
Oxidative Stress-Induced Hypertension of Developmental Origins: Preventive Aspects of Antioxidant Therapy



Abstract
:1. Introduction
2. Oxidative Stress and Hypertension
2.1. ROS/NO Disequilibrium
2.2. Oxidative Stress and Hypertension
2.2.1. Cardiovascular System
2.2.2. Renal System
2.2.3. Central Nervous System
2.2.4. The Regulatory Hormones
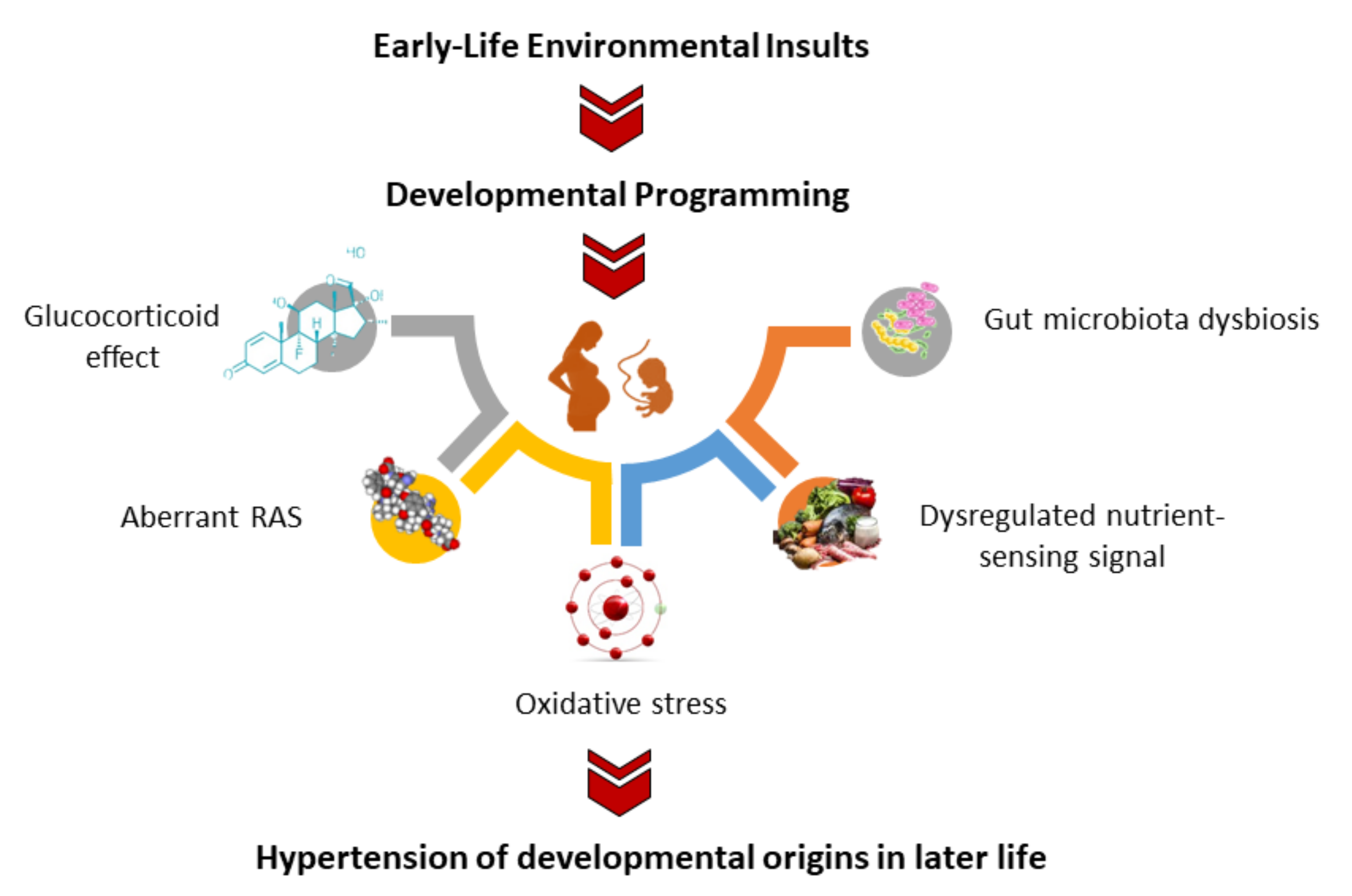
3. Oxidative-Stress-Related Hypertension of Developmental Origins
3.1. Oxidative Stress during Pregnancy
3.2. Evidence from Human Studies
3.3. Evidence from Animal Studies
3.4. Mechanisms Underpinning Oxidative Stress in Hypertension of Developmental Origins
3.5. Oxidative-Stress-Induced Renal Programming
3.6. Oxidative-Stress-Induced Cardiovascular Programming
3.7. Other Mechanisms Related to Oxidative Stress Programming
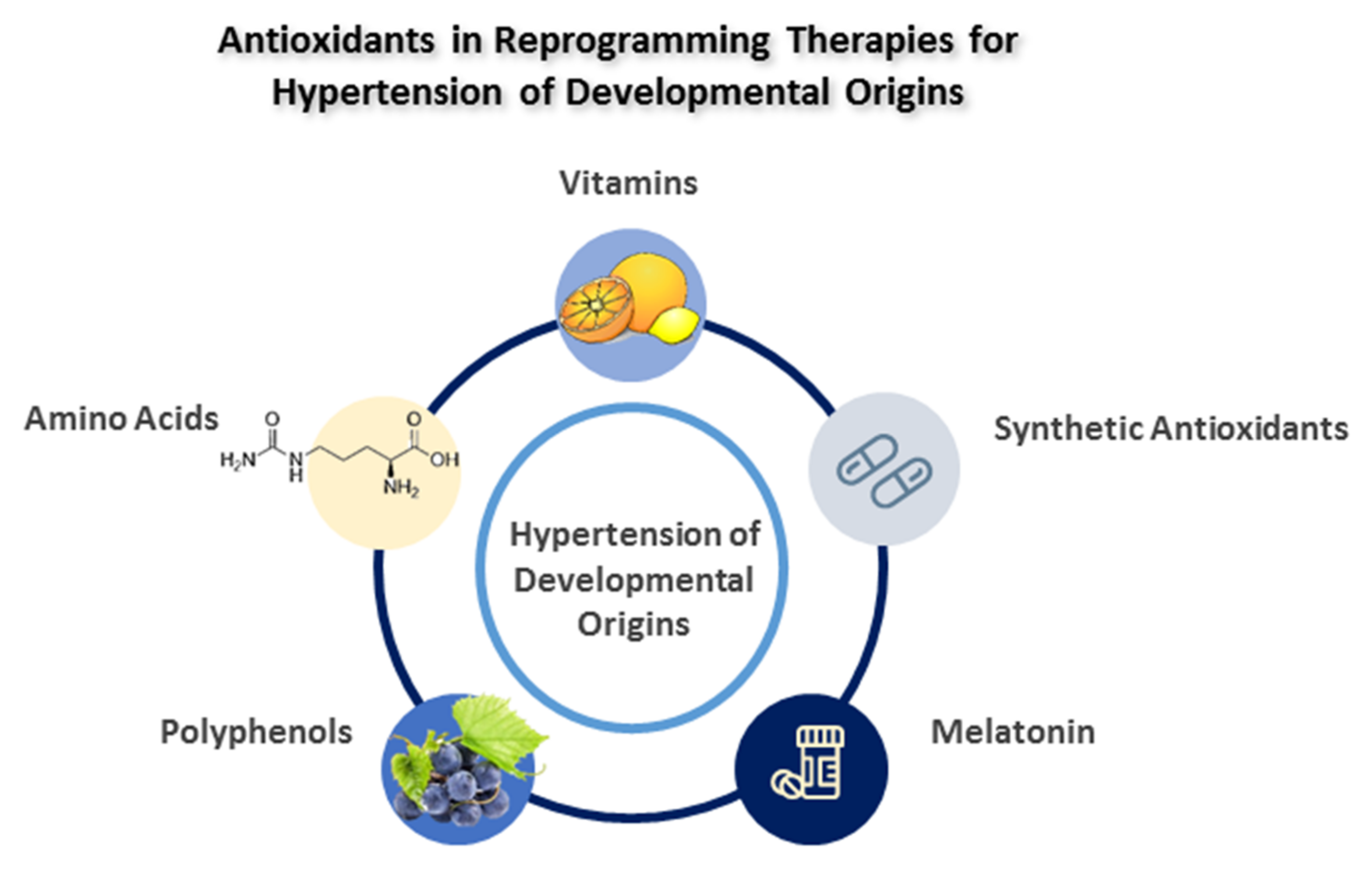
4. Antioxidants as Reprogramming Strategies
4.1. Vitamins
4.2. Amino Acids
4.3. Polyphenols
4.4. Melatonin
4.5. Synthetic Antioxidants
5. Concluding Remarks and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- World Health Organization. Hypertension. 2019. Available online: https://www.who.int/news-room/fact-sheets/detail/hypertension (accessed on 10 January 2022).
