
Iron Dysregulation in Mitochondrial Dysfunction and Alzheimer’s Disease


Abstract
:1. Introduction
2. Iron Chemistry and Oxidation States
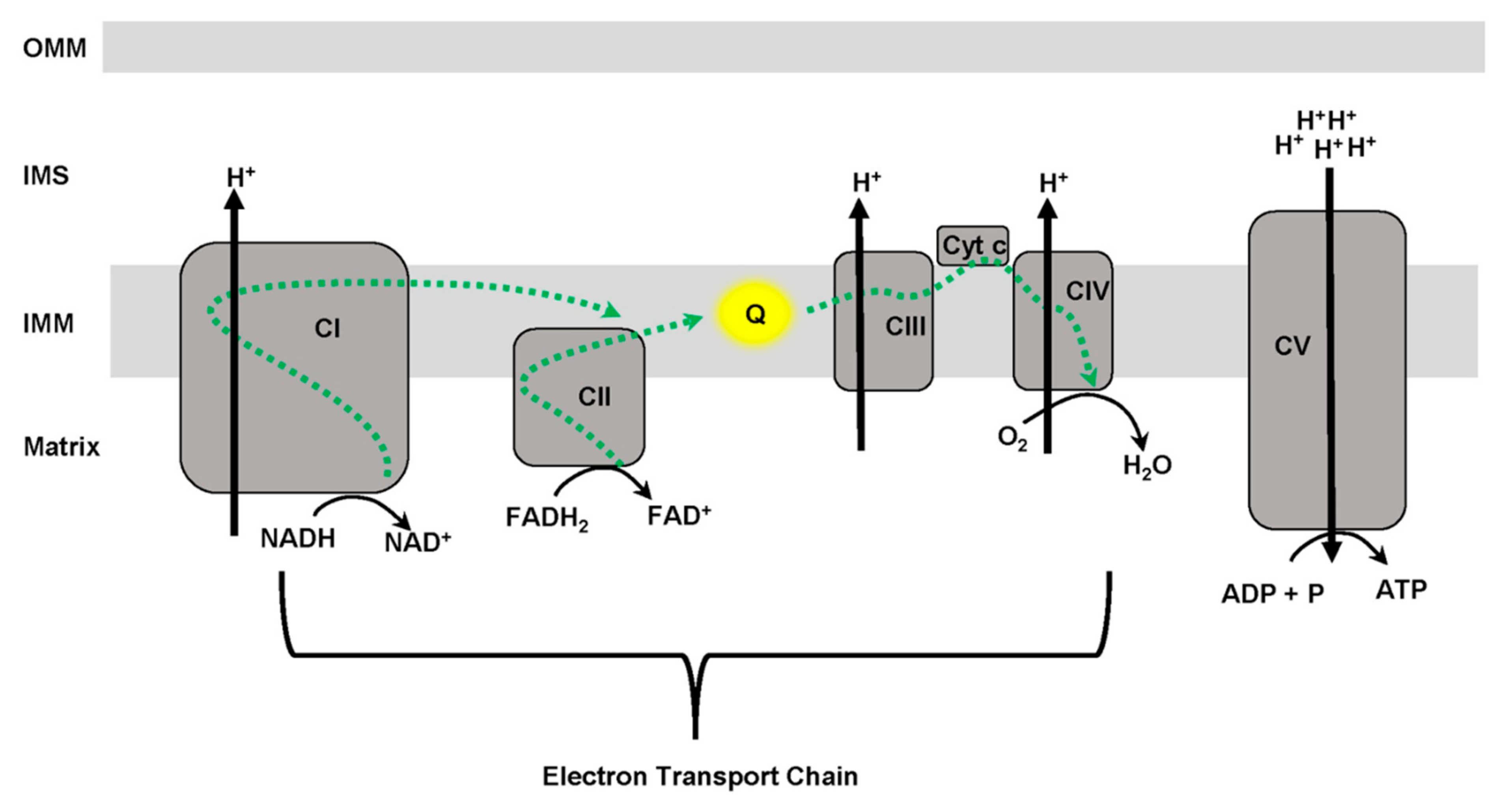
