
Metabolic Stress and Mitochondrial Dysfunction in Ataxia-Telangiectasia

 , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
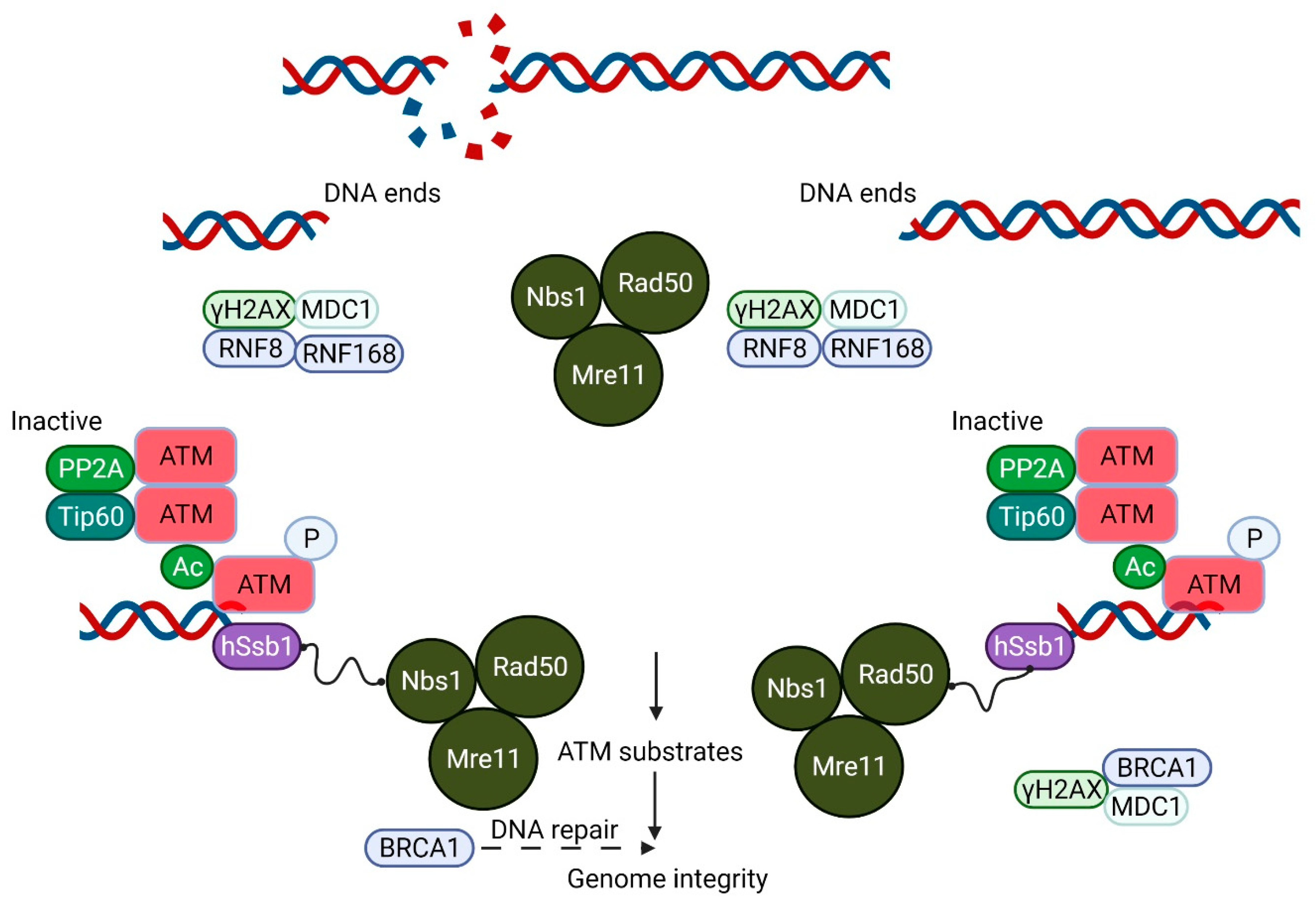
2. ATM Is Present in the Nucleus and Associated with Organelles in the Cytoplasm
3. Impaired Intracellular Signaling in A-T
4. ATM and Inflammation
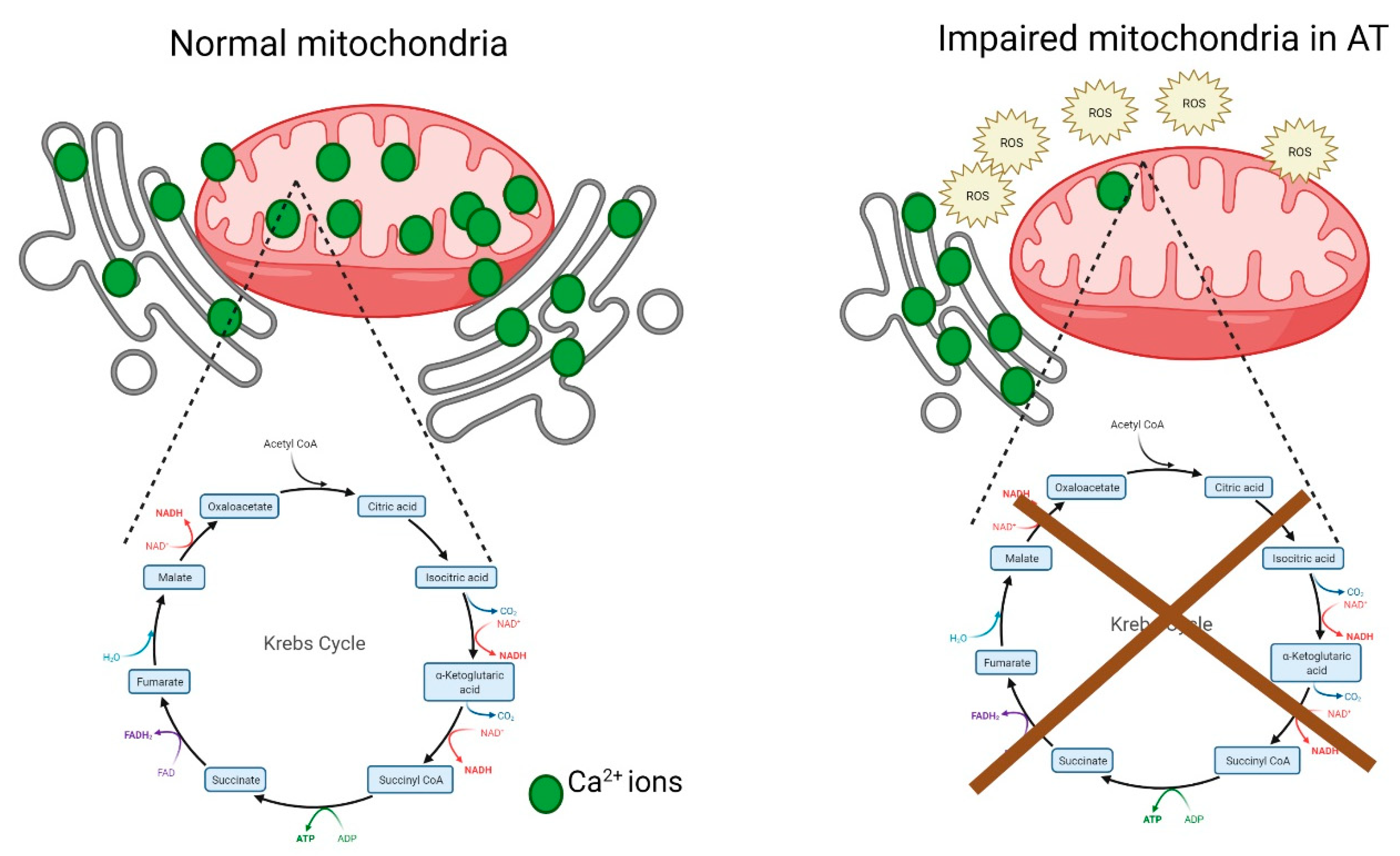
5. Mitochondrial Dysfunction in A-T
6. Metabolic Stress and A-T
7. Endoplasmic Reticulum (ER) Stress
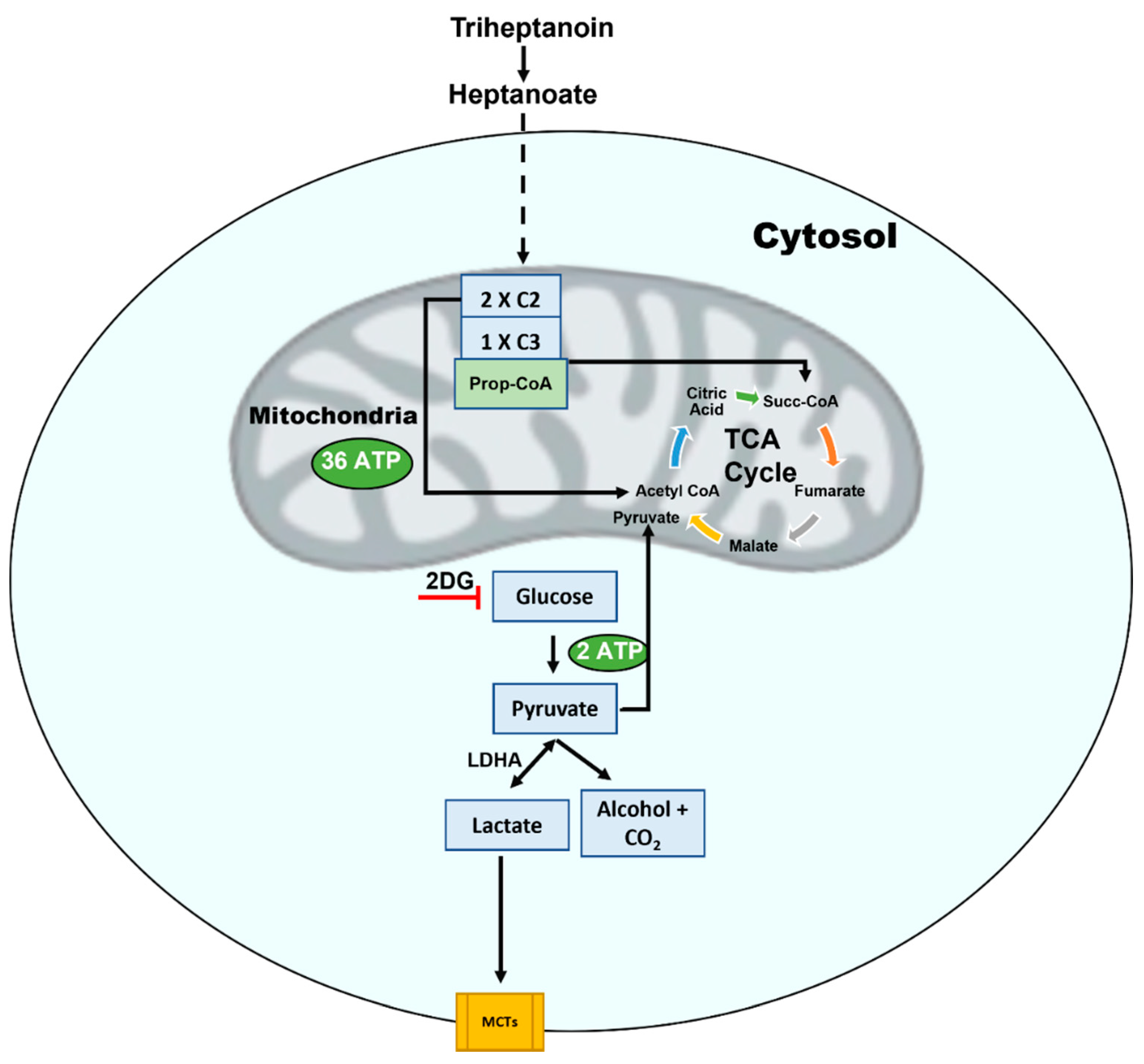
8. Focusing on a Therapy for A-T
9. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Lavin, M.F. Ataxia-telangiectasia: From a rare disorder to a paradigm for cell signalling and cancer. Nat. Rev. Mol. Cell Biol. 2008, 9, 759–769. [Google Scholar] [CrossRef] [PubMed]
- McKinnon, P.J. ATM and the Molecular Pathogenesis of Ataxia Telangiectasia. Annu. Rev. Pathol. Mech. Dis. 2012, 7, 303–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savitsky, K.; Bar-Shira, A.; Gilad, S.; Rotman, G.; Ziv, Y.; Vanagaite, L.; Tagle, D.A.; Smith, S.; Uziel, T.; Sfez, S.; et al. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science 1995, 268, 1749–1753. [Google Scholar] [CrossRef]
- Khanna, K.K.; Jackson, S.P. DNA double-strand breaks: Signaling, repair and the cancer connection. Nat. Genet. 2001, 27, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.M.R.; Harnden, D.G.; Arlett, C.F.; Harcourt, S.A.; Lehmann, A.R.; Stevens, S.; Bridges, B.A. Ataxia telangiectasia: A human mutation with abnormal radiation sensitivity. Nature 1975, 258, 427–429. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.C.; Lavin, M.; Kidson, C.; Moss, D. Identification of ataxia telangiectasia heterozygotes, a cancer prone population. Nature 1978, 274, 484–486. [Google Scholar] [CrossRef]
- Shiloh, Y.; Ziv, Y. The ATM protein kinase: Regulating the cellular response to genotoxic stress, and more. Nat. Rev. Mol. Cell Biol. 2013, 14, 197–210. [Google Scholar] [CrossRef]
- Mirzoeva, O.K.; Petrini, J.H.J. DNA replication-dependent nuclear dynamics of the Mre11 complex. Mol. Cancer Res. 2003, 1, 207–218. [Google Scholar]
- Uziel, T.; Lerenthal, Y.; Moyal, L.; Andegeko, Y.; Mittelman, L.; Shiloh, Y. Requirement of the MRN complex for ATM activation by DNA damage. EMBO J. 2003, 22, 5612–5621. [Google Scholar] [CrossRef] [Green Version]
- Bakkenist, C.J.; Kastan, M.B. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 2003, 421, 499–506. [Google Scholar] [CrossRef]
- Lavin, M.F.; Kozlov, S.; Gatei, M.; Kijas, A.W. ATM-dependent phosphorylation of all three members of the MRN complex: From sensor to adaptor. Biomolecules 2015, 5, 2877–2902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Z.; Kozlov, S.; Lavin, M.F.; Person, M.D.; Paull, T.T. ATM activation by oxidative stress. Science 2010, 330, 517–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sampadi, B.; Pines, A.; Munk, S.; Mišovic, B.; De Groot, A.J.; Van De Water, B.; Olsen, J.V.; Mullenders, L.H.F.; Vrieling, H. Quantitative phosphoproteomics to unravel the cellular response to chemical stressors with different modes of action. Arch. Toxicol. 2020, 94, 1655–1671. [Google Scholar] [CrossRef] [Green Version]