- Bromfield, S.; Muntner, P. High blood pressure: The leading global burden of disease risk factor and the need for worldwide prevention programs. Curr. Hypertens. Rep. 2013, 15, 134–136. [Google Scholar] [CrossRef] [PubMed]
- Ojeda, N.B.; Grigore, D.; Alexander, B.T. Developmental programming of hypertension: Insight from animal models of nutritional manipulation. Hypertension 2008, 52, 44–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chong, E.; Yosypiv, I.V. Developmental programming of hypertension and kidney disease. Int. J. Nephrol. 2012, 2012, 760580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paixão, A.D.; Alexander, B.T. How the kidney is impacted by the perinatal maternal environment to develop hypertension. Biol. Reprod. 2013, 89, 144. [Google Scholar] [CrossRef] [PubMed]
- Hanson, M. The birth and future health of DOHaD. J. Dev. Orig. Health Dis. 2015, 6, 434–437. [Google Scholar] [CrossRef]
- Hsu, C.N.; Tain, Y.L. The double-edged sword effects of maternal nutrition in the developmental programming of hypertension. Nutrients 2018, 10, 1917. [Google Scholar] [CrossRef] [Green Version]
- Guarner-Lans, V.; Ramírez-Higuera, A.; Rubio-Ruiz, M.E.; Castrejón-Téllez, V.; Soto, M.E.; Pérez-Torres, I. Early Programming of Adult Systemic Essential Hypertension. Int. J. Mol. Sci. 2020, 21, 1203. [Google Scholar] [CrossRef] [Green Version]
- Hsu, C.N.; Tain, Y.L. Amino Acids and Developmental Origins of Hypertension. Nutrients 2020, 12, 1763. [Google Scholar] [CrossRef]
- Dasinger, J.H.; Davis, G.K.; Newsome, A.D.; Alexander, B.T. Developmental Programming of Hypertension: Physiological Mechanisms. Hypertension 2016, 68, 826–831. [Google Scholar] [CrossRef]
- Tain, Y.L.; Joles, J.A. Reprogramming: A Preventive Strategy in Hypertension Focusing on the Kidney. Int. J. Mol. Sci. 2015, 17, 23. [Google Scholar] [CrossRef] [Green Version]
- Sinha, N.; Dabla, P.K. Oxidative stress and antioxidants in hypertension-a current review. Curr. Hypertens. Rev. 2015, 11, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Palm, F.; Nordquist, L. Renal oxidative stress, oxygenation, and hypertension. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 301, R1229–R1241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Touyz, R.M. Reactive oxygen species, vascular oxidative stress, and redox signaling in hypertension: What is the clinical significance? Hypertension 2004, 44, 248–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurr, C.; Young, C.N. Neural Control of Non-vasomotor Organs in Hypertension. Curr. Hypertens. Rep. 2016, 18, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sies, H. Oxidative stress: A concept in redox biology and medicine. Redox Biol. 2015, 4, 180–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, K.A.; Yuan Yuan, D.; Nawaz, W.; Ze, H.; Zhuo, C.X.; Talal, B.; Taleb, A.; Mais, E.; Qilong, D. Antioxidant therapy for management of oxidative stress induced hypertension. Free Radic. Res. 2017, 51, 428–438. [Google Scholar] [CrossRef] [PubMed]
- Kizhakekuttu, T.J.; Widlansky, M.E. Natural antioxidants and hypertension: Promise and challenges. Cardiovasc. Ther. 2010, 28, e20–e32. [Google Scholar] [CrossRef]
- Wilcox, C.S. Reactive oxygen species: Roles in blood pressure and kidney function. Curr. Hypertens. Rep. 2002, 4, 160–166. [Google Scholar] [CrossRef]
- Radi, R. Peroxynitrite, a stealthy biological oxidant. J. Biol. Chem. 2013, 288, 26464–26472. [Google Scholar] [CrossRef] [Green Version]
- Vertuani, S.; Angusti, A.; Manfredini, S. The antioxidants and pro-antioxidants network: An overview. Curr. Pharm. Des. 2004, 10, 1677–1694. [Google Scholar] [CrossRef] [Green Version]
- Tain, Y.L.; Hsu, C.N. Toxic Dimethylarginines: Asymmetric Dimethylarginine (ADMA) and Symmetric Dimethylarginine (SDMA). Toxins 2017, 9, 92. [Google Scholar] [CrossRef] [Green Version]
- Tain, Y.L.; Hsu, C.N. Targeting on Asymmetric Dimethylarginine-Related Nitric Oxide-Reactive Oxygen Species Imbalance to Reprogram the Development of Hypertension. Int. J. Mol. Sci. 2016, 17, 2020. [Google Scholar] [CrossRef] [Green Version]
- Oparil, S.; Acelajado, M.C.; Bakris, G.L.; Berlowitz, D.R.; Cífková, R.; Dominiczak, A.F.; Grassi, G.; Jordan, J.; Poulter, N.R.; Rodgers, A.; et al. Hypertension. Nat. Rev. Dis. Primers 2018, 4, 18014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konukoglu, D.; Uzun, H. Endothelial Dysfunction and Hypertension. Adv. Exp. Med. Biol. 2017, 956, 511–540. [Google Scholar] [PubMed]
- Bengtsson, S.H.; Gulluyan, L.M.; Dusting, G.J.; Drummond, G.R. Novel isoforms of NADPH oxidase in vascular physiology and pathophysiology. Clin. Exp. Pharmacol. Physiol. 2003, 30, 849–854. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Youn, J.Y.; Cai, H. Mechanisms and consequences of endothelial nitric oxide synthase dysfunction in hypertension. J. Hypertens. 2015, 33, 1128–1136. [Google Scholar] [CrossRef] [Green Version]
- Cardounel, A.J.; Cui, H.; Samouilov, A.; Johnson, W.; Kearns, P.; Tsai, A.L.; Berka, V.; Zweier, J.L. Evidence for the pathophysiological role of endogenous methylarginines in regulation of endothelial NO production and vascular function. J. Biol. Chem. 2007, 282, 879–887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sena, C.M.; Leandro, A.; Azul, L.; Seiça, R.; Perry, G. Vascular Oxidative Stress: Impact and Therapeutic Approaches. Front. Physiol. 2018, 9, 1668. [Google Scholar] [CrossRef] [Green Version]
- Wilcox, C.S. Oxidative stress and nitric oxide deficiency in the kidney: A critical link to hypertension? Am. J.Physiol. Regul. Integr. Comp. Physiol. 2005, 289, R913–R935. [Google Scholar] [CrossRef]
- Tain, Y.L.; Hsu, C.N.; Huang, L.T.; Lau, Y.T. Apocynin attenuates oxidative stress and hypertension in young spontaneously hypertensive rats independent of ADMA/NO pathway. Free Radic. Res. 2012, 46, 68–76. [Google Scholar] [CrossRef]
