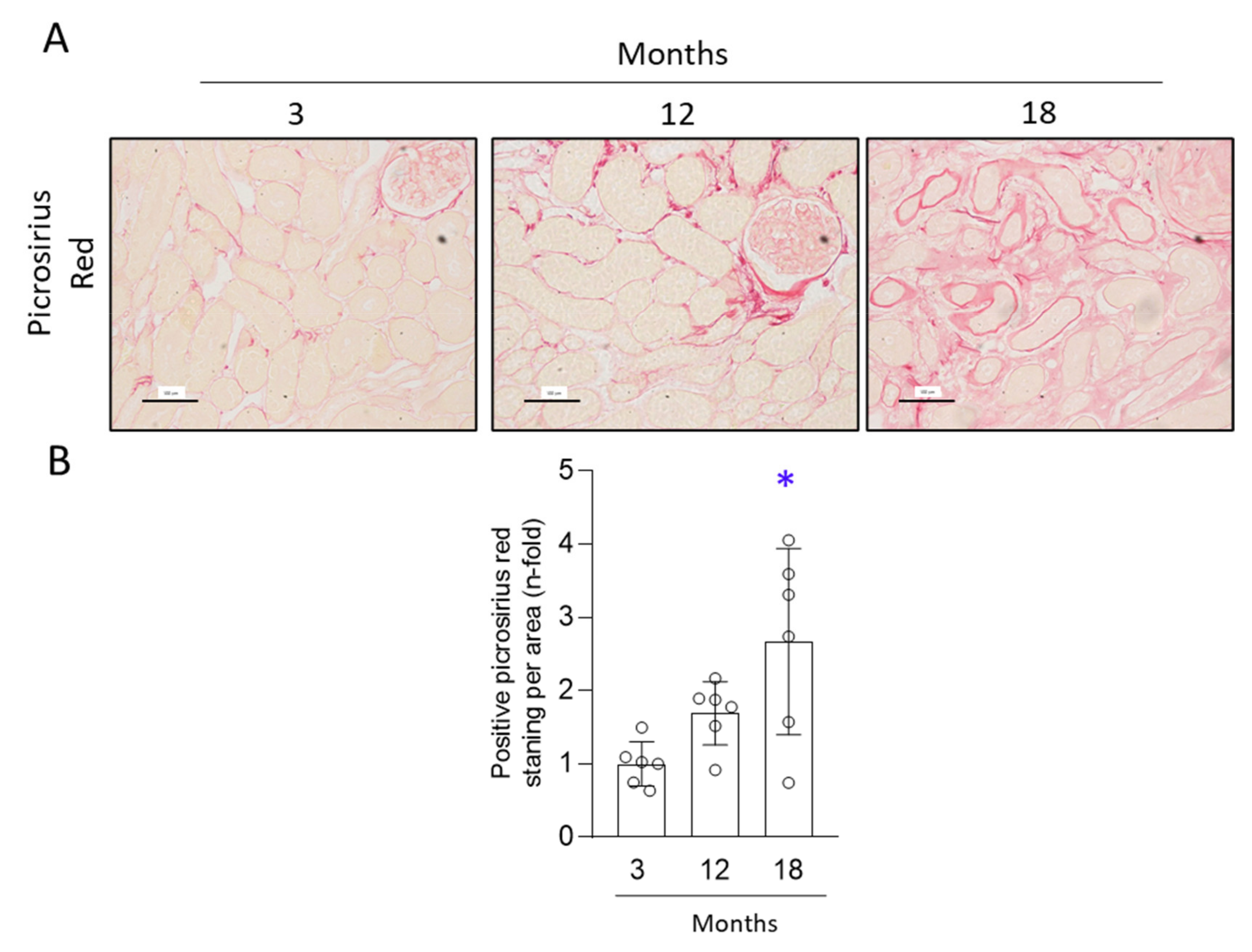
3.2. Aged Kidneys Display Increased Collagen Accumulation
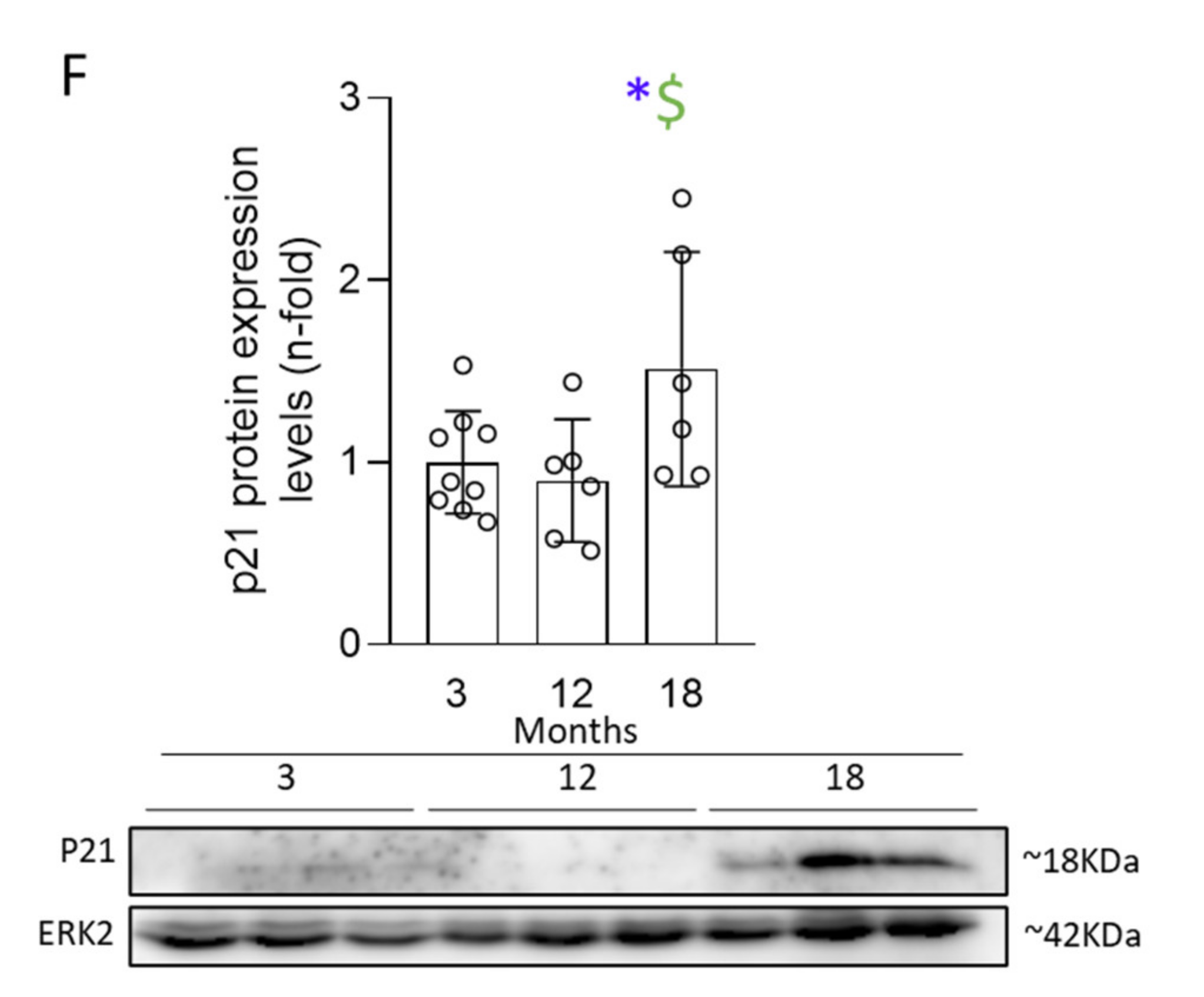
3.3. Activation of Senescent Mechanisms Associated with DNA-Damage Response (DDR) Precedes Cell-Cycle Arrest (CCA) and Induction of an Aberrant Secretome (SASP)
3.4. The Anti-Aging Factor Klotho Is Lost Early during Renal Aging
3.5. NRF2 Pathway Is Deregulated in the Aging Kidney
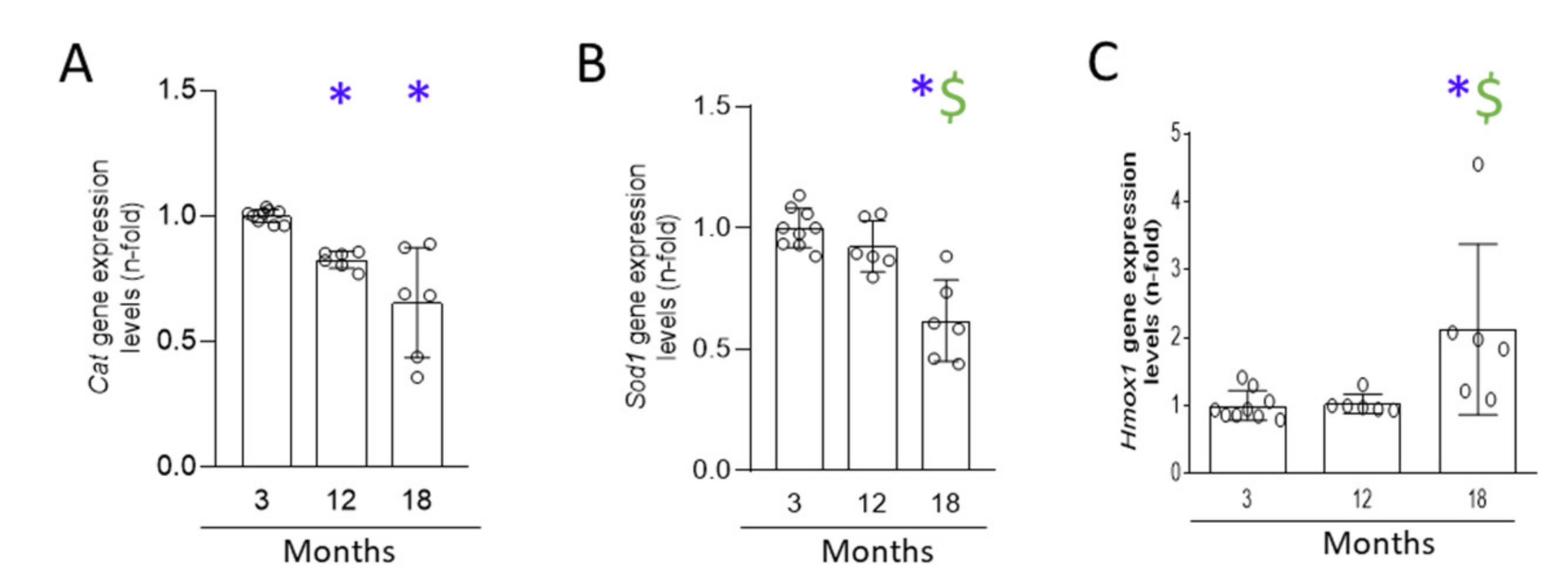
3.6. A Progressive Redox Imbalance Is Established in the Aging Kidney
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ortiz, A.; Roger, M.; Jiménez, V.M.; Perez, J.C.R.; Furlano, M.; Atxer, L.S.; Herranz, V.M. RICORS2040: The need for collaborative research in chronic kidney disease. Clin. Kidney J. 2021, 1, sfab170. [Google Scholar]
- Infante, B.; Franzin, R.; Madio, D.; Calvaruso, M.; Maiorano, A.; Sangregorio, F.; Netti, G.S.; Ranieri, E.; Gesualdo, L.; Castellano, G.; et al. Molecular Mechanisms of AKI in the Elderly: From Animal Models to Therapeutic Intervention. J. Clin. Med. 2020, 9, 2574. [Google Scholar] [CrossRef]
- Parmar, M.S.; Bashir, K. Crescentric Glomerulonephritis; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Stenvinkel, P.; Larsson, T.E. Chronic kidney disease: A clinical model of premature aging. Am. J. Kidney Dis. Off. J. Natl. Kidney Found. 2013, 62, 339–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sturmlechner, I.; Durik, M.; Sieben, C.J.; Baker, D.J.; Van Deursen, J.M. Cellular senescence in renal ageing and disease. Nat. Rev. Nephrol. 2017, 13, 77–89. [Google Scholar] [CrossRef]
- Banerjee, P.; Kotla, S.; Reddy Velatooru, L.; Abe, R.J.; Davis, E.A.; Cooke, J.P.; Schadler, K.; Deswal, A.; Herrmann, J.; Lin, S.H.; et al. Senescence-Associated Secretory Phenotype as a Hinge Between Cardiovascular Diseases and Cancer. Front. Cardiovasc. Med. 2021, 8, 763930. [Google Scholar] [CrossRef] [PubMed]
- Bonventre, J.V. Maladaptive proximal tubule repair: Cell cycle arrest. In Proceedings of the Nephron—Clinical Practice; S. Karger AG: Basel, Switzerland, 2014; Volume 127, pp. 61–64. [Google Scholar]
- Melk, A.; Schmidt, B.M.W.; Takeuchi, O.; Sawitzki, B.; Rayner, D.C.; Halloran, P.F. Expression of p16INK4a and other cell cycle regulator and senescence associated genes in aging human kidney. Kidney Int. 2004, 65, 510–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, B.; Wan, Y.; Chen, R.; Zhang, C.; Li, X.; Meng, F.; Glaser, S.; Wu, N.; Zhou, T.; Li, S.; et al. The emerging role of cellular senescence in renal diseases. J. Cell. Mol. Med. 2020, 24, 2087–2097. [Google Scholar] [CrossRef]