- Kozlov, S.V.; Waardenberg, A.; Engholm-Keller, K.; Arthur, J.W.; Graham, M.; Lavin, M. Reactive Oxygen Species (ROS)-activated ATM-dependent phosphorylation of cytoplasmic substrates identified by large-scale phosphoproteomics screen. Mol. Cell. Proteom. 2016, 15, 1032–1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlam-Babayov, S.; Bensimon, A.; Harel, M.; Geiger, T.; Aebersold, R.; Ziv, Y.; Shiloh, Y. Phosphoproteomics reveals novel modes of function and inter-relationships among PIKKs in response to genotoxic stress. EMBO J. 2020, 40, e104400. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial ROS-induced ROS release: An update and review. Biochim. Biophys. Acta 2006, 1757, 509–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeo, A.; Subramanian, G.; Chong, K.; Gatei, M.; Parton, R.; Coman, D.; Lavin, M. An anaplerotic approach to correct the mitochondrial dysfunction in ataxia-telangiectasia (A-T). Mol. Metab. 2021, 54, 101354. [Google Scholar] [CrossRef]
- Zhang, Y.; Lee, J.-H.; Paull, T.T.; Gehrke, S.; D’Alessandro, A.; Dou, Q.; Gladyshev, V.N.; Schroeder, E.A.; Steyl, S.K.; Christian, B.E.; et al. Mitochondrial redox sensing by the kinase ATM maintains cellular antioxidant capacity. Sci. Signal. 2018, 11, eaaq0702. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.E.; Chung, W.-H. Functional interplay between the oxidative stress response and DNA damage checkpoint signaling for genome maintenance in aerobic organisms. J. Microbiol. 2020, 58, 81–91. [Google Scholar] [CrossRef]
- Yeo, A.J.; Chong, K.L.; Gatei, M.; Zou, D.; Stewart, R.; Withey, S.; Wolvetang, E.; Parton, R.G.; Brown, A.D.; Kastan, M.B.; et al. Impaired endoplasmic reticulum-mitochondrial signaling in ataxia-telangiectasia. iScience 2021, 24, 101972. [Google Scholar] [CrossRef]
- Lee, J.-H.; Paull, T.T. Cellular functions of the protein kinase ATM and their relevance to human disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 796–814. [Google Scholar] [CrossRef] [PubMed]
- Baretić, D.; Pollard, H.K.; Fisher, D.I.; Johnson, C.M.; Santhanam, B.; Truman, C.M.; Kouba, T.; Fersht, A.R.; Phillips, C.; Williams, R.L. Structures of closed and open conformations of dimeric human ATM. Sci. Adv. 2017, 3, e1700933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watters, D.; Khanna, K.K.; Beamish, H.; Birrell, G.; Spring, K.; Kedar, P.; Gatei, M.; Stenzel, D.; Hobson, K.; Kozlov, S.; et al. Cellular localisation of the ataxia-telangiectasia (ATM) gene product and discrimination