- Chen, X.; Touyz, R.M.; Park, J.B.; Schiffrin, E.L. Antioxidant effects of vitamins C and E are associated with altered activation of vascular NADPH oxidase and superoxide dismutase in stroke-prone SHR. Hypertension 2001, 38, 606–611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erdely, A.; Freshour, G.; Tain, Y.L.; Engels, K.; Baylis, C. DOCA/NaCl-induced chronic kidney disease: A comparison of renal nitric oxide production in resistant and susceptible rat strains. Am. J. Physiol. Renal Physiol. 2007, 292, F192–F196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, S.; Roberts, L.J.; Cason, G.W.; Curry, T.S.; Manning, R.D., Jr. Superoxide dismutase and oxidative stress in Dahl salt-sensitive and -resistant rats. Am. J. Physiol. 2002, 283, R732–R738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tain, Y.L.; Freshour, G.; Dikalova, A.; Griendling, K.; Baylis, C. Vitamin E reduces glomerulosclerosis, restores renal neuronal NOS, and suppresses oxidative stress in the 5/6 nephrectomized rat. Am. J. Physiol. Renal Physiol. 2007, 292, F1404–F1410. [Google Scholar] [CrossRef] [PubMed]
- Welch, W.J.; Chabrashvili, T.; Solis, G.; Chen, Y.; Gill, P.S.; Aslam, S.; Wang, X.; Ji, H.; Sandberg, K.; Jose, P.; et al. Role of extracellular superoxide dismutase in the mouse angiotensin slow pressor response. Hypertension 2006, 48, 934–941. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Wang, X.; Cui, Y.; Jiang, S.; Wei, J.; Chan, J.; Thalakola, A.; Le, T.; Xu, L.; Zhao, L.; et al. Knockout of Macula Densa Neuronal Nitric Oxide Synthase Increases Blood Pressure in db/db Mice. Hypertension 2021, 78, 1760–1770. [Google Scholar] [CrossRef]
- Tain, Y.L.; Huang, L.T.; Lin, I.C.; Lau, Y.T.; Lin, C.Y. Melatonin prevents hypertension and increased asymmetric dimethylarginine in young spontaneous hypertensive rats. J Pineal Res. 2010, 49, 390–398. [Google Scholar] [CrossRef]
- Hisaki, R.; Fujita, H.; Saito, F.; Kushiro, T. Tempol attenuates the development of hypertensive renal injury in Dahl salt-sensitive rats. Am. J. Hypertens. 2005, 18, 707–713. [Google Scholar] [CrossRef] [Green Version]
- Tain, Y.L.; Lin, Y.J.; Sheen, J.M.; Yu, H.R.; Tiao, M.M.; Chen, C.C.; Tsai, C.C.; Huang, L.T.; Hsu, C.N. High fat diets sex-specifically affect the renal transcriptome and program obesity, kidney injury, and hypertension in the offspring. Nutrients 2017, 9, 357. [Google Scholar] [CrossRef] [Green Version]
- Tain, Y.L.; Wu, K.L.H.; Lee, W.C.; Leu, S.; Chan, J.Y.H. Prenatal Metformin Therapy Attenuates Hypertension of Developmental Origin in Male Adult Offspring Exposed to Maternal High-Fructose and Post-Weaning High-Fat Diets. Int. J. Mol. Sci. 2018, 19, 1066. [Google Scholar] [CrossRef] [Green Version]
- Siti, H.N.; Kamisah, Y.; Kamsiah, J. The role of oxidative stress, antioxidants and vascular inflammation in cardiovascular disease (a review). Vascul. Pharmacol. 2015, 71, 40–56. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.N.; Tain, Y.L. Early Origins of Hypertension: Should Prevention Start Before Birth Using Natural Antioxidants? Antioxidants 2020, 9, 1034. [Google Scholar] [CrossRef] [PubMed]
- Manning, R.D., Jr.; Tian, N.; Meng, S. Oxidative stress and antioxidant treatment invhypertension and the associated renal damage. Am. J. Nephrol. 2005, 25, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Lin, Y.J.; Chan, J.Y.H.; Lee, C.T.; Hsu, C.N. Maternal melatonin or agomelatine therapy prevents programmed hypertension in male offspring of mother exposed to continuous light. Biol. Reprod. 2017, 97, 636–643. [Google Scholar] [CrossRef]
- Tain, Y.L.; Lee, C.T.; Chan, J.Y.; Hsu, C.N. Maternal melatonin or N-acetylcysteine therapy regulates hydrogen sulfide-generating pathway and renal transcriptome to prevent prenatal N(G)-Nitro-l-arginine-methyl ester (L-NAME)-induced fetal programming of hypertension in adult male offspring. Am. J. Obstet. Gynecol. 2016, 215, 636. [Google Scholar] [CrossRef]
- Aicher, S.A.; Milner, T.A.; Pickel, V.M.; Reis, D.J. Anatomical substrates for baroreflex sympathoinhibition in the rat. Brain Res. Bull. 2000, 51, 107–110. [Google Scholar] [CrossRef]
- Chan, S.H.; Chan, J.Y. Brain stem NOS and ROS in neural mechanisms of hypertension. Antioxid. Redox Signal. 2014, 20, 146–163. [Google Scholar] [CrossRef]
- Chan, J.Y.H.; Chan, S.H.H. Differential impacts of brain stem oxidative stress and nitrosative stress on sympathetic vasomotor tone. Pharmacol. Ther. 2019, 201, 120–136. [Google Scholar] [CrossRef]
- Kishi, T.; Hirooka, Y.; Konno, S.; Sunagawa, K. Sympathoinhibition induced by centrally administered atorvastatin is associated with alteration of NAD(P)H and Mn superoxide dismutase activity in rostral ventrolateral medulla of stroke-prone spontaneously hypertensive rats. J. Cardiovasc. Pharmacol. 2010, 55, 184–190. [Google Scholar] [CrossRef]
- Chan, S.H.H.; Wu, C.A.; Wu, K.L.H.; Ho, Y.H.; Chang, A.Y.W.; Chan, J.Y.H. Transcriptional upregulation of mitochondrial uncoupling protein 2 protects against oxidative stress-associated neurogenic hypertension. Circ. Res. 2009, 105, 886–896. [Google Scholar] [CrossRef] [Green Version]
- Zanzinger, J. Mechanisms of action of nitric oxide in the brain stem: Role of oxidative stress. Auton. Neurosci. 2002, 98, 24–27. [Google Scholar] [CrossRef]
- Li, P.; Zhang, F.; Zhou, Y.B.; Cui, B.P.; Han, Y. Superoxide anions modulate the effects of angiotensin-(1–7) in the rostral ventrolateral medulla on cardiac sympathetic afferent reflex and sympathetic activity in rats. Neuroscience 2012, 223, 388–398. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.H.H.; Wu, K.L.H.; Chang, A.Y.W.; Tai, M.H.; Chan, J.Y.H. Oxidative impairment of mitochondrial electron transport chain complexes in rostral ventrolateral medulla contributes to neurogenic hypertension. Hypertension 2009, 53, 217–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, S.H.H.; Tai, M.H.; Li, C.Y.; Chan, J.Y.H. Reduction in molecular synthesis or enzyme activity of superoxide dismutases and catalase contributes to oxidative stress and neurogenic hypertension in spontaneously hypertensive rats. Free Radic. Biol. Med. 2006, 40, 2028–2039. [Google Scholar] [CrossRef] [PubMed]
- Schlaich, M.P.; Sobotka, P.A.; Krum, H.; Lambert, E.; Esler, M.D. Renal sympathetic-nerve ablation for uncontrolled hypertension. N. Engl. J. Med. 2009, 361, 932–934. [Google Scholar] [CrossRef] [PubMed]