3. Mitochondrial Function
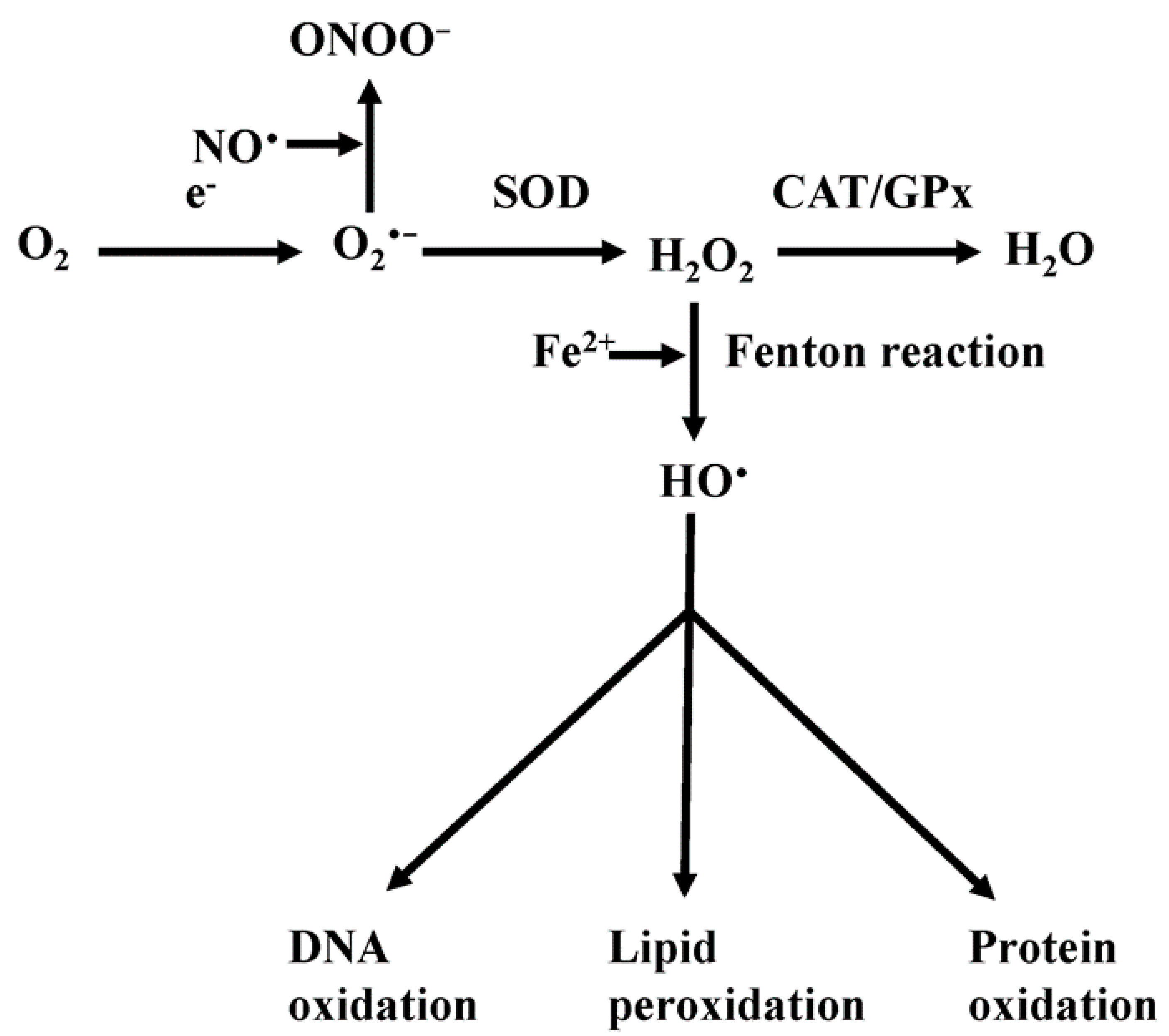
4. Mitochondrial Reactive Oxygen Species (ROS)
5. Mitochondrial Iron Metabolism and Homeostasis