between mutated and normal forms. Oncogene 1997, 14, 1911–1921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watters, D.; Kedar, P.; Spring, K.; Bjorkman, J.; Chen, P.; Gatei, M.; Birrell, G.; Garrone, B.; Srinivasa, P.; Crane, D.I.; et al. Localization of a Portion of Extranuclear ATM to Peroxisomes. J. Biol. Chem. 1999, 274, 34277–34282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, D.S.; Kirsch, D.G.; Canman, C.E.; Ahn, J.H.; Ziv, Y.; Newman, L.S.; Darnell, R.B.; Shiloh, Y.; Kastan, M.B. ATM binds to beta-adaptin in cytoplasmic vesicles. Proc. Natl. Acad. Sci. USA 1998, 95, 10146–10151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oka, A.; Takashima, S. Expression of the ataxia-telangiectasia gene (ATM) product in human cerebellar neurons during development. Neurosci. Lett. 1998, 252, 195–198. [Google Scholar] [CrossRef]
- Barlow, C.; Ribaut-Barassin, C.; Zwingman, T.A.; Pope, A.J.; Brown, K.D.; Owens, J.W.; Larson, D.; Harrington, E.A.; Haeberle, A.-M.; Mariani, J.; et al. ATM is a cytoplasmic protein in mouse brain required to prevent lysosomal accumulation. Proc. Natl. Acad. Sci. USA 2000, 97, 871–876. [Google Scholar] [CrossRef] [Green Version]
- Hensey, C.; Robertson, K.; Gautier, J. Expression and subcellular localization of X-ATM during early Xenopus development. Dev. Genes Evol. 2000, 210, 467–469. [Google Scholar] [CrossRef]
- Valentin-Vega, Y.A.; MacLean, K.H.; Tait-Mulder, J.; Milasta, S.; Steeves, M.; Dorsey, F.C.; Cleveland, J.L.; Green, D.; Kastan, M.B. Mitochondrial dysfunction in ataxia-telangiectasia. Blood 2012, 119, 1490–1500. [Google Scholar] [CrossRef] [Green Version]
- Morita, A.; Tanimoto, K.; Murakami, T.; Morinaga, T.; Hosoi, Y. Mitochondria are required for ATM activation by extranuclear oxidative stress in cultured human hepatoblastoma cell line Hep G2 cells. Biochem. Biophys. Res. Commun. 2014, 443, 1286–1290. [Google Scholar] [CrossRef]
- Blignaut, M.; Loos, B.; Botchway, S.W.; Parker, A.W.; Huisamen, B. Ataxia-Telangiectasia Mutated is located in cardiac mitochondria and impacts oxidative phosphorylation. Sci. Rep. 2019, 9, 4782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Tripathi, D.; Jing, J.; Alexander, A.; Kim, J.; Powell, R.T.; Dere, R.; Tait-Mulder, J.; Lee, J.-H.; Paull, T.T.; et al. ATM functions at the peroxisome to induce pexophagy in response to ROS. Nat. Cell Biol. 2015, 17, 1259–1269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiloh, Y.; Tabor, E.; Becker, Y. Colony-forming ability of ataxia-telangiectasia skin fibroblasts is an indicator of their early senescence and increased demand for growth factors. Exp. Cell Res. 1982, 140, 191–199. [Google Scholar] [CrossRef]
- O’Connor, R.D.; Linthicum, D. Mitogen receptor redistribution defects and concomitant absence of blastogenesis in ataxia-telangiectasia T lymphocytes. Clin. Immunol. Immunopathol. 1980, 15, 66–75. [Google Scholar] [CrossRef]
