
Superoxide Radicals in the Execution of Cell Death


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
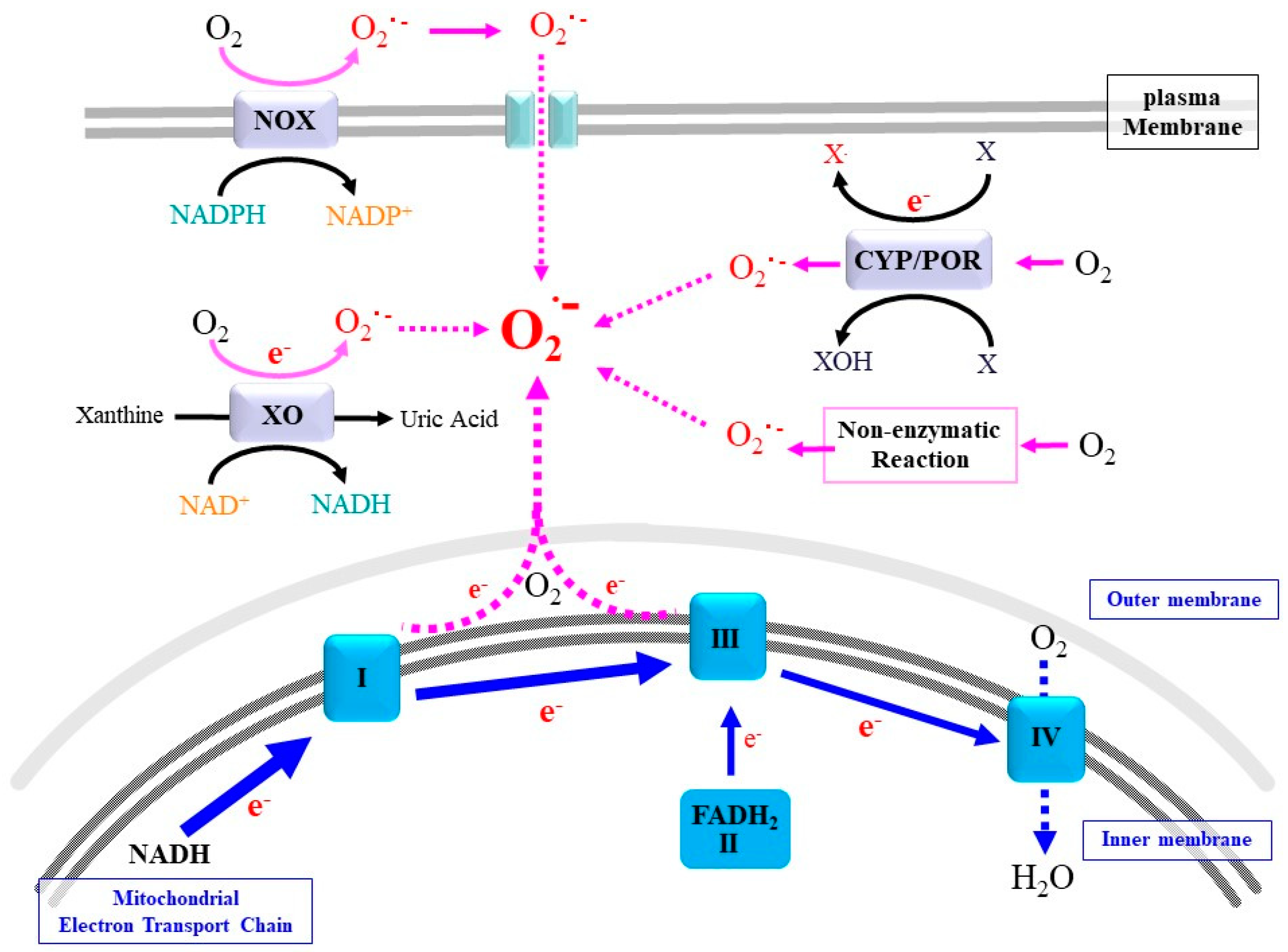
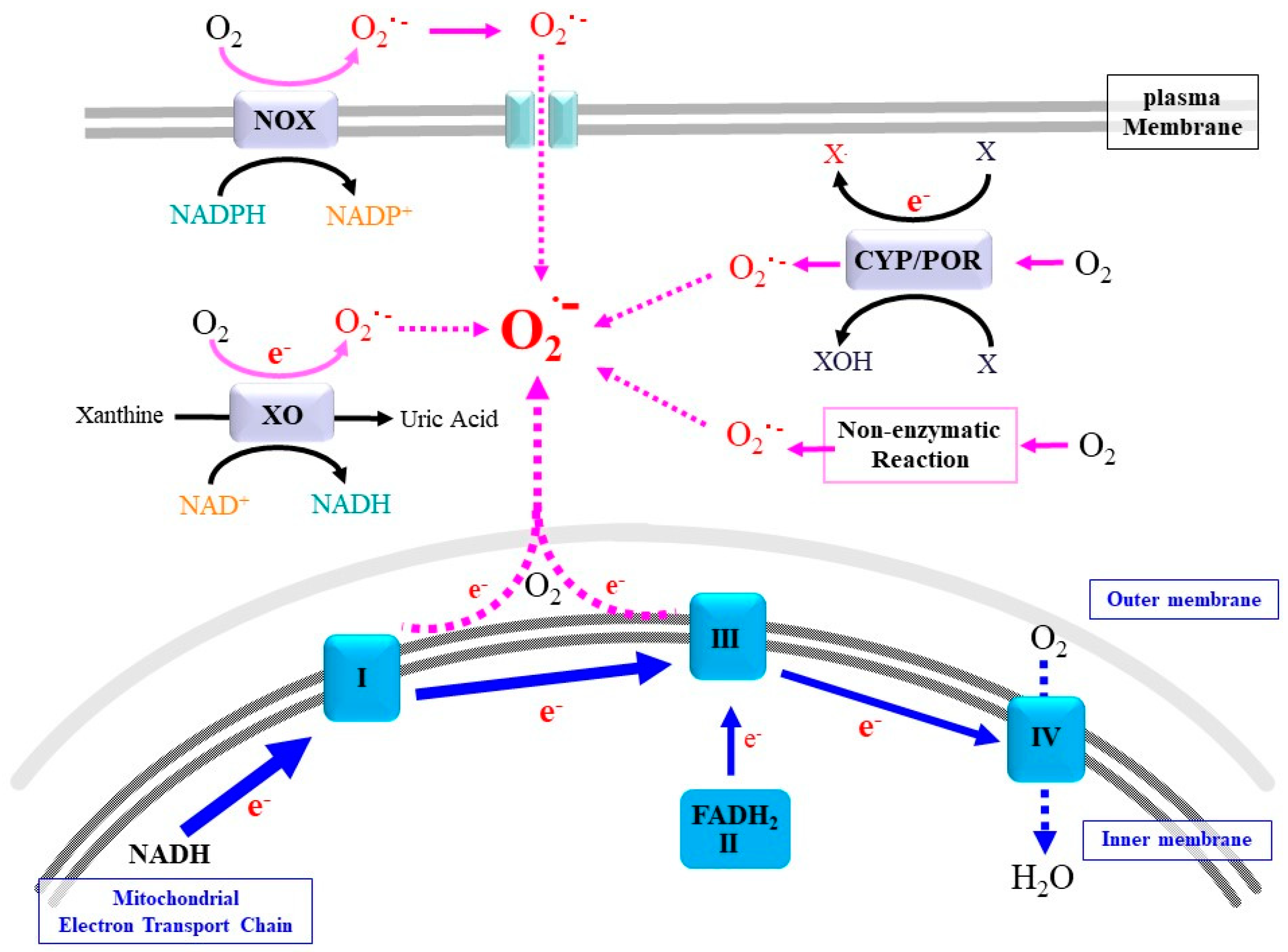
2. Sources and Actions of Superoxide and Related ROS
2.1. Mitochondrial Respiratory Chain
2.2. NADPH Oxidases
2.3. Cytochrome P450 (CYP)/Cytochrome P450 Reductase (POR) System
2.4. Xanthine Oxidoreductase
2.5. Non-Enzymatic Production of Superoxide
3. Antioxidative Enzymes and Compounds to Protect against ROS-Induced Oxidative Damage
3.1. Dismutation of Superoxide to Hydrogen Peroxide Abolishes Radical Electrons
3.1.1. SOD1
3.1.2. SOD2
3.1.3. SOD3
3.2. Peroxidases Complete Radical Detoxification
3.2.1. Catalase
3.2.2. GSH-GPX System
3.2.3. TRX-PRDX System
4. Cell Death Associated with an Oxidative Insult
4.1. Roles of ROS in Apoptosis
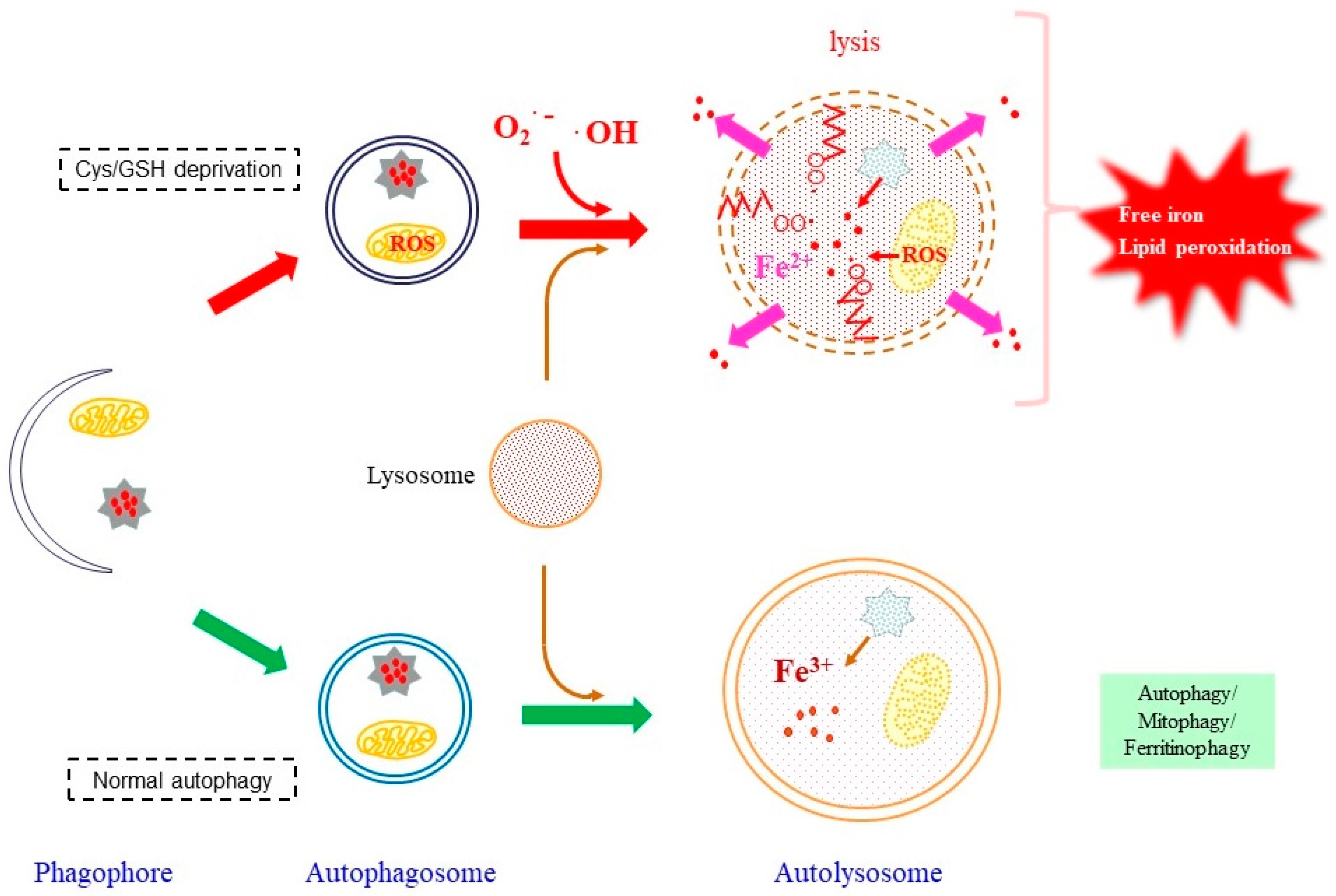
4.2. Ferroptosis as a Radical-Associated Cell Death
4.2.1. Roles of Autophagy in Providing Free Iron
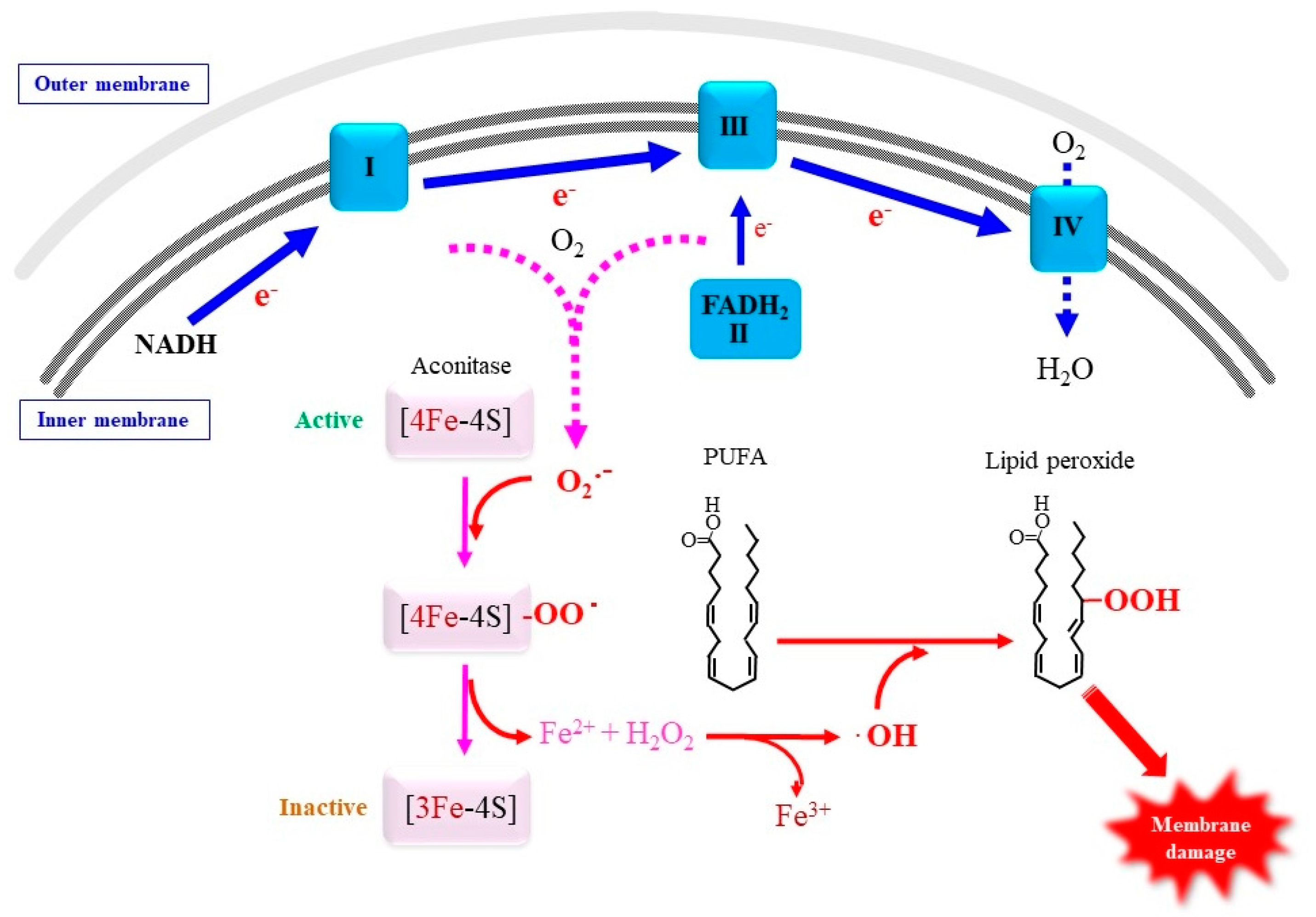
4.2.2. Involvement of Mitochondrial Superoxide in Ferroptosis
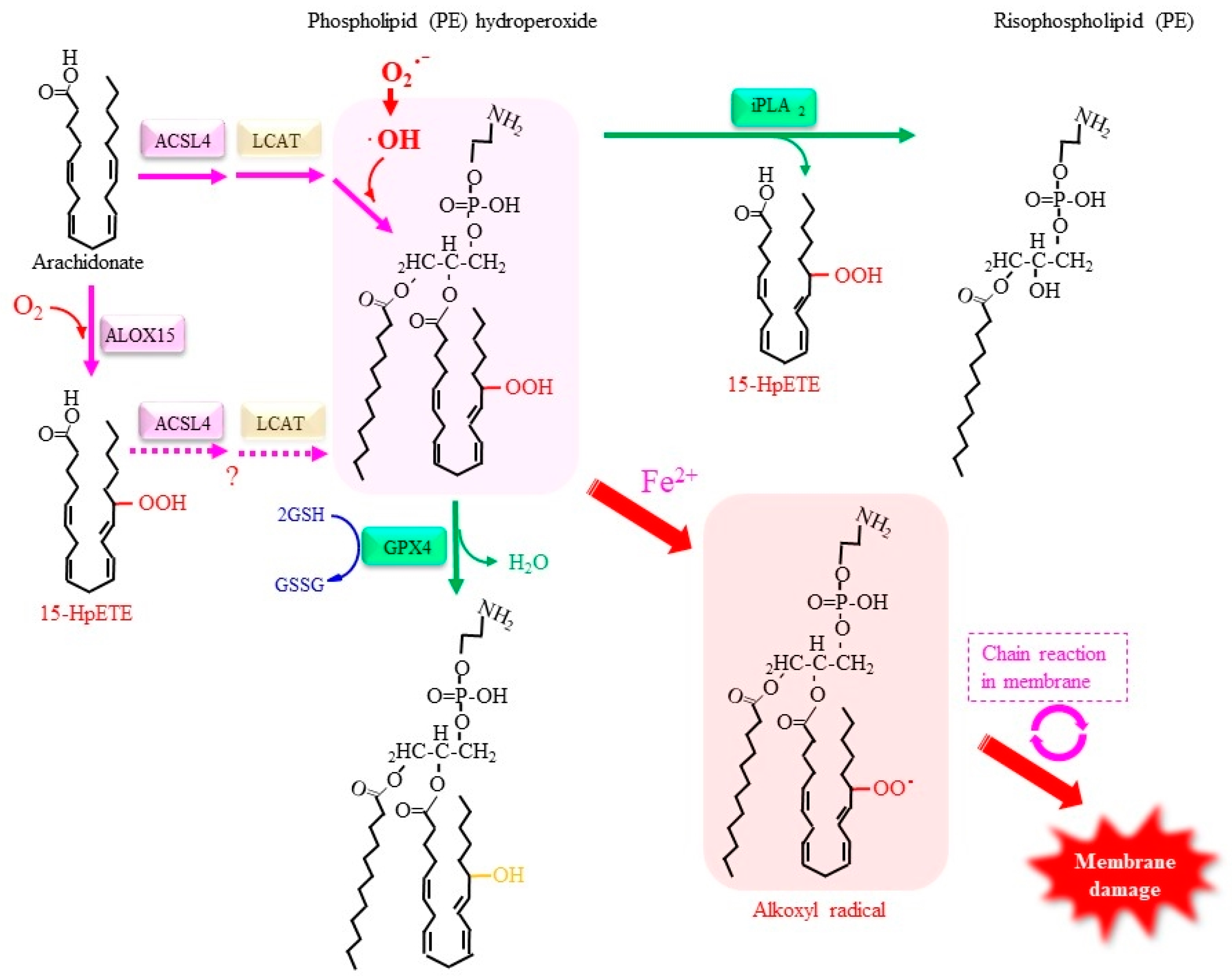
4.2.3. Roles of Lipid Peroxidation in Ferroptosis
4.2.4. Other Factors Involved in the Regulation of Ferroptosis
5. Antioxidative Metabolites to Combat Ferroptosis
5.1. Vitamin E Suppresses Lipid Peroxidation
5.2. Vitamin C Is a Hydrophilic Antioxidant but Suppresses Lipid Peroxidation
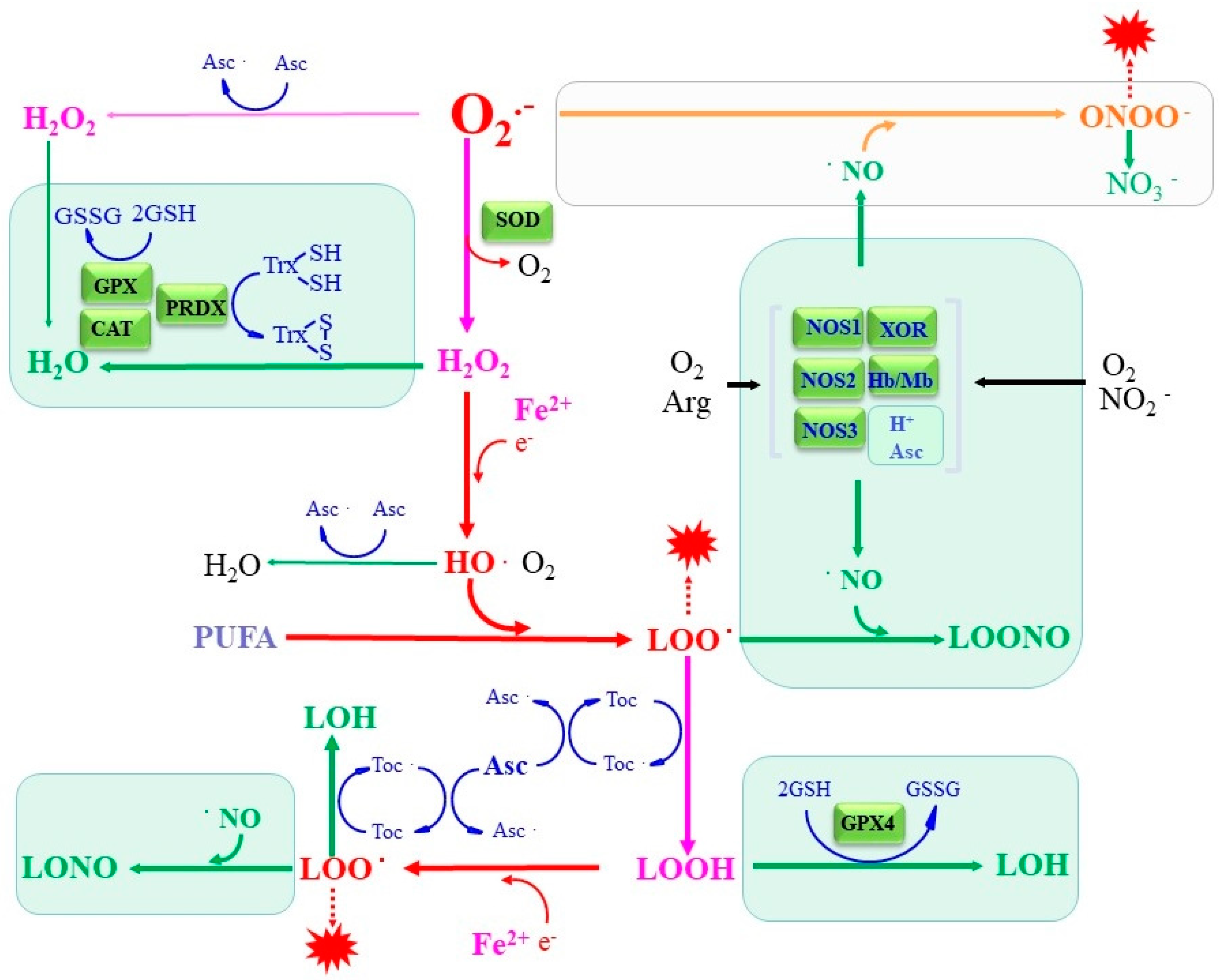
5.3. Nitric Oxide as Both Pro-Oxidant and Antioxidant
5.3.1. NO as a Potent Superoxide Scavenger
5.3.2. NO in Termination of Radical Chain Reactions in Lipid Peroxidation
6. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Fridovich, I. Superoxide radical and superoxide dismutases. Annu. Rev. Biochem. 1995, 64, 97–112. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.G. Cell signaling. H2O2, a necessary evil for cell signaling. Science 2006, 312, 1882–1883. [Google Scholar] [CrossRef] [PubMed]
- Finkel, T. Signal transduction by reactive oxygen species. J. Cell Biol. 2011, 194, 7–15. [Google Scholar] [CrossRef] [Green Version]
- Lennicke, C.; Cochemé, H.M. Redox metabolism: ROS as specific molecular regulators of cell signaling and function. Mol. Cell 2021, 81, 3691–3707. [Google Scholar] [CrossRef] [PubMed]
- Janssen-Heininger, Y.M.; Mossman, B.T.; Heintz, N.H.; Forman, H.J.; Kalyanaraman, B.; Finkel, T.; Stamler, J.S.; Rhee, S.G.; van der Vliet, A. Redox-based regulation of signal transduction: Principles, pitfalls, and promises. Free Radic. Biol. Med. 2008, 45, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Lundberg, J.O.; Weitzberg, E.; Gladwin, M.T. The nitrate-nitrite-nitric oxide pathway in physiology and therapeutics. Nat. Rev. Drug Discov. 2008, 7, 156–167. [Google Scholar] [CrossRef]
- Tejero, J.; Shiva, S.; Gladwin, M.T. Sources of Vascular Nitric Oxide and Reactive Oxygen Species and Their Regulation. Physiol. Rev. 2019, 99, 311–379. [Google Scholar] [CrossRef]
- Radi, R. Oxygen radicals, nitric oxide, and peroxynitrite: Redox pathways in molecular medicine. Proc. Natl. Acad. Sci. USA 2018, 115, 5839–5848. [Google Scholar] [CrossRef] [Green Version]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [Green Version]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef]
- Chen, Y.R.; Zweier, J.L. Cardiac mitochondria and reactive oxygen species generation. Circ. Res. 2014, 114, 524–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, H.S.; Dighe, P.A.; Mezera, V.; Monternier, P.A.; Brand, M.D. Production of superoxide and hydrogen peroxide from specific mitochondrial sites under different bioenergetic conditions. J. Biol. Chem. 2017, 292, 16804–16809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinlan, C.L.; Goncalves, R.L.; Hey-Mogensen, M.; Yadava, N.; Bunik, V.I.; Brand, M.D. The 2-oxoacid dehydrogenase complexes in mitochondria can produce superoxide/hydrogen peroxide at much higher rates than complex I. J. Biol. Chem. 2014, 289, 8312–8325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Burdon, R.H. Superoxide and hydrogen peroxide in relation to mammalian cell proliferation. Free Radic. Biol. Med. 1995, 18, 775–794. [Google Scholar] [CrossRef]
- Nordzieke, D.E.; Medraño-Fernandez, I. The Plasma Membrane: A Platform for Intra- and Intercellular Redox Signaling. Antioxidants 2018, 7, 168. [Google Scholar] [CrossRef] [Green Version]
- Nauseef, W.M. The phagocyte NOX2 NADPH oxidase in microbial killing and cell signaling. Curr. Opin. Immunol. 2019, 60, 130–140. [Google Scholar] [CrossRef]
- Suh, Y.A.; Arnold, R.S.; Lassegue, B.; Shi, J.; Xu, X.; Sorescu, D.; Chung, A.B.; Griendling, K.K.; Lambeth, J.D. Cell transformation by the superoxide-generating oxidase Mox1. Nature 1999, 401, 79–82. [Google Scholar] [CrossRef]
- Lassègue, B.; Sorescu, D.; Szöcs, K.; Yin, Q.; Akers, M.; Zhang, Y.; Grant, S.L.; Lambeth, J.D.; Griendling, K.K. Novel gp91(phox) homologues in vascular smooth muscle cells: Nox1 mediates angiotensin II-induced superoxide formation and redox-sensitive signaling pathways. Circ. Res. 2001, 88, 888–894. [Google Scholar] [CrossRef] [Green Version]
- Dustin, C.M.; Heppner, D.E.; Lin, M.J.; van der Vliet, A. Redox regulation of tyrosine kinase signalling: More than meets the eye. J. Biochem. 2020, 167, 151–163. [Google Scholar] [CrossRef] [Green Version]
- Cadenas, S. ROS and redox signaling in myocardial ischemia-reperfusion injury and cardioprotection. Free Radic. Biol. Med. 2018, 117, 76–89. [Google Scholar] [CrossRef] [PubMed]
- Vermot, A.; Petit-Härtlein, I.; Smith, S.M.E.; Fieschi, F. NADPH Oxidases (NOX): An Overview from Discovery, Molecular Mechanisms to Physiology and Pathology. Antioxidants 2021, 10, 890. [Google Scholar] [CrossRef] [PubMed]
- Tsutsumi, R.; Harizanova, J.; Stockert, R.; Schröder, K.; Bastiaens, P.I.H.; Neel, B.G. Assay to visualize specific protein oxidation reveals spatio-temporal regulation of SHP2. Nat. Commun. 2017, 8, 466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denu, J.M.; Dixon, J.E. Protein tyrosine phosphatases: Mechanisms of catalysis and regulation. Curr. Opin. Chem. Biol. 1998, 2, 633–641. [Google Scholar] [CrossRef]
- Goldstein, B.J.; Mahadev, K.; Wu, X. Redox paradox: Insulin action is facilitated by insulin-stimulated reactive oxygen species with multiple potential signaling targets. Diabetes 2005, 54, 311–321. [Google Scholar] [CrossRef] [Green Version]
- Sivaganesh, V.; Sivaganesh, V.; Scanlon, C.; Iskander, A.; Maher, S.; Lê, T.; Peethambaran, B. Protein Tyrosine Phosphatases: Mechanisms in Cancer. Int. J. Mol. Sci. 2021, 22, 12865. [Google Scholar] [CrossRef]
- Esteban-Villarrubia, J.; Soto-Castillo, J.J.; Pozas, J.; San Román-Gil, M.; Orejana-Martín, I.; Torres-Jiménez, J.; Carrato, A.; Alonso-Gordoa, T.; Molina-Cerrillo, J. Tyrosine Kinase Receptors in Oncology. Int. J. Mol. Sci. 2020, 21, 8529. [Google Scholar] [CrossRef]
- Yehia, L.; Keel, E.; Eng, C. The Clinical Spectrum of PTEN Mutations. Annu. Rev. Med. 2020, 71, 103–116. [Google Scholar] [CrossRef] [Green Version]
- Nguyen Huu, T.; Park, J.; Zhang, Y.; Park, I.; Yoon, H.J.; Woo, H.A.; Lee, S.R. Redox Regulation of PTEN by Peroxiredoxins. Antioxidants 2021, 10, 302. [Google Scholar] [CrossRef]
- Verbon, E.H.; Post, J.A.; Boonstra, J. The influence of reactive oxygen species on cell cycle progression in mammalian cells. Gene 2012, 511, 1–6. [Google Scholar] [CrossRef]