- Acosta, J.C.; Banito, A.; Wuestefeld, T.; Georgilis, A.; Janich, P.; Morton, J.P.; Athineos, D.; Kang, T.-W.; Lasitschka, F.; Andrulis, M.; et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat. Cell Biol. 2013, 15, 978–990. [Google Scholar] [CrossRef]
- Freund, A.; Orjalo, A.V.; Desprez, P.-Y.; Campisi, J. Inflammatory networks during cellular senescence: Causes and consequences. Trends Mol. Med. 2010, 16, 238–246. [Google Scholar] [CrossRef] [Green Version]
- Hoenicke, L.; Zender, L. Immune surveillance of senescent cells--biological significance in cancer- and non-cancer pathologies. Carcinogenesis 2012, 33, 1123–1126. [Google Scholar] [CrossRef] [Green Version]
- Krizhanovsky, V.; Xue, W.; Zender, L.; Yon, M.; Hernando, E.; Lowe, S.W. Implications of cellular senescence in tissue damage response, tumor suppression, and stem cell biology. Cold Spring Harb. Symp. Quant. Biol. 2008, 73, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Valentijn, F.A.; Falke, L.L.; Nguyen, T.Q.; Goldschmeding, R. Cellular senescence in the aging and diseased kidney. J. Cell Commun. Signal. 2018, 12, 69–82. [Google Scholar] [CrossRef] [Green Version]
- Knoppert, S.N.; Valentijn, F.A.; Nguyen, T.Q.; Goldschmeding, R.; Falke, L.L. Cellular Senescence and the Kidney: Potential Therapeutic Targets and Tools. Front. Pharmacol. 2019, 10, 770. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Espín, D.; Serrano, M. Cellular senescence: From physiology to pathology. Nat. Rev. Mol. Cell Biol. 2014, 15, 482–496. [Google Scholar] [CrossRef] [PubMed]
- Kirkland, J.L.; Tchkonia, T.; Zhu, Y.; Niedernhofer, L.J.; Robbins, P.D. The Clinical Potential of Senolytic Drugs. J. Am. Geriatr. Soc. 2017, 65, 2297–2301. [Google Scholar] [CrossRef]
- Kirkland, J.L.; Tchkonia, T. Cellular Senescence: A Translational Perspective. EBioMedicine 2017, 21, 21–28. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.-H.; Park, G.-T.; Lim, Y.-B.; Rue, S.-W.; Jung, J.-C.; Sonn, J.-K.; Bae, Y.-S.; Park, J.-W.; Lee, Y.-S. Expression of connective tissue growth factor, a biomarker in senescence of human diploid fibroblasts, is up-regulated by a transforming growth factor-beta-mediated signaling pathway. Biochem. Biophys. Res. Commun. 2004, 318, 819–825. [Google Scholar] [CrossRef]
- Ungvari, Z.; Valcarcel-Ares, M.N.; Tarantini, S.; Yabluchanskiy, A.; Fülöp, G.A.; Kiss, T.; Csiszar, A. Connective tissue growth factor (CTGF) in age-related vascular pathologies. GeroScience 2017, 39, 491–498. [Google Scholar] [CrossRef]