- Kondo, N.; Inoue, R.; Nishimura, S.; Kasahara, K.; Kameyama, T.; Miwa, Y.; Lorenzo, P.R.; Orii, T. Defective calcium-dependent signal transduction in T lymphocytes of ataxia-telangiectasia. Scand. J. Immunol. 1993, 38, 45–48. [Google Scholar] [CrossRef]
- Khanna, K.K.; Yan, J.; Watters, D.; Hobson, K.; Beamish, H.; Spring, K.; Shiloh, Y.; Gatti, R.A.; Lavin, M.F. Defective Signaling through the B Cell Antigen Receptor in Epstein-Barr Virus-transformed Ataxia-Telangiectasia Cells. J. Biol. Chem. 1997, 272, 9489–9495. [Google Scholar] [CrossRef] [Green Version]
- Khan, N.; Rothstein, T.L. The alternate pathway for BCR signaling induced by IL-4 requires Lyn tyrosine kinase. J. Mol. Biol. 2021, 433, 166667. [Google Scholar] [CrossRef]
- Speck, P.; Ikeda, M.; Ikeda, A.; Lederman, H.M.; Longnecker, R. Signal transduction through the B cell antigen receptor is normal int ataxia-telangiectasia B lymphocytes. J. Biol. Chem. 2002, 277, 4123–4127. [Google Scholar] [CrossRef] [Green Version]
- Stagni, V.; Di Bari, M.G.; Cursi, S.; Condò, I.; Cencioni, M.T.; Testi, R.; Lerenthal, Y.; Cundari, E.; Barila’, D. ATM kinase activity modulates Fas sensitivity through the regulation of FLIP in lymphoid cells. Blood 2008, 111, 829–837. [Google Scholar] [CrossRef] [Green Version]
- Hofseth Lorne, J.; Saito, S.I.; Hussain, S.P.; Espey, M.G.; Miranda, K.M.; Araki, Y.; Harris, C.C. Nitric oxide-induced cellular stress and p53 activation in chronic inflammation. Proc. Natl. Acad. Sci. USA 2003, 100, 143–148. [Google Scholar] [CrossRef] [Green Version]
- McCool, K.W.S.; Miyamoto, D.N.A. Damage-dependent NF-κB activation: NEMO turns nuclear signaling inside out. Immunol. Rev. 2012, 246, 311–326. [Google Scholar] [CrossRef] [PubMed]
- Sabatel, H.; Di Valentin, E.; Gloire, G.; Dequiedt, F.; Piette, J.; Habraken, Y. Phosphorylation of p65(RelA) on Ser547 by ATM Represses NF-κB-Dependent Transcription of Specific Genes after Genotoxic Stress. PLoS ONE 2012, 7, e38246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeo, A.J.; Henningham, A.; Fantino, E.; Galbraith, S.; Krause, L.; Wainwright, C.E.; Sly, P.D.; Lavin, M.F. Increased susceptibility of airway epithelial cells from ataxia-telangiectasia to S. pneumoniae infection due to oxidative damage and impaired innate immunity. Sci. Rep. 2019, 9, 2627. [Google Scholar] [CrossRef] [PubMed]
- Hartlova, A.S.; Erttmann, S.F.; Raffi, F.A.; Schmalz, A.M.; Resch, U.; Anugula, S.; Lienenklaus, S.; Nilsson, L.M.; Kröger, A.; Nilsson, J.A.; et al. DNA damage primes the type I interferon system via the cytosolic DNA sensor STING to promote anti-microbial innate immunity. Immunity 2015, 42, 332–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguado, J.; Chaggar, H.K.; Gomez-Inclan, C.; Shaker, M.R.; Leeson, H.C.; Mackay-Sim, A.; Wolvetang, E.J. Inhibition of the cGAS-STING pathway ameliorates the premature senescence hallmarks of Ataxia-Telangiectasia brain organoids. Aging Cell 2021, 20, e13468. [Google Scholar] [CrossRef] [PubMed]