- Savitsky, P.A.; Finkel, T. Redox regulation of Cdc25C. J. Biol. Chem. 2002, 277, 20535–20540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brisson, M.; Nguyen, T.; Wipf, P.; Joo, B.; Day, B.W.; Skoko, J.S.; Schreiber, E.M.; Foster, C.; Bansal, P.; Lazo, J.S. Redox regulation of Cdc25B by cell-active quinolinediones. Mol. Pharmacol. 2005, 68, 1810–1820. [Google Scholar] [CrossRef] [PubMed]
- Yamaura, M.; Mitsushita, J.; Furuta, S.; Kiniwa, Y.; Ashida, A.; Goto, Y.; Shang, W.H.; Kubodera, M.; Kato, M.; Takata, M.; et al. NADPH oxidase 4 contributes to transformation phenotype of melanoma cells by regulating G2-M cell cycle progression. Cancer Res. 2009, 69, 2647–2654. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Fan, M.; Candas, D.; Zhang, T.Q.; Qin, L.; Eldridge, A.; Wachsmann-Hogiu, S.; Ahmed, K.M.; Chromy, B.A.; Nantajit, D.; et al. Cyclin B1/Cdk1 coordinates mitochondrial respiration for cell-cycle G2/M progression. Dev. Cell 2014, 29, 217–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patterson, J.C.; Joughin, B.A.; van de Kooij, B.; Lim, D.C.; Lauffenburger, D.A.; Yaffe, M.B. ROS and Oxidative Stress Are Elevated in Mitosis during Asynchronous Cell Cycle Progression and Are Exacerbated by Mitotic Arrest. Cell Syst. 2019, 8, 163–167.e2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yasui, H.; Hayashi, S.; Sakurai, H. Possible involvement of singlet oxygen species as multiple oxidants in p450 catalytic reactions. Drug Metab. Pharmacokinet. 2005, 20, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Pandey, A.V.; Flück, C.E. NADPH P450 oxidoreductase: Structure, function, and pathology of diseases. Pharmacol. Ther. 2013, 138, 229–254. [Google Scholar] [CrossRef]
- Hrycay, E.G.; Bandiera, S.M. Monooxygenase, peroxidase and peroxygenase properties and reaction mechanisms of cytochrome P450 enzymes. Adv. Exp. Med. Biol. 2015, 851, 1–61. [Google Scholar] [CrossRef]
- Zeeshan, H.M.; Lee, G.H.; Kim, H.R.; Chae, H.J. Endoplasmic Reticulum Stress and Associated ROS. Int. J. Mol. Sci. 2016, 17, 327. [Google Scholar] [CrossRef] [Green Version]
- Harjumäki, R.; Pridgeon, C.S.; Ingelman-Sundberg, M. CYP2E1 in Alcoholic and Non-Alcoholic Liver Injury. Roles of ROS, Reactive Intermediates and Lipid Overload. Int. J. Mol. Sci. 2021, 22, 8221. [Google Scholar] [CrossRef]
- Kuwabara, Y.; Nishino, T.; Okamoto, K.; Matsumura, T.; Eger, B.T.; Pai, E.F.; Nishino, T. Unique amino acids cluster for switching from the dehydrogenase to oxidase form of xanthine oxidoreductase. Proc. Natl. Acad. Sci. USA 2003, 100, 8170–8175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishino, T.; Okamoto, K.; Eger, B.T.; Pai, E.F.; Nishino, T. Mammalian xanthine oxidoreductase—Mechanism of transition from xanthine dehydrogenase to xanthine oxidase. FEBS J. 2008, 275, 3278–3289. [Google Scholar] [CrossRef] [PubMed]
- Granger, D.N.; Kvietys, P.R. Reperfusion injury and reactive oxygen species: The evolution of a concept. Redox Biol. 2015, 6, 524–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bortolotti, M.; Polito, L.; Battelli, M.G.; Bolognesi, A. Xanthine oxidoreductase: One enzyme for multiple physiological tasks. Redox Biol. 2021, 41, 101882. [Google Scholar] [CrossRef]
- Ahmad, S.; Khan, M.Y.; Rafi, Z.; Khan, H.; Siddiqui, Z.; Rehman, S.; Shahab, U.; Khan, M.S.; Saeed, M.; Alouffi, S.; et al. Oxidation, glycation and glycoxidation-The vicious cycle and lung cancer. Semin. Cancer Biol. 2018, 49, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Mujtaba, S.F.; Masih, A.P.; Alqasmi, I.; Alsulimani, A.; Khan, F.H.; Haque, S. Oxidative-Stress-Induced Cellular Toxicity and Glycoxidation of Biomolecules by Cosmetic Products under Sunlight Exposure. Antioxidants 2021, 10, 1008. [Google Scholar] [CrossRef]
- Moldogazieva, N.T.; Mokhosoev, I.M.; Mel’nikova, T.I.; Porozov, Y.B.; Terentiev, A.A. Oxidative Stress and Advanced Lipoxidation and Glycation End Products (ALEs and AGEs) in Aging and Age-Related Diseases. Oxidative Med. Cell Longev. 2019, 2019, 3085756. [Google Scholar] [CrossRef] [Green Version]
- Getzoff, E.D.; Tainer, J.A.; Weiner, P.K.; Kollman, P.A.; Richardson, J.S.; Richardson, D.C. Electrostatic recognition between superoxide and copper, zinc superoxide dismutase. Nature 1983, 306, 287–290. [Google Scholar] [CrossRef]
- Tainer, J.A.; Getzoff, E.D.; Richardson, J.S.; Richardson, D.C. Structure and mechanism of copper, zinc superoxide dismutase. Nature 1983, 306, 284–287. [Google Scholar] [CrossRef]
- Ślesak, I.; Kula, M.; Ślesak, H.; Miszalski, Z.; Strzałka, K. How to define obligatory anaerobiosis? An evolutionary view on the antioxidant response system and the early stages of the evolution of life on Earth. Free Radic. Biol. Med. 2019, 140, 61–73. [Google Scholar] [CrossRef]
- Zelko, I.N.; Mariani, T.J.; Folz, R.J. Superoxide dismutase multigene family: A comparison of the CuZn-SOD (SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3) gene structures, evolution, and expression. Free Radic. Biol. Med. 2002, 33, 337–349. [Google Scholar] [CrossRef]
- Okado-Matsumoto, A.; Fridovich, I. Subcellular distribution of superoxide dismutases (SOD) in rat liver: Cu, Zn-SOD in mitochondria. J. Biol. Chem. 2001, 276, 38388–38393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Culotta, V.C.; Klomp, L.W.; Strain, J.; Casareno, R.L.; Krems, B.; Gitlin, J.D. The copper chaperone for superoxide dismutase. J. Biol. Chem. 1997, 272, 23469–23472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, P.C.; Waggoner, D.; Subramaniam, J.R.; Tessarollo, L.; Bartnikas, T.B.; Culotta, V.C.; Price, D.L.; Rothstein, J.; Gitlin, J.D. Copper chaperone for superoxide dismutase is essential to activate mammalian Cu/Zn superoxide dismutase. Proc. Natl. Acad. Sci. USA 2000, 97, 2886–2891. [Google Scholar] [CrossRef] [Green Version]
- Leitch, J.M.; Yick, P.J.; Culotta, V.C. The right to choose: Multiple pathways for activating copper, zinc superoxide dismutase. J. Biol. Chem. 2009, 284, 24679–24683. [Google Scholar] [CrossRef] [Green Version]
- Iñarrea, P.; Moini, H.; Rettori, D.; Han, D.; Martínez, J.; García, I.; Fernández-Vizarra, E.; Iturralde, M.; Cadenas, E. Redox activation of mitochondrial intermembrane space Cu, Zn-superoxide dismutase. Biochem. J. 2005, 387, 203–209. [Google Scholar] [CrossRef] [Green Version]
- Iñarrea, P.; Moini, H.; Han, D.; Rettori, D.; Aguiló, I.; Alava, M.A.; Iturralde, M.; Cadenas, E. Mitochondrial respiratory chain and thioredoxin reductase regulate intermembrane Cu, Zn-superoxide dismutase activity: Implications for mitochondrial energy metabolism and apoptosis. Biochem. J. 2007, 405, 173–179. [Google Scholar] [CrossRef] [Green Version]
- Ferrari, M.; Stagi, S. Oxidative Stress in Down and Williams-Beuren Syndromes: An Overview. Molecules 2021, 26, 3139. [Google Scholar] [CrossRef]
- Avraham, K.B.; Schickler, M.; Sapoznikov, D.; Yarom, R.; Groner, Y. Down’s syndrome: Abnormal neuromuscular junction in tongue of transgenic mice with elevated levels of human Cu/Zn-superoxide dismutase. Cell 1988, 54, 823–829. [Google Scholar] [CrossRef]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.X.; et al. Mutations, in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef]