- Stenvinkel, P.; Chertow, G.M.; Devarajan, P.; Levin, A.; Andreoli, S.P.; Bangalore, S.; Warady, B.A. Chronic Inflammation in Chronic Kidney Disease Progression: Role of Nrf2. Kidney Int. Rep. 2021, 6, 1775–1787. [Google Scholar] [CrossRef]
- Ucar, B.I.; Ucar, G.; Saha, S.; Buttari, B.; Profumo, E.; Saso, L. Pharmacological Protection against Ischemia-Reperfusion Injury by Regulating the Nrf2-Keap1-ARE Signaling Pathway. Antioxidants 2021, 10, 823. [Google Scholar] [CrossRef]
- González-Bosch, C.; Boorman, E.; Zunszain, P.A.; Mann, G.E. Short-chain fatty acids as modulators of redox signaling in health and disease. Redox Biol. 2021, 47, 102165. [Google Scholar] [CrossRef] [PubMed]
- Sharpless, N.E.; Sherr, C.J. Forging a signature of in vivo senescence. Nat. Rev. Cancer 2015, 15, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Segura, A.; Nehme, J.; Demaria, M. Hallmarks of Cellular Senescence. Trends Cell Biol. 2018, 28, 436–453. [Google Scholar] [CrossRef] [PubMed]
- Hudgins, A.D.; Tazearslan, C.; Tare, A.; Zhu, Y.; Huffman, D.; Suh, Y. Age- and Tissue-Specific Expression of Senescence Biomarkers in Mice. Front. Genet. 2018, 9, 59. [Google Scholar] [CrossRef] [Green Version]
- Kadota, Y.; Yano, A.; Kawakami, T.; Sato, M.; Suzuki, S. Metabolomic profiling of plasma from middle-aged and advanced-age male mice reveals the metabolic abnormalities of carnitine biosynthesis in metallothionein gene knockout mice. Aging 2021, 13, 24963–24988. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.K.; Shin, K.K.; Kim, H.; Hong, Y.H.; Choi, W.; Kwak, Y.-S.; Han, C.-K.; Hyun, S.H.; Cho, J.Y. Korean Red Ginseng exerts anti-inflammatory and autophagy-promoting activities in aged mice. J. Ginseng Res. 2021, 45, 717–725. [Google Scholar] [CrossRef]
- Marquez-Exposito, L.; Tejedor-Santamaria, L.; Santos-Sanchez, L.; Valentijn, F.A.; Cantero-Navarro, E.; Rayego-Mateos, S.; Rodrigues-Diez, R.R.; Tejera-Muñoz, A.; Marchant, V.; Sanz, A.B.; et al. Acute Kidney Injury is Aggravated in Aged Mice by the Exacerbation of Proinflammatory Processes. Front. Pharmacol. 2021, 12, 662020. [Google Scholar] [CrossRef]
- Zoja, C.; Corna, D.; Camozzi, D.; Cattaneo, D.; Rottoli, D.; Batani, C.; Zanchi, C.; Abbate, M.; Remuzzi, G. How to fully protect the kidney in a severe model of progressive nephropathy: A multidrug approach. J. Am. Soc. Nephrol. 2002, 13, 2898–2908. [Google Scholar] [CrossRef] [Green Version]
- Amin, R.P.; Vickers, A.E.; Sistare, F.; Thompson, K.L.; Roman, R.J.; Lawton, M.; Kramer, J.; Hamadeh, H.K.; Collins, J.; Grissom, S.; et al. Identification of putative gene-based markers of renal toxicity. Environ. Health Perspect. 2004, 112, 465–479. [Google Scholar] [CrossRef]