- Scheibye-Knudsen, M.; Scheibye-Alsing, K.; Canugovi, C.; Croteau, D.L.; Bohr, V.A. A novel diagnostic tool reveals mitochondrial pathology in human diseases and aging. Aging 2013, 5, 192–208. [Google Scholar] [CrossRef] [Green Version]
- Ambrose, M.; Goldstine, J.V.; Gatti, R.A. Intrinsic mitochondrial dysfunction in ATM-deficient lymphoblastoid cells. Hum. Mol. Genet. 2007, 16, 2154–2164. [Google Scholar] [CrossRef]
- Qi, Y.; Qiu, Q.; Gu, X.; Tian, Y.; Zhang, Y. ATM mediates spermidine-induced mitophagy via PINK1 and Parkin regulation in human fibroblasts. Sci. Rep. 2016, 6, 24700. [Google Scholar] [CrossRef] [Green Version]
- Fang, E.F.; Kassahun, H.; Croteau, D.L.; Scheibye-Knudsen, M.; Marosi, K.; Lu, H.; Shamanna, R.A.; Kalyanasundaram, S.; Bollineni, R.C.; Wilson, M.A.; et al. NAD+ replenishment improves lifespan and healthspan in ataxia telangiectasia models via mitophagy and DNA repair. Cell Metab. 2016, 24, 566–581. [Google Scholar] [CrossRef] [Green Version]
- Blignaut, M.; Harries, S.; Lochner, A.; Huisamen, B. Ataxia telangiectasia mutated protein kinase: A potential master puppeteer of oxidative stress-induced metabolic recycling. Oxidative Med. Cell. Longev. 2021, 2021, 8850708. [Google Scholar] [CrossRef]
- Sunderland, P.; Augustyniak, J.; Lenart, J.; Bużańska, L.; Carlessi, L.; Delia, D.; Sikora, E. ATM-deficient neural precursors develop senescence phenotype with disturbances in autophagy. Mech. Ageing Dev. 2020, 190, 111296. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, A.; Gandhi, V. Activation of ATM kinase by ROS generated during ionophore-induced mitophagy in human T and B cell malignancies. Mol. Cell. Biochem. 2021, 476, 2887. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, A.; Stellrecht, C.M.; Vangapandu, H.V.; Ayres, M.; Kaipparettu, B.A.; Park, J.H.; Balakrishnan, K.; Burks, J.K.; Pandita, T.K.; Hittelman, W.N.; et al. Ataxia-telangiectasia mutated interacts with Parkin and induces mitophagy independent of kinase activity. Evidence from mantle cell lymphoma. Haematologica 2020, 106, 495–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, N.K.; Lebedeva, M.; Thomas, T.; Kovalenko, O.A.; Stumpf, J.D.; Shadel, G.S.; Santos, J.H. Intrinsic mitochondrial DNA repair defects in Ataxia Telangiectasia. DNA Repair 2014, 13, 22–31. [Google Scholar] [CrossRef]
- Gao, Y.; Katyal, S.; Lee, Y.; Zhao, J.; Rehg, J.E.; Russell, H.R.; McKinnon, P.J. DNA ligase III is critical for mtDNA integrity but not Xrcc1-mediated nuclear DNA repair. Nature 2011, 471, 240–244. [Google Scholar] [CrossRef] [Green Version]