- Kaur, S.J.; McKeown, S.R.; Rashid, S. Mutant SOD1 mediated pathogenesis of Amyotrophic Lateral Sclerosis. Gene 2016, 577, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.; Gautier, O.; Tassoni-Tsuchida, E.; Ma, X.R.; Gitler, A.D. ALS Genetics: Gains, Losses, and Implications for Future Therapies. Neuron 2020, 108, 822–842. [Google Scholar] [CrossRef] [PubMed]
- Reaume, A.G.; Elliott, J.L.; Hoffman, E.K.; Kowall, N.W.; Ferrante, R.J.; Siwek, D.F.; Wilcox, H.M.; Flood, D.G.; Beal, M.F.; Brown, R.H., Jr.; et al. Motor neurons in Cu/Zn superoxide dismutase-deficient mice develop normally but exhibit enhanced cell death after axonal injury. Nat. Genet. 1996, 13, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Matzuk, M.M.; Dionne, L.; Guo, Q.; Kumar, T.R.; Lebovitz, R.M. Ovarian function in superoxide dismutase 1 and 2 knockout mice. Endocrinology 1998, 139, 4008–4011. [Google Scholar] [CrossRef]
- Ho, Y.S.; Gargano, M.; Cao, J.; Bronson, R.T.; Heimler, I.; Hutz, R.J. Reduced fertility in female mice lacking copper-zinc superoxide dismutase. J. Biol. Chem. 1998, 273, 7765–7769. [Google Scholar] [CrossRef] [Green Version]
- Andersen, P.M.; Nordström, U.; Tsiakas, K.; Johannsen, J.; Volk, A.E.; Bierhals, T.; Zetterström, P.; Marklund, S.L.; Hempel, M.; Santer, R. Phenotype in an Infant with SOD1 Homozygous Truncating Mutation. N. Engl. J. Med. 2019, 381, 486–488. [Google Scholar] [CrossRef]
- Park, J.H.; Elpers, C.; Reunert, J.; McCormick, M.L.; Mohr, J.; Biskup, S.; Schwartz, O.; Rust, S.; Grüneberg, M.; Seelhöfer, A.; et al. SOD1 deficiency: A novel syndrome distinct from amyotrophic lateral sclerosis. Brain 2019, 142, 2230–2237. [Google Scholar] [CrossRef]
- Subramaniam, J.R.; Lyons, W.E.; Liu, J.; Bartnikas, T.B.; Rothstein, J.; Price, D.L.; Cleveland, D.W.; Gitlin, J.D.; Wong, P.C. Mutant SOD1 causes motor neuron disease independent of copper chaperone-mediated copper loading. Nat. Neurosci. 2002, 5, 301–307. [Google Scholar] [CrossRef]
- Flynn, J.M.; Melov, S. SOD2 in mitochondrial dysfunction and neurodegeneration. Free Radic. Biol. Med. 2013, 62, 4–12. [Google Scholar] [CrossRef] [Green Version]
- Culotta, V.C.; Yang, M.; O’Halloran, T.V. Activation of superoxide dismutases: Putting the metal to the pedal. Biochim. Biophys. Acta 2006, 1763, 747–758. [Google Scholar] [CrossRef] [Green Version]
- Luk, E.E.; Culotta, V.C. Manganese superoxide dismutase in Saccharomyces cerevisiae acquires its metal co-factor through a pathway involving the Nramp metal transporter, Smf2p. J. Biol. Chem. 2001, 276, 47556–47562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itahana, K.; Campisi, J.; Dimri, G.P. Mechanisms of cellular senescence in human and mouse cells. Biogerontology 2004, 5, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.; Brown, K.; Hirschey, M.D.; Verdin, E.; Chen, D. Calorie restriction reduces oxidative stress by SIRT3-mediated SOD2 activation. Cell Metab. 2010, 12, 662–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, Y.C.; Pérez, V.I.; Song, W.; Lustgarten, M.S.; Salmon, A.B.; Mele, J.; Qi, W.; Liu, Y.; Liang, H.; Chaudhuri, A.; et al. Overexpression of Mn superoxide dismutase does not increase life span in mice. Biol. Sci. Med. Sci. 2009, 64, 1114–1125. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Huang, T.T.; Carlson, E.J.; Melov, S.; Ursell, P.C.; Olson, J.L.; Noble, L.J.; Yoshimura, M.P.; Berger, C.; Chan, P.H.; et al. Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nat. Genet. 1995, 11, 376–381. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Siu, B.; Ho, Y.S.; Vincent, R.; Chua, C.C.; Hamdy, R.C.; Chua, B.H. Overexpression of MnSOD protects against myocardial ischemia/reperfusion injury in transgenic mice. J. Mol. Cell. Cardiol. 1998, 30, 2281–2289. [Google Scholar] [CrossRef]
- Sharma, S.; Bhattarai, S.; Ara, H.; Sun, G.; St Clair, D.K.; Bhuiyan, M.S.; Kevil, C.; Watts, M.N.; Dominic, P.; Shimizu, T.; et al. SOD2 deficiency in cardiomyocytes defines defective mitochondrial bioenergetics as a cause of lethal dilated cardiomyopathy. Redox Biol. 2020, 37, 101740. [Google Scholar] [CrossRef]
- Lebovitz, R.M.; Zhang, H.; Vogel, H.; Cartwright, J., Jr.; Dionne, L.; Lu, N.; Huang, S.; Matzuk, M.M. Neurodegeneration, myocardial injury, and perinatal death in mitochondrial superoxide dismutase-deficient mice. Proc. Natl. Acad. Sci. USA 1996, 93, 9782–9787. [Google Scholar] [CrossRef] [Green Version]
- Kurahashi, T.; Hamashima, S.; Shirato, T.; Lee, J.; Homma, T.; Kang, E.S.; Fujii, J. An SOD1 deficiency enhances lipid droplet accumulation in the fasted mouse liver by aborting lipophagy. Biochem. Biophys. Res. Commun. 2015, 467, 866–871. [Google Scholar] [CrossRef]
- Lee, J.; Homma, T.; Kobayashi, S.; Ishii, N.; Fujii, J. Unveiling systemic organ disorders associated with impaired lipid catabolism in fasted SOD1-deficient mice. Arch. Biochem. Biophys. 2018, 654, 163–171. [Google Scholar] [CrossRef]
- Tsan, M.F.; White, J.E.; Caska, B.; Epstein, C.J.; Lee, C.Y. Susceptibility of heterozygous MnSOD gene-knockout mice to oxygen toxicity. Am. J. Respir. Cell Mol. Biol. 1998, 19, 114–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsan, M.F.; Cao, X.; White, J.E.; Sacco, J.; Lee, C.Y. Pertussis toxin-induced lung edema. Role of manganese superoxide dismutase and protein kinase C. Am. J. Respir. Cell Mol. Biol. 1999, 20, 465–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madamanchi, N.R.; Moon, S.K.; Hakim, Z.S.; Clark, S.; Mehrizi, A.; Patterson, C.; Runge, M.S. Differential activation of mitogenic signaling pathways in aortic smooth muscle cells deficient in superoxide dismutase isoforms. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 950–956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, M.D.; Van Remmen, H.; Conrad, C.C.; Huang, T.T.; Epstein, C.J.; Richardson, A. Increased oxidative damage is correlated to altered mitochondrial function in heterozygous manganese superoxide dismutase knockout mice. J. Biol. Chem. 1998, 273, 28510–28515. [Google Scholar] [CrossRef] [Green Version]
- Asimakis, G.K.; Lick, S.; Patterson, C. Postischemic recovery of contractile function is impaired in SOD2(+/−) but not SOD1(+/−) mouse hearts. Circulation 2002, 105, 981–986. [Google Scholar] [CrossRef] [Green Version]
- Marklund, S.L. Extracellular superoxide dismutase. Methods Enzymol. 2002, 349, 74–80. [Google Scholar] [CrossRef]
- Fukai, T.; Ushio-Fukai, M. Superoxide dismutases: Role in redox signaling, vascular function, and diseases. Antioxid. Redox Signal. 2011, 15, 1583–1606. [Google Scholar] [CrossRef] [Green Version]
- Kwon, M.J.; Jeon, Y.J.; Lee, K.Y.; Kim, T.Y. Superoxide dismutase 3 controls adaptive immune responses and contributes to the inhibition of ovalbumin-induced allergic airway inflammation in mice. Antioxid. Redox Signal. 2012, 17, 1376–1392. [Google Scholar] [CrossRef] [Green Version]
- Jeney, V.; Itoh, S.; Wendt, M.; Gradek, Q.; Ushio-Fukai, M.; Harrison, D.G.; Fukai, T. Role of antioxidant-1 in extracellular superoxide dismutase function and expression. Circ. Res. 2005, 96, 723–729. [Google Scholar] [CrossRef] [Green Version]
- Qin, Z.; Itoh, S.; Jeney, V.; Ushio-Fukai, M.; Fukai, T. Essential role for the Menkes ATPase in activation of extracellular superoxide dismutase: Implication for vascular oxidative stress. FASEB J. 2006, 20, 334–336. [Google Scholar] [CrossRef]