- Prozialeck, W.C.; Vaidya, V.S.; Liu, J.; Waalkes, M.P.; Edwards, J.R.; Lamar, P.C.; Bernard, A.M.; Dumont, X.; Bonventre, J.V. Kidney injury molecule-1 is an early biomarker of cadmium nephrotoxicity. Kidney Int. 2007, 72, 985–993. [Google Scholar] [CrossRef] [Green Version]
- Beker, B.M.; Corleto, M.G.; Fieiras, C.; Musso, C.G. Novel acute kidney injury biomarkers: Their characteristics, utility and concerns. Int. Urol. Nephrol. 2018, 50, 705–713. [Google Scholar] [CrossRef] [PubMed]
- Griffin, B.R.; Faubel, S.; Edelstein, C.L. Biomarkers of drug-induced kidney toxicity. Ther. Drug Monit. 2019, 41, 213–226. [Google Scholar] [CrossRef]
- Gohda, T.; Kamei, N.; Koshida, T.; Kubota, M.; Tanaka, K.; Yamashita, Y.; Adachi, E.; Ichikawa, S.; Murakoshi, M.; Ueda, S.; et al. Circulating kidney injury molecule-1 as a biomarker of renal parameters in diabetic kidney disease. J. Diabetes Investig. 2020, 11, 435–440. [Google Scholar] [CrossRef] [PubMed]
- Moreno, J.A.; Izquierdo, M.C.; Sanchez-Niño, M.D.; Suárez-Alvarez, B.; Lopez-Larrea, C.; Jakubowski, A.; Blanco, J.; Ramirez, R.; Selgas, R.; Ruiz-Ortega, M.; et al. The inflammatory cytokines TWEAK and TNFα reduce renal klotho expression through NFκB. J. Am. Soc. Nephrol. 2011, 22, 1315–1325. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Fernandez, B.; Izquierdo, M.C.; Valiño-Rivas, L.; Nastou, D.; Sanz, A.B.; Ortiz, A.; Sanchez-Niño, M.D. Albumin downregulates Klotho in tubular cells. Nephrol. Dial. Transplant. 2018, 33, 1712–1722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Niño, M.D.; Fernandez-Fernandez, B.; Ortiz, A. Klotho, the elusive kidney-derived anti-ageing factor. Clin. Kidney J. 2020, 13, 125–127. [Google Scholar] [CrossRef]
- Fernández-Fernández, B.; Valiño-Rivas, L.; Sánchez-Niño, M.D.; Ortiz, A. Albuminuria Downregulation of the Anti-Aging Factor Klotho: The Missing Link Potentially Explaining the Association of Pathological Albuminuria with Premature Death. Adv. Ther. 2020, 37, 62–72. [Google Scholar] [CrossRef]
- Guo, Y.; Hu, M.; Ma, J.; Chinnathambi, A.; Alharbi, S.A.; Shair, O.H.M.; Ge, P. Protective effect of panaxydol against repeated administration of aristolochic acid on renal function and lipid peroxidation products via activating Keap1-Nrf2/ARE pathway in rat kidney. J. Biochem. Mol. Toxicol. 2021, 35, e22619. [Google Scholar] [CrossRef]