- Guo, Q.; Wang, S.; Xu, H.; Li, X.; Guan, Y.; Yi, F.; Zhou, T.; Jiang, B.; Bai, N.; Ma, M.; et al. ATM-CHK 2-Beclin 1 axis promotes autophagy to maintain ROS homeostasis under oxidative stress. EMBO J. 2020, 39, e103111. [Google Scholar] [CrossRef]
- D’Souza, A.D.; Parish, I.A.; Krause, D.S.; Kaech, S.M.; Shadel, G.S. Reducing mitochondrial ROS improves disease-related pathology in a mouse model of ataxia-telangiectasia. Mol. Ther. 2013, 21, 42–48. [Google Scholar] [CrossRef] [Green Version]
- Schneider, J.G.; Finck, B.N.; Ren, J.; Standley, K.N.; Takagi, M.; Maclean, K.H.; Bernal-Mizrachi, C.; Muslin, A.J.; Kastan, M.B.; Semenkovich, C.F. ATM-dependent suppression of stress signaling reduces vascular disease in metabolic syndrome. Cell Metab. 2006, 4, 377–389. [Google Scholar] [CrossRef] [Green Version]
- Donath, H.; Hess, U.; Kieslich, M.; Theis, M.; Ohlenschläger, U.; Schubert, R.; Woelke, S.; Zielen, S. Diabetes in patients With ataxia telangiectasia: A national cohort study. Front. Pediatr. 2020, 8, 317. [Google Scholar] [CrossRef]
- Halaby, M.-J.; Hibma, J.C.; He, J.; Yang, D.-Q. ATM protein kinase mediates full activation of Akt and regulates glucose transporter 4 translocation by insulin in muscle cells. Cell. Signal. 2008, 20, 1555–1563. [Google Scholar] [CrossRef]
- Armata, H.L.; Golebiowski, D.; Jung, D.Y.; Ko, H.J.; Kim, J.K.; Sluss, H.K. Requirement of the ATM/p53 tumor suppressor pathway for glucose homeostasis. Mol. Cell. Biol. 2010, 30, 5787–5794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, X.Q.; Luo, Y.; Chen, H.; Liu, D. Mitochondria, endothelial cell function, and vascular diseases. Front. Physiol. 2014, 5, 175–192. [Google Scholar] [CrossRef] [PubMed]
- Howarth, C.; Gleeson, P.; Attwell, D. Updated energy budgets for neural computation in the neocortex and cerebellum. J. Cereb. Blood Flow Metab. 2012, 32, 1222–1232. [Google Scholar] [CrossRef] [PubMed]
- Chow, H.-M.; Cheng, A.; Song, X.; Swerdel, M.R.; Hart, R.P.; Herrup, K. ATM is activated by ATP depletion and modulates mitochondrial function through NRF1. J. Cell Biol. 2019, 218, 909–928. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, S.; Mishra, A.K.; Peer, G.D.G.; Bagabir, S.A.; Haque, S.; Pandey, R.P.; Raj, V.S.; Jain, N.; Pandey, A.; Kar, S.K. The interplay of the unfolded protein response in neurodegenerative diseases: A therapeutic role of curcumin. Front. Aging Neurosci. 2021, 13, 767493. [Google Scholar] [CrossRef]
- Jiang, Y.; Tao, Z.; Chen, H.; Xia, S. Endoplasmic reticulum quality control in immune cells. Front. Cell Dev. Biol. 2021, 9, 740653. [Google Scholar] [CrossRef]
- Yan, M.; Shen, J.; Person, M.D.; Kuang, X.; Lynn, W.S.; Atlas, D.; Wong, P.K. Endoplasmic reticulum stress and unfolded protein response in atm-deficient thymocytes and thymic lymphoma cells are attributable to oxidative stress. Neoplasia 2008, 10, 160–167. [Google Scholar] [CrossRef] [Green Version]