- Itoh, S.; Ozumi, K.; Kim, H.W.; Nakagawa, O.; McKinney, R.D.; Folz, R.J.; Zelko, I.N.; Ushio-Fukai, M.; Fukai, T. Novel mechanism for regulation of extracellular SOD transcription and activity by copper: Role of antioxidant-1. Free Radic. Biol. Med. 2009, 46, 95–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strålin, P.; Karlsson, K.; Johansson, B.O.; Marklund, S.L. The interstitium of the human arterial wall contains very large amounts of extracellular superoxide dismutase. Arterioscler. Thromb. Vasc. Biol. 1995, 15, 2032–2036. [Google Scholar] [CrossRef]
- Konkalmatt, P.R.; Beyers, R.J.; O’Connor, D.M.; Xu, Y.; Seaman, M.E.; French, B.A. Cardiac-selective expression of extracellular superoxide dismutase after systemic injection of adeno-associated virus 9 protects the heart against post-myocardial infarction left ventricular remodeling. Circ. Cardiovasc. Imaging 2013, 6, 478–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foresman, E.L.; Miller, F.J., Jr. Extracellular but not cytosolic superoxide dismutase protects against oxidant-mediated endothelial dysfunction. Redox Biol. 2013, 1, 292–296. [Google Scholar] [CrossRef] [Green Version]
- Schneider, M.P.; Sullivan, J.C.; Wach, P.F.; Boesen, E.I.; Yamamoto, T.; Fukai, T.; Harrison, D.G.; Pollock, D.M.; Pollock, J.S. Protective role of extracellular superoxide dismutase in renal ischemia/reperfusion injury. Kidney Int. 2010, 78, 374–381. [Google Scholar] [CrossRef] [Green Version]
- Jonsson, L.M.; Rees, D.D.; Edlund, T.; Marklund, S.L. Nitric oxide and blood pressure in mice lacking extracellular-superoxide dismutase. Free Radic. Res. 2002, 36, 755–758. [Google Scholar] [CrossRef] [PubMed]
- van Deel, E.D.; Lu, Z.; Xu, X.; Zhu, G.; Hu, X.; Oury, T.D.; Bache, R.J.; Duncker, D.J.; Chen, Y. Extracellular superoxide dismutase protects the heart against oxidative stress and hypertrophy after myocardial infarction. Free Radic. Biol. Med. 2008, 44, 1305–1313. [Google Scholar] [CrossRef] [Green Version]
- Kwon, M.J.; Han, J.; Kim, B.H.; Lee, Y.S.; Kim, T.Y. Superoxide dismutase 3 suppresses hyaluronic acid fragments mediated skin inflammation by inhibition of toll-like receptor 4 signaling pathway: Superoxide dismutase 3 inhibits reactive oxygen species-induced trafficking of toll-like receptor 4 to lipid rafts. Antioxid. Redox Signal. 2012, 16, 297–313. [Google Scholar] [CrossRef]
- Break, T.J.; Jun, S.; Indramohan, M.; Carr, K.D.; Sieve, A.N.; Dory, L.; Berg, R.E. Extracellular superoxide dismutase inhibits innate immune responses and clearance of an intracellular bacterial infection. J. Immunol. 2012, 188, 3342–3350. [Google Scholar] [CrossRef]
- Kliment, C.R.; Oury, T.D. Extracellular superoxide dismutase protects cardiovascular syndecan-1 from oxidative shedding. Free Radic. Biol. Med. 2011, 50, 1075–1080. [Google Scholar] [CrossRef] [Green Version]
- Sentman, M.L.; Granström, M.; Jakobson, H.; Reaume, A.; Basu, S.; Marklund, S.L. Phenotypes of mice lacking extracellular superoxide dismutase and copper- and zinc-containing superoxide dismutase. J. Biol. Chem. 2006, 281, 6904–6909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oberley-Deegan, R.E.; Regan, E.A.; Kinnula, V.L.; Crapo, J.D. Extracellular superoxide dismutase and risk of COPD. COPD J. Chronic Obstr. Pulm. Dis. 2009, 6, 307–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, M.J.; Kim, B.; Lee, Y.S.; Kim, T.Y. Role of superoxide dismutase 3 in skin inflammation. J. Dermatol. Sci. 2012, 67, 81–87. [Google Scholar] [CrossRef]
- Gongora, M.C.; Lob, H.E.; Landmesser, U.; Guzik, T.J.; Martin, W.D.; Ozumi, K.; Wall, S.M.; Wilson, D.S.; Murthy, N.; Gravanis, M.; et al. Loss of extracellular superoxide dismutase leads to acute lung damage in the presence of ambient air: A potential mechanism underlying adult respiratory distress syndrome. Am. J. Pathol. 2008, 173, 915–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castaldi, P.J.; Cho, M.H.; Cohn, M.; Langerman, F.; Moran, S.; Tarragona, N.; Moukhachen, H.; Venugopal, R.; Hasimja, D.; Kao, E.; et al. The COPD genetic association compendium: A comprehensive online database of COPD genetic associations. Hum. Mol. Genet. 2010, 19, 526–534. [Google Scholar] [CrossRef]
- Tang, W.; Bentley, A.R.; Kritchevsky, S.B.; Harris, T.B.; Newman, A.B.; Bauer, D.C.; Meibohm, B.; Cassano, P.A. Health ABC study. Genetic variation in antioxidant enzymes, cigarette smoking, and longitudinal change in lung function. Free Radic. Biol. Med. 2013, 63, 304–312. [Google Scholar] [CrossRef] [Green Version]
- Yao, H.; Arunachalam, G.; Hwang, J.W.; Chung, S.; Sundar, I.K.; Kinnula, V.L.; Crapo, J.D.; Rahman, I. Extracellular superoxide dismutase protects against pulmonary emphysema by attenuating oxidative fragmentation of ECM. Proc. Natl. Acad. Sci. USA 2010, 107, 15571–15576. [Google Scholar] [CrossRef] [Green Version]
- Toyokuni, S.; Ito, F.; Yamashita, K.; Okazaki, Y.; Akatsuka, S. Iron and thiol redox signaling in cancer: An exquisite balance to escape ferroptosis. Free Radic. Biol. Med. 2017, 108, 610–626. [Google Scholar] [CrossRef]
- Góth, L.; Nagy, T. Inherited catalase deficiency: Is it benign or a factor in various age related disorders? Mutat. Res. 2013, 753, 147–154. [Google Scholar] [CrossRef]
- Zou, Y.; Henry, W.S.; Ricq, E.L.; Graham, E.T.; Phadnis, V.V.; Maretich, P.; Paradkar, S.; Boehnke, N.; Deik, A.A.; Reinhardt, F.; et al. Plasticity of ether lipids promotes ferroptosis susceptibility and evasion. Nature 2020, 585, 603–608. [Google Scholar] [CrossRef]
- Muzio, G.; Barrera, G.; Pizzimenti, S. Peroxisome Proliferator-Activated Receptors (PPARs) and Oxidative Stress in Physiological Conditions and in Cancer. Antioxidants 2021, 10, 1734. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.S.; Kim, E.; Lee, H.G.; Lee, W.J.; Won, J.P.; Hur, J.; Fujii, J.; Seo, H.G. Peroxisome proliferator-activated receptor delta rescues xCT-deficient cells from ferroptosis by targeting peroxisomes. Biomed. Pharmacother. 2021, 143, 112223. [Google Scholar] [CrossRef] [PubMed]
- Meister, A.; Anderson, M.E. Glutathione. Annu. Rev. Biochem. 1983, 52, 711–760. [Google Scholar] [CrossRef]
- Fujii, J.; Homma, T.; Kobayashi, S. Ferroptosis caused by cysteine insufficiency and oxidative insult. Free Radic. Res. 2020, 54, 969–980. [Google Scholar] [CrossRef] [PubMed]
- Griffith, O.W. Biologic and pharmacologic regulation of mammalian glutathione synthesis. Free Radic. Biol. Med. 1999, 27, 922–935. [Google Scholar] [CrossRef]
- Homma, T.; Kobayashi, S.; Fujii, J. Cysteine preservation confers resistance to glutathione-depleted cells against ferroptosis via CDGSH iron sulphur domain-containing proteins (CISDs). Free Radic. Res. 2020, 54, 397–407. [Google Scholar] [CrossRef]
- Homma, T.; Takeda, Y.; Nakano, T.; Akatsuka, S.; Kinoshita, D.; Kurahashi, T.; Saitoh, S.; Yamada, K.I.; Miyata, S.; Asao, H.; et al. Defective biosynthesis of ascorbic acid in Sod1-deficient mice results in lethal damage to lung tissue. Free Radic. Biol. Med. 2021, 162, 255–265. [Google Scholar] [CrossRef]
- Kobayashi, S.; Ikeda, Y.; Shigeno, Y.; Konno, H.; Fujii, J. γ-Glutamylcysteine synthetase and γ-glutamyl transferase as differential enzymatic sources of γ-glutamylpeptides in mice. Amino Acids 2020, 52, 555–566. [Google Scholar] [CrossRef]
