
The Antioxidant Selenoprotein T Mimetic, PSELT, Induces Preconditioning-like Myocardial Protection by Relieving Endoplasmic-Reticulum Stress

 , , , , , and
, , , , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Peptides and Drugs
2.2. Animals
2.3. Langendorff Isolated Rat Heart Perfusion
2.4. Experimental Protocols
2.4.1. Ischemia/Reperfusion (I/R) Protocol
2.4.2. Experimental Groups
- (a).
- Sham group: hearts underwent only 190 min of perfusion;
- (b).
- I/R Control group: hearts were exposed to I/R protocol;
- (c).
- PSELT group, pre-conditioning with PSELT: after stabilization, PSELT was infused for 20 min before I/R at the concentration of 5 nM according to our prior ex vivo study [17], where PSELT elicited cardioprotection as post-conditioning agent;
- (d).
- TM group, pre-conditioning with tunicamycin (TM): after stabilization, TM (2.5 µg/mL) was infused for 5 min before I/R [23];
- (e).
- TM + PSELT group, pre-conditioning with TM + PSELT: after stabilization, TM was infused for 5 min followed by a 20 min 5 nM PSELT infusion before I/R;
- (f).
- I-PSELT group, pre-conditioning with inert PSELT (I-PSELT): after stabilization, 5 nM I-PSELT was infused for 20 min before I/R [17].
2.5. Assessment of Myocardial Injury
2.5.1. Lactate Dehydrogenase Activity in Coronary Effluent
2.5.2. Infarct Size
2.6. Biochemical Analyses
2.6.1. Superoxide Dismutase (SOD) Assay
2.6.2. Catalase (CAT) Assay
2.6.3. Thiobarbituric Acid Reactive Substances (TBARS) Assay
2.6.4. Protein Carbonyl Content Assay
2.7. Western Blot Analyses on Cardiac Tissues
2.8. Cell Culture and Treatments
2.8.1. Cell Viability by 3-(4,5-Dimethylthiazol-)2,5-diphenyl Tetrazolium Bromide (MTT) Assay
2.8.2. Western Blot Analysis on H9c2 Cells
2.8.3. Short Interfering RNA (siRNA) Transfection for SELENOT Silencing
2.8.4. Immunofluorescence Analysis
2.9. Statistical Analyses
3. Results
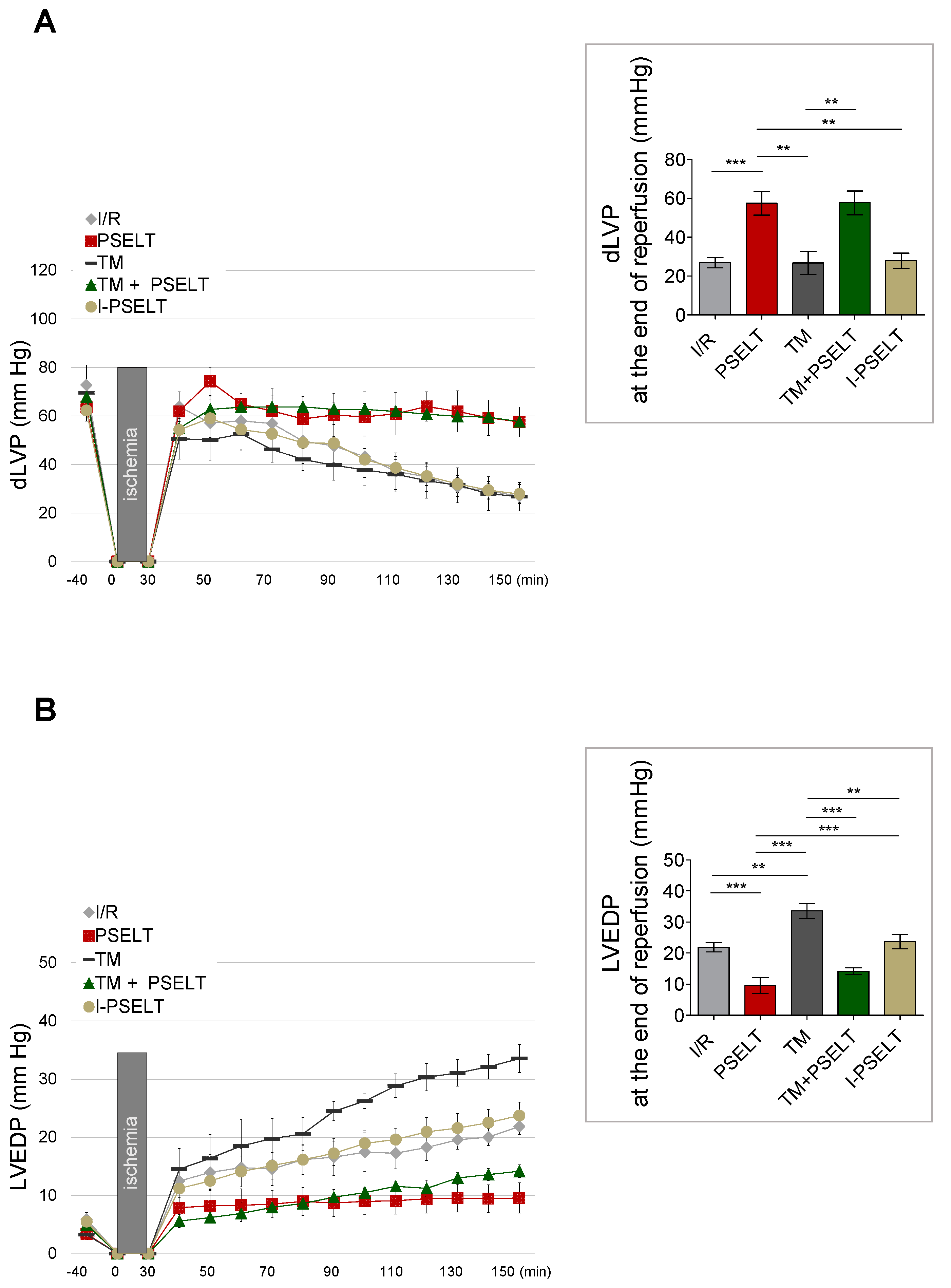
3.1. Action of PSELT on Post-Ischemic Systolic and Diastolic Recovery
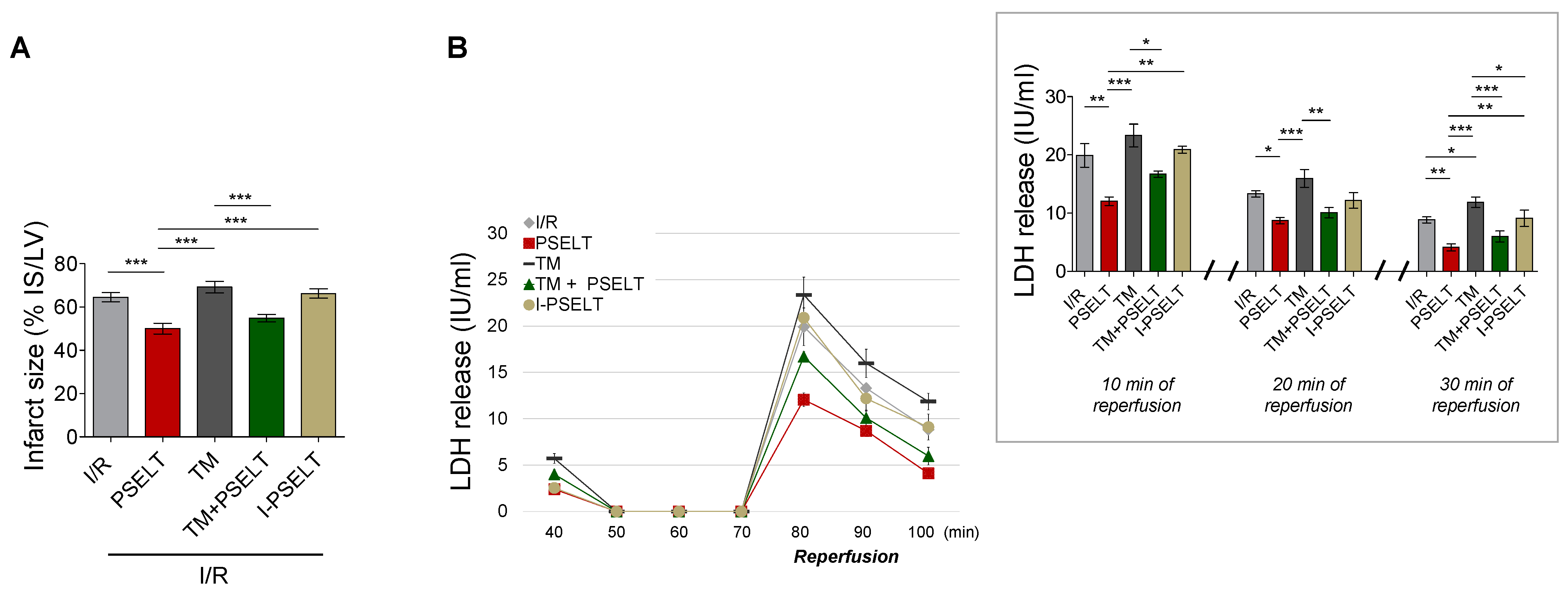
3.2. Effect of PSELT on Infarct Size (IS) and Lactate Dehydrogenase Release (LDH)
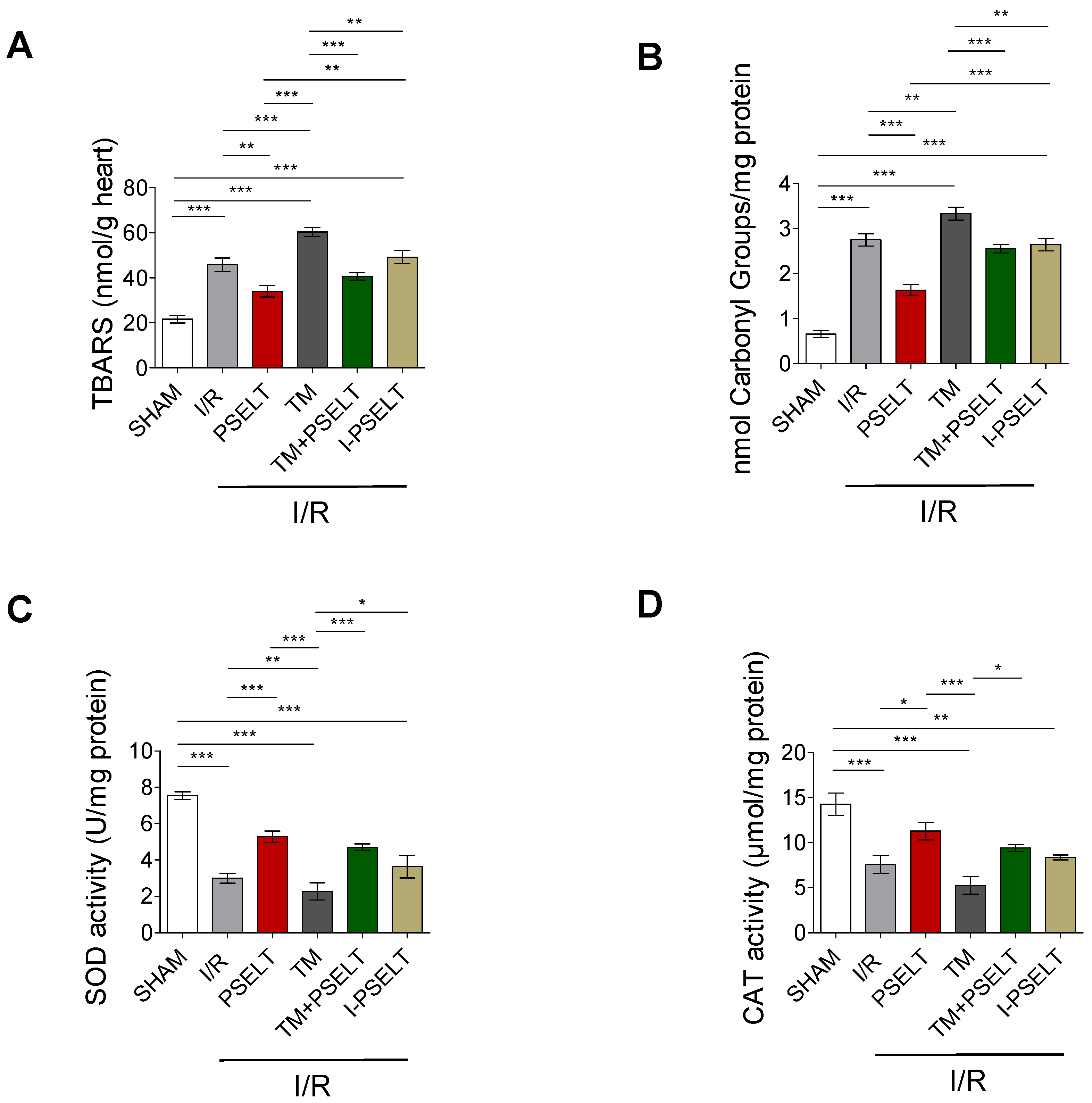
3.3. Effect of PSELT on Cardiac Oxidative Stress and the Activity of Endogenous Antioxidant Enzymes
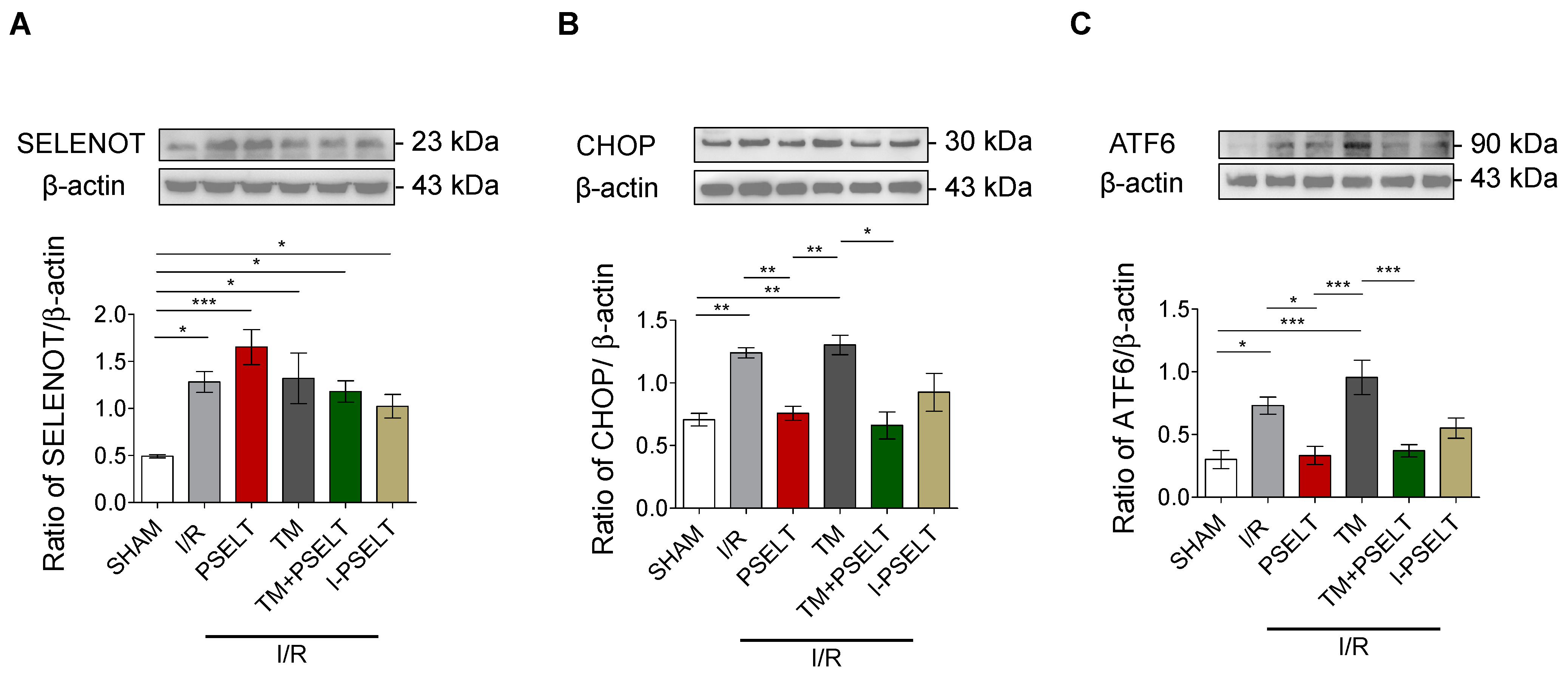
3.4. Influence of PSELT on the Expression of SELENOT and ER Stress Response Markers in I/R Hearts