- Poletto, M.; Yang, D.; Fletcher, S.; Vendrell, I.; Fischer, R.; Legrand, A.; Dianov, G.L. Modulation of proteostasis counteracts oxidative stress and affects DNA base excision repair capacity in ATM-deficient cells. Nucleic Acids Res. 2017, 45, 10042–10055. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.-H.; Mand, M.R.; Kao, C.-H.; Zhou, Y.; Ryu, S.W.; Richards, A.L.; Coon, J.J.; Paull, T.T. ATM directs DNA damage responses and proteostasis via genetically separable pathways. Sci. Signal. 2018, 11, eaan5598. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.-H.; Ryu, S.W.; Ender, N.A.; Paull, T.T. Poly-ADP-ribosylation drives loss of protein homeostasis in ATM and Mre11 deficiency. Mol. Cell 2021, 81, 1515–1533.e5. [Google Scholar] [CrossRef]
- Yan, J.; Khanna, K.K.; Lavin, M.F. Defective radiation signal transduction in ataxia-telangiectasia cells. Int. J. Radiat. Biol. 2000, 76, 1025–1035. [Google Scholar] [PubMed]
- Cárdenas, C.; Miller, R.A.; Smith, I.; Bui, T.; Molgó, J.; Müller, M.; Vais, H.; Cheung, K.-H.; Yang, J.; Parker, I.; et al. Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell 2010, 142, 270–283. [Google Scholar] [CrossRef] [Green Version]
- McGrath-Morrow, S.A.; Rothblum-Oviatt, C.C.; Wright, J.; Schlechter, H.; Lefton-Greif, M.A.; Natale, V.A.; Crawford, T.O.; Lederman, H.M. Multidisciplinary management of ataxia telangiectasia: Current perspectives. J. Multidiscip. Healthc. 2021, 14, 1637–1643. [Google Scholar] [CrossRef]
- De Oliveira, B.S.P.; Putti, S.; Naro, F.; Pellegrini, M. Bone marrow transplantation as therapy for ataxia-telangiectasia: A systematic review. Cancers 2020, 12, 3207. [Google Scholar] [CrossRef] [PubMed]
- Yi, M.; Rosin, M.P.; Anderson, C.K. Response of fibroblast cultures from ataxia-telangiectasia patients to oxidative stress. Cancer Lett 1990, 54, 43–50. [Google Scholar] [CrossRef]
- Yeo, A.J.; Fantino, E.; Czovek, D.; Wainwright, C.; Sly, P.D.; Lavin, M.F. Loss of ATM in airway epithelial cells is associated with susceptibility to oxidative stress. Am. J. Respir. Crit. Care Med. 2017, 196, 391–393. [Google Scholar] [CrossRef]
- Paulino, T.L.; Rafael, M.N.; Hix, S.; Shigueoka, D.C.; Ajzen, S.A.; Kochi, C.; Suano-Souza, F.I.; Da Silva, R.; Costa-Carvalho, B.T.; Sarni, R.O.S. Is age a risk factor for liver disease and metabolic alterations in ataxia Telangiectasia patients? Orphanet J. Rare Dis. 2017, 12, 1–7. [Google Scholar] [CrossRef]
- Caballero, T.; Caba-Molina, M.; Salmerón, J.; Gómez-Morales, M. Nonalcoholic steatohepatitis in a patient with ataxia-telangiectasia. Case Rep. Hepatol. 2014, 2014, 761250. [Google Scholar] [CrossRef] [Green Version]
- Aksoy, Y.; Sanal, O.; Metin, A.; Tezcan, I.; Ersoy, F.; Oğüş, H.; Ozer, N. Antioxidant enzymes in red blood cells and lymphocytes of ataxia-telangiectasia patients. Turk. J. Pediatr. 2004, 46, 204–207. [Google Scholar]