- Soga, T.; Baran, R.; Suematsu, M.; Ueno, Y.; Ikeda, S.; Sakurakawa, T.; Kakazu, Y.; Ishikawa, T.; Robert, M.; Nishioka, T.; et al. Differential metabolomics reveals ophthalmic acid as an oxidative stress biomarker indicating hepatic glutathione consumption. J. Biol. Chem. 2006, 281, 16768–16776. [Google Scholar] [CrossRef] [Green Version]
- Kang, Y.P.; Mockabee-Macias, A.; Jiang, C.; Falzone, A.; Prieto-Farigua, N.; Stone, E.; Harris, I.S.; DeNicola, G.M. Non-canonical Glutamate-Cysteine Ligase Activity Protects against Ferroptosis. Cell Metab. 2021, 33, 174–189.e7. [Google Scholar] [CrossRef]
- Brigelius-Flohé, R.; Flohé, L. Regulatory Phenomena in the Glutathione Peroxidase Superfamily. Antioxid. Redox Signal. 2020, 33, 498–516. [Google Scholar] [CrossRef] [PubMed]
- Toppo, S.; Flohé, L.; Ursini, F.; Vanin, S.; Maiorino, M. Catalytic mechanisms and specificities of glutathione peroxidases: Variations of a basic scheme. Biochim. Biophys. Acta. 2009, 1790, 1486–1500. [Google Scholar] [CrossRef] [PubMed]
- Dalla Tiezza, M.; Bickelhaupt, F.M.; Flohé, L.; Maiorino, M.; Ursini, F.; Orian, L. A dual attack on the peroxide bond. The common principle of peroxidatic cysteine or selenocysteine residues. Redox Biol. 2020, 34, 101540. [Google Scholar] [CrossRef] [PubMed]
- Cho, C.S.; Lee, S.; Lee, G.T.; Woo, H.A.; Choi, E.J.; Rhee, S.G. Irreversible inactivation of glutathione peroxidase 1 and reversible inactivation of peroxiredoxin II by H2O2 in red blood cells. Antioxid. Redox Signal. 2010, 12, 1235–1246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bulleid, N.J.; Ellgaard, L. Multiple ways to make disulfides. Trends Biochem. Sci. 2011, 36, 485–492. [Google Scholar] [CrossRef] [PubMed]
- Yant, L.J.; Ran, Q.; Rao, L.; Van Remmen, H.; Shibatani, T.; Belter, J.G.; Motta, L.; Richardson, A.; Prolla, T.A. The selenoprotein GPX4 is essential for mouse development and protects from radiation and oxidative damage insults. Free Radic. Biol. Med. 2003, 34, 496–502. [Google Scholar] [CrossRef]
- Imai, H.; Hirao, F.; Sakamoto, T.; Sekine, K.; Mizukura, Y.; Saito, M.; Kitamoto, T.; Hayasaka, M.; Hanaoka, K.; Nakagawa, Y. Early embryonic lethality caused by targeted disruption of the mouse PHGPx gene. Biochem. Biophys. Res. Commun. 2003, 305, 278–286. [Google Scholar] [CrossRef]
- Seibt, T.M.; Proneth, B.; Conrad, M. Role of GPX4 in ferroptosis and its pharmacological implication. Free Radic. Biol. Med. 2019, 133, 144–152. [Google Scholar] [CrossRef]
- Ingold, I.; Berndt, C.; Schmitt, S.; Doll, S.; Poschmann, G.; Buday, K.; Roveri, A.; Peng, X.; Porto Freitas, F.; Seibt, T.; et al. Selenium Utilization by GPX4 Is Required to Prevent Hydroperoxide-Induced Ferroptosis. Cell 2018, 172, 409–422.e21. [Google Scholar] [CrossRef] [Green Version]
- Buday, K.; Conrad, M. Emerging roles for non-selenium containing ER-resident glutathione peroxidases in cell signaling and disease. Biol. Chem. 2020, 402, 271–287. [Google Scholar] [CrossRef]
- Lu, J.; Holmgren, A. The thioredoxin antioxidant system. Free Radic. Biol. Med. 2014, 66, 75–87. [Google Scholar] [CrossRef]
- Rhee, S.G.; Kil, I.S. Multiple Functions and Regulation of Mammalian Peroxiredoxins. Annu. Rev. Biochem. 2017, 86, 749–775. [Google Scholar] [CrossRef] [PubMed]
- Tamura, T.; Stadtman, T.C. Mammalian thioredoxin reductases. Methods. Enzymol. 2002, 347, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Biteau, B.; Labarre, J.; Toledano, M.B. ATP-dependent reduction of cysteine-sulphinic acid by S. cerevisiae sulphiredoxin. Nature 2003, 425, 980–984. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.S.; Jeong, W.; Woo, H.A.; Lee, S.M.; Park, S.; Rhee, S.G. Characterization of mammalian sulfiredoxin and its reactivation of hyperoxidized peroxiredoxin through reduction of cysteine sulfinic acid in the active site to cysteine. J. Biol. Chem. 2004, 279, 50994–51001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.J. Knockout Mouse Models for Peroxiredoxins. Antioxidants 2020, 9, 182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujii, T.; Fujii, J.; Taniguchi, N. Augmented expression of peroxiredoxin VI in rat lung and kidney after birth implies an antioxidative role. Eur. J. Biochem. 2001, 268, 218–225. [Google Scholar] [CrossRef]
- Zhou, S.; Sorokina, E.M.; Harper, S.; Li, H.; Ralat, L.; Dodia, C.; Speicher, D.W.; Feinstein, S.I.; Fisher, A.B. Peroxiredoxin 6 homodimerization and heterodimerization with glutathione S-transferase pi are required for its peroxidase but not phospholipase A2 activity. Free Radic. Biol. Med. 2016, 94, 145–156. [Google Scholar] [CrossRef] [Green Version]
- Fisher, A.B.; Dodia, C.; Manevich, Y.; Chen, J.W.; Feinstein, S.I. Phospholipid hydroperoxides are substrates for non-selenium glutathione peroxidase. J. Biol. Chem. 1999, 274, 21326–21334. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.W.; Dodia, C.; Feinstein, S.I.; Jain, M.K.; Fisher, A.B. 1-Cys peroxiredoxin, a bifunctional enzyme with glutathione peroxidase and phospholipase A2 activities. J. Biol. Chem. 2000, 275, 28421–28427. [Google Scholar] [CrossRef] [Green Version]
- Fisher, A.B. The phospholipase A2 activity of peroxiredoxin 6. J. Lipid Res. 2018, 59, 1132–1147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuchs, Y.; Steller, H. Programmed cell death in animal development and disease. Cell 2011, 147, 742–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Kuang, F.; Kroemer, G.; Klionsky, D.J.; Kang, R.; Tang, D. Autophagy-Dependent Ferroptosis: Machinery and Regulation. Cell Chem. Biol. 2020, 27, 420–435. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Kang, R.; Berghe, T.V.; Vandenabeele, P.; Kroemer, G. The molecular machinery of regulated cell death. Cell Res. 2019, 29, 347–364. [Google Scholar] [CrossRef] [Green Version]
- Nagata, S. Apoptosis and Clearance of Apoptotic Cells. Annu. Rev. Immunol. 2018, 36, 489–517. [Google Scholar] [CrossRef]
- Reubold, T.F.; Eschenburg, S. A molecular view on signal transduction by the apoptosome. Cell Signal. 2012, 24, 1420–1425. [Google Scholar] [CrossRef]
- Suto, D.; Sato, K.; Ohba, Y.; Yoshimura, T.; Fujii, J. Suppression of the pro-apoptotic function of cytochrome c by singlet oxygen via a haem redox state-independent mechanism. Biochem. J. 2005, 392, 399–406. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.C.; Bratton, S.B. Regulation of the intrinsic apoptosis pathway by reactive oxygen species. Antioxid. Redox Signal. 2013, 19, 546–558. [Google Scholar] [CrossRef] [Green Version]
- Kagan, V.E.; Bayir, H.A.; Belikova, N.A.; Kapralov, O.; Tyurina, Y.Y.; Tyurin, V.A.; Jiang, J.; Stoyanovsky, D.A.; Wipf, P.; Kochanek, P.M.; et al. Cytochrome c/cardiolipin relations in mitochondria: A kiss of death. Free Radic. Biol. Med. 2009, 46, 1439–1453. [Google Scholar] [CrossRef] [Green Version]
- Nagaoka, Y.; Otsu, K.; Okada, F.; Sato, K.; Ohba, Y.; Kotani, N.; Fujii, J. Specific inactivation of cysteine protease-type cathepsin by singlet oxygen generated from naphthalene endoperoxides. Biochem. Biophys. Res. Commun. 2005, 331, 215–223. [Google Scholar] [CrossRef]
- Meissner, F.; Molawi, K.; Zychlinsky, A. Superoxide dismutase 1 regulates caspase-1 and endotoxic shock. Nat. Immunol. 2008, 9, 866–872. [Google Scholar] [CrossRef] [PubMed]