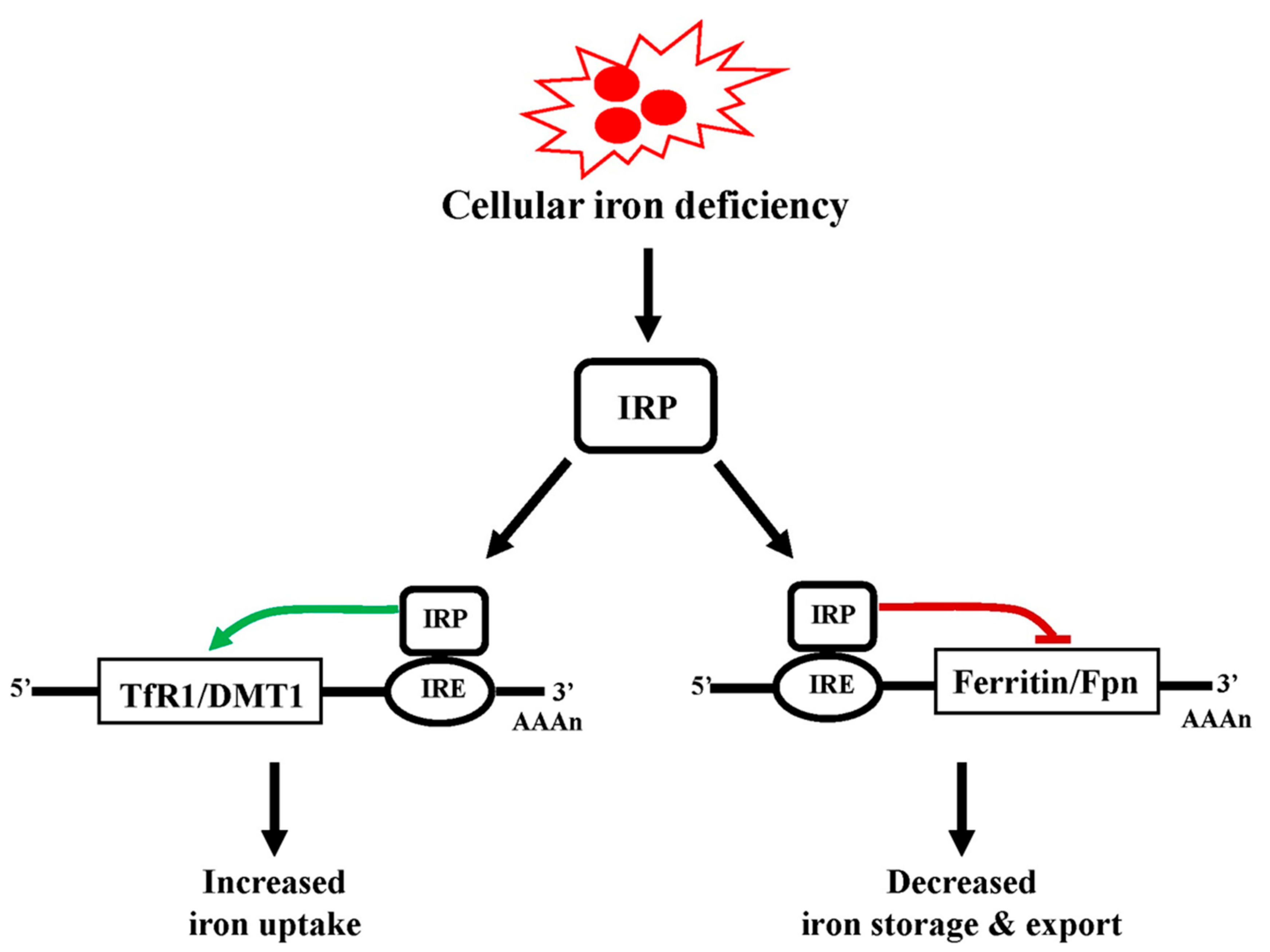
5.1. Iron Homeostasis
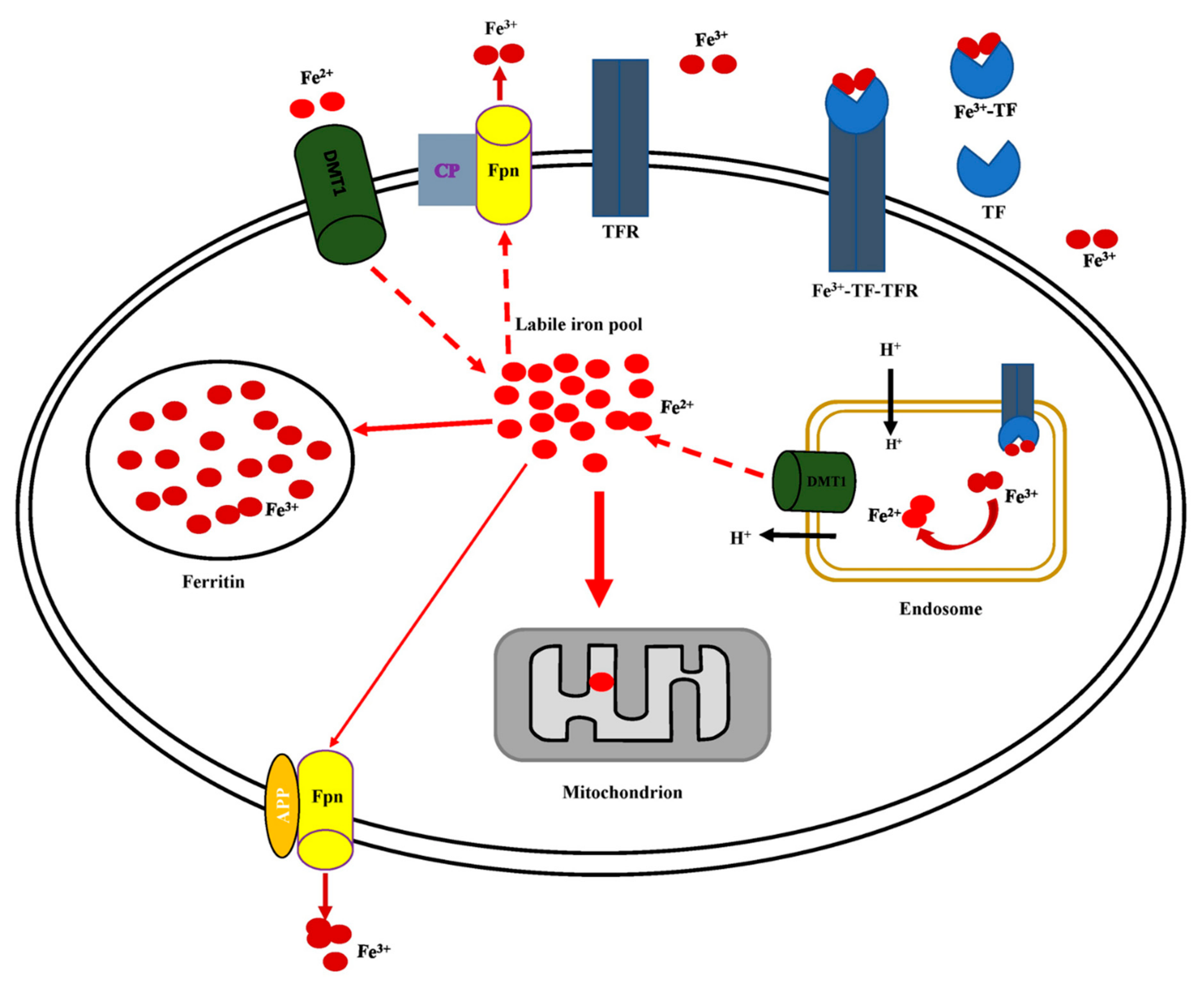
5.2. Cellular Iron Uptake, Storage, Utilization, and Efflux