- Rodrigo, R.; González, J.; Paoletto, F. The role of oxidative stress in the pathophysiology of hypertension. Hypertens. Res. 2011, 34, 431–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sata, Y.; Head, G.A.; Denton, K.; May, C.N.; Schlaich, M.P. Role of the Sympathetic Nervous System and Its Modulation in Renal Hypertension. Front. Med. (Lausanne) 2018, 5, 82. [Google Scholar] [CrossRef] [Green Version]
- Hsu, C.N.; Tain, Y.L. Targeting the Renin-Angiotensin-Aldosterone System to Prevent Hypertension and Kidney Disease of Developmental Origins. Int. J. Mol. Sci. 2021, 22, 2298. [Google Scholar] [CrossRef]
- Harrison, D.G. The immune system in hypertension. Trans. Am. Clin. Climatol. Assoc. 2014, 125, 130–138. [Google Scholar]
- Oparil, S.; Chen, Y.F.; Peng, N.; Wyss, J.M. Anterior hypothalamic norepinephrine, atrial natriuretic peptide, and hypertension. Front. Neuroendocrinol. 1996, 17, 212–246. [Google Scholar] [CrossRef]
- Rautureau, Y.; Schiffrin, E.L. Endothelin in hypertension: An update. Curr. Opin. Nephrol. Hypertens. 2012, 21, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Brito, R.; Castillo, G.; González, J.; Valls, N.; Rodrigo, R. Oxidative stress in hypertension: Mechanisms and therapeutic opportunities. Exp. Clin. Endocrinol. Diabetes 2015, 123, 325–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baltatu, O.C.; Amaral, F.G.; Campos, L.A.; Cipolla-Neto, J. Melatonin, mitochondria and hypertension. Cell Mol. Life Sci. 2017, 74, 3955–3964. [Google Scholar] [CrossRef] [PubMed]
- Kelly, J.J.; Mangos, G.; Williamson, P.M.; Whitworth, J.A. Cortisol and hypertension. Clin. Exp. Pharmacol. Physiol. Suppl. 1998, 25, S51–S56. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.N.; Tain, Y.L. Hydrogen Sulfide in Hypertension and Kidney Disease of Developmental Origins. Int. J. Mol. Sci. 2018, 19, 1438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khodor, S.A.; Reichert, B.; Shatat, I.F. The microbiome and blood pressure: Can microbes regulate our blood pressure? Front. Pediatr. 2017, 5, 138. [Google Scholar] [CrossRef]
- Dennery, P.A. Oxidative stress in development: Nature or nurture? Free Radic. Biol. Med. 2010, 49, 1147–1151. [Google Scholar] [CrossRef]
- Carter, A.M. Placental oxygen consumption. Part I. In vivo studies—A review. Placenta 2000, 21, S31–S37. [Google Scholar] [CrossRef]
- Jenkins, C.; Wilson, R.; Roberts, J.; Miller, H.; McKillop, J.H.; Walker, J.J. Antioxidants: Their role in pregnancy and miscarriage. Antioxid. Redox Signal. 2000, 2, 623–628. [Google Scholar] [CrossRef]
- Thompson, L.P.; Al-Hasan, Y. Impact of oxidative stress in fetal programming. J. Pregnancy 2012, 2012, 582748. [Google Scholar] [CrossRef]
- Hsu, C.N.; Tain, Y.L. Developmental Origins of Kidney Disease: Why Oxidative Stress Matters? Antioxidants 2020, 10, 33. [Google Scholar] [CrossRef] [PubMed]
- Barker, D.J.; Osmond, C.; Golding, J.; Kuh, D.; Wadsworth, M.E. Growth in utero, blood pressure in childhood and adult life, and mortality from cardiovascular disease. BMJ 1989, 298, 564–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Painter, R.C.; Roseboom, T.J.; Bleker, O.P. Prenatal exposure to the Dutch famine and disease in later life: An overview. Reprod. Toxicol. 2005, 20, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Oken, E.; Huh, S.Y.; Taveras, E.M.; Rich-Edwards, J.W.; Gillman, M.W. Associations of maternal prenatal smoking with child adiposity and blood pressure. Obes. Res. 2005, 13, 2021–2028. [Google Scholar] [CrossRef] [Green Version]
- Filler, G.; Yasin, A.; Kesarwani, P.; Garg, A.X.; Lindsay, R.; Sharma, A.P. Big mother or small baby: Which predicts hypertension? J. Clin. Hypertens. 2011, 13, 35–41. [Google Scholar] [CrossRef]
- Hrudey, E.J.; Reynolds, R.M.; Oostvogels, A.J.; Brouwer, I.A.; Vrijkotte, T.G. The association between maternal 25-hydroxyvitamin D concentration during gestation and early childhood cardio-metabolic outcomes: Is there interaction with pre-pregnancy BMI? PLoS ONE 2015, 10, e0133313. [Google Scholar] [CrossRef]
- Fraser, A.; Nelson, S.M.; Macdonald-Wallis, C.; Sattar, N.; Lawlor, D.A. Hypertensive disorders of pregnancy and cardiometabolic health in adolescent offspring. Hypertension 2013, 62, 614–620. [Google Scholar] [CrossRef] [Green Version]
- Keijzer-Veen, M.G.; Finken, M.J.; Nauta, J.; Dekker, F.W.; Hille, E.T.; Frölich, M.; Wit, J.M.; van der Heijden, A.J. Dutch POPS-19 collaborative study group. Is blood pressure increased 19 years after intrauterine growth restriction and preterm birth? A prospective follow-up study in The Netherlands. Pediatrics 2005, 116, 725–731. [Google Scholar] [CrossRef]
- Hosaka, M.; Asayama, K.; Staessen, J.A.; Ohkubo, T.; Hayashi, K.; Tatsuta, N.; Kurokawa, N.; Satoh, M.; Hashimoto, T.; Hirose, T.; et al. Breastfeeding leads to lower blood pressure in 7-year-old Japanese children: Tohoku study of child development. Hypertens. Res. 2013, 36, 117–122. [Google Scholar] [CrossRef] [Green Version]
- Hsu, C.N.; Tain, Y.L. Animal Models for DOHaD Research: Focus on Hypertension of Developmental Origins. Biomedicines 2021, 9, 623. [Google Scholar] [CrossRef]
- McMullen, S.; Mostyn, A. Animal models for the study of the developmental origins of health and disease. Proc. Nutr. Soc. 2009, 68, 306–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tain, Y.L.; Hsieh, C.S.; Lin, I.C.; Chen, C.C.; Sheen, J.M.; Huang, L.T. Effects of maternal l-citrulline supplementation on renal function and blood pressure in offspring exposed to maternal caloric restriction: The impact of nitric oxide pathway. Nitric Oxide 2010, 23, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Huang, L.T.; Hsu, C.N.; Lee, C.T. Melatonin therapy prevents programmed hypertension and nitric oxide deficiency in offspring exposed to maternal caloric restriction. Oxid. Med. Cell Longev. 2014, 2014, 283180. [Google Scholar] [CrossRef] [PubMed]