- Murao, A.; Aziz, M.; Wang, H.; Brenner, M.; Wang, P. Release mechanisms of major DAMPs. Apoptosis 2021, 26, 152–162. [Google Scholar] [CrossRef] [PubMed]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascón, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin-Sanchez, D.; Ruiz-Andres, O.; Poveda, J.; Carrasco, S.; Cannata-Ortiz, P.; Sanchez-Niño, M.D.; Ruiz Ortega, M.; Egido, J.; Linkerman, N.A.; Ortiz, A.; et al. Ferroptosis, but Not Necroptosis, Is Important in Nephrotoxic Folic Acid-Induced AKI. J. Am. Soc. Nephrol. 2017, 28, 218–229. [Google Scholar] [CrossRef]
- Tang, L.J.; Zhou, Y.J.; Xiong, X.M.; Li, N.S.; Zhang, J.J.; Luo, X.J.; Peng, J. Ubiquitin-specific protease 7 promotes ferroptosis via activation of the p53/TfR1 pathway in the rat hearts after ischemia/reperfusion. Free Radic. Biol. Med. 2021, 162, 339–352. [Google Scholar] [CrossRef]
- Gao, M.; Monian, P.; Quadri, N.; Ramasamy, R.; Jiang, X. Glutaminolysis and Transferrin Regulate Ferroptosis. Mol. Cell 2015, 59, 298–308. [Google Scholar] [CrossRef] [Green Version]
- Torii, S.; Shintoku, R.; Kubota, C.; Yaegashi, M.; Torii, R.; Sasaki, M.; Suzuki, T.; Mori, M.; Yoshimoto, Y.; Takeuchi, T.; et al. An essential role for functional lysosomes in ferroptosis of cancer cells. Biochem. J. 2016, 473, 769–777. [Google Scholar] [CrossRef] [Green Version]
- Latunde-Dada, G.O. Ferroptosis: Role of lipid peroxidation, iron and ferritinophagy. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 1893–1900. [Google Scholar] [CrossRef] [Green Version]
- Hou, W.; Xie, Y.; Song, X.; Sun, X.; Lotze, M.T.; Zeh, H.J., 3rd; Kang, R.; Tang, D. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 2016, 12, 1425–1428. [Google Scholar] [CrossRef]
- Wu, Z.; Geng, Y.; Lu, X.; Shi, Y.; Wu, G.; Zhang, M.; Shan, B.; Pan, H.; Yuan, J. Chaperone-mediated autophagy is involved in the execution of ferroptosis. Proc. Natl. Acad. Sci. USA 2019, 116, 2996–3005. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Quinn, P.J. Vitamin E and its function in membranes. Prog. Lipid Res. 1999, 38, 309–336. [Google Scholar] [CrossRef]
- Chiang, S.K.; Chen, S.E.; Chang, L.C. A Dual Role of Heme Oxygenase-1 in Cancer Cells. Int. J. Mol. Sci. 2018, 20, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adedoyin, O.; Boddu, R.; Traylor, A.; Lever, J.M.; Bolisetty, S.; George, J.F.; Agarwal, A. Heme oxygenase-1 mitigates ferroptosis in renal proximal tubule cells. Am. J. Physiol. Ren. Physiol. 2018, 314, F702–F714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, M.Y.; Park, E.; Lee, S.J.; Chung, S.W. Heme oxygenase-1 accelerates erastin-induced ferroptotic cell death. Oncotarget 2015, 6, 24393–24403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Kang, R.; Kroemer, G.; Tang, D. Organelle-specific regulation of ferroptosis. Cell Death Differ. 2021, 28, 2843–2856. [Google Scholar] [CrossRef]
- Bayır, H.; Anthonymuthu, T.S.; Tyurina, Y.Y.; Patel, S.J.; Amoscato, A.A.; Lamade, A.M.; Yang, Q.; Vladimirov, G.K.; Philpott, C.C.; Kagan, V.E. Achieving Life through Death: Redox Biology of Lipid Peroxidation in Ferroptosis. Cell Chem. Biol. 2020, 27, 387–408. [Google Scholar] [CrossRef]
- Friedmann Angeli, J.P.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef] [Green Version]
- Gao, M.; Yi, J.; Zhu, J.; Minikes, A.M.; Monian, P.; Thompson, C.B.; Jiang, X. Role of Mitochondria in Ferroptosis. Mol. Cell 2019, 73, 354–363.e3. [Google Scholar] [CrossRef] [Green Version]
- Brand, M.D.; Goncalves, R.L.; Orr, A.L.; Vargas, L.; Gerencser, A.A.; Borch Jensen, M.; Wang, Y.T.; Melov, S.; Turk, C.N.; Matzen, J.T.; et al. Suppressors of Superoxide-H2O2 Production at Site IQ of Mitochondrial Complex I Protect against Stem Cell Hyperplasia and Ischemia-Reperfusion Injury. Cell Metab. 2016, 24, 582–592. [Google Scholar] [CrossRef] [Green Version]
- Orr, A.L.; Varga, L.; Turk, C.N.; Baaten, J.E.; Matzen, J.T.; Dardov, V.J.; Attle, S.J.; Li, J.; Quackenbush, D.C.; Goncalves, R.L.; et al. Suppressors of superoxide production from mitochondrial complex III. Nat. Chem. Biol. 2015, 11, 834–836. [Google Scholar] [CrossRef] [Green Version]
- Wong, H.S.; Monternier, P.A.; Brand, M.D. S1QELs suppress mitochondrial superoxide/hydrogen peroxide production from site I(Q) without inhibiting reverse electron flow through Complex, I. Free Radic. Biol. Med. 2019, 143, 545–559. [Google Scholar] [CrossRef] [PubMed]
- Homma, T.; Kobayashi, S.; Sato, H.; Fujii, J. Superoxide produced by mitochondrial complex III plays a pivotal role in the execution of ferroptosis induced by cysteine starvation. Arch. Biochem. Biophys. 2021, 700, 108775. [Google Scholar] [CrossRef] [PubMed]
- Rouault, T.A. The role of iron regulatory proteins in mammalian iron homeostasis and disease. Nat. Chem. Biol. 2006, 2, 406–414. [Google Scholar] [CrossRef] [PubMed]
- Gardner, P.R.; Fridovich, I. Superoxide sensitivity of the Escherichia coli aconitase. J. Biol. Chem. 1991, 266, 19328–19333. [Google Scholar] [CrossRef]
- Gardner, P.R.; Nguyen, D.D.; White, C.W. Aconitase is a sensitive and critical target of oxygen poisoning in cultured mammalian cells and in rat lungs. Proc. Natl. Acad. Sci. USA 1994, 91, 12248–12252. [Google Scholar] [CrossRef] [Green Version]
- Vasquez-Vivar, J.; Kalyanaraman, B.; Kennedy, M.C. Mitochondrial aconitase is a source of hydroxyl radical. An electron spin resonance investigation. J. Biol. Chem. 2000, 275, 14064–14069. [Google Scholar] [CrossRef] [Green Version]
- Castro, L.; Tórtora, V.; Mansilla, S.; Radi, R. Aconitases: Non-redox Iron-Sulfur Proteins Sensitive to Reactive Species. Acc. Chem. Res. 2019, 52, 2609–2619. [Google Scholar] [CrossRef]
- Karmi, O.; Marjault, H.B.; Pesce, L.; Carloni, P.; Onuchic, J.N.; Jennings, P.A.; Mittler, R.; Nechushtai, R. The unique fold and lability of the [2Fe-2S] clusters of NEET proteins mediate their key functions in health and disease. J. Biol. Inorg. Chem. 2018, 23, 599–612. [Google Scholar] [CrossRef] [Green Version]
- Karmi, O.; Holt, S.H.; Song, L.; Tamir, S.; Luo, Y.; Bai, F.; Adenwalla, A.; Darash-Yahana, M.; Sohn, Y.S.; Jennings, P.A.; et al. Interactions between mitoNEET and NAF-1 in cells. PLoS ONE 2017, 12, e0175796. [Google Scholar] [CrossRef] [Green Version]
- Yuan, H.; Li, X.; Zhang, X.; Kang, R.; Tang, D. CISD1 inhibits ferroptosis by protection against mitochondrial lipid peroxidation. Biochem. Biophys. Res. Commun. 2016, 478, 838–844. [Google Scholar] [CrossRef]
- Li, Y.; Wang, X.; Huang, Z.; Zhou, Y.; Xia, J.; Hu, W.; Wang, X.; Du, J.; Tong, X.; Wang, Y. CISD3 inhibition drives cystine-deprivation induced ferroptosis. Cell Death Dis. 2021, 12, 839. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.H.; Shin, D.; Lee, J.; Jung, A.R.; Roh, J.L. CISD2 inhibition overcomes resistance to sulfasalazine-induced ferroptotic cell death in head and neck cancer. Cancer Lett. 2018, 432, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Wang, T.; Li, Y.; Zhou, Y.; Wang, X.; Yu, X.; Ren, X.; An, Y.; Wu, Y.; Sun, W.; et al. DHA inhibits proliferation and induces ferroptosis of leukemia cells through autophagy dependent degradation of ferritin. Free Radic. Biol. Med. 2019, 131, 356–369. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, S.W.; Sviderskiy, V.O.; Terzi, E.M.; Papagiannakopoulos, T.; Moreira, A.L.; Adams, S.; Sabatini, D.M.; Birsoy, K.; Possemato, R. NFS1 undergoes positive selection in lung tumours and protects cells from ferroptosis. Nature 2017, 551, 639–643. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Zhou, Y.; Li, Y.; Xia, J.; Chen, Y.; Chen, S.; Wang, X.; Sun, W.; Wang, T.; Ren, X.; et al. Identification of Frataxin as a regulator of ferroptosis. Redox Biol. 2020, 32, 101483. [Google Scholar] [CrossRef]