- Soulage, C.O.; Pelletier, C.C.; Florens, N.; Lemoine, S.; Dubourg, L.; Juillard, L.; Guebre-Egziabher, F. Two Toxic Lipid Aldehydes, 4-hydroxy-2-hexenal (4-HHE) and 4-hydroxy-2-nonenal (4-HNE), Accumulate in Patients with Chronic Kidney Disease. Toxins 2020, 12, 567. [Google Scholar] [CrossRef]
- Wetzels, J.F.M.; Kiemeney, L.A.L.M.; Swinkels, D.W.; Willems, H.L.; den Heijer, M. Age- and gender-specific reference values of estimated GFR in Caucasians: The Nijmegen Biomedical Study. Kidney Int. 2007, 72, 632–637. [Google Scholar] [CrossRef] [Green Version]
- Hommos, M.S.; Glassock, R.J.; Rule, A.D. Structural and Functional Changes in Human Kidneys with Healthy Aging. J. Am. Soc. Nephrol. 2017, 28, 2838–2844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, J.; Zeng, M.; Shu, Y.; Guo, D.; Sun, Y.; Guo, Z.; Wang, Y.; Liu, Z.; Zhou, H.; Zhang, W. Aging increases the susceptibility of cisplatin-induced nephrotoxicity. AGE 2015, 37, 112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nath, K.A.; Grande, J.P.; Farrugia, G.; Croatt, A.J.; Belcher, J.D.; Hebbel, R.P.; Vercellotti, G.M.; Katusic, Z.S. Age sensitizes the kidney to heme protein-induced acute kidney injury. Am. J. Physiol. Renal Physiol. 2013, 304, F317–F325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maddens, B.; Vandendriessche, B.; Demon, D.; Vanholder, R.; Chiers, K.; Cauwels, A.; Meyer, E. Severity of sepsis-induced acute kidney injury in a novel mouse model is age dependent. Crit. Care Med. 2012, 40, 2638–2646. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.A.; Buehner, G.; Chang, Y.; Harper, J.M.; Sigler, R.; Smith-Wheelock, M. Methionine-deficient diet extends mouse lifespan, slows immune and lens aging, alters glucose, T4, IGF-I and insulin levels, and increases hepatocyte MIF levels and stress resistance. Aging Cell 2005, 4, 119–125. [Google Scholar] [CrossRef]
- Turturro, A.; Witt, W.W.; Lewis, S.; Hass, B.S.; Lipman, R.D.; Hart, R.W. Growth curves and survival characteristics of the animals used in the Biomarkers of Aging Program. J. Gerontol. A Biol. Sci. Med. Sci. 1999, 54, B492–B501. [Google Scholar] [CrossRef] [Green Version]
- Miao, J.; Liu, J.; Niu, J.; Zhang, Y.; Shen, W.; Luo, C.; Liu, Y.; Li, C.; Li, H.; Yang, P.; et al. Wnt/β-catenin/RAS signaling mediates age-related renal fibrosis and is associated with mitochondrial dysfunction. Aging Cell 2019, 18, e13004. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Fogo, A.B. Cell senescence in the aging kidney. J. Am. Soc. Nephrol. 2010, 21, 1436–1439. [Google Scholar] [CrossRef] [Green Version]
- Melk, A. Senescence of renal cells: Molecular basis and clinical implications. Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transpl. Assoc.-Eur. Ren. Assoc. 2003, 18, 2474–2478. [Google Scholar] [CrossRef] [Green Version]