- Franco Mdo, C.; Fortes, Z.B.; Akamine, E.H.; Kawamoto, E.M.; Scavone, C.; de Britto, L.R.; Muscara, M.N.; Teixeira, S.A.; Tostes, R.C.; Carvalho, M.H.; et al. Tetrahydrobiopterin improves endothelial dysfunction and vascular oxidative stress in microvessels of intrauterine undernourished rats. J. Physiol. 2004, 558, 239–248. [Google Scholar] [CrossRef]
- Rodríguez-Rodríguez, P.; López de Pablo, A.L.; García-Prieto, C.F.; Somoza, B.; Quintana-Villamandos, B.; Gómez de Diego, J.J.; Gutierrez-Arzapalo, P.Y.; Ramiro-Cortijo, D.; González, M.C.; Arribas, S.M. Long term effects of fetal undernutrition on rat heart. Role of hypertension and oxidative stress. PLoS ONE 2017, 12, e0171544. [Google Scholar] [CrossRef]
- Cambonie, G.; Comte, B.; Yzydorczyk, C.; Ntimbane, T.; Germain, N.; Lê, N.L.; Pladys, P.; Gauthier, C.; Lahaie, I.; Abran, D.; et al. Antenatal antioxidant prevents adult hypertension, vascular dysfunction, and microvascular rarefaction associated with in utero exposure to a low-protein diet. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 292, R1236–R1245. [Google Scholar] [CrossRef]
- Hsu, C.N.; Hou, C.Y.; Chang-Chien, G.P.; Lin, S.; Chan, J.Y.H.; Lee, C.T.; Tain, Y.L. Maternal resveratrol therapy protected adult rat offspring against hypertension programmed by combined exposures to asymmetric dimethylarginine and trimethylamine-N-oxide. J. Nutr. Biochem. 2021, 93, 108630. [Google Scholar] [CrossRef]
- Tain, Y.L.; Lee, W.C.; Hsu, C.N.; Lee, W.C.; Huang, L.T.; Lee, C.T.; Lin, C.Y. Asymmetric dimethylarginine is associated with developmental programming of adult kidney disease and hypertension in offspring of streptozotocin-treated mothers. PLoS ONE 2013, 8, e55420. [Google Scholar] [CrossRef]
- Martínez Gascón, L.E.; Ortiz, M.C.; Galindo, M.; Sanchez, J.M.; Sancho-Rodriguez, N.; Albaladejo Otón, M.D.; Rodriguez Mulero, M.D.; Rodriguez, F. Role of heme oxygenase in the regulation of the renal hemodynamics in a model of sex dependent programmed hypertension by maternal diabetes. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2022. [Google Scholar] [CrossRef]
- Yu, C.; Chen, S.; Wang, X.; Wu, G.; Zhang, Y.; Fu, C.; Hu, C.; Liu, Z.; Luo, X.; Wang, J.; et al. Exposure to maternal diabetes induces endothelial dysfunction and hypertension in adult male rat offspring. Microvasc. Res. 2021, 133, 104076. [Google Scholar] [CrossRef]
- Tain, Y.L.; Hsu, C.N.; Lee, C.T.; Lin, Y.J.; Tsai, C.C. N-Acetylcysteine prevents programmed hypertension in male rat offspring born to suramin-treated mothers. Biol. Reprod. 2016, 95, 8. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Lee, W.C.; Wu, K.L.H.; Leu, S.; Chan, J.Y.H. Targeting arachidonic acid pathway to prevent programmed hypertension in maternal fructose-fed male adult rat offspring. J. Nutr. Biochem. 2016, 38, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Chao, Y.M.; Wu, K.L.H.; Tsai, P.C.; Tain, Y.L.; Leu, S.; Lee, W.C.; Chan, J.Y.H. Anomalous AMPK-regulated angiotensin AT1R expression and SIRT1-mediated mitochondrial biogenesis at RVLM in hypertension programming of offspring to maternal high fructose exposure. J. Biomed. Sci. 2020, 27, 68. [Google Scholar] [CrossRef] [PubMed]
- Tsai, P.C.; Chao, Y.M.; Chan, J.Y.H. Sympathetic activation of splenic T-lymphocytes in hypertension of adult offspring programmed by maternal high fructose exposure. Chin. J. Physiol. 2020, 63, 263–275. [Google Scholar]
- Tain, Y.L.; Lee, W.C.; Wu, K.L.H.; Leu, S.; Chan, J.Y.H. Resveratrol Prevents the Development of Hypertension Programmed by Maternal Plus Post-Weaning High-Fructose Consumption through Modulation of Oxidative Stress, Nutrient-Sensing Signals, and Gut Microbiota. Mol. Nutr. Food Res. 2018, 30, e1800066. [Google Scholar] [CrossRef]
- Tain, Y.L.; Chan, J.Y.H.; Lee, C.T.; Hsu, C.N. Maternal Melatonin Therapy Attenuates Methyl-Donor Diet-Induced Programmed Hypertension in Male Adult Rat Offspring. Nutrients 2018, 10, 1407. [Google Scholar] [CrossRef] [Green Version]
- Hsu, C.N.; Yang, H.W.; Hou, C.Y.; Chang-Chien, G.P.; Lin, S.; Tain, Y.L. Maternal Adenine-Induced Chronic Kidney Disease Programs Hypertension in Adult Male Rat Offspring: Implications of Nitric Oxide and Gut Microbiome Derived Metabolites. Int. J. Mol. Sci. 2020, 21, 7237. [Google Scholar] [CrossRef]
- Hsu, C.N.; Hou, C.Y.; Chang-Chien, G.P.; Lin, S.; Yang, H.W.; Tain, Y.L. Perinatal Resveratrol Therapy Prevents Hypertension Programmed by Maternal Chronic Kidney Disease in Adult Male Offspring: Implications of the Gut Microbiome and Their Metabolites. Biomedicines 2020, 8, 567. [Google Scholar] [CrossRef]
- Do Nascimento, L.C.P.; Neto, J.P.R.C.; de Andrade Braga, V.; Lagranha, C.J.; de Brito Alves, J.L. Maternal exposure to high-fat and high-cholesterol diet induces arterial hypertension and oxidative stress along the gut-kidney axis in rat offspring. Life Sci. 2020, 261, 118367. [Google Scholar] [CrossRef]
- Lamothe, J.; Khurana, S.; Tharmalingam, S.; Williamson, C.; Byrne, C.J.; Lees, S.J.; Khaper, N.; Kumar, A.; Tai, T.C. Oxidative Stress Mediates the Fetal Programming of Hypertension by Glucocorticoids. Antioxidants 2021, 10, 531. [Google Scholar] [CrossRef]
- Tain, Y.L.; Sheen, J.M.; Chen, C.C.; Yu, H.R.; Tiao, M.M.; Kuo, H.C.; Huang, L.T. Maternal citrulline supplementation prevents prenatal dexamethasone-induced programmed hypertension. Free Radic. Res. 2014, 48, 580–586. [Google Scholar] [CrossRef] [PubMed]
- Tai, I.H.; Sheen, J.M.; Lin, Y.J.; Yu, H.R.; Tiao, M.M.; Chen, C.C.; Huang, L.T.; Tain, Y.L. Maternal N-acetylcysteine therapy regulates hydrogen sulfide-generating pathway and prevents programmed hypertension in male offspring exposed to prenatal dexamethasone and postnatal high-fat diet. Nitric Oxide 2016, 53, 6–12. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.N.; Lin, Y.J.; Yu, H.R.; Lin, I.C.; Sheen, J.M.; Huang, L.T.; Tain, Y.L. Protection of Male Rat Offspring against Hypertension Programmed by Prenatal Dexamethasone Administration and Postnatal High-Fat Diet with the Nrf2 Activator Dimethyl Fumarate during Pregnancy. Int. J. Mol. Sci. 2019, 20, 3957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, C.N.; Lin, Y.J.; Lu, P.C.; Tain, Y.L. Maternal resveratrol therapy protects male rat offspring against programmed hypertension induced by TCDD and dexamethasone exposures: Is it relevant to aryl hydrocarbon receptor? Int. J. Mol. Sci. 2018, 19, 2459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, C.N.; Lin, Y.J.; Tain, Y.L. Maternal exposure to bisphenol A combined with high-fat diet-induced programmed hypertension in adult male rat offspring: Effects of resveratrol. Int. J. Mol. Sci. 2019, 20, 4382. [Google Scholar] [CrossRef] [Green Version]