- Skouta, R.; Dixon, S.J.; Wang, J.; Dunn, D.E.; Orman, M.; Shimada, K.; Rosenberg, P.A.; Lo, D.C.; Weinberg, J.M.; Linkermann, A.; et al. Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models. J. Am. Chem. Soc. 2014, 136, 4551–4556. [Google Scholar] [CrossRef]
- Yang, W.S.; Stockwell, B.R. Ferroptosis: Death by Lipid Peroxidation. Trends Cell Biol. 2016, 26, 165–176. [Google Scholar] [CrossRef] [Green Version]
- Stoyanovsky, D.A.; Tyurina, Y.Y.; Shrivastava, I.; Bahar, I.; Tyurin, V.A.; Protchenko, O.; Jadhav, S.; Bolevich, S.B.; Kozlov, A.V.; Vladimirov, Y.A.; et al. Iron catalysis of lipid peroxidation in ferroptosis: Regulated enzymatic or random free radical reaction? Free Radic. Biol. Med. 2019, 133, 153–161. [Google Scholar] [CrossRef]
- Homma, T.; Kobayashi, S.; Sato, H.; Fujii, J. Edaravone, a free radical scavenger, protects against ferroptotic cell death in vitro. Exp. Cell Res. 2019, 384, 111592. [Google Scholar] [CrossRef]
- Bersuker, K.; Hendricks, J.M.; Li, Z.; Magtanong, L.; Ford, B.; Tang, P.H.; Roberts, M.A.; Tong, B.; Maimone, T.J.; Zoncu, R.; et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 2019, 575, 688–692. [Google Scholar] [CrossRef]
- Doll, S.; Freitas, F.P.; Shah, R.; Aldrovandi, M.; da Silva, M.C.; Ingold, I.; Grocin, A.G.; Xavier da Silva, T.N.; Panzilius, E.; Scheel, C.H.; et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 2019, 575, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Niki, E. Lipid peroxidation: Physiological levels and dual biological effects. Free Radic. Biol. Med. 2009, 47, 469–484. [Google Scholar] [CrossRef] [PubMed]
- Doll, S.; Conrad, M. Iron and ferroptosis: A still ill-defined liaison. IUBMB Life 2017, 69, 423–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doll, S.; Proneth, B.; Tyurina, Y.Y.; Panzilius, E.; Kobayashi, S.; Ingold, I.; Irmler, M.; Beckers, J.; Aichler, M.; Walch, A.; et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 2017, 13, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Kagan, V.E.; Mao, G.; Qu, F.; Angeli, J.P.; Doll, S.; Croix, C.S.; Dar, H.H.; Liu, B.; Tyurin, V.A.; Ritov, V.B.; et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol. 2017, 13, 81–90. [Google Scholar] [CrossRef]
- Zhang, H.L.; Hu, B.X.; Li, Z.L.; Du, T.; Shan, J.L.; Ye, Z.P.; Peng, X.D.; Li, X.; Huang, Y.; Zhu, X.Y.; et al. PKCβII phosphorylates ACSL4 to amplify lipid peroxidation to induce ferroptosis. Nat. Cell Biol. 2022, 24, 88–98. [Google Scholar] [CrossRef]
- Feng, H.; Stockwell, B.R. Unsolved mysteries: How does lipid peroxidation cause ferroptosis? PLoS Biol. 2018, 16, e2006203. [Google Scholar] [CrossRef]
- Chu, B.; Kon, N.; Chen, D.; Li, T.; Liu, T.; Jiang, L.; Song, S.; Tavana, O.; Gu, W. ALOX12 is required for p53-mediated tumour suppression through a distinct ferroptosis pathway. Nat. Cell Biol. 2019, 21, 579–591. [Google Scholar] [CrossRef]
- Chen, D.; Chu, B.; Yang, X.; Liu, Z.; Jin, Y.; Kon, N.; Rabadan, R.; Jiang, X.; Stockwell, B.R.; Gu, W. iPLA2beta-mediated lipid detoxification controls p53-driven ferroptosis independent of GPX4. Nat. Commun. 2021, 12, 3644. [Google Scholar] [CrossRef]
- Sun, W.Y.; Tyurin, V.A.; Mikulska-Ruminska, K.; Shrivastava, I.H.; Anthonymuthu, T.S.; Zhai, Y.J.; Pan, M.H.; Gong, H.B.; Lu, D.H.; Sun, J.; et al. Phospholipase iPLA(2)beta averts ferroptosis by eliminating a redox lipid death signal. Nat. Chem. Biol. 2021, 17, 465–476. [Google Scholar] [CrossRef]
- Lu, B.; Chen, X.B.; Hong, Y.C.; Zhu, H.; He, Q.J.; Yang, B.; Ying, M.D.; Cao, J. Identification of PRDX6 as a regulator of ferroptosis. Acta Pharmacol. Sin. 2019, 40, 1334–1342. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, A.; Schmitz, W.; Hufnagel, A.; Schartl, M.; Meierjohann, S. Peroxiredoxin 6 triggers melanoma cell growth by increasing arachidonic acid-dependent lipid signalling. Biochem. J. 2015, 471, 267–279. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Yu, C.; Kang, R.; Tang, D. Iron Metabolism in Ferroptosis. Front. Cell Dev. Biol. 2020, 8, 590226. [Google Scholar] [CrossRef] [PubMed]
- Kang, R.; Kroemer, G.; Tang, D. The tumor suppressor protein p53 and the ferroptosis network. Free Radic. Biol. Med. 2019, 133, 162–168. [Google Scholar] [CrossRef]
- Jiang, L.; Kon, N.; Li, T.; Wang, S.J.; Su, T.; Hibshoosh, H.; Baer, R.; Gu, W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 2015, 520, 57–62. [Google Scholar] [CrossRef] [Green Version]
- Kang, R.; Tang, D. Autophagy and Ferroptosis—What’s the Connection? Curr. Pathobiol. Rep. 2017, 5, 153–159. [Google Scholar] [CrossRef]
- Xie, Y.; Zhu, S.; Song, X.; Sun, X.; Fan, Y.; Liu, J.; Zhong, M.; Yuan, H.; Zhang, L.; Billiar, T.R.; et al. The Tumor Suppressor p53 Limits Ferroptosis by Blocking DPP4 Activity. Cell Rep. 2017, 20, 1692–1704. [Google Scholar] [CrossRef] [Green Version]
- Shin, D.; Lee, J.; You, J.H.; Kim, D.; Roh, J.L. Dihydrolipoamide dehydrogenase regulates cystine deprivation-induced ferroptosis in head and neck cancer. Redox Biol. 2020, 30, 101418. [Google Scholar] [CrossRef]
- Vučković, A.M.; Venerando, R.; Tibaldi, E.; Bosello Travain, V.; Roveri, A.; Bordin, L.; Miotto, G.; Cozza, G.; Toppo, S.; Maiorino, M.; et al. Aerobic pyruvate metabolism sensitizes cells to ferroptosis primed by GSH depletion. Free Radic. Biol. Med. 2021, 167, 45–53. [Google Scholar] [CrossRef]
- Zou, Y.; Li, H.; Graham, E.T.; Deik, A.A.; Eaton, J.K.; Wang, W.; Sandoval-Gomez, G.; Clish, C.B.; Doench, J.G.; Schreiber, S.L. Cytochrome P450 oxidoreductase contributes to phospholipid peroxidation in ferroptosis. Nat. Chem. Biol. 2020, 16, 302–309. [Google Scholar] [CrossRef]
- Yan, B.; Ai, Y.; Sun, Q.; Ma, Y.; Cao, Y.; Wang, J.; Zhang, Z.; Wang, X. Membrane Damage during Ferroptosis Is Caused by Oxidation of Phospholipids Catalyzed by the Oxidoreductases POR and CYB5R1. Mol. Cell 2021, 81, 355–369.e10. [Google Scholar] [CrossRef] [PubMed]
- Fujii, J. Ascorbate is a multifunctional micronutrient whose synthesis is lacking in primates. J. Clin. Biochem. Nutr. 2021, 69, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Buettner, G.R. The pecking order of free radicals and antioxidants: Lipid peroxidation, α-tocopherol, and ascorbate. Arch. Biochem. Biophys. 1993, 300, 535–543. [Google Scholar] [CrossRef] [PubMed]
- Njus, D.; Kelley, P.M.; Tu, Y.J.; Schlegel, H.B. Ascorbic acid: The chemistry underlying its antioxidant properties. Free Radic. Biol. Med. 2020, 159, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Badu-Boateng, C.; Naftalin, R.J. Ascorbate and ferritin interactions: Consequences for iron release in vitro and in vivo and implications for inflammation. Free Radic. Biol. Med. 2019, 133, 75–87. [Google Scholar] [CrossRef]
- Smirnoff, N. Ascorbic acid metabolism and functions: A comparison of plants and mammals. Free Radic. Biol. Med. 2018, 122, 116–129. [Google Scholar] [CrossRef]