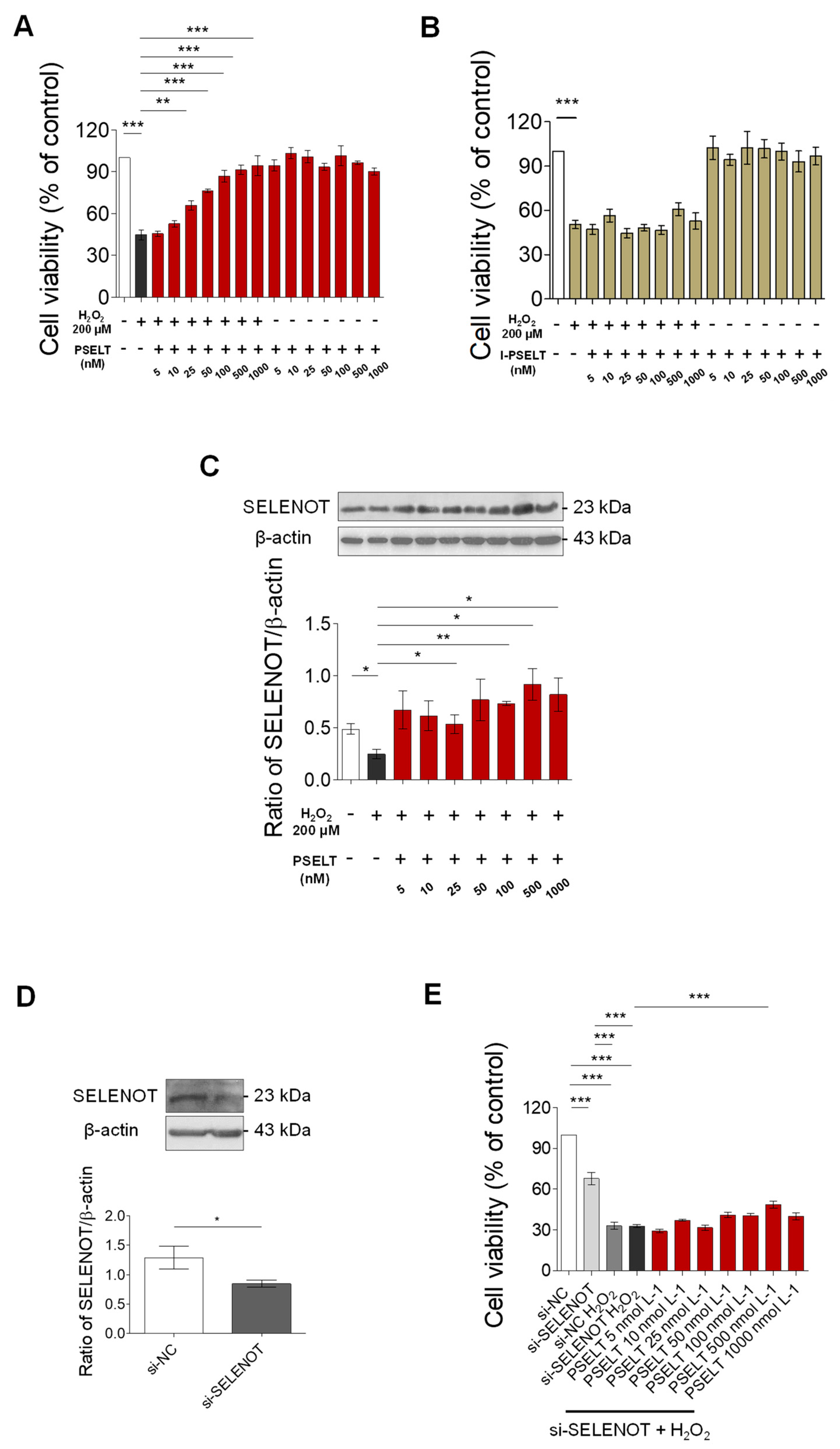
3.5. Effect of PSELT against Oxidative Stress in H9c2 Cells
3.6. Role of Endogenous SELENOT in PSELT-Dependent H9c2 Cell Protection following H2O2 Exposure
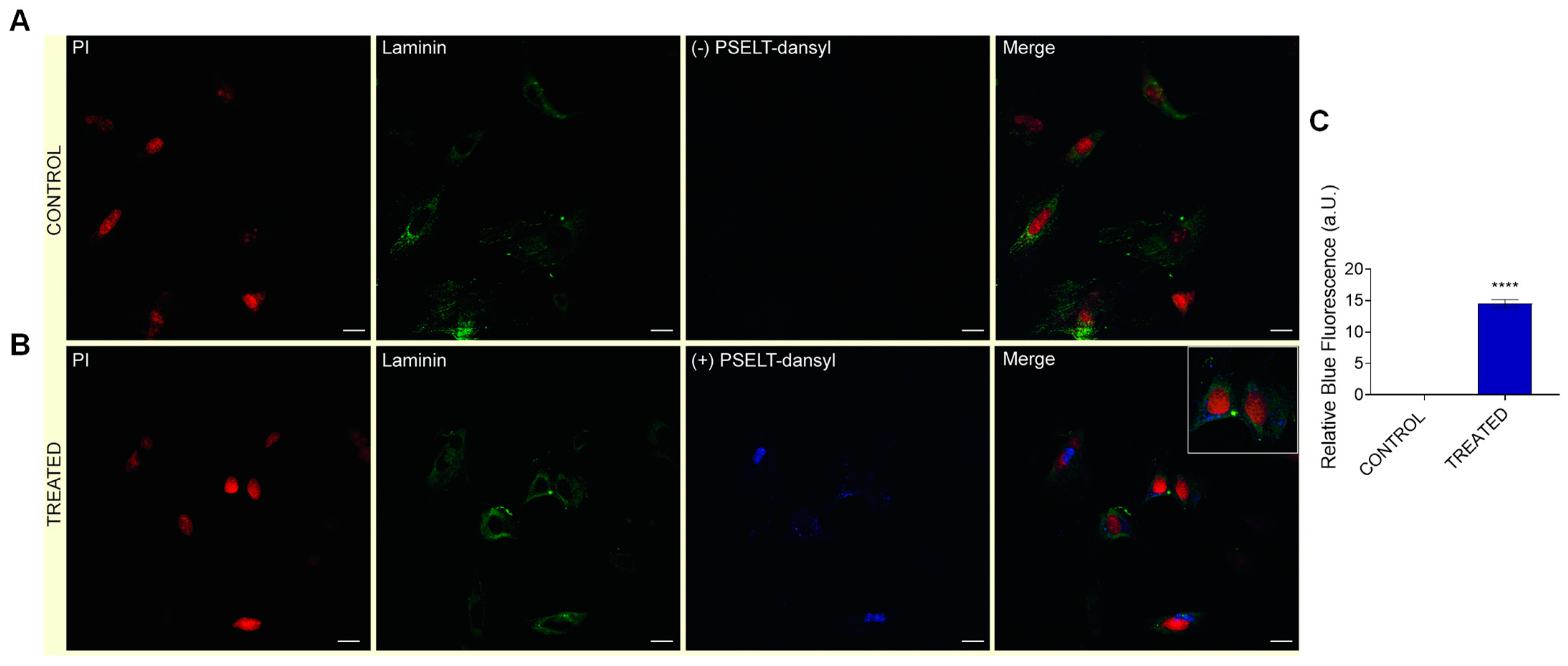
3.7. Evaluation of Cardiomyocyte-Penetrating Capacity and Cell Distribution of PSELT
4. Discussion
4.1. PSELT Exerts Preconditioning-like Myocardial Protective Effects against I/R Injury and TM-Induced ERS through the Sec Residue
4.2. PSELT Reduces the Cardiac Expression of the ERS Markers CHOP and ATF6 and Increases Endogenous SELENOT Protein Expression
4.3. PSELT Reduces Oxidative Stress and Improves the Endogenous Antioxidant Defence System to Promote Cardioprotection
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Virani, S.S.; Alonso, A.; Aparicio, H.J.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Cheng, S.; Delling, F.N.; et al. Heart Disease and Stroke Statistics-2021 Update: A Report from the American Heart Association. Circulation 2021, 143, e254–e743. [Google Scholar] [CrossRef] [PubMed]
- Hausenloy, D.J.; Yellon, D.M. Myocardial ischemia-reperfusion injury: A neglected therapeutic target. J. Clin. Investig. 2013, 123, 92–100. [Google Scholar] [CrossRef]
- Heusch, G. Myocardial ischaemia-reperfusion injury and cardioprotection in perspective. Nat. Rev. Cardiol. 2020, 17, 773–789. [Google Scholar] [CrossRef] [PubMed]
- Hausenloy, D.J.; Barrabes, J.A.; Bøtker, H.E.; Davidson, S.M.; Di Lisa, F.; Downey, J.; Engstrom, T.; Ferdinandy, P.; Carbrera-Fuentes, H.A.; Heusch, G.; et al. Ischaemic conditioning and targeting reperfusion injury: A 30 year voyage of discovery. Basic Res. Cardiol. 2016, 111, 70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kloner, R.A.; Jennings, R.B. Consequences of brief ischemia: Stunning, preconditioning, and their clinical implications: Part 2. Circulation 2001, 104, 3158–3167. [Google Scholar] [CrossRef] [Green Version]
- Yellon, D.M.; Downey, J.M. Preconditioning the myocardium: From cellular physiology to clinical cardiology. Physiol. Rev. 2003, 83, 1113–1151. [Google Scholar] [CrossRef]
- Yellon, D.M.; Hausenloy, D.J. Myocardial reperfusion injury. N. Engl. J. Med. 2007, 357, 1121–1135. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Bi, Y.; Sowers, J.R.; Hetz, C.; Zhang, Y. Endoplasmic reticulum stress and unfolded protein response in cardiovascular diseases. Nat. Rev. Cardiol. 2021, 18, 499–521. [Google Scholar] [CrossRef]