- Van Deursen, J.M. The role of senescent cells in ageing. Nature 2014, 509, 439–446. [Google Scholar] [CrossRef] [Green Version]
- Ovadya, Y.; Krizhanovsky, V. Senescent cells: SASPected drivers of age-related pathologies. Biogerontology 2014, 15, 627–642. [Google Scholar] [CrossRef] [PubMed]
- Franzin, R.; Stasi, A.; Ranieri, E.; Netti, G.S.; Cantaluppi, V.; Gesualdo, L.; Stallone, G.; Castellano, G. Targeting Premature Renal Aging: From Molecular Mechanisms of Cellular Senescence to Senolytic Trials. Front. Pharmacol. 2021, 12, 630419. [Google Scholar] [CrossRef] [PubMed]
- Goronzy, J.J.; Weyand, C.M. Understanding immunosenescence to improve responses to vaccines. Nat. Immunol. 2013, 14, 428–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montecino-Rodriguez, E.; Berent-Maoz, B.; Dorshkind, K. Causes, consequences, and reversal of immune system aging. J. Clin. Investig. 2013, 123, 958–965. [Google Scholar] [CrossRef]
- Franceschi, C.; Campisi, J. Chronic inflammation (Inflammaging) and its potential contribution to age-associated diseases. J. Gerontol.-Ser. A Biol. Sci. Med. Sci. 2014, 69, S4–S9. [Google Scholar] [CrossRef]
- Rodwell, G.E.J.; Sonu, R.; Zahn, J.M.; Lund, J.; Wilhelmy, J.; Wang, L.; Xiao, W.; Mindrinos, M.; Crane, E.; Segal, E.; et al. A transcriptional profile of aging in the human kidney. PLoS Biol. 2004, 2, e427. [Google Scholar] [CrossRef] [Green Version]
- Øien, C.M.; Reisæter, A.V.; Leivestad, T.; Dekker, F.W.; Line, P.D.; Os, I. Living donor kidney transplantation: The effects of donor age and gender on short- and long-term outcomes. Transplantation 2007, 83, 600–606. [Google Scholar] [CrossRef]
- Aiello, A.; Farzaneh, F.; Candore, G.; Caruso, C.; Davinelli, S.; Gambino, C.M.; Ligotti, M.E.; Zareian, N.; Accardi, G. Immunosenescence and Its Hallmarks: How to Oppose Aging Strategically? A Review of Potential Options for Therapeutic Intervention. Front. Immunol. 2019, 10, 2247. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, I.J.; Lalinde Ruiz, N.; Llano León, M.; Martínez Enríquez, L.; Montilla Velásquez, M.D.P.; Ortiz Aguirre, J.P.; Rodríguez Bohórquez, O.M.; Velandia Vargas, E.A.; Hernández, E.D.; Parra López, C.A. Immunosenescence Study of T Cells: A Systematic Review. Front. Immunol. 2020, 11, 604591. [Google Scholar] [CrossRef]