- Takahashi, M.; Miyata, S.; Fujii, J.; Inai, Y.; Ueyama, S.; Araki, M.; Soga, T.; Fujinawa, R.; Nishitani, C.; Ariki, S.; et al. In vivo role of aldehyde reductase. Biochim. Biophys. Acta. 2012, 1820, 1787–1796. [Google Scholar] [CrossRef]
- Kondo, Y.; Masutomi, H.; Noda, Y.; Ozawa, Y.; Takahashi, K.; Handa, S.; Maruyama, N.; Shimizu, T.; Ishigami, A. Senescence marker protein-30/superoxide dismutase 1 double knockout mice exhibit increased oxidative stress and hepatic steatosis. FEBS Open Bio 2014, 4, 522–532. [Google Scholar] [CrossRef] [Green Version]
- Furchgott, R.F.; Zawadzki, J.V. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature 1980, 288, 373–376. [Google Scholar] [CrossRef]
- Bruckdorfer, R. The basics about nitric oxide. Mol. Asp. Med. 2005, 26, 3–31. [Google Scholar] [CrossRef]
- Benjamin, N.; O’Driscoll, F.; Dougall, H.; Duncan, C.; Smith, L.; Golden, M.; McKenzie, H. Stomach NO synthesis. Nature 1994, 368, 502. [Google Scholar] [CrossRef] [PubMed]
- Kim-Shapiro, D.B.; Schechter, A.N.; Gladwin, M.T. Unraveling the reactions of nitric oxide, nitrite, and hemoglobin in physiology and therapeutics. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 697–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, K.; Piknova, B.; Pittman, R.N.; Schechter, A.N.; Popel, A.S. Nitric oxide from nitrite reduction by hemoglobin in the plasma and erythrocytes. Nitric Oxide 2008, 18, 47–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Umbreit, J. Methemoglobin-it’s not just blue: A concise review. Am. J. Hematol. 2007, 82, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Millar, T.M.; Stevens, C.R.; Benjamin, N.; Eisenthal, R.; Harrison, R.; Blake, D.R. Xanthine oxidoreductase catalyses the reduction of nitrates and nitrite to nitric oxide under hypoxic conditions. FEBS Lett. 1998, 427, 225–228. [Google Scholar] [CrossRef] [Green Version]
- Godber, B.L.; Doel, J.J.; Sapkota, G.P.; Blake, D.R.; Stevens, C.R.; Eisenthal, R.; Harrison, R. Reduction of nitrite to nitric oxide catalyzed by xanthine oxidoreductase. J. Biol. Chem. 2000, 275, 7757–7763. [Google Scholar] [CrossRef] [Green Version]
- Kina-Tanada, M.; Sakanashi, M.; Tanimoto, A.; Kaname, T.; Matsuzaki, T.; Noguchi, K.; Uchida, T.; Nakasone, J.; Kozuka, C.; Ishida, M.; et al. Long-term dietary nitrite and nitrate deficiency causes the metabolic syndrome, endothelial dysfunction and cardiovascular death in mice. Diabetologia 2017, 60, 1138–1151. [Google Scholar] [CrossRef] [Green Version]
- Huie, R.E.; Padmaja, S. The reaction of no with superoxide. Free Radic. Res. Commun. 1993, 18, 195–199. [Google Scholar] [CrossRef]
- Schildknecht, S.; Ullrich, V. Peroxynitrite as regulator of vascular prostanoid synthesis. Arch. Biochem. Biophys. 2009, 484, 183–189. [Google Scholar] [CrossRef]
- Zeidler, P.C.; Roberts, J.R.; Castranova, V.; Chen, F.; Butterworth, L.; Andrew, M.E.; Robinson, V.A.; Porter, D.W. Response of alveolar macrophages from inducible nitric oxide synthase knockout or wild-type mice to an in vitro lipopolysaccharide or silica exposure. J. Toxicol. Environ. Health A 2003, 66, 995–1013. [Google Scholar] [CrossRef]
- Niu, X.L.; Xia, Y.; Hoshiai, K.; Tanaka, K.; Sawamura, S.; Nakazawa, H. Inducible nitric oxide synthase knockout mouse macrophages disclose prooxidant effect of interferon-gamma on low-density lipoprotein oxidation. Nitric Oxide 2000, 4, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Laubach, V.E.; Foley, P.L.; Shockey, K.S.; Tribble, C.G.; Kron, I.L. Protective roles of nitric oxide and testosterone in endotoxemia: Evidence from NOS-2-deficient mice. Am. J. Physiol. 1998, 275, H2211–H2218. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.; Homma, T.; Fujii, J. Nitric oxide produced by NOS2 copes with the cytotoxic effects of superoxide in macrophages. Biochem. Biophys. Rep. 2021, 26, 100942. [Google Scholar] [CrossRef]
- Sies, H.; Sharov, V.S.; Klotz, L.O.; Briviba, K. Glutathione peroxidase protects against peroxynitrite-mediated oxidations. A new function for selenoproteins as peroxynitrite reductase. J. Biol. Chem. 1997, 272, 27812–27817. [Google Scholar] [CrossRef] [Green Version]
- Bryk, R.; Griffin, P.; Nathan, C. Peroxynitrite reductase activity of bacterial peroxiredoxins. Nature 2000, 407, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Manta, B.; Hugo, M.; Ortiz, C.; Ferrer-Sueta, G.; Trujillo, M.; Denicola, A. The peroxidase and peroxynitrite reductase activity of human erythrocyte peroxiredoxin 2. Arch. Biochem. Biophys. 2009, 484, 146–154. [Google Scholar] [CrossRef]
- Ursini, F.; Maiorino, M. Lipid peroxidation and ferroptosis: The role of GSH and GPx4. Free Radic. Biol. Med. 2020, 152, 175–185. [Google Scholar] [CrossRef]
- O’Donnell, V.B.; Chumley, P.H.; Hogg, N.; Bloodsworth, A.; Darley-Usmar, V.M.; Freeman, B.A. Nitric oxide inhibition of lipid peroxidation: Kinetics of reaction with lipid peroxyl radicals and comparison with alpha-tocopherol. Biochemistry 1997, 36, 15216–15223. [Google Scholar] [CrossRef]
- Padmaja, S.; Huie, R.E. The reaction of nitric oxide with organic peroxyl radicals. Biochem. Biophys. Res. Commun. 1993, 195, 539–544. [Google Scholar] [CrossRef]
- Rubbo, H.; Radi, R.; Trujillo, M.; Telleri, R.; Kalyanaraman, B.; Barnes, S.; Kirk, M.; Freeman, B.A. Nitric oxide regulation of superoxide and peroxynitrite-dependent lipid peroxidation. Formation of novel nitrogen-containing oxidized lipid derivatives. J. Biol. Chem. 1994, 269, 26066–26075. [Google Scholar] [CrossRef]
- Kotamraju, S.; Hogg, N.; Joseph, J.; Keefer, L.K.; Kalyanaraman, B. Inhibition of oxidized low-density lipoprotein-induced apoptosis in endothelial cells by nitric oxide. Peroxyl radical scavenging as an antiapoptotic mechanism. J. Biol. Chem. 2001, 76, 17316–17323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wink, D.A.; Miranda, K.M.; Espey, M.G.; Pluta, R.M.; Hewett, S.J.; Colton, C.; Vitek, M.; Feelisch, M.; Grisham, M.B. Mechanisms of the antioxidant effects of nitric oxide. Antioxid. Redox Signal. 2001, 3, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Kapralov, A.A.; Yang, Q.; Dar, H.H.; Tyurina, Y.Y.; Anthonymuthu, T.S.; Kim, R.; St Croix, C.M.; Mikulska-Ruminska, K.; Liu, B.; Shrivastava, I.H.; et al. Redox lipid reprogramming commands susceptibility of macrophages and microglia to ferroptotic death. Nat. Chem. Biol. 2020, 16, 278–290. [Google Scholar] [CrossRef] [PubMed]
- Homma, T.; Kobayashi, S.; Conrad, M.; Konno, H.; Yokoyama, C.; Fujii, J. Nitric oxide protects against ferroptosis by aborting the lipid peroxidation chain reaction. Nitric Oxide 2021, 115, 34–43. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fujii, J.; Homma, T.; Osaki, T. Superoxide Radicals in the Execution of Cell Death. Antioxidants 2022, 11, 501. https://doi.org/10.3390/antiox11030501
Fujii J, Homma T, Osaki T. Superoxide Radicals in the Execution of Cell Death. Antioxidants. 2022; 11(3):501. https://doi.org/10.3390/antiox11030501
Chicago/Turabian StyleFujii, Junichi, Takujiro Homma, and Tsukasa Osaki. 2022. "Superoxide Radicals in the Execution of Cell Death" Antioxidants 11, no. 3: 501. https://doi.org/10.3390/antiox11030501
APA StyleFujii, J., Homma, T., & Osaki, T. (2022). Superoxide Radicals in the Execution of Cell Death. Antioxidants, 11(3), 501. https://doi.org/10.3390/antiox11030501