- Hetz, C.; Zhang, K.; Kaufman, R.J. Mechanisms, regulation and functions of the unfolded protein response. Nat. Rev. Mol. Cell Biol. 2020, 21, 421–438. [Google Scholar] [CrossRef]
- Xu, C.; Bailly-Maitre, B.; Reed, J.C. Endoplasmic reticulum stress: Cell life and death decisions. J. Clin. Investig. 2005, 115, 2656–2664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amen, O.M.; Sarker, S.D.; Ghildyal, R.; Arya, A. Endoplasmic Reticulum Stress Activates Unfolded Protein Response Signaling and Mediates Inflammation, Obesity, and Cardiac Dysfunction: Therapeutic and Molecular Approach. Front. Pharmacol. 2019, 10, 977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, J.; Holmgren, A. Selenoproteins. J. Biol. Chem. 2009, 284, 723–727. [Google Scholar] [CrossRef] [Green Version]
- Pitts, M.W.; Hoffmann, P.R. Endoplasmic reticulum-resident selenoproteins as regulators of calcium signaling and homeostasis. Cell Calcium 2018, 70, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Kalogeris, T.; Baines, C.P.; Krenz, M.; Korthuis, R.J. Cell biology of ischemia/reperfusion injury. Int. Rev. Cell Mol. Biol. 2012, 298, 229–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rocca, C.; Pasqua, T.; Boukhzar, L.; Anouar, Y.; Angelone, T. Progress in the emerging role of selenoproteins in cardiovascular disease: Focus on endoplasmic reticulum-resident selenoproteins. Cell. Mol. Life Sci. 2019, 76, 3969–3985. [Google Scholar] [CrossRef] [PubMed]
- Anouar, Y.; Lihrmann, I.; Falluel-Morel, A.; Boukhzar, L. Selenoprotein T is a key player in ER proteostasis, endocrine homeostasis and neuroprotection. Free Radic. Biol. Med. 2018, 127, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Rocca, C.; Boukhzar, L.; Granieri, M.C.; Alsharif, I.; Mazza, R.; Lefranc, B.; Tota, B.; Leprince, J.; Cerra, M.C.; Anouar, Y.; et al. A selenoprotein T-derived peptide protects the heart against ischaemia/reperfusion injury through inhibition of apoptosis and oxidative stress. Acta Physiol. 2018, 223, e13067. [Google Scholar] [CrossRef]
- Hamieh, A.; Cartier, D.; Abid, H.; Calas, A.; Burel, C.; Bucharles, C.; Jehan, C.; Grumolato, L.; Landry, M.; Lerouge, P.; et al. Selenoprotein T is a novel OST subunit that regulates UPR signaling and hormone secretion. EMBO Rep. 2017, 18, 1935–1946. [Google Scholar] [CrossRef] [PubMed]
- Alsharif, I.; Boukhzar, L.; Lefranc, B.; Godefroy, D.; Aury-Landas, J.; Rego, J.D.; Rego, J.D.; Naudet, F.; Arabo, A.; Chagraoui, A.; et al. Cell-penetrating, antioxidant SELENOT mimetic protects dopaminergic neurons and ameliorates motor dysfunction in Parkinson’s disease animal models. Redox Biol. 2021, 40, 101839. [Google Scholar] [CrossRef]
- Rocca, C.; Scavello, F.; Colombo, B.; Gasparri, A.M.; Dallatomasina, A.; Granieri, M.C.; Amelio, D.; Pasqua, T.; Cerra, M.C.; Tota, B.; et al. Physiological levels of chromogranin A prevent doxorubicin-induced cardiotoxicity without impairing its anticancer activity. FASEB J. 2019, 33, 7734–7747. [Google Scholar] [CrossRef]
- Penna, C.; Tullio, F.; Femminò, S.; Rocca, C.; Angelone, T.; Cerra, M.C.; Gallo, M.P.; Gesmundo, I.; Fanciulli, A.; Brizzi, M.F.; et al. Obestatin regulates cardiovascular function and promotes cardioprotection through the nitric oxide pathway. J. Cell. Mol. Med. 2017, 21, 3670–3678. [Google Scholar] [CrossRef] [PubMed]
- Rocca, C.; Scavello, F.; Granieri, M.C.; Pasqua, T.; Amodio, N.; Imbrogno, S.; Gattuso, A.; Mazza, R.; Cerra, M.C.; Angelone, T. Phoenixin-14: Detection and novel physiological implications in cardiac modulation and cardioprotection. Cell. Mol. Life Sci. 2018, 75, 743–756. [Google Scholar] [CrossRef]
- Wu, H.; Ye, M.; Yang, J.; Ding, J.; Yang, J.; Dong, W.; Wang, X. Nicorandil Protects the Heart from Ischemia/Reperfusion Injury by Attenuating Endoplasmic Reticulum Response-induced Apoptosis Through PI3K/Akt Signaling Pathway. Cell. Physiol. Biochem. 2015, 35, 2320–2332. [Google Scholar] [CrossRef]
- Morrison, R.R.; Jones, R.; Byford, A.M.; Stell, A.R.; Peart, J.; Headrick, J.P.; Matherne, G.P. Transgenic overexpression of cardiac A(1) adenosine receptors mimics ischemic preconditioning. Am. J. Physiol. Heart Circ. Physiol. 2000, 279, H1071–H1078. [Google Scholar] [CrossRef] [PubMed]
- McQueen, M.J. Optimal Assay of LDH and a-HBD at 37 °C. Ann. Clin. Biochem. 1972, 9, 21–25. [Google Scholar] [CrossRef] [Green Version]
- Marklund, S.; Marklund, G. Involvement of the superoxide anion radical in the autoxidation of pyrogallol and a convenient assay for superoxide dismutase. Eur. J. Biochem. 1974, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Claiborne, A. Catalase activity. In CRC Handbook of Methods for Oxygen Radical Research; Greenwald, R.A., Ed.; CRC Press: Boca Raton, FL, USA, 1985; pp. 283–284. [Google Scholar]
- Li, T.; Danelisen, I.; Belló-Klein, A.; Singal, P.K. Effects of probucol on changes of antioxidant enzymes in adriamycin-induced cardiomyopathy in rats. Cardiovasc. Res. 2000, 46, 523–530. [Google Scholar] [CrossRef] [Green Version]
- Aust, S.D. Lipid peroxidation. In Handbook of Methods for Oxygen Radical Research; Greenwald, R.A., Ed.; CRC Press: Boca Raton, FL, USA, 1985; pp. 203–207. [Google Scholar]
- Reznick, A.Z.; Packer, L. Oxidative damage to proteins: Spectrophotometric method for carbonyl assay. Methods Enzymol. 1994, 233, 357–363. [Google Scholar] [CrossRef]
- Assimakopoulos, S.F.; Vagianos, C.E.; Zervoudakis, G.; Filos, K.S.; Georgiou, C.; Nikolopoulou, V.; Scopa, C.D. Gut regulatory peptides bombesin and neurotensin reduce hepatic oxidative stress and histological alterations in bile duct ligated rats. Regul. Pept. 2004, 120, 185–193. [Google Scholar] [CrossRef]
- Pasqua, T.; Rocca, C.; Lupi, F.R.; Baldino, N.; Amelio, D.; Parisi, O.I.; Granieri, M.C.; De Bartolo, A.; Lauria, A.; Dattilo, M.; et al. Cardiac and Metabolic Impact of Functional Foods with Antioxidant Properties Based on Whey Derived Proteins Enriched with Hemp Seed Oil. Antioxidants 2020, 9, 1066. [Google Scholar] [CrossRef]
- Rocca, C.; Grande, F.; Granieri, M.C.; Colombo, B.; De Bartolo, A.; Giordano, F.; Rago, V.; Amodio, N.; Tota, B.; Cerra, M.C.; et al. The chromogranin A1-373 fragment reveals how a single change in the protein sequence exerts strong cardioregulatory effects by engaging neuropilin-1. Acta Physiol. 2021, 231, e13570. [Google Scholar] [CrossRef] [PubMed]
- Rocca, C.; De Bartolo, A.; Grande, F.; Rizzuti, B.; Pasqua, T.; Giordano, F.; Granieri, M.C.; Occhiuzzi, M.A.; Garofalo, A.; Amodio, N.; et al. Cateslytin abrogates lipopolysaccharide-induced cardiomyocyte injury by reducing inflammation and oxidative stress through toll like receptor 4 interaction. Int. Immunopharmacol. 2021, 94, 107487. [Google Scholar] [CrossRef] [PubMed]
- Grande, F.; De Bartolo, A.; Occhiuzzi, M.A.; Caruso, A.; Rocca, C.; Pasqua, T.; Carocci, A.; Rago, V.; Angelone, T.; Sinicropi, M.S. Carbazole and Simplified Derivatives: Novel Tools toward b-Adrenergic Receptors Targeting. Appl. Sci. 2021, 11, 5486. [Google Scholar] [CrossRef]
- Rocca, C.; Femminò, S.; Aquila, G.; Granieri, M.C.; De Francesco, E.M.; Pasqua, T.; Rigiracciolo, D.C.; Fortini, F.; Cerra, M.C.; Maggiolini, M.; et al. Notch1 Mediates Preconditioning Protection Induced by GPER in Normotensive and Hypertensive Female Rat Hearts. Front. Physiol. 2018, 9, 521. [Google Scholar] [CrossRef]
- Nettore, I.C.; Rocca, C.; Mancino, G.; Albano, L.; Amelio, D.; Grande, F.; Puoci, F.; Pasqua, T.; Desiderio, S.; Mazza, R.; et al. Quercetin and its derivative Q2 modulate chromatin dynamics in adipogenesis and Q2 prevents obesity and metabolic disorders in rats. J. Nutr. Biochem. 2019, 69, 151–162. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Liu, Y.; Li, J.Y.; Li, L.Z.; Zhang, Y.L.; Gong, H.Y.; Cui, Y. Protective Effect of Rosamultin against H2O2-Induced Oxidative Stress and Apoptosis in H9c2 Cardiomyocytes. Oxid. Med. Cell. Longev. 2018, 2018, 8415610. [Google Scholar] [CrossRef] [Green Version]
- Murry, C.E.; Jennings, R.B.; Reimer, K.A. Preconditioning with ischemia: A delay of lethal cell injury in ischemic myocardium. Circulation 1986, 74, 1124–1136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Z.Q.; Corvera, J.S.; Halkos, M.E.; Kerendi, F.; Wang, N.P.; Guyton, R.A.; Vinten-Johansen, J. Inhibition of myocardial injury by ischemic postconditioning during reperfusion: Comparison with ischemic preconditioning. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H579–H588. [Google Scholar] [CrossRef]
- Przyklenk, K.; Bauer, B.; Ovize, M.; Kloner, R.A.; Whittaker, P. Regional ischemic ‘preconditioning’ protects remote virgin myocardium from subsequent sustained coronary occlusion. Circulation 1993, 87, 893–899. [Google Scholar] [CrossRef] [Green Version]
- Hausenloy, D.J.; Garcia-Dorado, D.; Bøtker, H.E.; Davidson, S.M.; Downey, J.; Engel, F.B.; Jennings, R.; Lecour, S.; Leor, J.; Madonna, R.; et al. Novel targets and future strategies for acute cardioprotection: Position Paper of the European Society of Cardiology Working Group on Cellular Biology of the Heart. Cardiovasc. Res. 2017, 113, 564–585. [Google Scholar] [CrossRef] [Green Version]
- Pothion, H.; Jehan, C.; Tostivint, H.; Cartier, D.; Bucharles, C.; Falluel-Morel, A.; Boukhzar, L.; Anouar, Y.; Lihrmann, I. Selenoprotein T: An Essential Oxidoreductase Serving as a Guardian of Endoplasmic Reticulum Homeostasis. Antioxid. Redox Signal. 2020, 33, 1257–1275. [Google Scholar] [CrossRef]
- Heitz, F.; Morris, M.C.; Divita, G. Twenty years of cell-penetrating peptides: From molecular mechanisms to therapeutics. Br. J. Pharmacol. 2009, 157, 195–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boukhzar, L.; Hamieh, A.; Cartier, D.; Tanguy, Y.; Alsharif, I.; Castex, M.; Arabo, A.; El Hajji, S.; Bonnet, J.J.; Errami, M.; et al. Selenoprotein T Exerts an Essential Oxidoreductase Activity That Protects Dopaminergic Neurons in Mouse Models of Parkinson’s Disease. Antioxid. Redox Signal. 2016, 24, 557–574. [Google Scholar] [CrossRef] [PubMed]
- Prevost, G.; Arabo, A.; Jian, L.; Quelennec, E.; Cartier, D.; Hassan, S.; Falluel-Morel, A.; Tanguy, Y.; Gargani, S.; Lihrmann, I.; et al. The PACAP-regulated gene selenoprotein T is abundantly expressed in mouse and human β-cells and its targeted inactivation impairs glucose tolerance. Endocrinology 2013, 154, 3796–3806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanguy, Y.; Falluel-Morel, A.; Arthaud, S.; Boukhzar, L.; Manecka, D.L.; Chagraoui, A.; Prevost, G.; Elias, S.; Dorval-Coiffec, I.; Lesage, J.; et al. The PACAP-regulated gene selenoprotein T is highly induced in nervous, endocrine, and metabolic tissues during ontogenetic and regenerative processes. Endocrinology 2011, 152, 4322–4335. [Google Scholar] [CrossRef] [Green Version]
- Kim, I.; Xu, W.; Reed, J.C. Cell death and endoplasmic reticulum stress: Disease relevance and therapeutic opportunities. Nat. Rev. Drug Discov. 2008, 7, 1013–1030. [Google Scholar] [CrossRef]
- Bell, R.M.; Mocanu, M.M.; Yellon, D.M. Retrograde heart perfusion: The Langendorff technique of isolated heart perfusion. J. Mol. Cell. Cardiol. 2011, 50, 940–950. [Google Scholar] [CrossRef] [PubMed]
- Hakulinen, J.K.; Hering, J.; Brändén, G.; Chen, H.; Snijder, A.; Ek, M.; Johansson, P. MraY-antibiotic complex reveals details of tunicamycin mode of action. Nat. Chem. Biol. 2017, 13, 265–267. [Google Scholar] [CrossRef] [PubMed]
- Gibbons, R.J.; Valeti, U.S.; Araoz, P.A.; Jaffe, A.S. The quantification of infarct size. J. Am. Coll. Cardiol. 2004, 44, 1533–1542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doroudgar, S.; Thuerauf, D.J.; Marcinko, M.C.; Belmont, P.J.; Glembotski, C.C. Ischemia activates the ATF6 branch of the endoplasmic reticulum stress response. J. Biol. Chem. 2009, 284, 29735–29745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glembotski, C.C. Roles for ATF6 and the sarco/endoplasmic reticulum protein quality control system in the heart. J. Mol. Cell. Cardiol. 2014, 71, 11–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szegezdi, E.; Logue, S.E.; Gorman, A.M.; Samali, A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 2006, 7, 880–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glembotski, C.C. Endoplasmic reticulum stress in the heart. Circ. Res. 2007, 101, 975–984. [Google Scholar] [CrossRef]
- Kurian, G.A.; Rajagopal, R.; Vedantham, S.; Rajesh, M. The Role of Oxidative Stress in Myocardial Ischemia and Reperfusion Injury and Remodeling: Revisited. Oxid. Med. Cell. Longev. 2016, 2016, 1656450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forman, H.J.; Zhang, H. Targeting oxidative stress in disease: Promise and limitations of antioxidant therapy. Nat. Rev. Drug Discov. 2021, 20, 689–709. [Google Scholar] [CrossRef]
- Hescheler, J.; Meyer, R.; Plant, S.; Krautwurst, D.; Rosenthal, W.; Schultz, G. Morphological, biochemical, and electrophysiological characterization of a clonal cell (H9c2) line from rat heart. Circ. Res. 1991, 69, 1476–1486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Branco, A.F.; Pereira, S.P.; Gonzalez, S.; Gusev, O.; Rizvanov, A.A.; Oliveira, P.J. Gene Expression Profiling of H9c2 Myoblast Differentiation towards a Cardiac-Like Phenotype. PLoS ONE 2015, 10, e0129303. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rocca, C.; De Bartolo, A.; Granieri, M.C.; Rago, V.; Amelio, D.; Falbo, F.; Malivindi, R.; Mazza, R.; Cerra, M.C.; Boukhzar, L.; et al. The Antioxidant Selenoprotein T Mimetic, PSELT, Induces Preconditioning-like Myocardial Protection by Relieving Endoplasmic-Reticulum Stress. Antioxidants 2022, 11, 571. https://doi.org/10.3390/antiox11030571
Rocca C, De Bartolo A, Granieri MC, Rago V, Amelio D, Falbo F, Malivindi R, Mazza R, Cerra MC, Boukhzar L, et al. The Antioxidant Selenoprotein T Mimetic, PSELT, Induces Preconditioning-like Myocardial Protection by Relieving Endoplasmic-Reticulum Stress. Antioxidants. 2022; 11(3):571. https://doi.org/10.3390/antiox11030571
Chicago/Turabian StyleRocca, Carmine, Anna De Bartolo, Maria Concetta Granieri, Vittoria Rago, Daniela Amelio, Flavia Falbo, Rocco Malivindi, Rosa Mazza, Maria Carmela Cerra, Loubna Boukhzar, and et al. 2022. "The Antioxidant Selenoprotein T Mimetic, PSELT, Induces Preconditioning-like Myocardial Protection by Relieving Endoplasmic-Reticulum Stress" Antioxidants 11, no. 3: 571. https://doi.org/10.3390/antiox11030571