- Baar, M.P.; Brandt, R.M.C.; Putavet, D.A.; Klein, J.D.D.; Derks, K.W.J.; Bourgeois, B.R.M.; Stryeck, S.; Rijksen, Y.; van Willigenburg, H.; Feijtel, D.A.; et al. Targeted Apoptosis of Senescent Cells Restores Tissue Homeostasis in Response to Chemotoxicity and Aging. Cell 2017, 169, 132–147.e16. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Jiang, X.; Dong, F.; Zhang, F.; Ji, X.; Xue, M.; Yang, F.; Chen, J.; Hu, X.; Bao, Z. Lipid accumulation-induced hepatocyte senescence regulates the activation of hepatic stellate cells through the Nrf2-antioxidant response element pathway. Exp. Cell Res. 2021, 405, 112689. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Zaman, T.; Fahad, T.M.; Akther, T.; Hasan, M.F.; Naz, T.; Kishi, S. Carbofuran affects cellular autophagy and developmental senescence through the impairment of Nrf2 signalling. J. Cell. Mol. Med. 2021, 26, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Ungvari, Z.; Tarantini, S.; Nyúl-Tóth, Á.; Kiss, T.; Yabluchanskiy, A.; Csipo, T.; Balasubramanian, P.; Lipecz, A.; Benyo, Z.; Csiszar, A. Nrf2 dysfunction and impaired cellular resilience to oxidative stressors in the aged vasculature: From increased cellular senescence to the pathogenesis of age-related vascular diseases. GeroScience 2019, 41, 727–738. [Google Scholar] [CrossRef]
- Kim, H.J.; Vaziri, N.D. Contribution of impaired Nrf2-Keap1 pathway to oxidative stress and inflammation in chronic renal failure. Am. J. Physiol. Ren. Physiol. 2010, 298, F662–F671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stenvinkel, P.; Meyer, C.J.; Block, G.A.; Chertow, G.M.; Shiels, P.G. Understanding the role of the cytoprotective transcription factor nuclear factor erythroid 2-related factor 2-lessons from evolution, the animal kingdom and rare progeroid syndromes. Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transpl. Assoc.-Eur. Ren. Assoc. 2020, 35, 2036–2045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuro-o, M.; Matsumura, Y.; Aizawa, H.; Kawaguchi, H.; Suga, T.; Utsugi, T.; Ohyama, Y.; Kurabayashi, M.; Kaname, T.; Kume, E.; et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature 1997, 390, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Kurosu, H.; Yamamoto, M.; Clark, J.D.; Pastor, J.V.; Nandi, A.; Gurnani, P.; McGuinness, O.P.; Chikuda, H.; Yamaguchi, M.; Kawaguchi, H.; et al. Physiology: Suppression of aging in mice by the hormone Klotho. Science 2005, 309, 1829–1833. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Niño, M.D.; Sanz, A.B.; Ortiz, A. Klotho to treat kidney fibrosis. J. Am. Soc. Nephrol. 2013, 24, 687–689. [Google Scholar] [CrossRef]