- Ojeda, N.B.; Hennington, B.S.; Williamson, D.T.; Hill, M.L.; Betson, N.E.; Sartori-Valinotti, J.C.; Reckelhoff, J.F.; Royals, T.P.; Alexander, B.T. Oxidative stress contributes to sex differences in blood pressure in adult growth-restricted offspring. Hypertension 2012, 60, 114–122. [Google Scholar] [CrossRef] [Green Version]
- Tsai, W.L.; Hsu, C.N.; Tain, Y.L. Whether AICAR in Pregnancy or Lactation Prevents Hypertension Programmed by High Saturated Fat Diet: A Pilot Study. Nutrients 2020, 12, 448. [Google Scholar] [CrossRef] [Green Version]
- Feng, X.; Guo, Q.; Xue, H.; Duan, X.; Jin, S.; Wu, Y. Hydrogen Sulfide Attenuated Angiotensin II-Induced Sympathetic Excitation in Offspring of Renovascular Hypertensive Rats. Front. Pharmacol. 2020, 11, 565726. [Google Scholar] [CrossRef]
- Svitok, P.; Okuliarova, M.; Varga, I.; Zeman, M. Renal impairment induced by prenatal exposure to angiotensin II in male rat offspring. Exp. Biol. Med. (Maywood) 2019, 244, 923–931. [Google Scholar] [CrossRef]
- Piecha, G.; Koleganova, N.; Ritz, E.; Müller, A.; Fedorova, O.V.; Bagrov, A.Y.; Lutz, D.; Schirmacher, P.; Gross-Weissmann, M.L. High salt intake causes adverse fetal programming--vascular effects beyond blood pressure. Nephrol. Dial. Transplant. 2012, 27, 3464–3476. [Google Scholar]
- Liu, Y.; Qi, L.; Wu, J.; Xu, T.; Yang, C.; Chen, X.; Lv, J.; Xu, Z. Prenatal high-salt diet impaired vasodilatation with reprogrammed renin-angiotensin system in offspring rats. J. Hypertens. 2018, 36, 2369–2379. [Google Scholar] [CrossRef] [PubMed]
- Vieira, L.D.; Farias, J.S.; de Queiroz, D.B.; Cabral, E.V.; Lima-Filho, M.M.; Sant’Helena, B.R.M.; Aires, R.S.; Ribeiro, V.S.; Santos-Rocha, J.; Xavier, F.E.; et al. Oxidative stress induced by prenatal LPS leads to endothelial dysfunction and renal haemodynamic changes through angiotensin II/NADPH oxidase pathway: Prevention by early treatment with α-tocopherol. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3577–3587. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.P.; Chen, L.; Wang, X.J.; Jiang, Q.H.; Bei, X.Y.; Sun, W.L.; Xia, S.J.; Jiang, J.T. Maternal exposure to di-n-butyl phthalate (DBP) induces renal fibrosis in adult rat offspring. Oncotarget 2017, 8, 31101–31111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hendricks, A.S.; Lawson, M.J.; Figueroa, J.P.; Chappell, M.C.; Diz, D.I.; Shaltout, H.A. Central ANG-(1-7) infusion improves blood pressure regulation in antenatal betamethasone-exposed sheep and reveals sex-dependent effects on oxidative stress. Am. J. Physiol. Heart Circ. Physiol. 2019, 316, H1458–H1467. [Google Scholar] [CrossRef] [PubMed]
- Gwathmey, T.M.; Shaltout, H.A.; Rose, J.C.; Diz, D.I.; Chappell, M.C. Glucocorticoid-induced fetal programming alters the functional complement of angiotensin receptor subtypes within the kidney. Hypertension 2011, 57, 620–626. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Zadi, Z.H.; Zhang, J.; Scharf, S.M.; Pae, E.K. Intermittent hypoxia in utero damages postnatal growth and cardiovascular function in rats. J. Appl. Physiol. 2018, 124, 821–830. [Google Scholar] [CrossRef]
- Skeffington, K.L.; Beck, C.; Itani, N.; Niu, Y.; Shaw, C.J.; Giussani, D.A. Hypertension Programmed in Adult Hens by Isolated Effects of Developmental Hypoxia In Ovo. Hypertension 2020, 76, 533–544. [Google Scholar] [CrossRef]
- Brain, K.L.; Allison, B.J.; Niu, Y.; Cross, C.M.; Itani, N.; Kane, A.D.; Herrera, E.A.; Skeffington, K.L.; Botting, K.J.; Giussani, D.A. Intervention against hypertension in the next generation programmed by developmental hypoxia. PLoS Biol. 2019, 17, e2006552. [Google Scholar] [CrossRef] [Green Version]
- Hsu, C.N.; Tain, Y.L. The Good, the Bad, and the Ugly of Pregnancy Nutrients and Developmental Programming of Adult Disease. Nutrients 2019, 11, 894. [Google Scholar] [CrossRef] [Green Version]
- Sengupta, P. The laboratory rat: Relating its age with human’s. Int. J. Prev. Med. 2013, 4, 624–630. [Google Scholar]
- Bandookwala, M.; Sengupta, P. 3-Nitrotyrosine: A versatile oxidative stress biomarker for major neurodegenerative diseases. Int. J. Neurosci. 2020, 130, 1047–1062. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Hsu, C.N. Regulation of Nitric Oxide Production in the Developmental Programming of Hypertension and Kidney Disease. Int. J. Mol. Sci. 2019, 20, 681. [Google Scholar]
- Marrocco, I.; Altieri, F.; Peluso, I. Measurement and Clinical Significance of Biomarkers of Oxidative Stress in Humans. Oxid. Med. Cell Longev. 2017, 2017, 6501046. [Google Scholar] [CrossRef] [PubMed]
- Kett, M.M.; Denton, K.M. Renal programming: Cause for concern? Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 300, R791–R803. [Google Scholar] [CrossRef]
- Tain, Y.L.; Hsu, C.N. Developmental Origins of Chronic Kidney Disease: Should We Focus on Early Life? Int. J. Mol. Sci. 2017, 18, 381. [Google Scholar] [CrossRef] [Green Version]
- Ortiz, L.A.; Quan, A.; Weinberg, A.; Baum, M. Effect of prenatal dexamethasone on rat renal development. Kidney Int. 2001, 59, 1663–1669. [Google Scholar] [CrossRef] [Green Version]
- Tain, Y.L.; Hsu, C.N.; Chan, J.Y.; Huang, L.T. Renal Transcriptome Analysis of Programmed Hypertension Induced by Maternal Nutritional Insults. Int. J. Mol. Sci. 2015, 16, 17826–17837. [Google Scholar] [CrossRef] [Green Version]
- Sheen, J.M.; Yu, H.R.; Tiao, M.M.; Chen, C.C.; Huang, L.T.; Chang, H.Y.; Tain, Y.L. Prenatal dexamethasone-induced programmed hypertension and renal programming. Life Sci. 2015, 132, 41–48. [Google Scholar] [CrossRef]