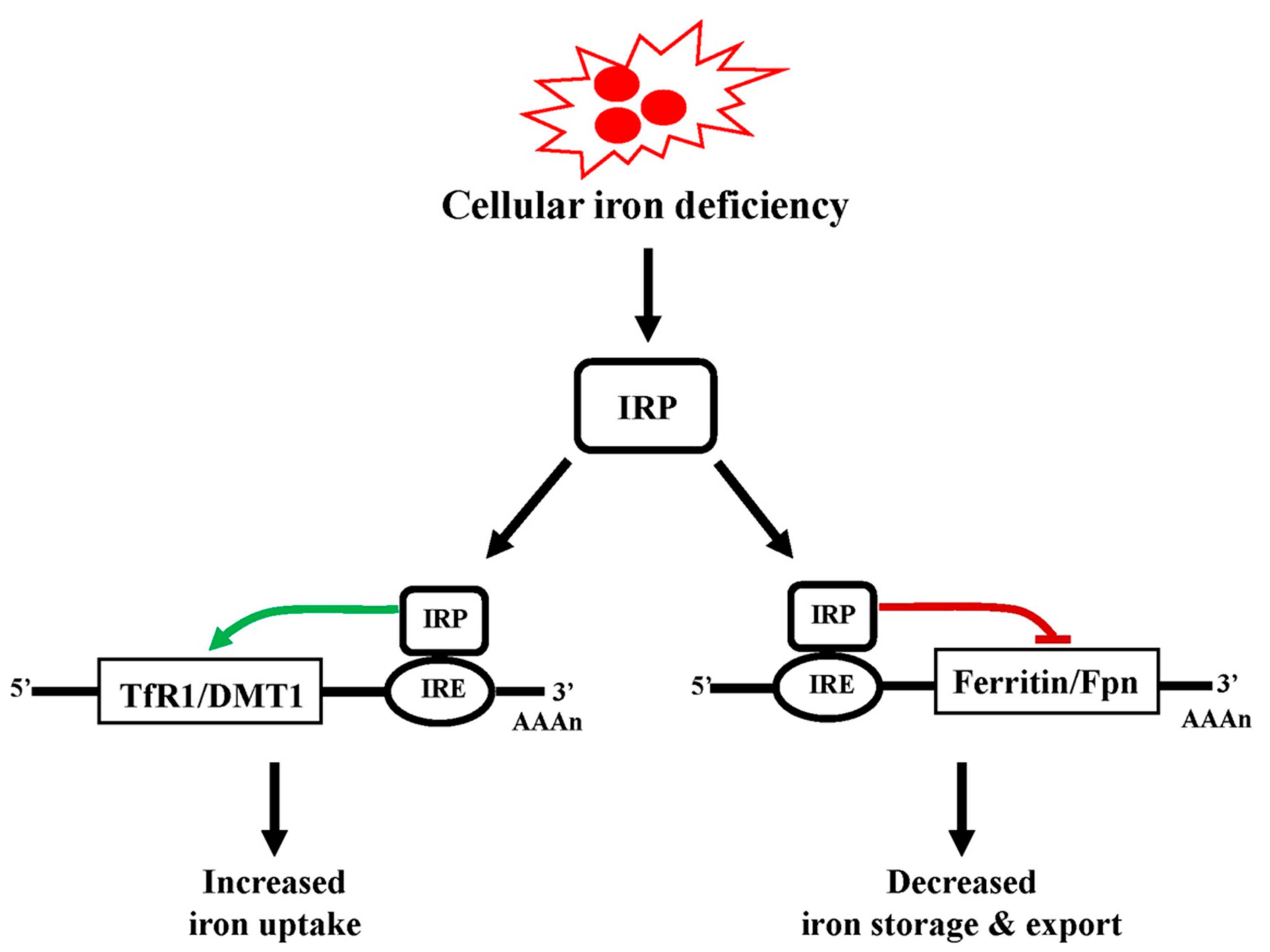
5.3. Regulation of Cellular Iron
6. Iron and Alzheimer’s Disease
7. Mitochondrial Iron Dysregulation and Alzheimer’s Disease