- Reichenbach, J.; Schubert, R.; Schwan, C.; Müller, K.; Böhles, H.J.; Zielen, S. Anti-oxidative capacity in patients with ataxia telangiectasia. Clin. Exp. Immunol. 1999, 117, 535–539. [Google Scholar] [CrossRef]
- Chen, P.; Peng, C.; Luff, J.; Spring, K.; Watters, D.; Bottle, S.; Furuya, S.; Lavin, M. Oxidative stress is responsible for deficient survival and dendritogenesis in purkinje neurons from ataxia-telangiectasia mutated mutant mice. J. Neurosci. 2003, 23, 11453–11460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, E.F.; Bohr, V.A. NAD+: The convergence of DNA repair and mitophagy. Autophagy 2017, 13, 442–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wehbe, Z.; Tucci, S. Therapeutic potential of triheptanoin in metabolic and neurodegenerative diseases. J. Inherit. Metab. Dis. 2020, 43, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Roe, D.; Yang, B.; Vianey-Saban, C.; Struys, E.; Sweetman, L.; Roe, C. Differentiation of long-chain fatty acid oxidation disorders using alternative precursors and acylcarnitine profiling in fibroblasts. Mol. Genet. Metab. 2006, 87, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Shirley, M. Triheptanoin: First Approval. Drugs 2020, 80, 1595–1600. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Wang, L.; Tandon, N.; Sun, H.; Tian, J.; Du, H.; Pascual, J.M.; Guo, L. Triheptanoin mitigates brain ATP depletion and mitochondrial dysfunction in a mouse model of Alzheimer’s disease. J. Alzheimer’s Dis. 2020, 78, 425–437. [Google Scholar] [CrossRef]
- Laemmle, A.; Steck, A.L.; Schaller, A.; Kurth, S.; Hoigné, E.P.; Felser, A.D.; Slavova, N.; Salvisberg, C.; Atencio, M.; Mochel, F.; et al. Triheptanoin–Novel therapeutic approach for the ultra-rare disease mitochondrial malate dehydrogenase deficiency. Mol. Genet. Metab. Rep. 2021, 29, 100814. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Subramanian, G.N.; Yeo, A.J.; Gatei, M.H.; Coman, D.J.; Lavin, M.F. Metabolic Stress and Mitochondrial Dysfunction in Ataxia-Telangiectasia. Antioxidants 2022, 11, 653. https://doi.org/10.3390/antiox11040653
Subramanian GN, Yeo AJ, Gatei MH, Coman DJ, Lavin MF. Metabolic Stress and Mitochondrial Dysfunction in Ataxia-Telangiectasia. Antioxidants. 2022; 11(4):653. https://doi.org/10.3390/antiox11040653
Chicago/Turabian StyleSubramanian, Goutham Narayanan, Abrey Jie Yeo, Magtouf Hnaidi Gatei, David John Coman, and Martin Francis Lavin. 2022. "Metabolic Stress and Mitochondrial Dysfunction in Ataxia-Telangiectasia" Antioxidants 11, no. 4: 653. https://doi.org/10.3390/antiox11040653
APA StyleSubramanian, G. N., Yeo, A. J., Gatei, M. H., Coman, D. J., & Lavin, M. F. (2022). Metabolic Stress and Mitochondrial Dysfunction in Ataxia-Telangiectasia. Antioxidants, 11(4), 653. https://doi.org/10.3390/antiox11040653