- Tain, Y.L.; Leu, S.; Wu, K.L.; Lee, W.C.; Chan, J.Y. Melatonin prevents maternal fructose intake-induced programmed hypertension in the offspring: Roles of nitric oxide and arachidonic acid metabolites. J. Pineal Res. 2014, 57, 80–89. [Google Scholar] [CrossRef]
- Thornburg, K.L. The programming of cardiovascular disease. J. Dev. Orig. Health Dis. 2015, 6, 366–376. [Google Scholar] [CrossRef]
- Tain, Y.-L.; Hsu, C.-N. Interplay between Oxidative Stress and Nutrient Sensing Signaling in the Developmental Origins of Cardiovascular Disease. Int. J. Mol. Sci. 2017, 18, 841. [Google Scholar] [CrossRef]
- Blackmore, H.L.; Ozanne, S.E. Programming of cardiovascular disease across the life-course. J. Mol. Cell. Cardiol. 2015, 83, 122–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itani, N.; Salinas, C.E.; Villena, M.; Skeffington, K.L.; Beck, C.; Villamor, E.; Blanco, C.E.; Giussani, D.A. The highs and lows of programmed cardiovascular disease by developmental hypoxia: Studies in the chicken embryo. J. Physiol. 2017, 596, 2991–3001. [Google Scholar] [CrossRef] [PubMed]
- Goto, K.; Ohtsubo, T.; Kitazono, T. Endothelium-Dependent Hyperpolarization (EDH) in Hypertension: The Role of Endothelial Ion Channels. Int. J. Mol. Sci. 2018, 19, 315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stead, R.; Musa, M.G.; Bryant, C.L.; Lanham, S.A.; Johnston, D.A.; Reynolds, R.; Torrens, C.; Fraser, P.A.; Clough, G.F. Developmental conditioning of endothelium-derived hyperpolarizing factor-mediated vasorelaxation. J. Hypertens. 2016, 34, 452–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tain, Y.L.; Hsu, C.N.; Lin, C.Y.; Huang, L.T.; Lau, Y.T. Aliskiren prevents hypertension and reduces asymmetric dimethylarginine in young spontaneously hypertensive rats. Eur. J. Pharmacol. 2011, 670, 561–565. [Google Scholar] [CrossRef]
- Stiemsma, L.T.; Michels, K.B. The role of the microbiome in the developmental origins of health and disease. Pediatrics 2018, 141, e20172437. [Google Scholar] [CrossRef] [Green Version]
- Hsu, C.N.; Hou, C.Y.; Hsu, W.H.; Tain, Y.L. Cardiovascular Diseases of Developmental Origins: Preventive Aspects of Gut Microbiota-Targeted Therapy. Nutrients 2021, 13, 2290. [Google Scholar] [CrossRef]
- Hsu, C.N.; Hou, C.Y.; Chang-Chien, G.P.; Lin, S.; Tain, Y.L. Maternal Garlic Oil Supplementation Prevents High-Fat Diet-Induced Hypertension in Adult Rat Offspring: Implications of H2S-Generating Pathway in the Gut and Kidneys. Mol. Nutr. Food Res. 2021, 65, e2001116. [Google Scholar] [CrossRef]
- Pisoschi, A.M.; Pop, A. The role of antioxidants in the chemistry of oxidative stress: A review. Eur. J. Med. Chem. 2015, 97, 55–74. [Google Scholar] [CrossRef]
- Nimse, S.B.; Palb, D. Free radicals, natural antioxidants, and their reaction mechanisms. RSC. Adv. 2015, 5, 27986–28006. [Google Scholar] [CrossRef] [Green Version]
- Azzi, A.; Ricciarelli, R.; Zingg, J.M. Non-antioxidant molecular functions of alpha-tocopherol (vitaminE). FEBS Lett. 2002, 519, 8–10. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Yin, N.; Deng, Y.; Wei, Y.; Huang, Y.; Pu, X.; Li, L.; Zheng, Y.; Guo, J.; Yu, J.; et al. Ascorbic Acid Protects against Hypertension through Downregulation of ACE1 Gene Expression Mediated by Histone Deacetylation in Prenatal Inflammation Induced Offspring. Sci. Rep. 2016, 6, 39469. [Google Scholar] [CrossRef] [PubMed]
- Koeners, M.P.; Racasan, S.; Koomans, H.A.; Joles, J.A.; Braam, B. Nitric oxide, superoxide and renal blood flow autoregulation in SHR after perinatal l-arginine and antioxidants. Acta Physiol. 2007, 190, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Racasan, S.; Braam, B.; van der Giezen, D.M.; Goldschmeding, R.; Boer, P.; Koomans, H.A.; Joles, J.A. Perinatal l-arginine and antioxidant supplements reduce adult blood pressure in spontaneously hypertensive rats. Hypertension 2004, 44, 83–88. [Google Scholar] [CrossRef] [Green Version]
- Schwedhelm, E.; Maas, R.; Freese, R.; Jung, D.; Lukacs, Z.; Jambrecina, A.; Spickler, W.; Schulze, F.; Boger, R.H. Pharmacokinetic and pharmacodynamics properties of oral l-citrulline and l-arginine: Impact on nitric oxide metabolism. Br. J. Clin. Pharmacol. 2008, 65, 51–59. [Google Scholar] [CrossRef]
- Hsu, C.N.; Tain, Y.L. Impact of Arginine Nutrition and Metabolism during Pregnancy on Offspring Outcomes. Nutrients 2019, 11, 1452. [Google Scholar] [CrossRef] [Green Version]
- Cynober, L.; Moinard, C.; De Bandt, J.P. The 2009 ESPEN Sir David Cuthbertson. Citrulline: A new major signaling molecule or just another player in the pharmaconutrition game? Clin. Nutr. 2010, 29, 545–551. [Google Scholar] [CrossRef]
- Fraga, C.G.; Croft, K.D.; Kennedy, D.O.; Tomás-Barberán, F.A. The effects of polyphenols and other bioactives on human health. Food Funct. 2019, 10, 514–528. [Google Scholar] [CrossRef] [Green Version]
- Urquiaga, I.; Leighton, F. Plant polyphenol antioxidants and oxidative stress. Biol. Res. 2000, 33, 55–64. [Google Scholar] [CrossRef]
- Rodrigo, R.; Gil, D.; Miranda-Merchak, A.; Kalantzidis, G. Antihypertensive role of polyphenols. Adv. Clin. Chem. 2012, 58, 22. [Google Scholar]
- Truong, V.L.; Jun, M.; Jeong, W.S. Role of resveratrol in regulation of cellular defense systems against oxidative stress. Biofactors 2018, 44, 36–49. [Google Scholar] [CrossRef] [PubMed]
- Xia, N.; Daiber, A.; Förstermann, U.; Li, H. Antioxidant effects of resveratrol in the cardiovascular system. Br. J. Pharmacol. 2017, 174, 1633–1646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Care, A.S.; Sung, M.M.; Panahi, S.; Gragasin, F.S.; Dyck, J.R.; Davidge, S.T.; Bourque, S.L. Perinatal Resveratrol Supplementation to Spontaneously Hypertensive Rat Dams Mitigates the Development of Hypertension in Adult Offspring. Hypertension 2016, 67, 1038–1044. [Google Scholar] [CrossRef] [Green Version]
- Reiter, R.J.; Mayo, J.C.; Tan, D.X.; Sainz, R.M.; Alatorre-Jimenez, M.; Qin, L. Melatonin as an antioxidant: Under promises but over delivers. J. Pineal Res. 2016, 61, 253–278. [Google Scholar] [CrossRef]
- Hsu, C.N.; Huang, L.T.; Tain, Y.L. Perinatal Use of Melatonin for Offspring Health: Focus on Cardiovascular and Neurological Diseases. Int. J. Mol. Sci. 2019, 20, 5681. [Google Scholar] [CrossRef] [Green Version]
- Šalamon, Š.; Kramar, B.; Marolt, T.P.; Poljšak, B.; Milisav, I. Medical and Dietary Uses of N-Acetylcysteine. Antioxidants 2019, 8, 111. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.H.; Leonard, S.; Shi, X.; Goins, M.R.; Vallyathan, V. Antioxidant activity of lazaroid (U-75412E) and its protective effects against crystalline silica-induced cytotoxicity. Free Radic. Biol. Med. 1998, 24, 529–536. [Google Scholar] [CrossRef]
- Vomund, S.; Schäfer, A.; Parnham, M.J.; Brüne, B.; Von Knethen, A. Nrf2, the Master Regulator of Anti-Oxidative Responses. Int. J. Mol. Sci. 2017, 18, 2772. [Google Scholar] [CrossRef] [Green Version]
- Wilcox, C.S. Effects of tempol and redox-cycling nitroxides in models of oxidative stress. Pharmacol. Ther. 2010, 126, 119–145. [Google Scholar] [CrossRef] [Green Version]
- Villanueva, C.; Kross, R.D. Antioxidant-induced stress. Int. J. Mol Sci. 2012, 13, 2091–2109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tain, Y.L.; Hsu, C.N. AMP-Activated Protein Kinase as a Reprogramming Strategy for Hypertension and Kidney Disease of Developmental Origin. Int. J. Mol. Sci. 2018, 19, 1744. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
| Animal Models | Species/ Gender | Age at Evaluation | Mechanisms of Oxidative Stress | Programmed Organ System | Ref. |
|---|---|---|---|---|---|
| Maternal caloric restriction diet | SD rat/M | 12 weeks | ↑ Renal 8-OHdG expression, ↑ ADMA, ↓ NO | Kidneys | [83,84] |
| Maternal caloric restriction diet | Wistar rat/M | 16 weeks | ↑ 3-NT, ↓ NO | Vessels | [85] |
| Maternal caloric restriction diet | SD rat/M | 6 months | ↑ Xanthine-oxidase expression | Heart | [86] |
| Maternal protein restriction diet | Wistar rat/M | 12 weeks | ↑ F2-isoprostane, ↓ glutathione | Kidneys | [87] |
| Maternal L-NAME administration | SD rat/M | 12 weeks | ↑ Renal F2-isoprostane | Kidneys | [46] |
| Maternal ADMA administration | SD rat/M | 12 weeks | ↓ NO | Kidneys | [88] |
| Streptozotocin-induced diabetes | SD rat/M | 12 weeks | ↑ ADMA, ↓ NO | Kidneys | [89] |
| Streptozotocin-induced diabetes | SD rat/M | 12 weeks | ↑ Renal TBARS and 3-NT | Kidneys, vessels | [90] |
| Streptozotocin-induced diabetes | SD rat/M | 24 weeks | ↑ ROS, ↓ NO, ↓ SOD activity | Vessels | [91] |
| Maternal suramin administration | SD rat/M | 12 weeks | ↑ ADMA, ↓ NO | Kidneys | [92] |
| Maternal high-fructose diet | SD rat/M | 12 weeks | ↑ Renal 8-OHdG expression, ↓ NO | Kidneys | [93] |
| Maternal high-fructose diet | SD rat/M | 12 weeks | ↑ NADPH-oxidase expression and MDA | Brain | [94] |
| Maternal high-fructose diet | SD rat/M | 24 weeks | ↑ ROS | Spleen | [95] |
| Maternal plus post-weaning high-fructose diet | SD rat/M | 12 weeks | ↑ Renal 8-OHdG expression | Kidneys | [96] |
| Maternal methyl-deficient diet | SD rat/M | 12 weeks | ↑ Renal 8-OHdG expression | Kidneys | [97] |
| Maternal high methyl-donor diet | SD rat/M | 12 weeks | ↑ Renal 8-OHdG expression | Kidneys | [97] |
| Maternal adenine-induced CKD | SD rat/M | 12 weeks | ↑ Renal 8-OHdG expression,↑ ADMA, ↓ NO | Kidneys | [98,99] |
| Maternal high-fat and high-cholesterol diet | SD rat/M & F | 90 days | ↓ SOD activity in M; ↑ Renal MDA level in F | Kidneys | [100] |
| Prenatal dexamethasone exposure | Wistar rat/M & F | 14 weeks | ↑ NADPH-oxidase, ↓ Gpx1 expression | Adrenal glands | [101] |
| Prenatal dexamethasone exposure | SD rat/M | 16 weeks | ↓ Renal NO | Kidneys | [102] |
| Prenatal dexamethasone exposure plus postnatal high-fat intake | SD rat/M | 16 weeks | ↑ Renal 8-OHdG expression, ↓ NO | Kidneys | [103,104] |
| Prenatal dexamethasone plus TCDD exposure | SD rat/M | 16 weeks | ↑ Renal 8-OHdG expression, ↑ ADMA | Kidneys | [105] |
| Prenatal bisphenol A exposure plus high-fat diet | SD rat/M | 16 weeks | ↑ Renal 8-OHdG expression, ↑ ADMA, ↓ NO | Kidneys | [106] |
| Reduced uterine perfusion | SD rat/M | 16 weeks | ↑ Urinary F2-isoprostane level & renal NADPH-oxidase-dependent superoxide | Kidneys | [107] |
| Maternal plus post-weaning high-fat diet | SD rat/M | 16 weeks | ↑ Renal 8-OHdG expression | Kidneys | [108] |
| Maternal 1K1C model | SD rat/M | 16 weeks | ↑ NADPH-oxidase expression, ↑ 3-NT | Brain | [109] |
| Maternal angiotensin II administration | Wistar rat/M | 18 weeks | ↑ Renal ROS | Kidneys | [110] |
| Maternal high-salt diet | SD rat/M | 12 weeks | ↑ 3-NT, ↑ ADMA | Vessels | [111] |
| Maternal high-salt diet | Wistar rat/M | 5 months | ↑ NADPH-oxidase expression, ↑ MDA level, ↓ Antioxidant activity | Vessels | [112] |
| Prenatal LPSExposure | Wistar rat/M | 28 weeks | ↑ Renal MDA | Kidneys | [113] |
| Maternal di-n-butyl phthalate exposure | SD rat/M & F | 18 months | ↑ Renal ROS | Kidneys | [114] |
| Prenatal betamethasone exposure | Sheep/M | 6 months | ↑ 4-HNE | Brain | [115] |
| Prenatal betamethasone exposure | Sheep/M & F | 18 months | ↑ ROS, ↓ NO | Kidneys | [116] |
| Prenatal hypoxia exposure | SD rat/M & F | 8 weeks | ↑ Lipid peroxidation | Heart | [117] |
| Prenatal hypoxia exposure | Chicken/M & F | 6 months | ↓ NO | Heart, vessels | [118] |
| Prenatal hypoxia exposure | Sheep/M & F | 9 months | ↓ NO | Vessels | [119] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tain, Y.-L.; Hsu, C.-N. Oxidative Stress-Induced Hypertension of Developmental Origins: Preventive Aspects of Antioxidant Therapy. Antioxidants 2022, 11, 511. https://doi.org/10.3390/antiox11030511
Tain Y-L, Hsu C-N. Oxidative Stress-Induced Hypertension of Developmental Origins: Preventive Aspects of Antioxidant Therapy. Antioxidants. 2022; 11(3):511. https://doi.org/10.3390/antiox11030511
Chicago/Turabian StyleTain, You-Lin, and Chien-Ning Hsu. 2022. "Oxidative Stress-Induced Hypertension of Developmental Origins: Preventive Aspects of Antioxidant Therapy" Antioxidants 11, no. 3: 511. https://doi.org/10.3390/antiox11030511
APA StyleTain, Y.-L., & Hsu, C.-N. (2022). Oxidative Stress-Induced Hypertension of Developmental Origins: Preventive Aspects of Antioxidant Therapy. Antioxidants, 11(3), 511. https://doi.org/10.3390/antiox11030511