7.1. Dysfunction in Mitochondrial Bioenergetics
7.2. Dysregulation in Mitochondrial Fission and Fusion
7.3. Linking Iron Induced Mitochondrial Dysfunction in Alzheimer’s Disease
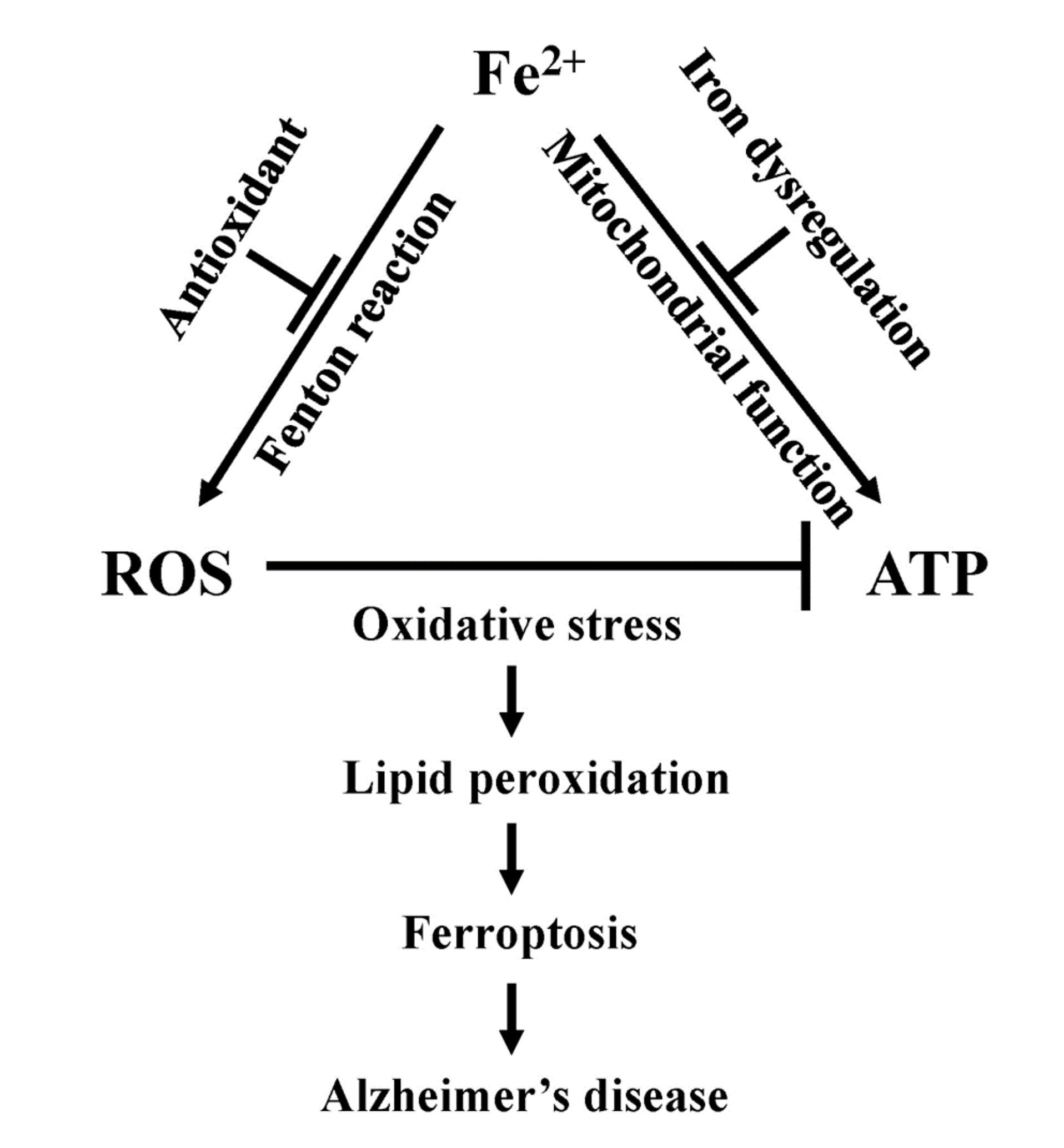
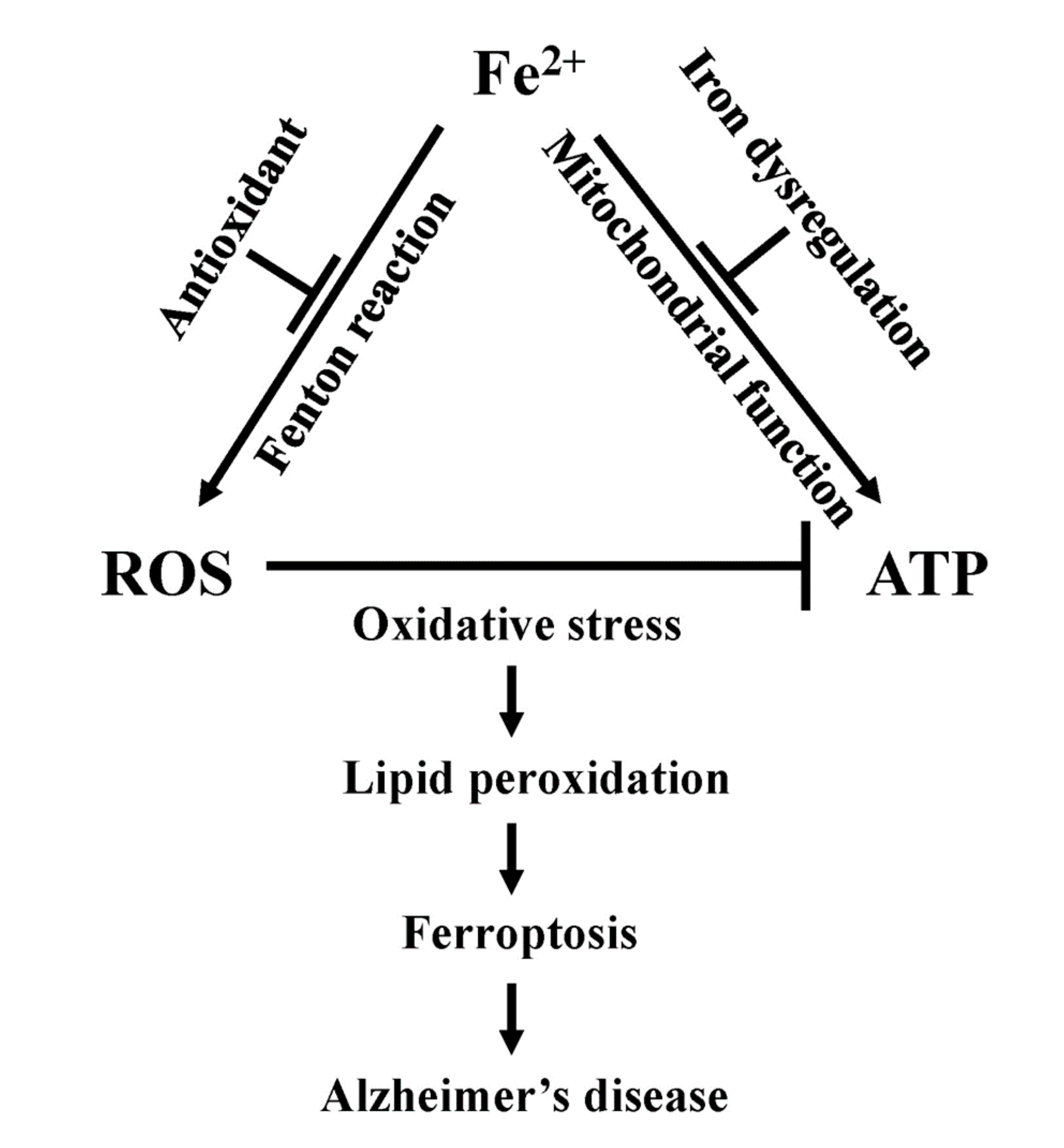
8. Ferroptosis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Small Molecules/Chemicals | Mechanisms/Target | Effects on Ferroptosis | Reference |
|---|---|---|---|
| Erastin | Inhibit system Xc | Induce | [147] |
| Sorafenib | Inhibit system Xc | Induce | [148] |
| