
Alpha-Tocopherol, a Powerful Molecule, Leads to the Formation of Oxylipins in Polyunsaturated Oils Differently to the Temperature Increase: A Detailed Study by Proton Nuclear Magnetic Resonance of Walnut Oil Oxidation

Abstract
:
1. Introduction
2. Materials and Methods
2.1. Subject Samples of Study
2.2. Storage Conditions
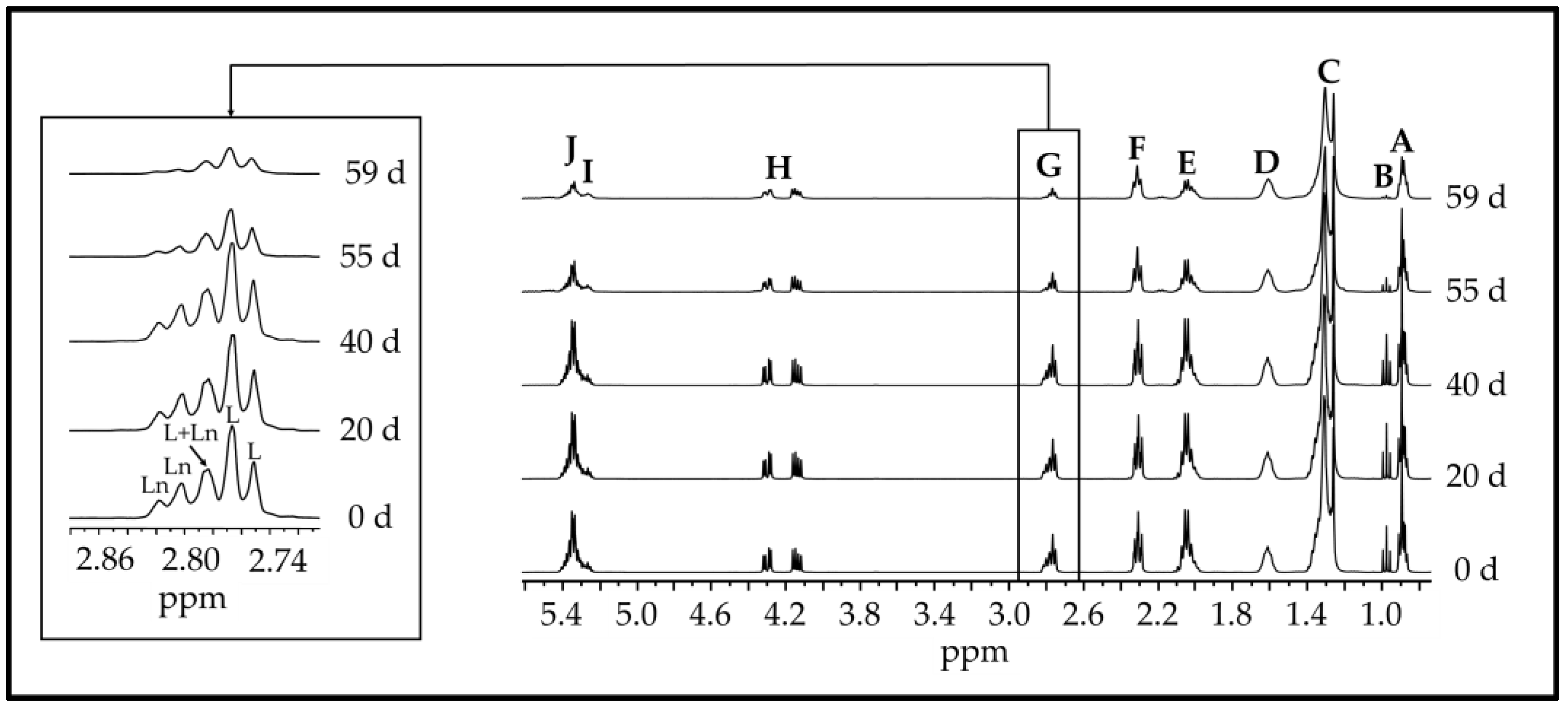
2.3. Study of the Samples Evolution by 1H NMR Spectroscopy
2.3.1. 1H NMR Spectroscopy Operating Conditions
2.3.2. Identification of Components
2.3.3. Quantification of the Components
2.4. Statistical Analysis
3. Results
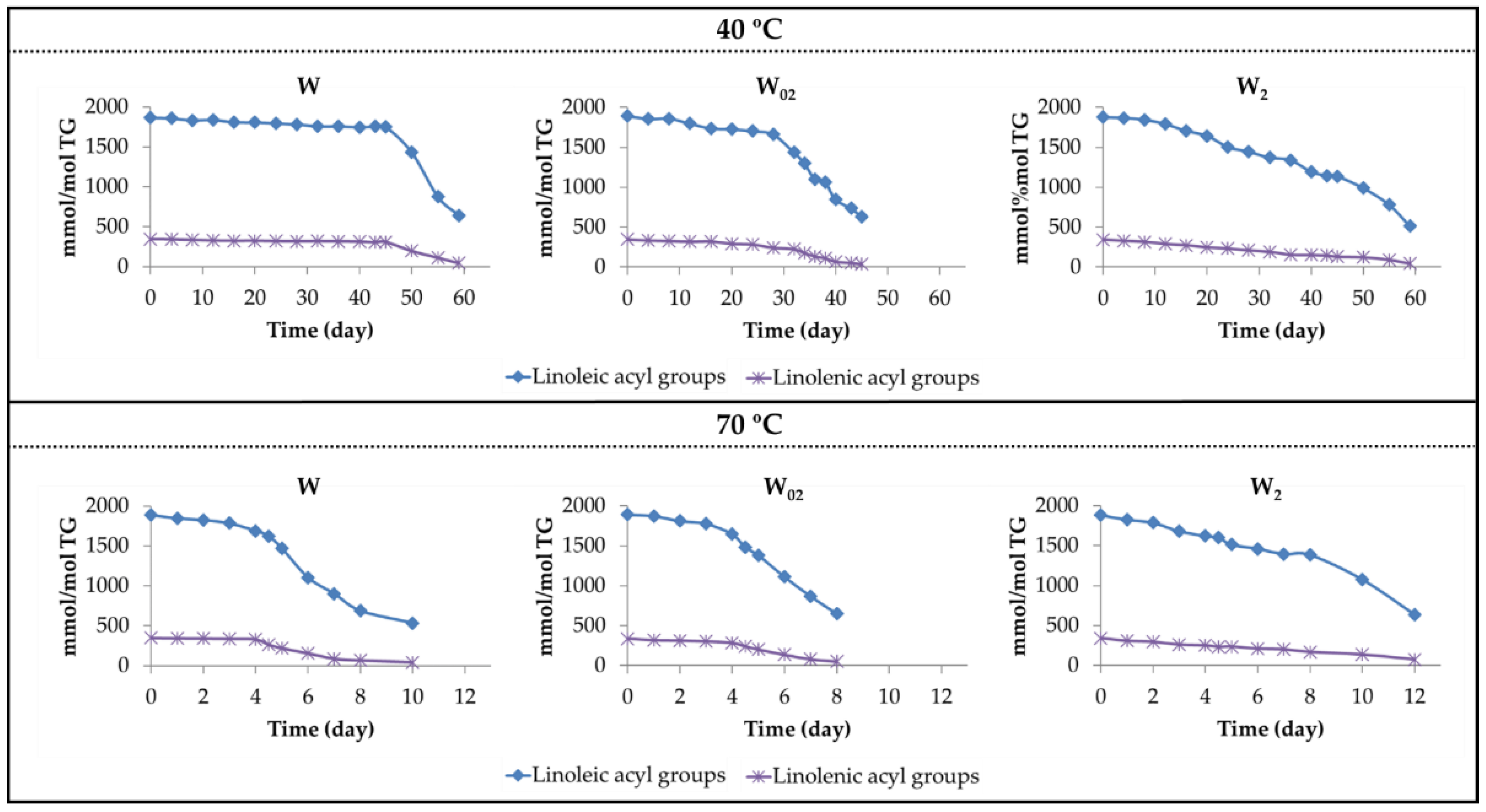
3.1. Evolution of Polyunsaturated Acyl Groups Concentration. Influence of Both Storage Temperature and Enrichment Degree of the Oil in αT
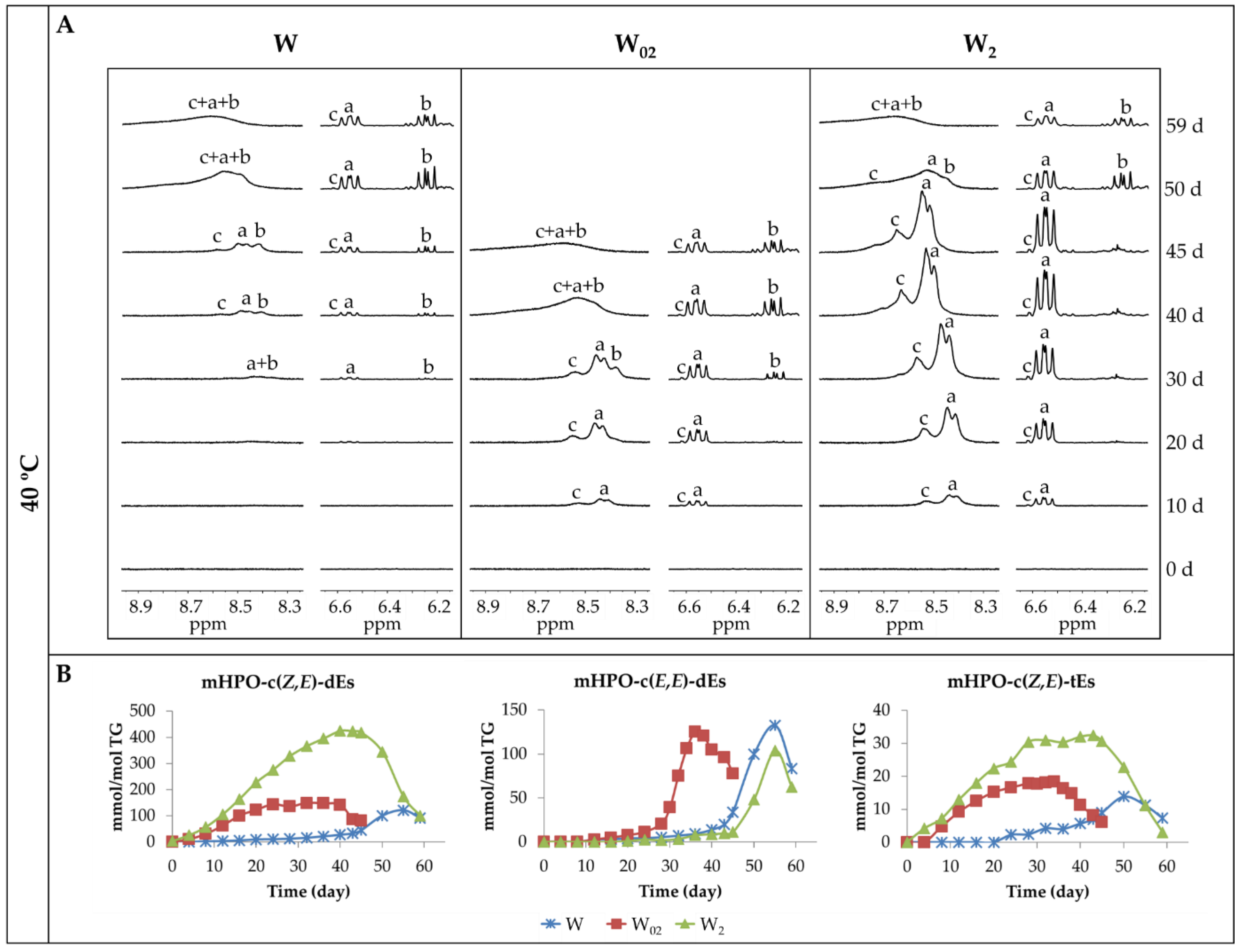
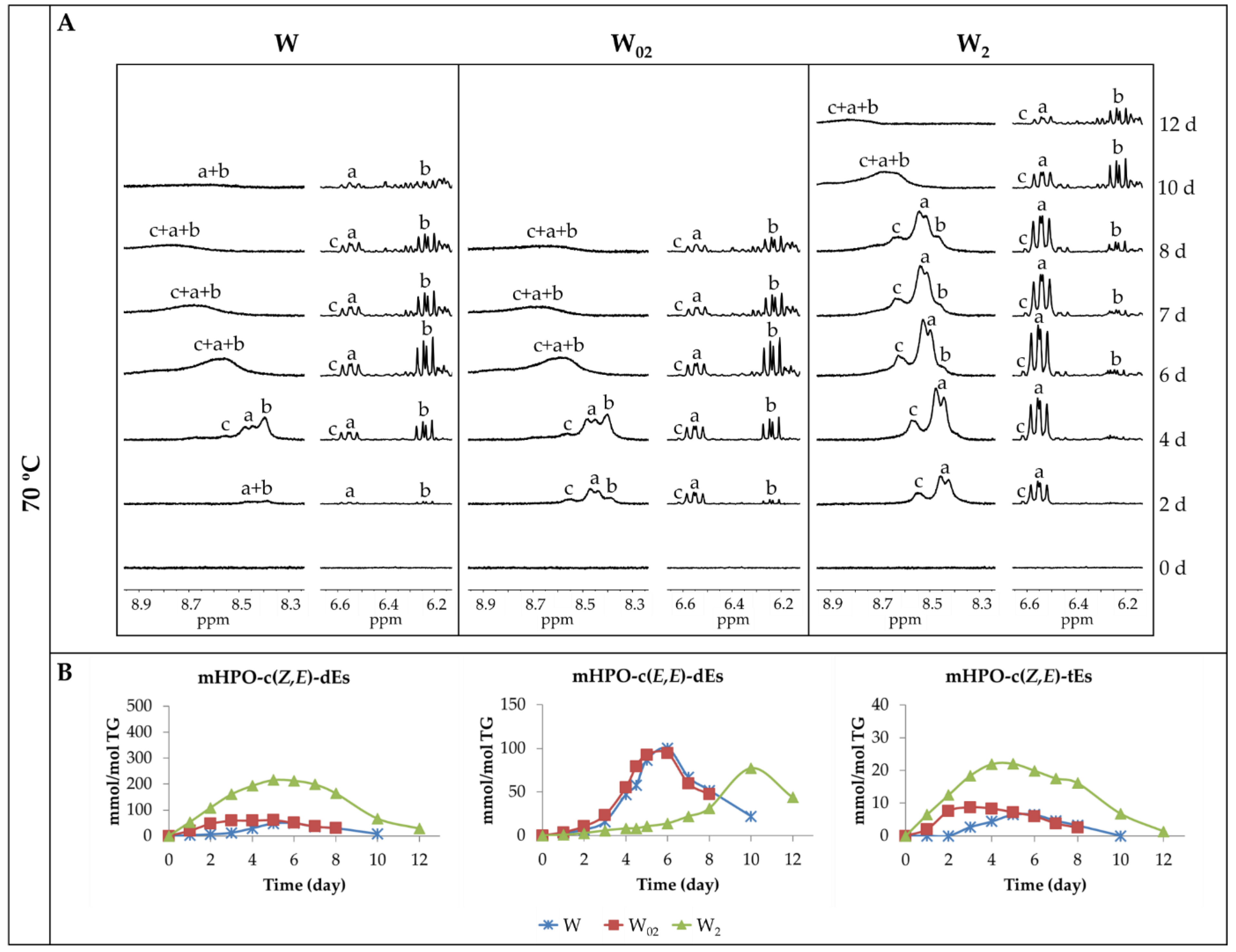
3.2. Formation and Evolution of the Concentration of Oxidation Products. Influence of Both Storage Temperature and Enrichment Degree of the Oil in αT
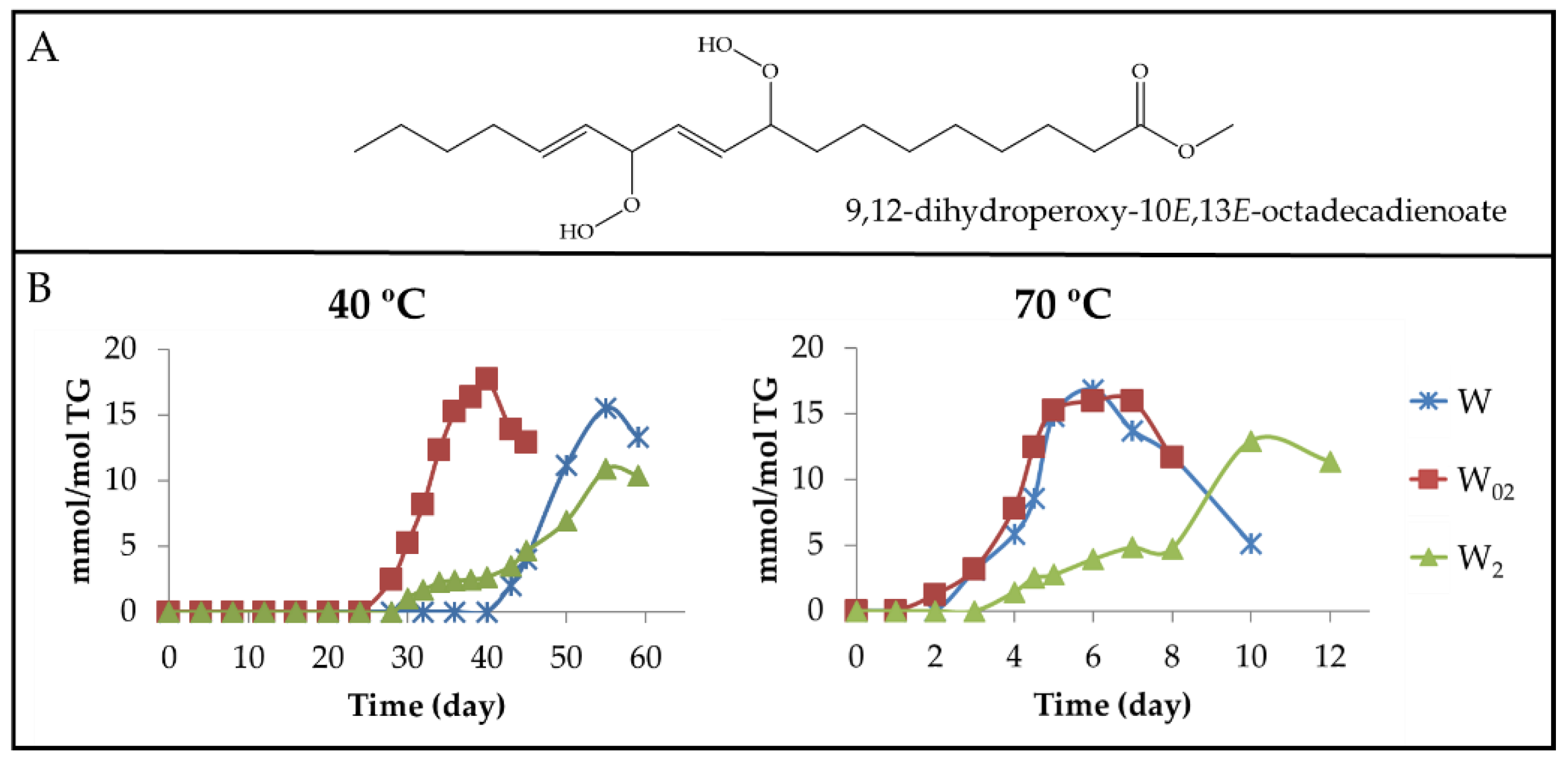
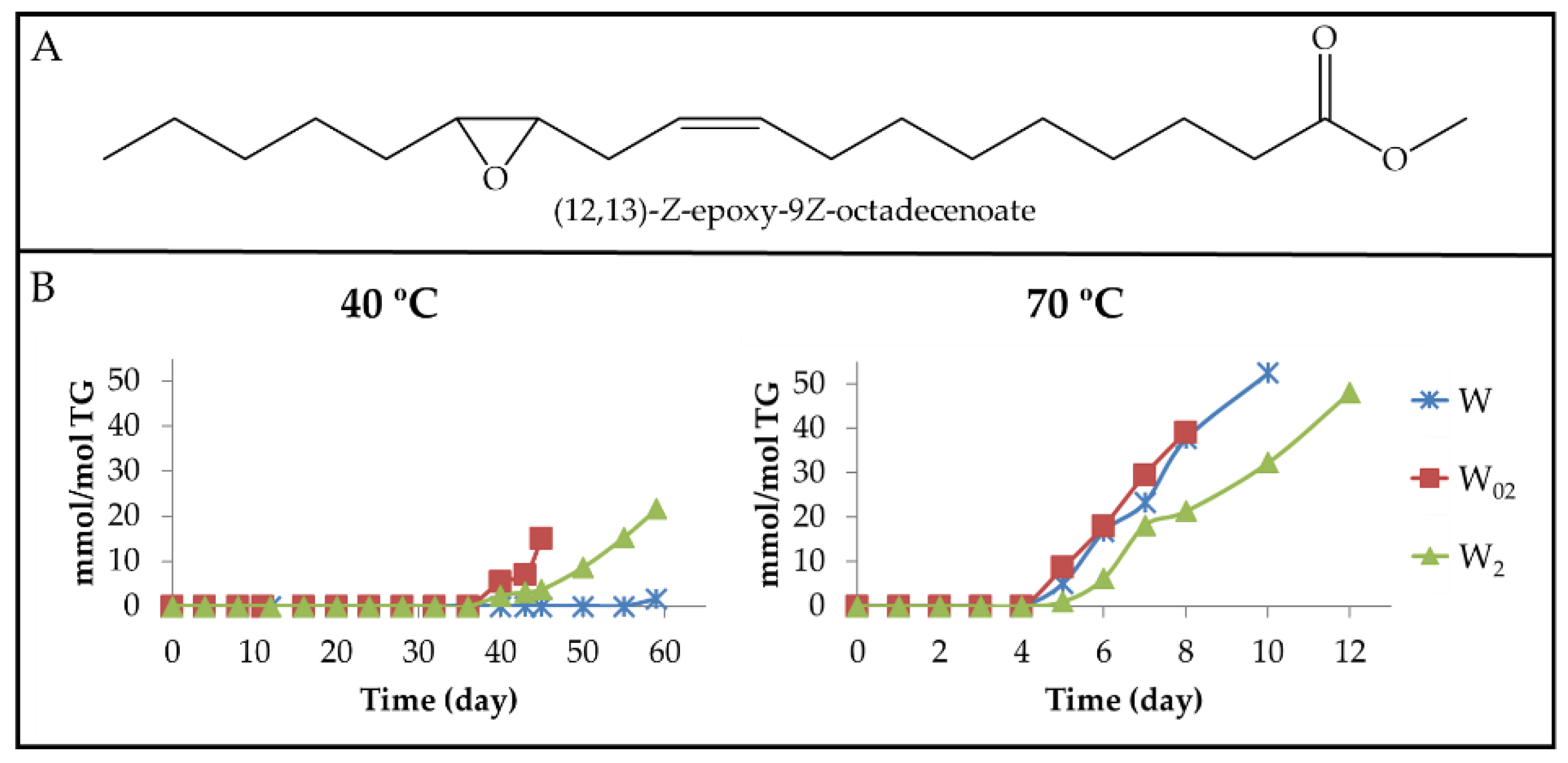
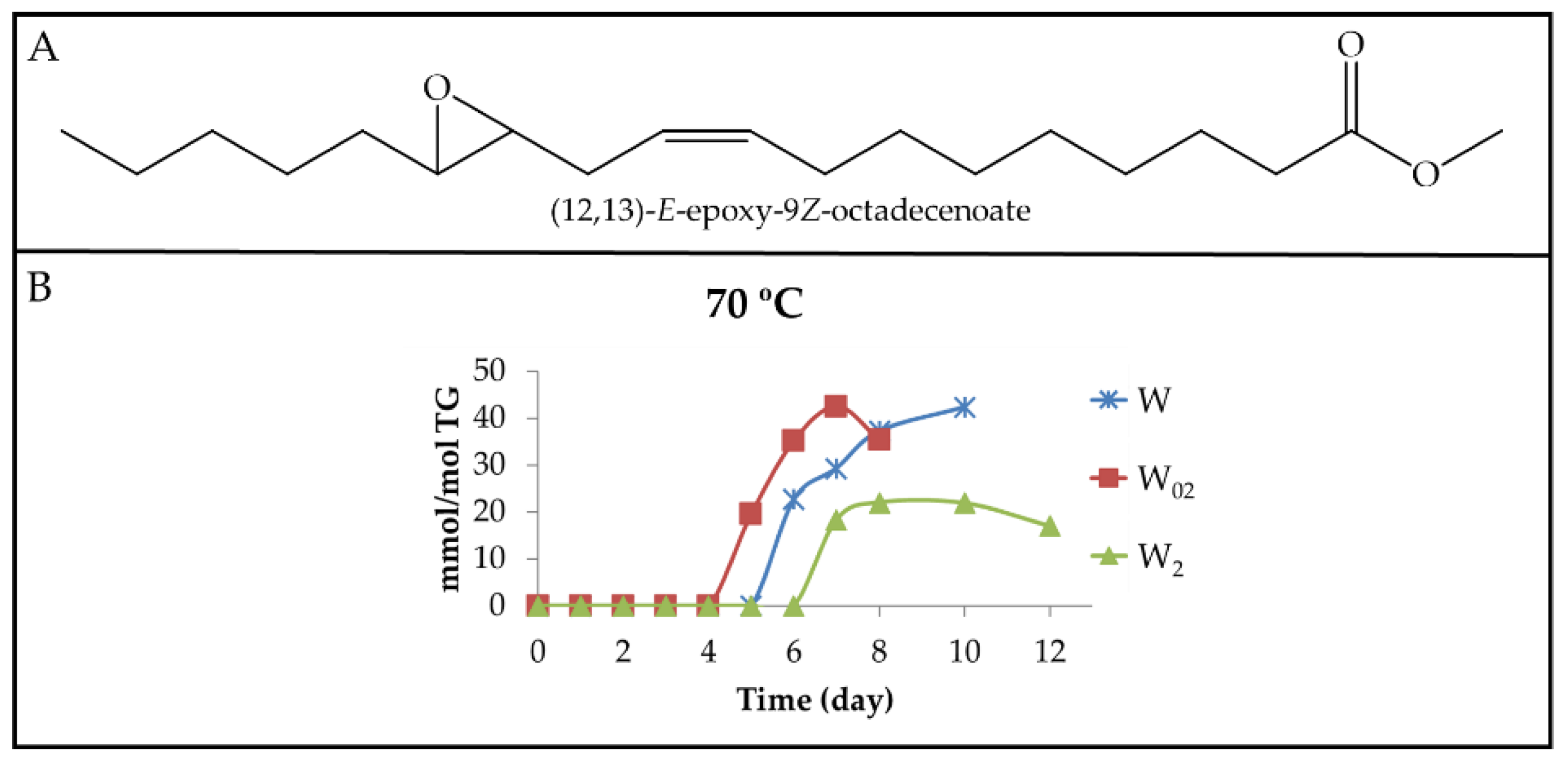
3.2.1. Primary Oxidation Compounds. Long Chains Supporting Hydroperoxy Groups and Conjugated Z,E- or E,E-dienes
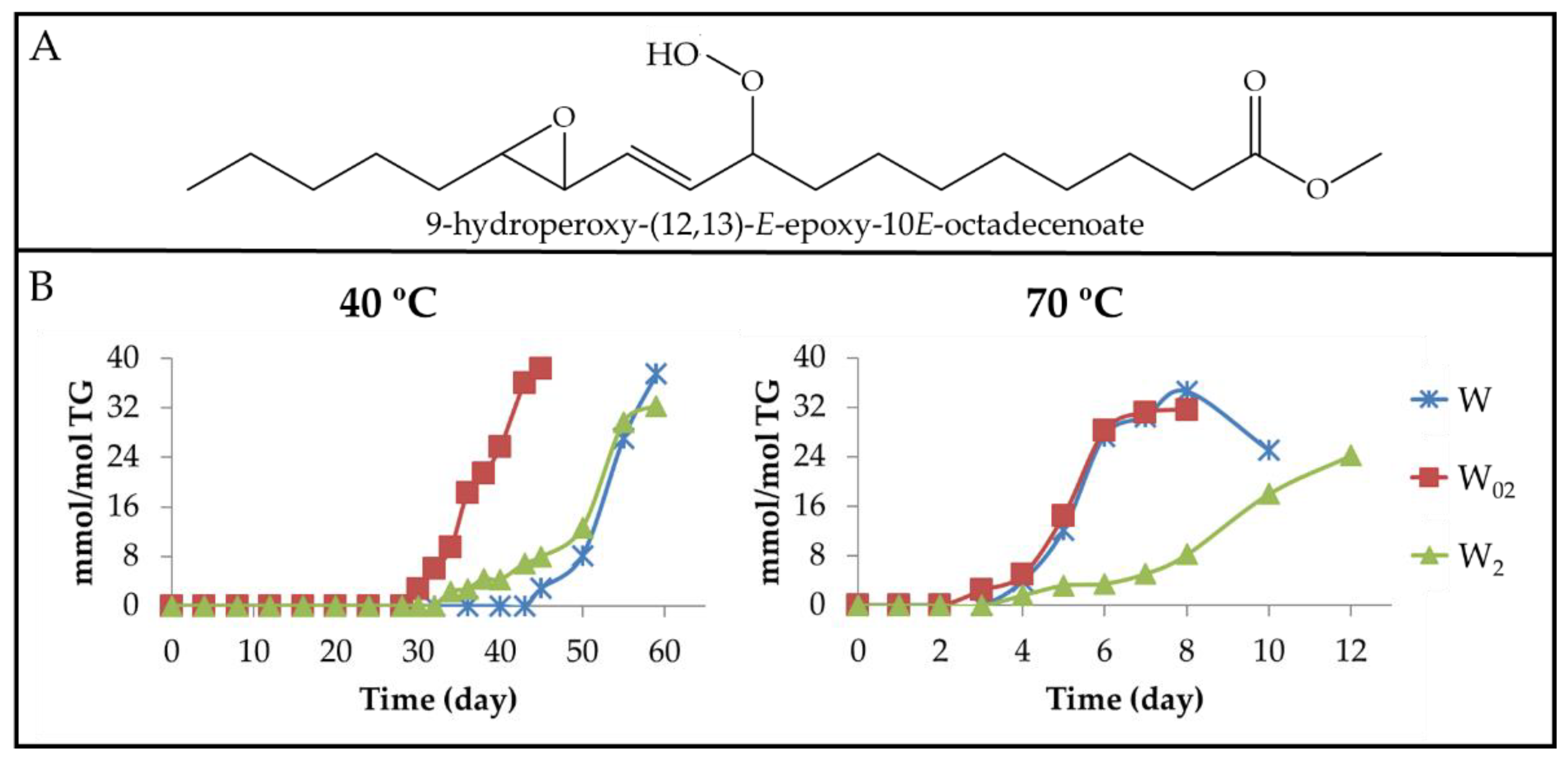
3.2.2. Long-Chain Secondary Oxidation Compounds Supporting Hydroperoxy Groups
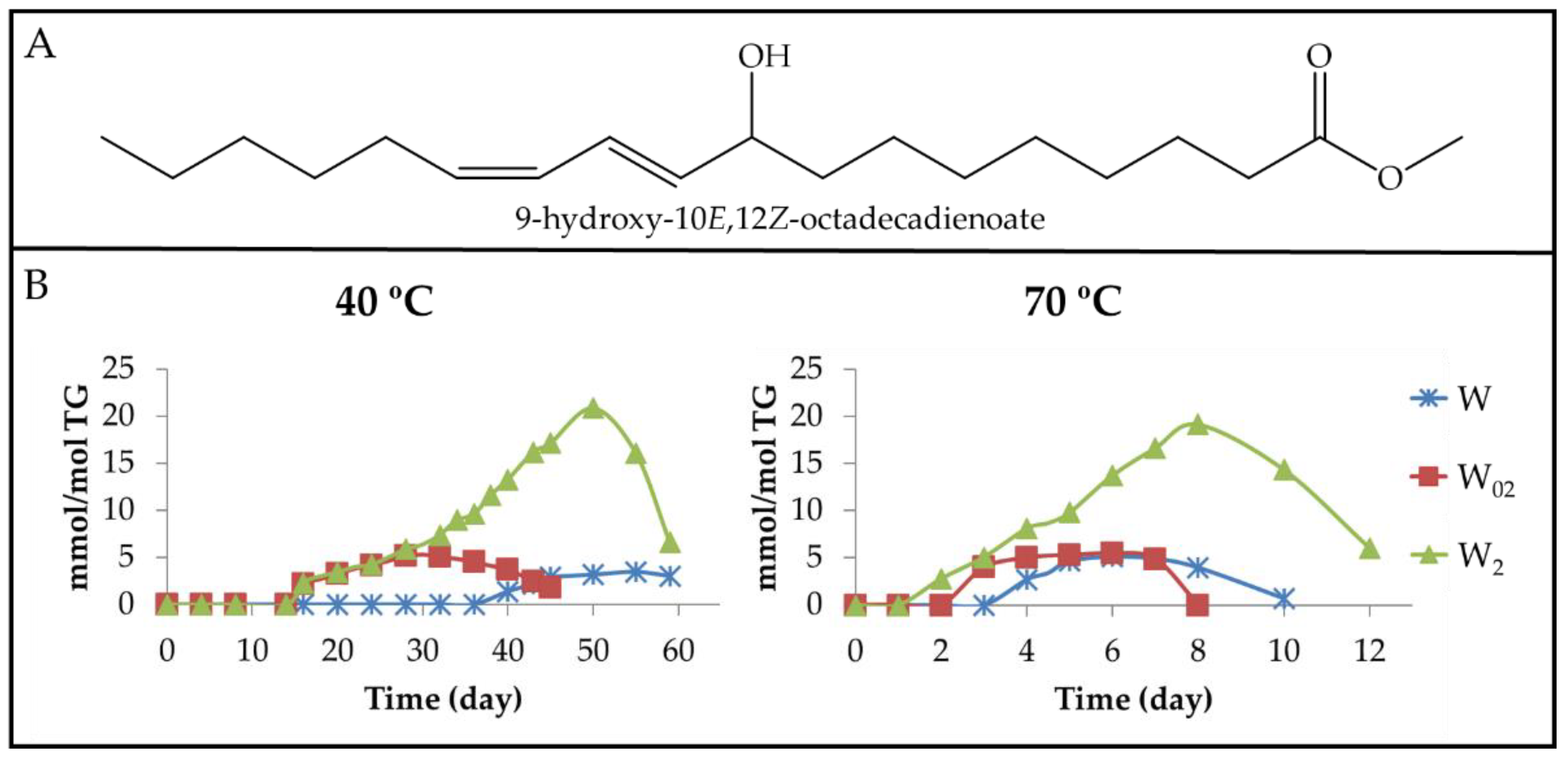
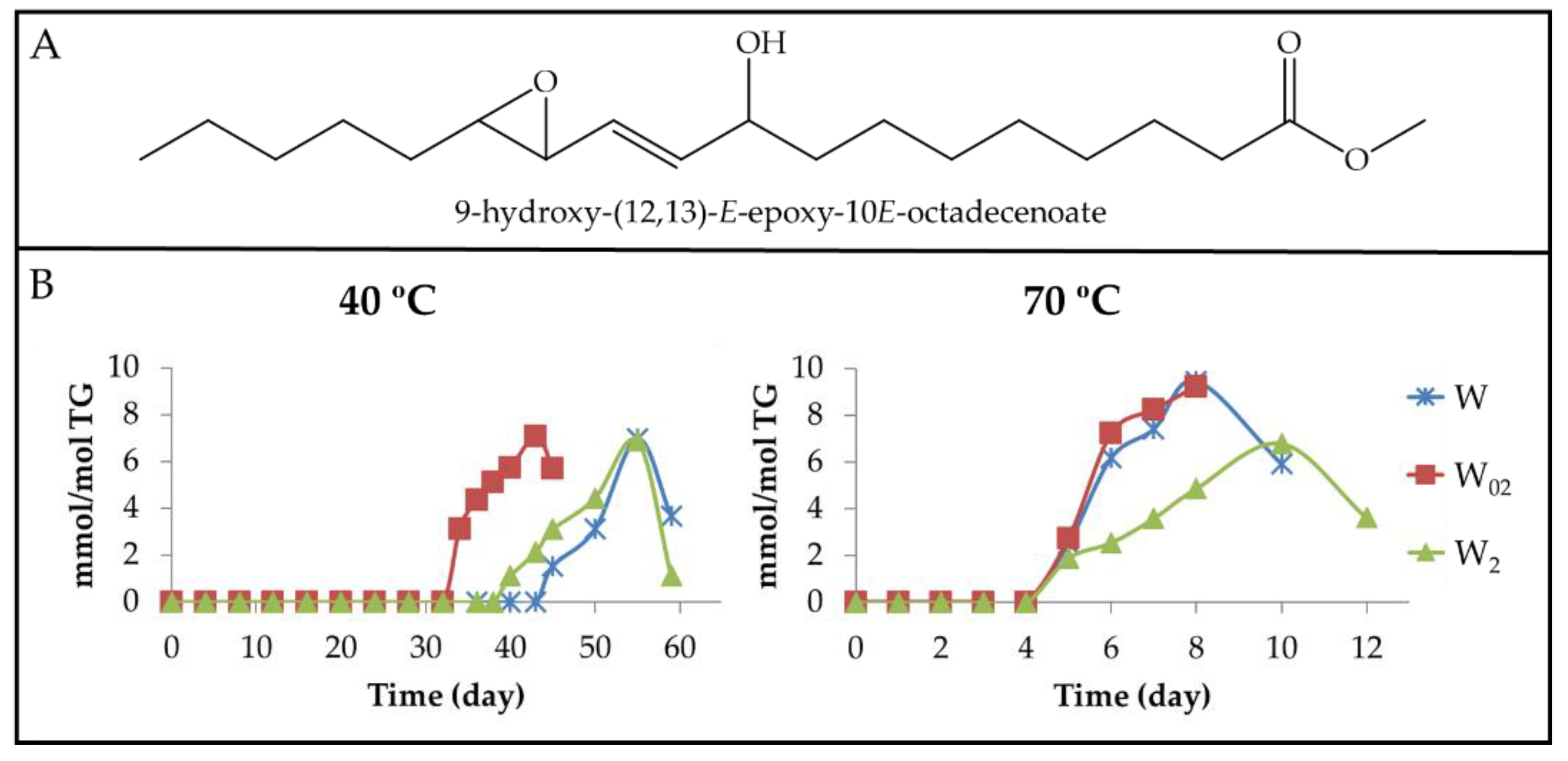
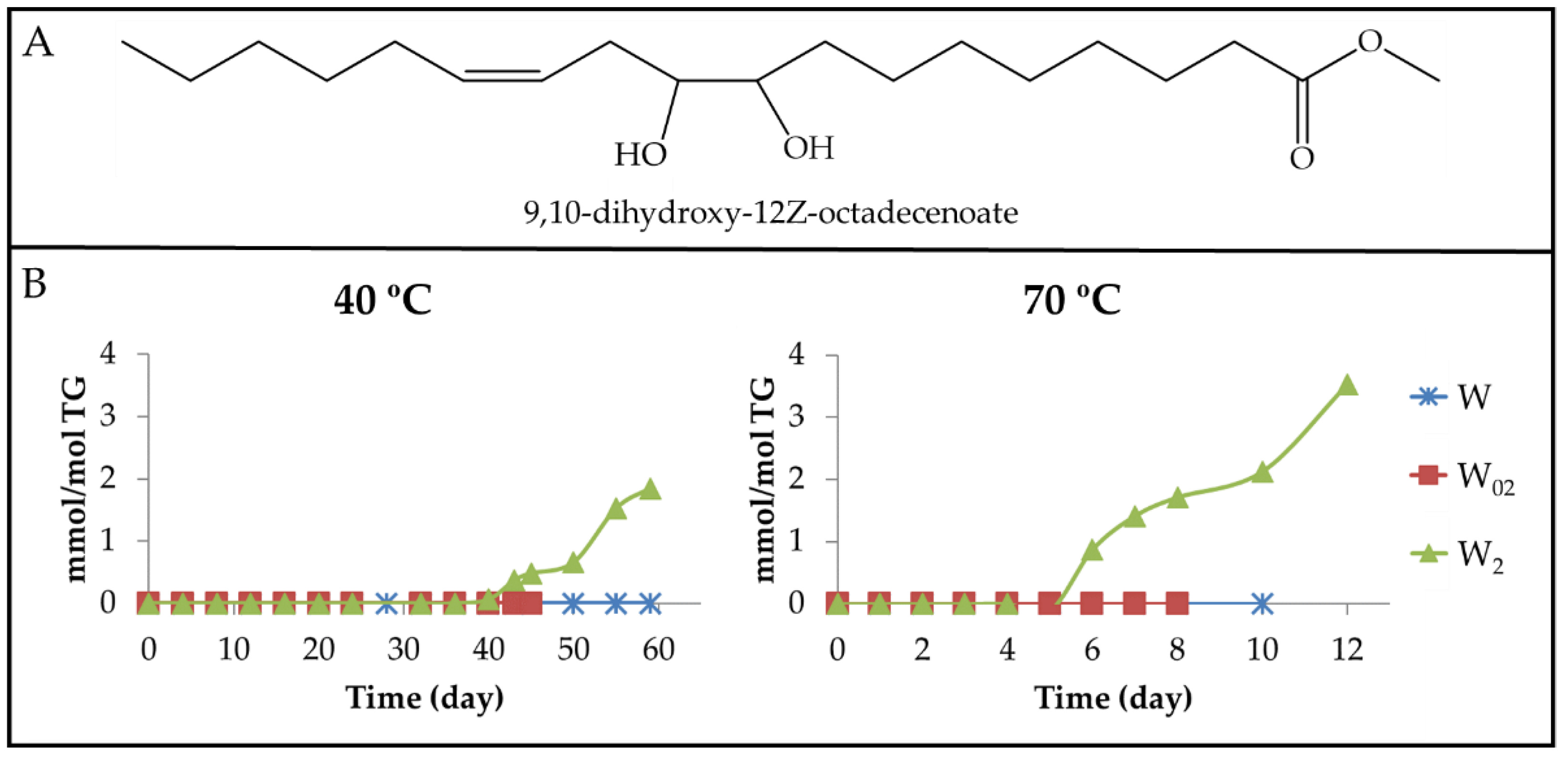
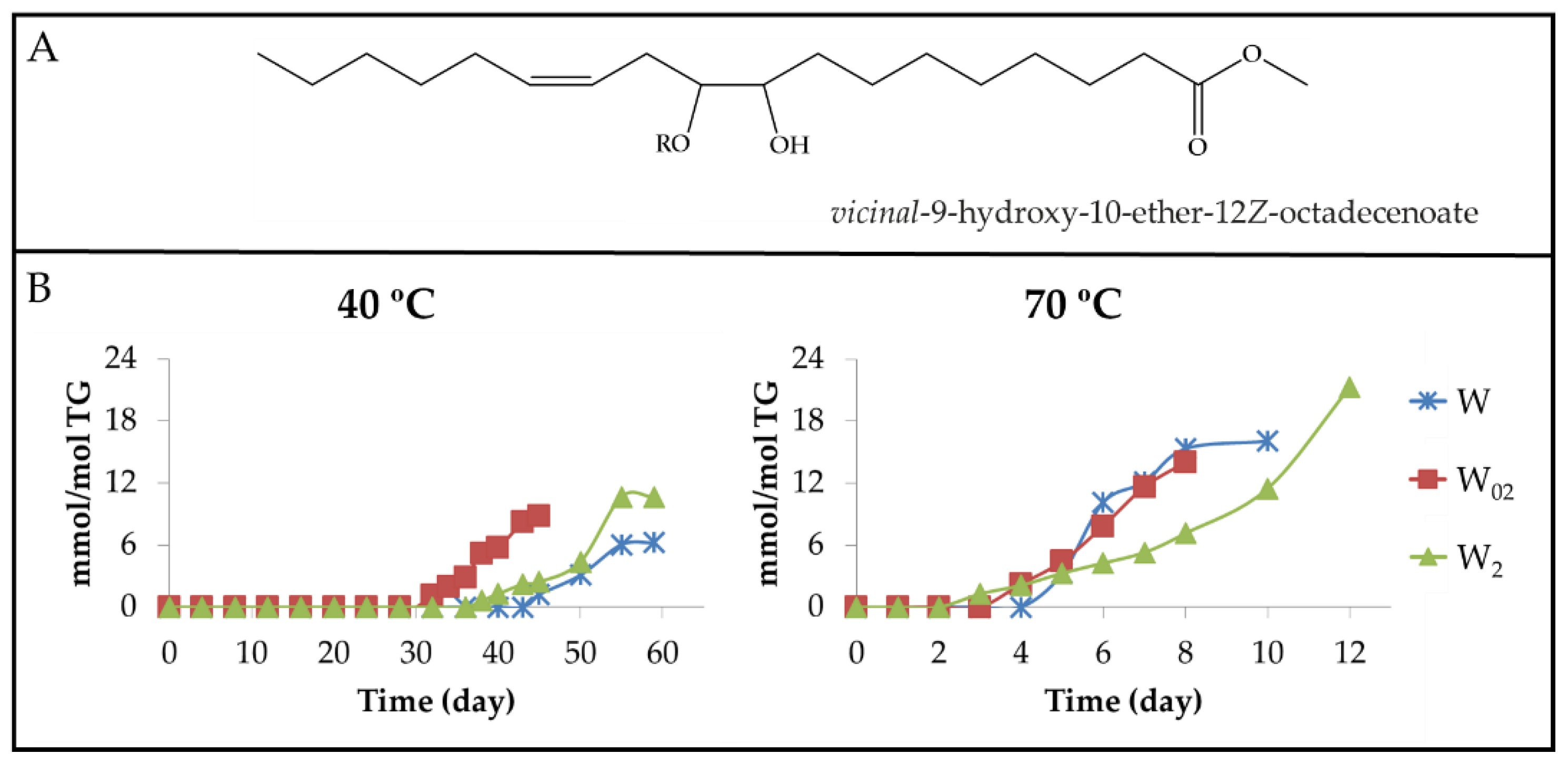
3.2.3. Long-Chain Secondary or Further Oxidation Compounds Supporting a Hydroxy Group
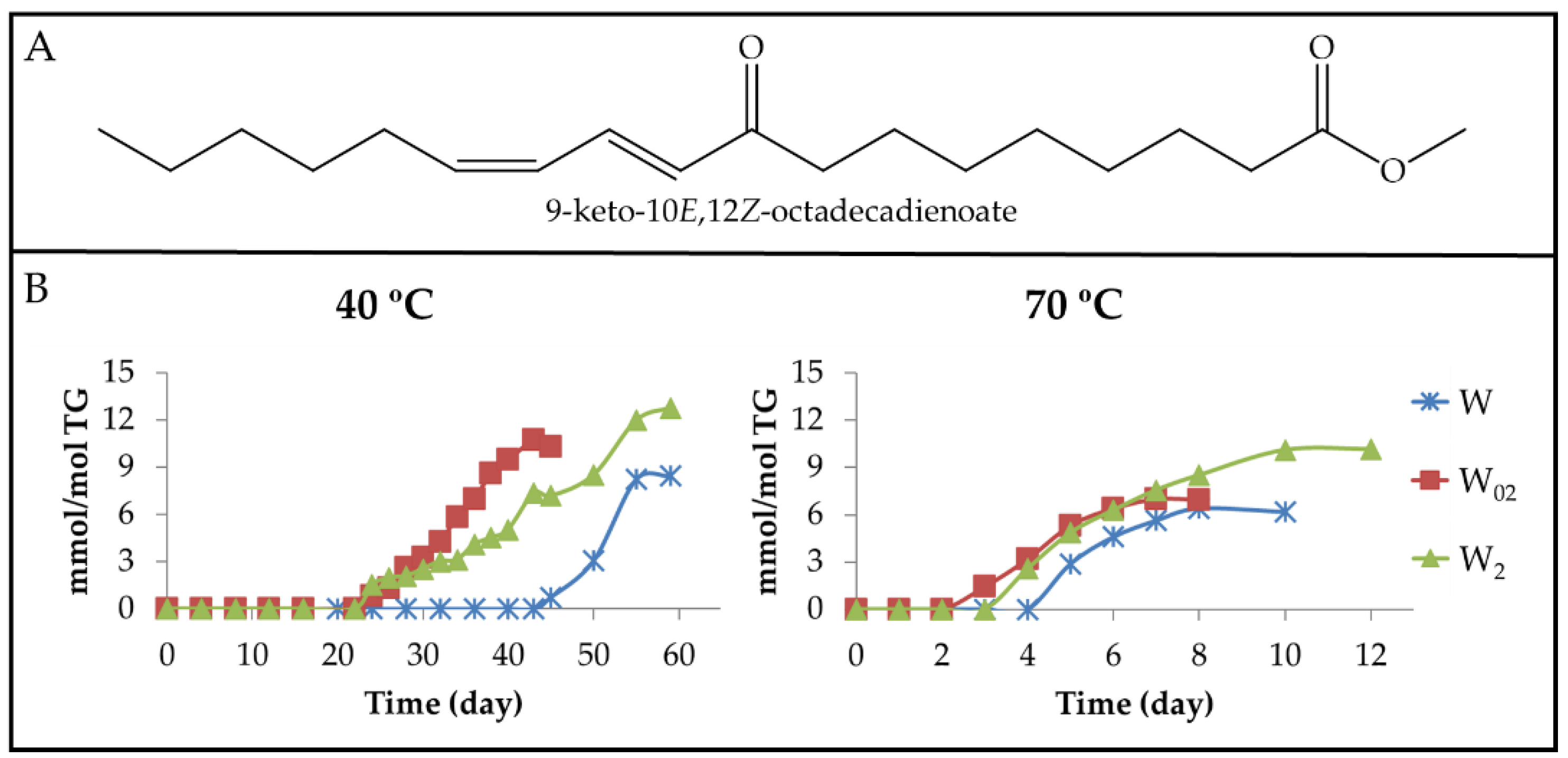
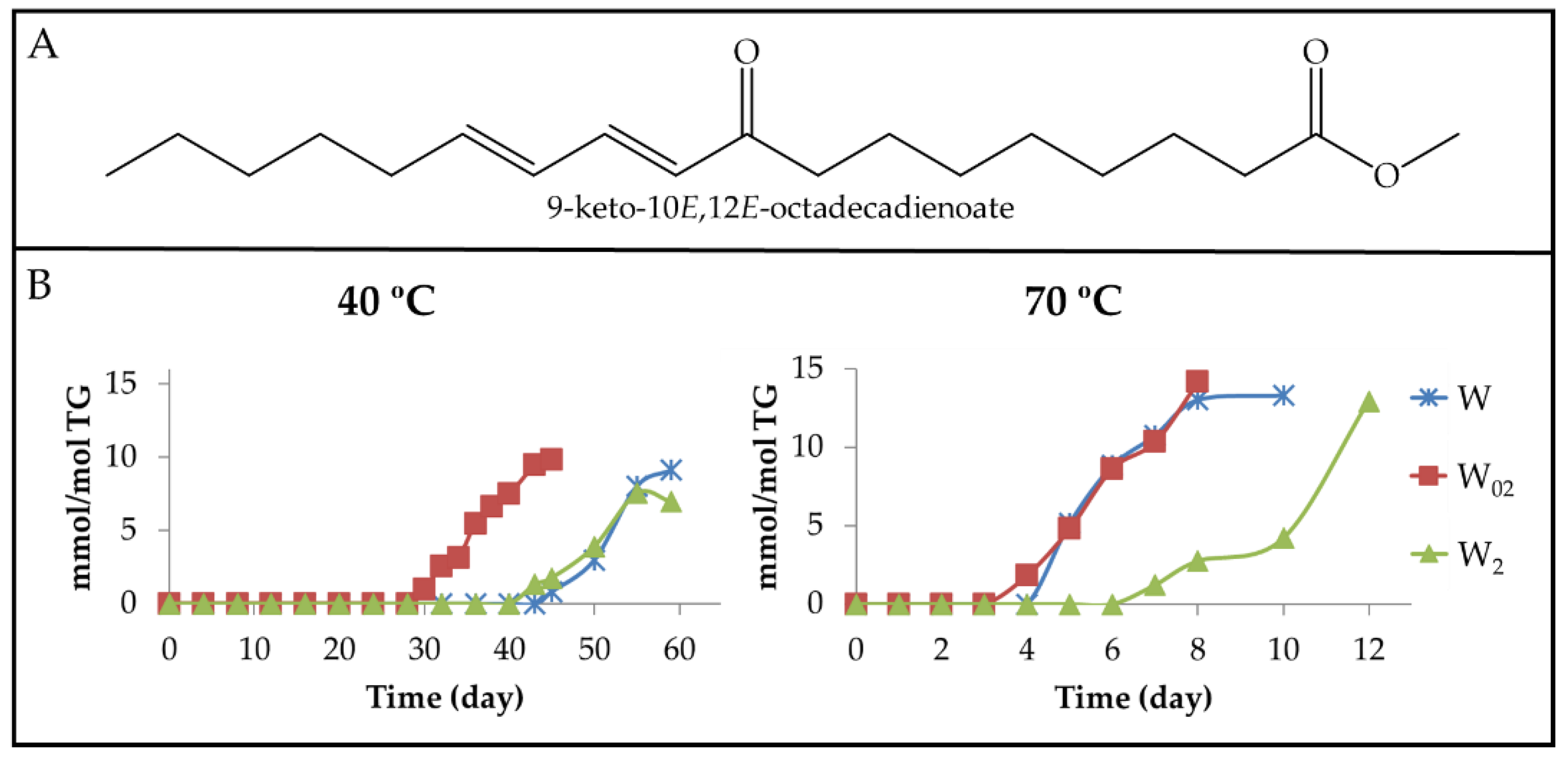
3.2.4. Long-Chain Secondary or Further Oxidation Compounds Supporting a Keto Group
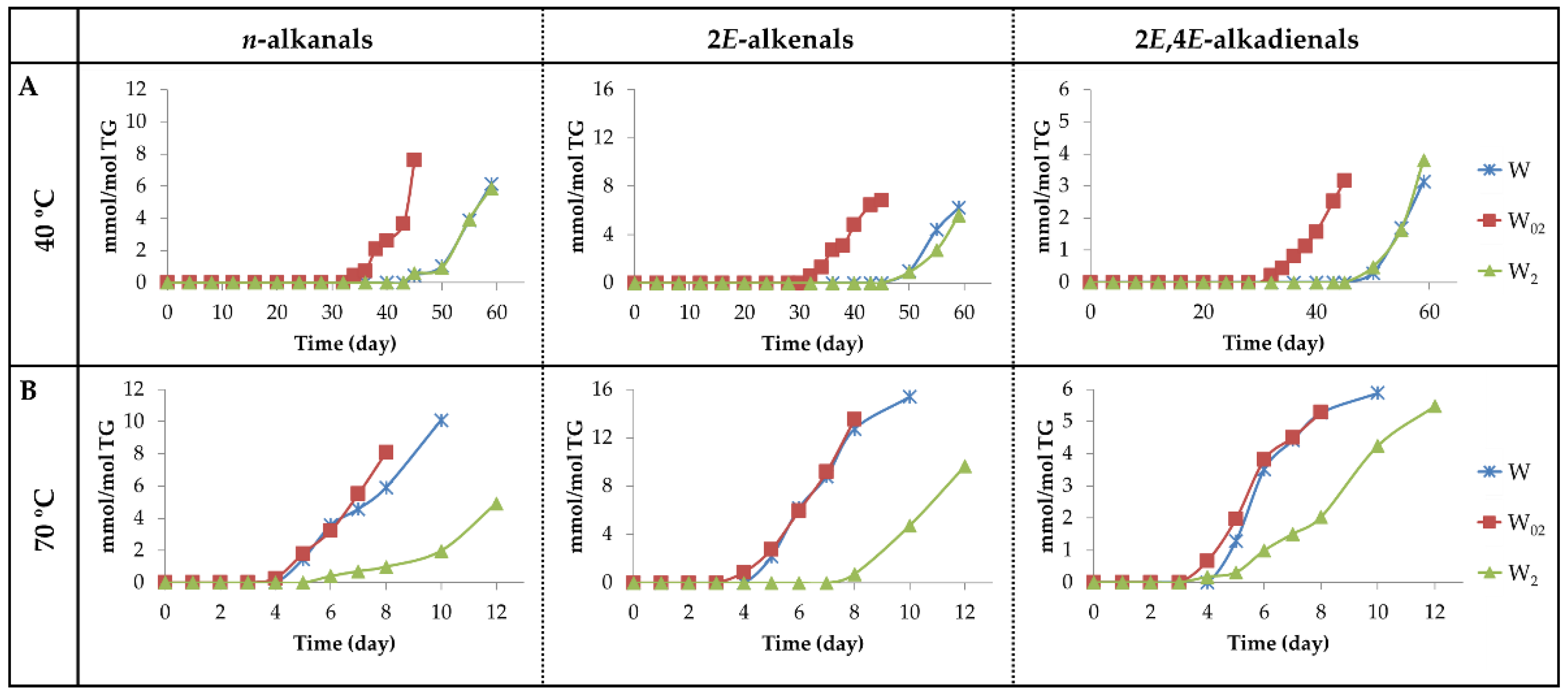
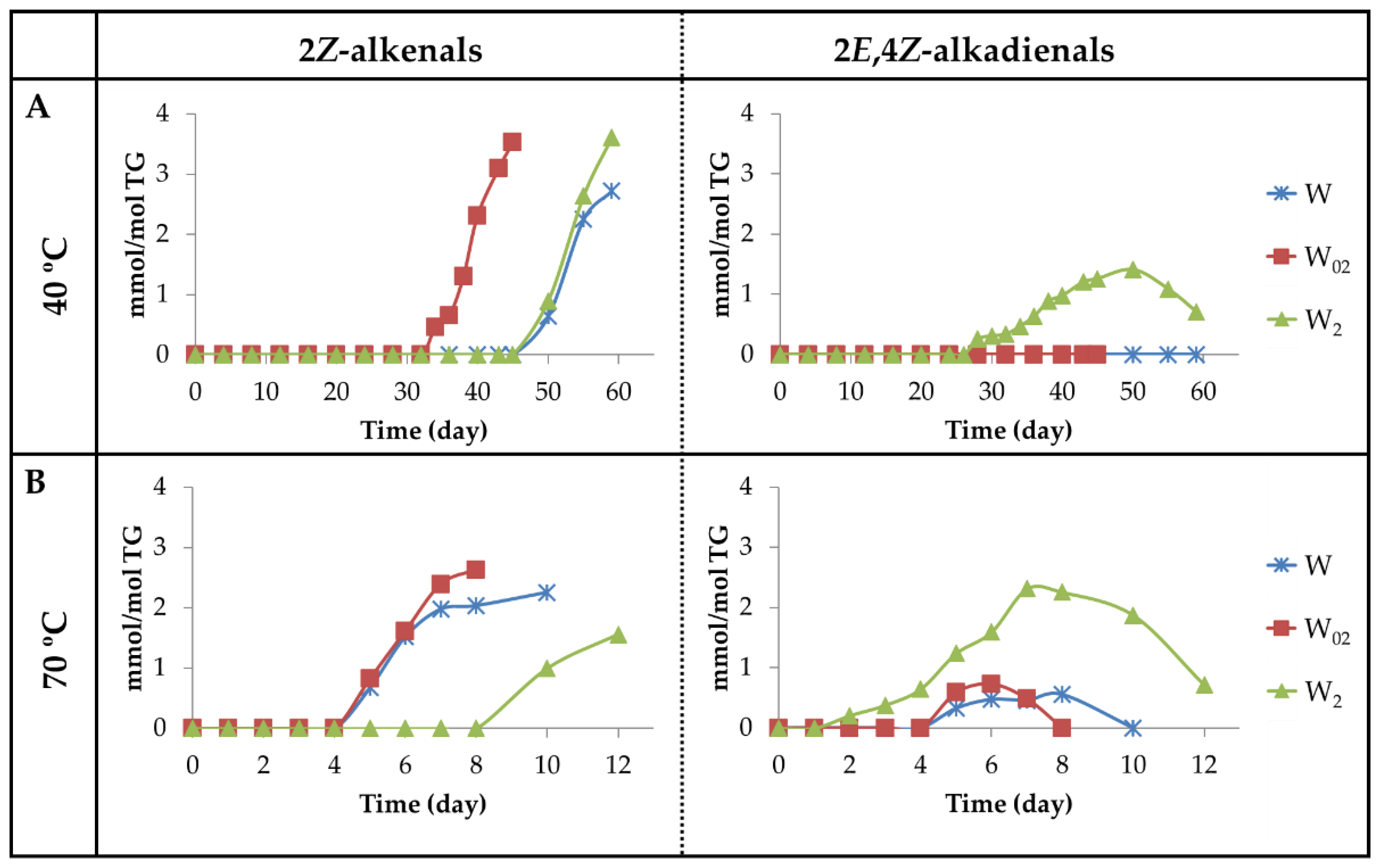
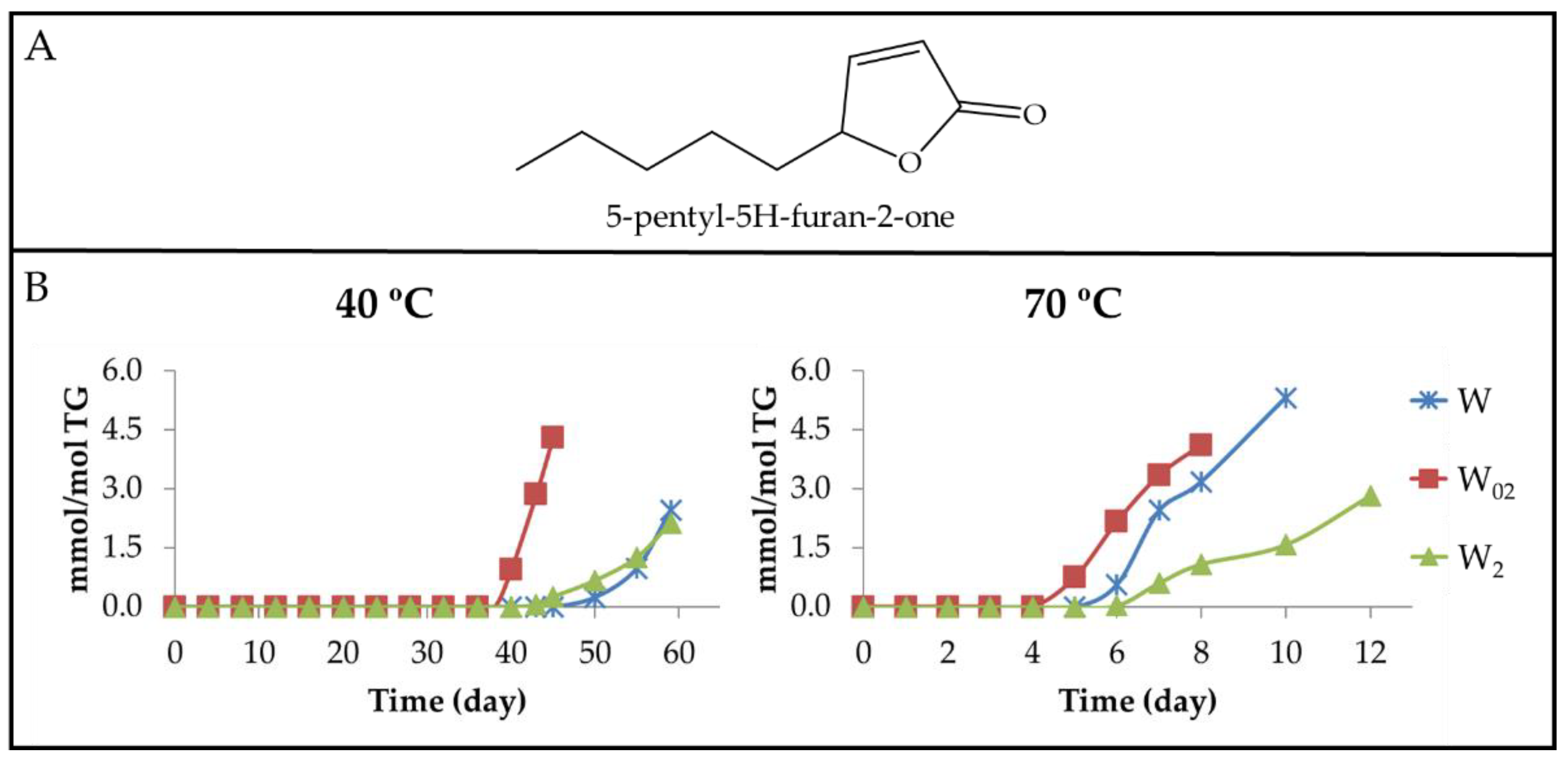
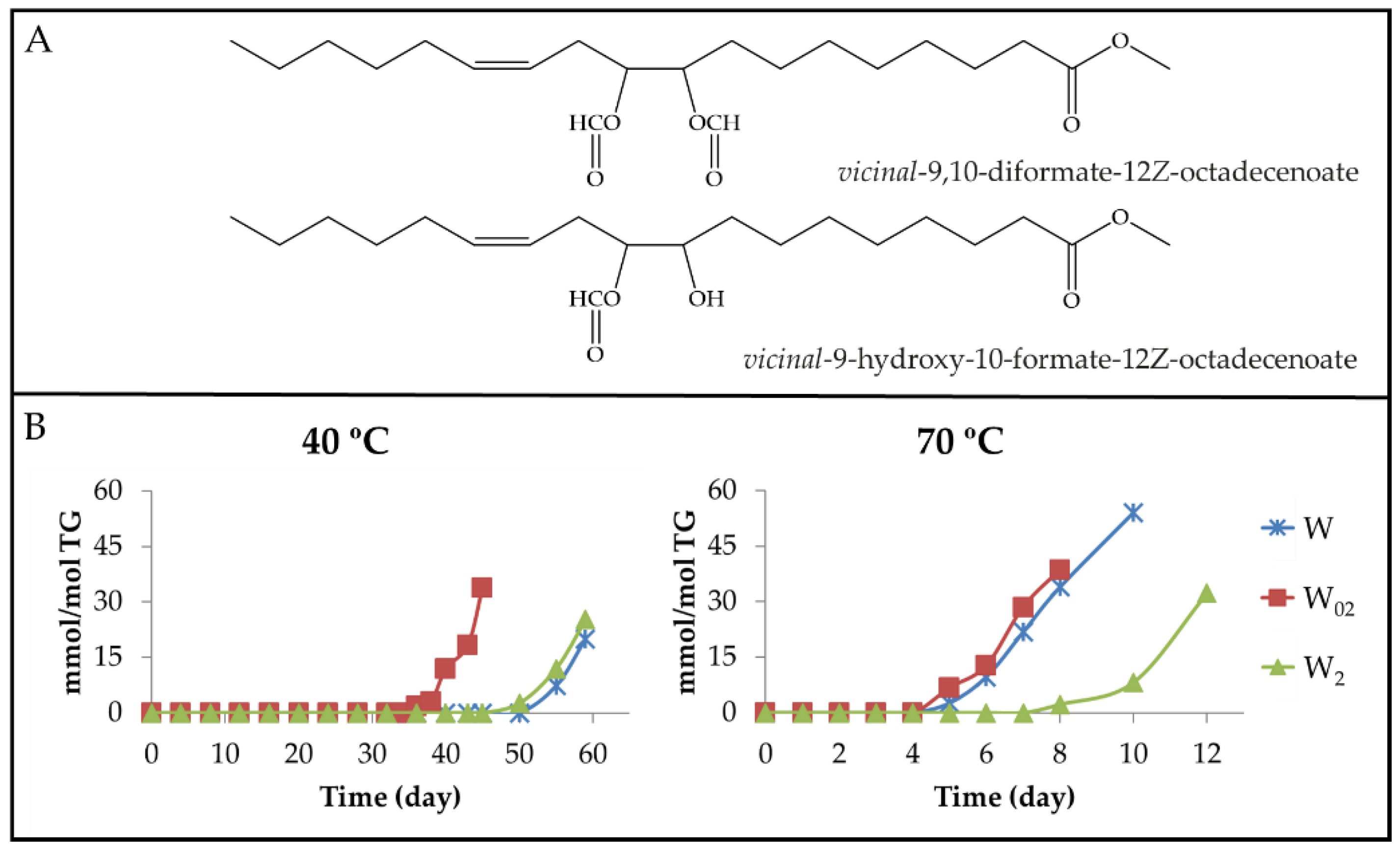
3.2.5. Aldehyde Groups, Furan Rings, and Acid Groups Joined Either to Small Molecules or to Truncated Acyl Groups
3.2.6. Other Oxygenated Functional Groups Attached to Long Chains Formed in Advanced Stages of the Oxidation Process
3.3. Evolution of αT Concentration in the Enriched Walnut Oil Samples throughout the Storage and Effect
3.4. Final Considerations Regarding the Factors That Govern the Walnut Oil Oxidation Process and Their Complex Effects
3.5. Importance of Oxylipins Detection and Quantification to Evaluate Biological Activities
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Esterbauer, H.; Schaur, R.J.; Zollner, H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic. Biol. Med. 1991, 11, 81–128. [Google Scholar] [CrossRef]
- Seppanen, C.M.; Csallany, A.S. Incorporation of the toxic aldehyde 4-hydroxy-2-trans-nonenal into food fried in thermally oxidized soybean oil. J. Am. Oil Chem. Soc. 2004, 81, 1137–1141. [Google Scholar] [CrossRef]
- Guillén, M.D.; Ruiz, A. Oxidation process of oils with high content of linoleic acyl groups and formation of toxic hydroperoxy- and hydroxyalkenals. A study by 1H nuclear magnetic resonance. J. Sci. Food Agric. 2005, 85, 2413–2420. [Google Scholar] [CrossRef]
- Han, I.H.; Csallany, A.S. Formation of Toxic α,β-Unsaturated 4-Hydroxy-Aldehydes in Thermally Oxidized Fatty Acid Methyl Esters. J. Am. Oil Chem. Soc. 2009, 86, 253–260. [Google Scholar] [CrossRef]
- Guillén, M.D.; Goicoechea, E. Toxic Oxygenated α,β-Unsaturated Aldehydes and their Study in Foods: A Review. Crit. Rev. Food Sci. Nutr. 2008, 48, 119–136. [Google Scholar] [CrossRef] [PubMed]
- Guillén, M.D.; Ruiz, A. Monitoring of heat-induced degradation of edible oils by proton NMR. Eur. J. Lipid Sci. Technol. 2008, 110, 52–60. [Google Scholar] [CrossRef]
- Connell, J.J. Control of Fish Quality, 1st ed.; Fishing News: Oxford, UK, 1975. [Google Scholar]
- Addis, P. Occurrence of lipid oxidation products in foods. Food Chem. Toxicol. 1986, 24, 1021–1030. [Google Scholar] [CrossRef]
- Devasagayam, T.P.A.; Boloor, K.K.; Ramasarma, T. Methods for estimating lipid peroxidation: An analysis of merits and demerits. Indian J. Biochem. Biophys. 2003, 40, 300–308. [Google Scholar]
- Frankel, E.N. Lipid Oxidation, 2nd ed.; Woodhead Publishing: Sawston, UK, 2005; pp. 1–470. [Google Scholar]
- Frankel, E.N. Review. Recent advances in lipid oxidation.zz. J. Sci. Food Agric. 1991, 54, 495–511. [Google Scholar] [CrossRef]
- Steenhorst-Slikkerveer, L.; Louter, A.; Janssen, H.-G.; Bauer-Plank, C. Analysis of nonvolatile lipid oxidation products in vegetable oils by normal-phase high-performance liquid chromatography with mass spectrometric detection. J. Am. Oil Chem. Soc. 2000, 77, 837–845. [Google Scholar] [CrossRef]
- Byrdwell, W.C.; Neff, W.E. Electrospray ionization MS of high M.W. TAG oligomers. J. Am. Oil Chem. Soc. 2004, 81, 13–26. [Google Scholar] [CrossRef]
- Velasco, J.; Barroso, A.M.; Ruiz-Méndez, M.V.; Márquez-Ruiz, G. Quantitative determination of major oxidation products in edible oils by direct NP-HPLC-DAD analysis. J. Chromatogr. A 2018, 1547, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Guillén, M.; Ruiz, A. Study of the oxidative stability of salted and unsalted salmon fillets by 1H nuclear magnetic resonance. Food Chem. 2004, 86, 297–304. [Google Scholar] [CrossRef]
- Guillén, M.D.; Ruiz, A. Study by means of 1H nuclear magnetic resonance of the oxidation process undergone by edible oils of different natures submitted to microwave action. Food Chem. 2006, 96, 665–674. [Google Scholar] [CrossRef]
- Guillén, M.D.; Uriarte, P.S. Contribution to Further Understanding of the Evolution of Sunflower Oil Submitted to Frying Temperature in a Domestic Fryer: Study by 1H Nuclear Magnetic Resonance. J. Agric. Food Chem. 2009, 57, 7790–7799. [Google Scholar] [CrossRef]
- Martínez-Yusta, A.; Guillén, M.D. Deep-frying food in extra virgin olive oil: A study by 1H nuclear magnetic resonance of the influence of food nature on the evolving composition of the frying medium. Food Chem. 2014, 150, 429–437. [Google Scholar] [CrossRef] [PubMed]
- Gardner, H.W.; Kleiman, R.; Weisleder, D. Homolytic decomposition of linoleic acid hydroperoxide: Identification of fatty acid products. Lipids 1974, 9, 696–706. [Google Scholar] [CrossRef]
- Gardner, H.W.; Weisleder, D.; Kleiman, R. Formation oftrans-12,13-epoxy-9-hydroperoxy-trans-10-octadecenoic acid from 13-L-hydroperoxy-cis-9, trans-11-octadecadienoic acid catalyzed by either a soybean extract or cysteine-FeC13. Lipids 1978, 13, 246–252. [Google Scholar] [CrossRef]
- Gardner, H.; Crawford, C. Degradation of linoleic acid hydroperoxides by a cysteine FeCl3 catalyst as a model for similar biochemical reactions: III. A novel product, trans-12,13-epoxy-11-oxo-trans-9-octadecenoic acid, from 13-l (S)-hydroperoxy-cis-9,trans-11-octadecadienoic acid. Biochim. Biophys. Acta Lipids Lipid Metab. 1981, 665, 126–133. [Google Scholar] [CrossRef]
- Gardner, H.; Kleiman, R. Degradation of linoleic acid hydroperoxides by a cysteine FeCl3 catalyst as a model for similar biochemical reactions: II. Specificity in formation of fatty acid epoxides. Biochim. Et Biophys. Acta Lipids Lipid Metab. 1981, 665, 113–125. [Google Scholar] [CrossRef]
- Schieberle, P.; Grosch, W. Detection of monohydroperoxides with unconjugated diene systems as minor products of the autoxidation of methyl linoleate. Eur. Food Res. Technol. 1981, 173, 199–203. [Google Scholar] [CrossRef]
- Neff, W.E.; Frankel, E.N.; Selke, E.; Weisleder, D. Photosensitized oxidation of methyl linoleate monohydroperoxides: Hydroperoxy cyclic peroxides, dihydroperoxides, keto esters and volatile thermal decomposition products. Lipids 1983, 18, 868–876. [Google Scholar] [CrossRef]
- Schieberle, P.; Trebert, Y.; Firl, J.; Grosch, W. Photolysis of unsaturated fatty acid hydroperoxides 4. Fatty acid products from the aerobic decomposition of methyl 13(S)-hydroperoxy-9(Z),11(E)-octadecadienoate dissolved in cyclohexane. Chem. Phys. Lipids 1988, 48, 281–288. [Google Scholar] [CrossRef]
- Kuklev, D.V.; Christie, W.W.; Durand, T.; Rossi, J.C.; Vidal, J.P.; Kasyanov, S.P.; Akulin, V.N.; Bezuglov, V.V. Synthesis of keto- and hydroxydienoic compounds from linoleic acid. Chem. Phys. Lipids 1997, 85, 125–134. [Google Scholar] [CrossRef]
- Schneider, C.; Tallman, K.A.; Porter, N.A.; Brash, A.R. Two Distinct Pathways of Formation of 4-Hydroxynonenal: Mechanisms of nonenzymatic transformation of the 9- and 13-hydroperoxides of linoleic acid to 4-hydroxyalkenals. J. Biol. Chem. 2001, 276, 20831–20838. [Google Scholar] [CrossRef] [Green Version]
- Schneider, C.; Boeglin, W.E.; Yin, H.; Porter, N.A.; Brash, A.R. Intermolecular Peroxyl Radical Reactions during Autoxidation of Hydroxy and Hydroperoxy Arachidonic Acids Generate a Novel Series of Epoxidized Products. Chem. Res. Toxicol. 2008, 21, 895–903. [Google Scholar] [CrossRef] [PubMed]
- Xia, W.; Budge, S.M.; Lumsden, M.D. 1H-NMR Characterization of Epoxides Derived from Polyunsaturated Fatty Acids. J. Am. Oil Chem. Soc. 2016, 93, 467–478. [Google Scholar] [CrossRef]
- Alberdi-Cedeño, J.; Ibargoitia, M.L.; Guillén, M.D. Oxylipins Associated to Current Diseases Detected for the First Time in the Oxidation of Corn Oil as a Model System of Oils Rich in Omega-6 Polyunsaturated Groups. A Global, Broad and in-Depth Study by 1H NMR Spectroscopy. Antioxidants 2020, 9, 544. [Google Scholar] [CrossRef]
- Brand-Williams, W.M.; Cuvelier, M.E.; Berset, C. Use of a free radical method to evaluate antioxidant activity. LWT Food Sci. Technol. 1995, 28, 25–30. [Google Scholar] [CrossRef]
- Miller, N.J.; Rice-Evans, C.A. Factors Influencing the Antioxidant Activity Determined by the ABTS+ Radical Cation Assay. Free Radic. Res. 1997, 26, 195–199. [Google Scholar] [CrossRef]
- Benzie, I.F.F.; Strain, J.J. Ferric reducing/antioxidant power assay: Direct measure of total antioxidant activity of biological fluids and modified version for simultaneous measurement of total antioxidant power and ascorbic acid concentration. Methods Enzymol. 1999, 299, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Cao, G.; Alessio, H.M.; Cutler, R.G. Oxygen-radical absorbance capacity assay for antioxidants. Free Radic. Biol. Med. 1993, 14, 303–311. [Google Scholar] [CrossRef] [Green Version]
- Commission Regulation (EU) No 1129/2011 of 11 November 2011 amending Annex II to Regulation (EC) No 1333/2008 of the European Parliament and of the Council by establishing a Union list of food additives. Off. J. Eur. Union 2011, L 295, 1–177.
- Koskas, J.P.; Cillard, J. Autoxidation of linoleic acid and behavior of its hydroperoxides with and without tocopherols. J. Am. Oil Chem. Soc. 1984, 61, 1466–1469. [Google Scholar] [CrossRef]
- Kinen, M.M.; Kamal-Eldin, A.; Lampi, A.-M.; Hopia, A. Effects of α- and γ-tocopherols on formation of hydroperoxides and two decomposition products from methyl linoleate. J. Am. Oil Chem. Soc. 2000, 77, 801–806. [Google Scholar] [CrossRef]
- Jung, M.Y.; Min, D.B. Effects of α-, γ-, and δ-Tocopherols on oxidative stability of soybean oil. J. Food Sci. 1990, 55, 1464–1465. [Google Scholar] [CrossRef]
- Evans, J.C.; Kodali, D.R.; Addis, P.B. Optimal tocopherol concentrations to inhibit soybean oil oxidation. J. Am. Oil Chem. Soc. 2002, 79, 47–51. [Google Scholar] [CrossRef]
- Marinova, E.M.; Yanishlieva, N.V. Effect of temperature on the antioxidative action of inhibitors in lipid autoxidation. J. Sci. Food Agric. 1992, 60, 313–318. [Google Scholar] [CrossRef]
- Mancebo-Campos, V.; Salvador, M.D.; Fregapane, G. Antioxidant capacity of individual and combined virgin olive oil minor compounds evaluated at mild temperature (25 and 40 °C) as compared to accelerated and antiradical assays. Food Chem. 2014, 150, 374–381. [Google Scholar] [CrossRef]
- Dolde, D.; Wang, T. Oxidation of corn oils with spiked tocols. J. Am. Oil Chem. Soc. 2011, 88, 1759–1765. [Google Scholar] [CrossRef]
- Guillén, M.D.; Ruiz, A. 1H nuclear magnetic resonance as a fast tool for determining the composition of acyl chains in acylglycerol mixtures. Eur. J. Lipid Sci. Technol. 2003, 105, 502–507. [Google Scholar] [CrossRef]
- Guillén, M.D.; Ruiz, A. Rapid simultaneous determination by proton NMR of unsaturation and composition of acyl groups in vegetable oils. Eur. J. Lipid Sci. Technol. 2003, 105, 688–696. [Google Scholar] [CrossRef]
- Guillén, M.D.; Uriarte, P.S. Study by 1H NMR spectroscopy of the evolution of extra virgin olive oil composition submitted to frying temperature in an industrial fryer for a prolonged period of time. Food Chem. 2012, 134, 162–172. [Google Scholar] [CrossRef]
- Guillén, M.D.; Ruiz, A. Monitoring the oxidation of unsaturated oils and formation of oxygenated aldehydes by proton NMR. Eur. J. Lipid Sci. Technol. 2005, 107, 36–47. [Google Scholar] [CrossRef]
- Guillén, M.D.; Ruiz, A. Study by proton nuclear magnetic resonance of the thermal oxidation of oils rich in oleic acyl groups. J. Am. Oil Chem. Soc. 2005, 82, 349–355. [Google Scholar] [CrossRef]
- Martin-Rubio, A.S.; Sopelana, P.; Guillén, M.D. A thorough insight into the complex effect of gamma-tocopherol on the oxidation process of soybean oil by means of 1H nuclear magnetic resonance. Comparison with alpha-tocopherol. Food Res. Int. 2018, 114, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Martin-Rubio, A.S.; Sopelana, P.; Ibargoitia, M.L.; Guillén, M.D. Prooxidant effect of α-tocopherol on soybean oil. Global monitoring of its oxidation process under accelerated storage conditions by 1H nuclear magnetic resonance. Food Chem. 2018, 245, 312–323. [Google Scholar] [CrossRef]
- Martínez-Yusta, A.; Goicoechea, E.; Guillén, M.D. A review of thermo-oxidative degradation of food lipids studied by 1H NMR spectroscopy: Influence of degradative conditions and food lipid nature. Compr. Rev. Food Sci. Food Saf. 2014, 13, 838–859. [Google Scholar] [CrossRef]
- Goicoechea, E.; Guillén, M.D. Analysis of hydroperoxides, aldehydes and epoxides by 1H nuclear magnetic resonance in sunflower oil oxidized at 70 and 100 °C. J. Agric. Food Chem. 2010, 58, 6234–6245. [Google Scholar] [CrossRef]
- Frankel, E.N.; Neff, W.E.; Miyashita, K. Autoxidation of polyunsaturated triacylglycerols. II. Trilinolenoylglycerol. Lipids 1990, 25, 40–47. [Google Scholar] [CrossRef]
- Huang, S.W.; Frankel, E.N.; German, J.B. Antioxidant activity of α-and γ-tocopherols in bulk oils and in oil-in-water emulsions. J. Agric. Food Chem. 1994, 42, 2108–2114. [Google Scholar] [CrossRef]
- Lampi, A.; Kataja, L.; Kamal-Eldin, A.; Vieno, P. Antioxidant activities of α- and γ-tocopherols in the oxidation of rapeseed oil triacylglycerols. J. Am. Oil Chem. Soc. 1999, 76, 749–755. [Google Scholar] [CrossRef]
- Makinen, E.M.; Hopia, A.I. Effects of α-tocopherol and ascorbyl palmitate on the isomerization and decomposition of methyl linoleate hydroperoxides. Lipids 2000, 35, 1215–1223. [Google Scholar] [CrossRef]
- Peers, K.E.; Coxon, D.T.; Chan, H.W.S. Autoxidation of methyl linolenate and methyl linoleate: The effect of α-tocopherol. J. Sci. Food Agric. 1981, 32, 898–904. [Google Scholar] [CrossRef]
- Schneider, C. An update on products and mechanisms of lipid peroxidation. Mol. Nutr. Food Res. 2009, 53, 315–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terao, J.; Matsushita, S. Geometrical isomers of monohydroperoxides formed by autoxidation of methyl linoleate. Agric. Biol. Chem. 1977, 41, 2401–2405. [Google Scholar] [CrossRef]
- Porter, N.A.; Weber, B.A.; Weenen, H.; Khan, J.A. Autoxidation of polyunsaturated lipids. Factors controlling the stereochemistry of product hydroperoxides. J. Am. Chem. Soc. 1980, 102, 5597–5601. [Google Scholar] [CrossRef]
- Porter, N.A.; Caldwell, S.E.; Mills, K.A. Mechanisms of free radical oxidation of unsaturated lipids. Lipids 1995, 30, 277–290. [Google Scholar] [CrossRef]
- Zhang, W. Synthesis and Fragmentation Reactions of Linoleic Acid-Derived Hydroperoxides. Ph.D. Thesis, Case Western Reserve University, Cleveland, OH, USA, January 2008. [Google Scholar]
- Schneider, C.; Boeglin, W.E.; Yin, H.; Stec, D.F.; Hachey, D.L.; Porter, N.A.; Brash, A.R. Synthesis of dihydroperoxides of linoleic and linolenic acids and studies on their transformation to 4-hydroperoxynonenal. Lipids 2005, 40, 1155–1162. [Google Scholar] [CrossRef] [PubMed]
- Onyango, A.N.; Kumura, N.; Tominaga, H.; Baba, N. Dihydroperoxidation facilitates the conversion of lipids to aldehydic products via alkoxyl radicals. Food Res. Int. 2010, 43, 925–929. [Google Scholar] [CrossRef]
- Gu, X.; Salomon, R.G. Fragmentation of a linoleate-derived γ-hydroperoxy-α,β-unsaturated epoxide to γ-hydroxy- and γ-oxo-alkenals involves a unique pseudo-symmetrical diepoxycarbinyl radical. Free Radic. Biol. Med. 2012, 52, 601–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gassenmeier, K.; Schieberle, P. Formation of the intense flavor compound trans-4,5-epoxy-(E)-2-decenal in thermally treated fats. J. Am. Oil Chem. Soc. 1994, 71, 1315–1319. [Google Scholar] [CrossRef]
- Schneider, C.; Porter, N.A.; Brash, A.R. Autoxidative transformation of chiral ω6 hydroxy linoleic and arachidonic acids to chiral 4-hydroxy-2E-nonenal. Chem. Res. Toxicol. 2004, 17, 937–941. [Google Scholar] [CrossRef] [PubMed]
- Schieberle, P.; Tsoukalas, B.; Grosch, W. Decomposition of linoleic acid hydroperoxides by radicals-I. structures of products of methyl 13-hydroperoxy-cis, trans-9, 11-octadecadienoate. Z. Lebensm. Unters. Forsch. 1979, 168, 448–456. [Google Scholar] [CrossRef]
- Lin, D.; Zhang, J.; Sayre, L.M. Synthesis of six epoxyketooctadecenoic acid (EKODE) isomers, their generation from nonenzymatic oxidation of linoleic acid, and their reactivity with imidazole nucleophiles. J. Org. Chem. 2007, 72, 9471–9480. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo, F.J.; Zamora, R.; Vioque, E. Syntheses and reactions of methyl (Z)-9,10-epoxy-13-oxo-(E)-11-octadecenoate and methyl (E)-9,10-epoxy-13-oxo-(E)-11-octadecenoate. Chem. Phys. Lipids 1992, 60, 225–233. [Google Scholar] [CrossRef]
- Ramsden, C.E.; Domenichiello, A.F.; Yuan, Z.X.; Sapio, M.R.; Keyes, G.S.; Mishra, S.K.; Iadarola, M.J. A systems approach for discovering linoleic acid derivatives that potentially mediate pain and itch. Sci. Signal. 2017, 10. [Google Scholar] [CrossRef] [Green Version]
- Guillén, M.D.; Cabo, N.; Ibargoitia, M.L.; Ruiz, A. Study of both sunflower oil and its headspace throughout the oxidation process. Occurrence in the headspace of toxic oxygenated aldehydes. J. Agric. Food Chem. 2005, 53, 1093–1101. [Google Scholar] [CrossRef]
- Zhang, W.; Sun, M.; Salomon, R.G. Preparative singlet oxygenation of linoleate provides doubly allylic dihydroperoxides: Putative intermediates in the generation of biologically active aldehydes in vivo. J. Org. Chem. 2006, 71, 5607–5615. [Google Scholar] [CrossRef]
- Shahidi, F.; Oh, W.Y. Lipid-derived flavor and off-flavor of traditional and functional foods: An overview. J. Food Bioact. 2020, 10, 20–31. [Google Scholar] [CrossRef]
- Grootveld, M.; Atherton, M.D.; Sheerin, A.N.; Hawkes, J.; Blake, D.R.; Richens, T.E.; Claxson, A.W.D. In vivo absorption, metabolism, and urinary excretion of α,β-unsaturated aldehydes in experimental animals. Relevance to the development of cardiovascular diseases by the dietary ingestion of thermally stressed polyunsaturate-rich culinary oils. J. Clin. Investig. 1998, 101, 1210–1218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paradisi, L.; Panagini, C.; Parola, M.; Barrera, G.; Dianzani, M.U. Effects of 4-hydroxynonenal on adenylate cyclase and 5′-nucleotidase activities in rat liver plasma membranes. Chem.-Biol. Interact. 1985, 53, 209–217. [Google Scholar] [CrossRef]
- Rossi, M.A.; Garramone, A.; Dianzani, M.U. Stimulation of phospholipase C activity by 4-hydroxynonenal; influence of GTP and calcium concentration. Int. J. Tissue React. 1988, 10, 321–325. [Google Scholar] [PubMed]
- Curzio, M.; Esterbauer, H.; Di Mauro, C.; Dianzani, M.U. Influence of the lipid peroxidation product 4-hydroxynonenal on human neutrophil migration. Int. J. Immunother. 1990, 6, 13–18. [Google Scholar]
- Barrera, G.; Di Mauro, C.; Muraca, R.; Ferrero, D.; Cavalli, G.; Fazio, V.M.; Dianzani, M.U. Induction of differentiation in human HL-60 cells by 4-hydroxynonenal, a product of lipid peroxidation. Exp. Cell Res. 1991, 197, 148–152. [Google Scholar] [CrossRef]
- Zarkovic, N.; Ilic, Z.; Jurin, M.; Schaur, R.J.; Puhl, H.; Esterbauer, H. Stimulation of HeLa cell growth by physiological concentrations of 4-hydroxynonenal. Cell Biochem. Funct. 1993, 11, 279–286. [Google Scholar] [CrossRef]
- Dianzani, M.U. 4-hydroxynonenal from pathology to physiology. Mol. Asp. Med. 2003, 24, 263–272. [Google Scholar] [CrossRef]
- Schwarzer, E.; Arese, P.; Skorokhod, O.A. Role of the lipoperoxidation product 4-hydroxynonenal in the pathogenesis of severe malaria anemia and malaria immunodepression. Oxid. Med. Cell. Longev. 2015. [Google Scholar] [CrossRef] [Green Version]
- Moumtaz, S.; Percival, B.C.; Parmar, D.; Grootveld, K.L.; Jansson, P.; Grootveld, M. Toxic aldehyde generation in and food uptake from culinary oils during frying practices: Peroxidative resistance of a monounsaturate-rich algae oil. Sci. Rep. 2019, 9. [Google Scholar] [CrossRef] [Green Version]
- Schieberle, P.; Grosch, W. Photolyse von 13(S)-Hydroperoxy-9(Z),11(E)-octadecadiensäuremethylester in gegenwart von sauerstoff—analyse der niedermolekularen reaktionsprodukte. Fette Seifen Anstrichm. 1985, 87, 76–80. [Google Scholar] [CrossRef]
- Zhang, Q.; Qin, W.; Lin, D.; Shen, Q.; Saleh, A.S.M. The changes in the volatile aldehydes formed during the deep-fat frying process. J. Food Sci. Technol. 2015, 52, 7683–7696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeoka, G.; Perrino, C., Jr.; Buttery, R. Volatile constituents of used frying oils. J. Agric. Food Chem. 1996, 44, 654–660. [Google Scholar] [CrossRef]
- Goicoechea, E.; Guillén, M.D. Volatile compounds generated in corn oil stored at room temperature. Presence of toxic compounds. Eur. J. Lipid Sci. Technol. 2014, 116, 395–406. [Google Scholar] [CrossRef]
- Uriarte, P.S.; Goicoechea, E.; Guillén, M.D. Volatile components of several virgin and refined oils differing in their botanical origin. J. Sci. Food Agric. 2011, 91, 1871–1884. [Google Scholar] [CrossRef] [PubMed]
- Loury, M. Possible mechanisms of autoxidative rancidity. Lipids 1972, 7, 671–675. [Google Scholar] [CrossRef]
- De Man, J.M.; Tie, F.; de Man, L. Formation of short chain volatile organic acids in the automated AOM method. J. Am. Oil Chem. Soc. 1987, 64, 993–996. [Google Scholar] [CrossRef]
- Guillén, M.D.; Ruiz, A. High resolution 1H nuclear magnetic resonance in the study of edible oils and fats. Trends Food Sci. Technol. 2001, 12, 328–338. [Google Scholar] [CrossRef]
- Gabbs, M.; Leng, S.; Devassy, J.G.; Monirujjaman, M.; Aukema, H.M. Advances in our understanding of oxylipins derived from dietary PUFAs 1,2. Adv. Nutr. 2015, 6, 513–540. [Google Scholar] [CrossRef] [Green Version]
- Alaiz, M.; Maza, M.P.; Zamora, R.; Hidalgo, F.J.; Millán, F.; Vioque, E. Epoxidation of ethyl (Z)-9-(Z)-12-(Z)-15-octadecatrienoate with m-chloroperbenzoic acid. Chem. Phys. Lipids 1989, 49, 221–224. [Google Scholar] [CrossRef]
- Aerts, H.A.J.; Jacobs, P.A. Epoxide yield determination of oils and fatty acid methyl esters using 1H NMR. J. Am. Oil Chem. Soc. 2004, 81, 841–846. [Google Scholar] [CrossRef]
- Anuar, S.T.; Zhao, Y.Y.; Mugo, S.M.; Curtis, J.M. Monitoring the epoxidation of canola oil by non-aqueous reversed phase liquid chromatography/mass spectrometry for process optimization and control. J. Am. Oil Chem. Soc. 2012, 89, 1951–1960. [Google Scholar] [CrossRef]
- Nilewski, C.; Chapelain, C.L.; Wolfrum, S.; Carreira, E.M. Synthesis and biological evaluation of chlorinated analogs of leukotoxin diol. Org. Lett. 2015, 17, 5602–5605. [Google Scholar] [CrossRef] [PubMed]
- De Dios Miguel, T.; Duc Vu, N.; Lemaire, M.; Duguet, N. Biobased aldehydes from fatty epoxides through thermal cleavage of β-hydroxy hydroperoxides. ChemSusChem 2021, 14, 379–386. [Google Scholar] [CrossRef]
- Peng, L.; Xie, Q.; Nie, Y.; Liu, X.; Lu, M.; Ji, J. Room-temperature production of bio-based aldehydes from vegetable oil-derived epoxide: Via H2WO4@Al-MCM-41 as recyclable catalyst. RSC Adv. 2019, 9, 23061–23070. [Google Scholar] [CrossRef] [Green Version]
- Soutelo-Maria, A.; Dubois, J.L.; Couturier, J.L.; Cravotto, G. Oxidative cleavage of fatty acid derivatives for monomer synthesis. Catalysts 2018, 8, 464. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; De Liedekerke Beaufort, M.; Gyurik, L.; Dorresteijn, J.; Otte, M.; Klein Gebbink, R.J.M. Highly efficient epoxidation of vegetable oils catalyzed by a manganese complex with hydrogen peroxide and acetic acid. Green Chem. 2019, 21, 2436–2447. [Google Scholar] [CrossRef] [Green Version]
- Moghaddam, M.F.; Grant, D.F.; Cheek, J.M.; Greene, J.F.; Williamson, K.C.; Hammock, B.D. Bioactivation of leukotoxins to their toxic diols by epoxide hydrolase. Nat. Med. 1997, 3, 562–566. [Google Scholar] [CrossRef]
- Ahmad, S.; Ashraf, S.M.; Sharmin, E.; Zafar, F.; Hasnat, A. Studies on ambient cured polyurethane modified epoxy coatings synthesized from a sustainable resource. Prog. Cryst. Growth Charact. Mater. 2002, 45, 83–88. [Google Scholar] [CrossRef]
- Harry-O’Kuru, R.E.; Carriere, C.J. Synthesis, rheological characterization, and constitutive modeling of polyhydroxy triglycerides derived from milkweed oil. J. Agric. Food Chem. 2002, 50, 3214–3221. [Google Scholar] [CrossRef]
- Yang, J.; Morton, M.D.; Hill, D.W.; Grant, D.F. NMR and HPLC-MS/MS analysis of synthetically prepared linoleic acid diol glucuronides. Chem. Phys. Lipids 2006, 140, 75–87. [Google Scholar] [CrossRef]
- Monteavaro, L.L.; Da Silva, E.O.; Costa, A.P.O.; Samios, D.; Gerbase, A.E.; Petzhold, C.L. Polyurethane networks from formiated soy polyols: Synthesis and mechanical characterization. J. Am. Oil Chem. Soc. 2005, 82, 365–371. [Google Scholar] [CrossRef]
- Caillol, S.; Desroches, M.; Boutevin, G.; Loubat, C.; Auvergne, R.; Boutevin, B. Synthesis of new polyester polyols from epoxidized vegetable oils and biobased acids. Eur. J. Lipid Sci. Technol. 2012, 114, 1447–1459. [Google Scholar] [CrossRef]
- Lopes, R.D.V.V.; Zamian, J.R.; Resck, I.S.; Sales, M.J.A.; Dos Santos, M.L.; Da Cunha, F.R. Physicochemical and rheological properties of passion fruit oil and its polyol. Eur. J. Lipid Sci. Technol. 2010, 112, 1253–1262. [Google Scholar] [CrossRef]
- Harry-O’kuru, R.E.; Biresaw, G.; Tisserat, B.; Evangelista, R. Synthesis of polyformate esters of vegetable oils: Milkweed, pennycress, and soy. J. Lipids 2016, 2016, 3128604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abraham, R.J.; Byrne, J.J.; Griffiths, L.; Perez, M. 1H chemical shifts in NMR: Part 23, the effect of dimethyl sulphoxide versus chloroform solvent on 1H chemical shifts. Magn. Reson. Chem. 2006, 44, 491–509. [Google Scholar] [CrossRef]
- Lligadas, G.; Ronda, J.C.; Galià, M.; Biermann, U.; Metzger, J.O. Synthesis and characterization of polyurethanes from epoxidized methyl oleate based polyether polyols as renewable resources. J. Polym. Sci. Part A Polym. Chem. 2006, 44, 634–645. [Google Scholar] [CrossRef]
- De Souza, V.H.R.; Silva, S.A.; Ramos, L.P.; Zawadzki, S.F. Synthesis and characterization of polyols derived from corn oil by epoxidation and ozonolysis. J. Am. Oil Chem. Soc. 2012, 89, 1723–1731. [Google Scholar] [CrossRef]
- Spector, A.A.; Kim, H.Y. Cytochrome P450 epoxygenase pathway of polyunsaturated fatty acid metabolism. Biochim. Biophys. Acta Mol. Cell Biol. Lipids. 2015, 1851, 356–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaschler, M.M.; Stockwell, B.R. Lipid peroxidation in cell death. Biochem. Biophys. Res. Commun. 2017, 482, 419–425. [Google Scholar] [CrossRef]
- Kaneda, T.; Sakai, H.; Ishii, S. Nutritive value or toxicity of highly unsaturated fatty acids. II. J. Biochem. 1955, 42, 561–573. [Google Scholar] [CrossRef]
- Watanabe, T.; Nakamura, T. The effects of linoleate hydroperoxide on respiration and oxidative phosphorylation of rat liver mitochondria. J. Biochem. 1979, 86, 1041–1047. [Google Scholar] [CrossRef] [PubMed]
- Shiotani, A.; Watanabe, T.; Matsuoka, I.; Nakamura, T. Comparative studies on the effects of linoleate and methyl linoleate and their hydroperoxides on the respiration and reactivities of rat heart mitochondria. J. Biochem. 1980, 88, 677–683. [Google Scholar] [CrossRef]
- De Meyer, G.R.Y.; Bult, H.; Verbeuren, T.J.; Herman, A.G. The role of endothelial cells in the relaxations induced by 13-hydroxy- and 13-hydroperoxylinoleic acid in canine arteries. Br. J. Pharmacol. 1992, 107, 597–603. [Google Scholar] [CrossRef] [Green Version]
- Rohr, M.; Narasimhulu, C.A.; Keewan, E.; Hamid, S.; Parthasarathy, S. The dietary peroxidized lipid, 13-HPODE, promotes intestinal inflammation by mediating granzyme B secretion from natural killer cells. Food Funct. 2020, 11, 9526–9534. [Google Scholar] [CrossRef]
- König, B.; Eder, K. Differential action of 13-HPODE on PPARα downstream genes in rat fao and human HepG2 hepatoma cell lines. J. Nutr. Biochem. 2006, 17, 410–418. [Google Scholar] [CrossRef] [PubMed]
- Corteselli, E.M.; Gibbs-Flournoy, E.; Simmons, S.O.; Bromberg, P.; Gold, A.; Samet, J.M. Long chain lipid hydroperoxides increase the glutathione redox potential through glutathione peroxidase 4. Biochim. Biophys. Acta Gen. Subj. 2019, 1863, 950–959. [Google Scholar] [CrossRef]
- Imagawa, T.; Kasai, S.; Matsui, K.; Nakamura, T. Methyl hydroperoxy-epoxy-octadecenoate as an autoxidation product of methyl linoleate: A new inhibitor-uncoupler of mitochondrial respiration. J. Biochem. 1982, 92, 1109–1121. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, Y.; Nishikawa, Y.; Mori, H.; Tamura, H.; Matsushita, Y.I.; Matsui, T. Antitumor activity of (10E,12Z)-9-hydroxy-10,12-octadecadienoic acid from rice bran. J. Ferment. Bioeng. 1998, 86, 149–153. [Google Scholar] [CrossRef]
- Jira, W.; Spitellera, G.; Richter, A. Increased levels of lipid oxidation products in rheumatically destructed bones of patients suffering from rheumatoid arthritis. Z. Naturforsch. C J. Biosci. 1998, 53, 1061–1071. [Google Scholar] [CrossRef] [PubMed]
- Altmann, R.; Hausmann, M.; Spöttl, T.; Gruber, M.; Bull, A.W.; Menzel, K.; Rogler, G. 13-oxo-ODE is an endogenous ligand for PPARγ in human colonic epithelial cells. Biochem. Pharmacol. 2007, 74, 612–622. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Sakurai, Y.; Shibata, K.; Kikkawa, F.; Tomoda, Y.; Mizukami, H. Cytotoxic fatty acid ketodienes from eggplants. Jpn. J. Food Chem. Saf. 2014, 21, 42–47. [Google Scholar]
- Zhao, B.; Tomoda, Y.; Mizukami, H.; Makino, T. 9-oxo-(10E,12E)-octadecadienoic acid, a cytotoxic fatty acid ketodiene isolated from eggplant calyx, induces apoptosis in human ovarian cancer (HRA) cells. J. Nat. Med. 2015, 69, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Doolen, S.; Keyes, G.S.; Ramsden, C.E. Hydroxy-epoxide and keto-epoxide derivatives of linoleic acid activate trigeminal neurons. Neurobiol. Pain 2020, 7, 100046. [Google Scholar] [CrossRef] [PubMed]
- Goodfriend, T.L.; Ball, D.L.; Gardner, H.W. An oxidized derivative of linoleic acid affects aldosterone secretion by adrenal cells in vitro. Prostaglandins Leukot. Essent. Fatty Acids. 2002, 67, 163–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, F.L.; Chen, H.J.; Guttenplan, J.B.; Nishikawa, A.; Hard, G.C. 2,3-epoxy-4-hydroxynonanal as a potential tumor-initiating agent of lipid peroxidation. Carcinogenesis 1993, 14, 2073–2077. [Google Scholar] [CrossRef]
- Eckl, P.M.; Ortner, A.; Esterbauer, H. Genotoxic properties of 4-hydroxyalkenals and analogous aldehydes. Mutat. Res. Fundam. Mol. Mech. Mutagen. 1993, 290, 183–192. [Google Scholar] [CrossRef]
- Guillén, M.D.; Goicoechea, E. Oxidation of corn oil at room temperature: Primary and secondary oxidation products and determination of their concentration in the oil liquid matrix from 1H nuclear magnetic resonance data. Food Chem. 2009, 116, 183–192. [Google Scholar] [CrossRef]
- Ozawa, T.; Hayakawa, M.; Kosaka, K.; Sugiyama, S.; Ogawa, T.; Yokoo, K.; Izawa, Y. Leukotoxin, 9,10-epoxy-12-octadecenoate, as a burn toxin causing adult respiratory distress syndrome. Adv. Prostaglandin Thromboxane Leukot. Res. 1991, 21 B, 569–572. [Google Scholar]
- Hanaki, Y.; Kamiya, H.; Ohno, M.; Hayakawa, M.; Sugiyama, S.; Ozawa, T. Leukotoxin, 9,10-epoxy-12-octadecenoate: A possible responsible factor in circulatory shock and disseminated intravascular coagulation. Jpn. J. Med. 1991, 30, 224–228. [Google Scholar] [CrossRef] [Green Version]
- Greene, J.F.; Newman, J.W.; Williamson, K.C.; Hammock, B.D. Toxicity of epoxy fatty acids and related compounds cells expressing human soluble epoxide hydrolase. Chem. Res. Toxicol. 2000, 13, 217–226. [Google Scholar] [CrossRef]
- Zheng, J.; Plopper, C.G.; Lakritz, J.; Storms, D.H.; Hammock, B.D. Leukotoxin-diol: A putative toxic mediator involved in acute respiratory distress syndrome. Am. J. Respir. Cell Mol. Biol. 2001, 25, 434–438. [Google Scholar] [CrossRef] [PubMed]
- Markaverich, B.M.; Crowley, J.R.; Alejandro, M.A.; Shoulars, K.; Casajuna, N.; Mani, S.; Sharp, J. Leukotoxin diols from ground corncob bedding disrupt estrous cyclicity in rats and stimulate MCF-7 breast cancer cell proliferation. Environ. Health Perspect. 2005, 113, 1698–1704. [Google Scholar] [CrossRef] [PubMed]
- Miyazawa, T. Lipid hydroperoxides in nutrition, health, and diseases. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2021, 37, 161–196. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Storage Conditions | Acyl Groups | Oil Samples | First Stage | Second Stage | ||
|---|---|---|---|---|---|---|
| Time (Days) | DR (mmol/mol TG Day) | Time (Days) | DR (mmol/mol TG Day) | |||
| 40 °C | Linoleic | W | 0–45 | −2.98 (0.95) | 45–59 | −82.98 (0.99) |
| W02 | 0–30 | −8.93 (0.92) | 30–45 | −64.97 (0.98) | ||
| W2 | 0–45 | −17.93 (0.97) | 45–59 | −43.92 (0.95) | ||
| Linolenic | W | 0–45 | −0.78 (0.91) | 45–59 | −18.34 (0.99) | |
| W02 | 0–30 | −3.34 (0.92) | 30–45 | −14.74 (0.97) | ||
| W2 | 0–59 | −4.94 (0.99) | --- | --- | ||
| 70 °C | Linoleic | W | 0–4 | −42.91(0.93) | 4–8 | −264.21 (0.94) |
| W02 | 0–4 | −57.15 (0.93) | 4–8 | −247.96 (0.99) | ||
| W2 | 0–8 | −66.61 (0.98) | 8–12 | −187.23 (0.99) | ||
| Linolenic | W | 0–4 | −3.93 (0.93) | 4–7 | −77.46 (0.98) | |
| W02 | 0–4 | −12.03 (0.93) | 4–8 | −58.90 (0.98) | ||
| W2 | 0–12 | −21.11 (0.99) | --- | --- | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caño-Ochoa, S.d.; Ruiz-Aracama, A.; Guillén, M.D. Alpha-Tocopherol, a Powerful Molecule, Leads to the Formation of Oxylipins in Polyunsaturated Oils Differently to the Temperature Increase: A Detailed Study by Proton Nuclear Magnetic Resonance of Walnut Oil Oxidation. Antioxidants 2022, 11, 604. https://doi.org/10.3390/antiox11040604
Caño-Ochoa Sd, Ruiz-Aracama A, Guillén MD. Alpha-Tocopherol, a Powerful Molecule, Leads to the Formation of Oxylipins in Polyunsaturated Oils Differently to the Temperature Increase: A Detailed Study by Proton Nuclear Magnetic Resonance of Walnut Oil Oxidation. Antioxidants. 2022; 11(4):604. https://doi.org/10.3390/antiox11040604
Chicago/Turabian StyleCaño-Ochoa, Sofía del, Ainhoa Ruiz-Aracama, and María D. Guillén. 2022. "Alpha-Tocopherol, a Powerful Molecule, Leads to the Formation of Oxylipins in Polyunsaturated Oils Differently to the Temperature Increase: A Detailed Study by Proton Nuclear Magnetic Resonance of Walnut Oil Oxidation" Antioxidants 11, no. 4: 604. https://doi.org/10.3390/antiox11040604