- Carracedo, J.; Buendía, P.; Merino, A.; Madueño, J.A.; Peralbo, E.; Ortiz, A.; Martín-Malo, A.; Aljama, P.; Rodríguez, M.; Ramírez, R. Klotho modulates the stress response in human senescent endothelial cells. Mech. Ageing Dev. 2012, 133, 647–654. [Google Scholar] [CrossRef]
- De Oliveira, R.M. Klotho RNAi induces premature senescence of human cells via a p53/p21 dependent pathway. FEBS Lett. 2006, 580, 5753–5758. [Google Scholar] [CrossRef] [Green Version]
- Kanbay, M.; Demiray, A.; Afsar, B.; Covic, A.; Tapoi, L.; Ureche, C.; Ortiz, A. Role of Klotho in the Development of Essential Hypertension. Hypertension 2021, 77, 740–750. [Google Scholar] [CrossRef] [PubMed]
- Izquierdo, M.C.; Perez-Gomez, M.V.; Sanchez-Niño, M.D.; Sanz, A.B.; Ruiz-Andres, O.; Poveda, J.; Moreno, J.A.; Egido, J.; Ortiz, A. Klotho, phosphate and inflammation/ageing in chronic kidney disease. Nephrol. Dial. Transplant. 2012, 27, iv6–iv10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maique, J.; Flores, B.; Shi, M.; Shepard, S.; Zhou, Z.; Yan, S.; Moe, O.W.; Hu, M.C. High Phosphate Induces and Klotho Attenuates Kidney Epithelial Senescence and Fibrosis. Front. Pharmacol. 2020, 11, 1273. [Google Scholar] [CrossRef] [PubMed]











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marquez-Exposito, L.; Tejedor-Santamaria, L.; Valentijn, F.A.; Tejera-Muñoz, A.; Rayego-Mateos, S.; Marchant, V.; Rodrigues-Diez, R.R.; Rubio-Soto, I.; Knoppert, S.N.; Ortiz, A.; et al. Oxidative Stress and Cellular Senescence Are Involved in the Aging Kidney. Antioxidants 2022, 11, 301. https://doi.org/10.3390/antiox11020301
Marquez-Exposito L, Tejedor-Santamaria L, Valentijn FA, Tejera-Muñoz A, Rayego-Mateos S, Marchant V, Rodrigues-Diez RR, Rubio-Soto I, Knoppert SN, Ortiz A, et al. Oxidative Stress and Cellular Senescence Are Involved in the Aging Kidney. Antioxidants. 2022; 11(2):301. https://doi.org/10.3390/antiox11020301
Chicago/Turabian StyleMarquez-Exposito, Laura, Lucia Tejedor-Santamaria, Floris A. Valentijn, Antonio Tejera-Muñoz, Sandra Rayego-Mateos, Vanessa Marchant, Raul R. Rodrigues-Diez, Irene Rubio-Soto, Sebastiaan N. Knoppert, Alberto Ortiz, and et al. 2022. "Oxidative Stress and Cellular Senescence Are Involved in the Aging Kidney" Antioxidants 11, no. 2: 301. https://doi.org/10.3390/antiox11020301
APA StyleMarquez-Exposito, L., Tejedor-Santamaria, L., Valentijn, F. A., Tejera-Muñoz, A., Rayego-Mateos, S., Marchant, V., Rodrigues-Diez, R. R., Rubio-Soto, I., Knoppert, S. N., Ortiz, A., Ramos, A. M., Goldschmeding, R., & Ruiz-Ortega, M. (2022). Oxidative Stress and Cellular Senescence Are Involved in the Aging Kidney. Antioxidants, 11(2), 301. https://doi.org/10.3390/antiox11020301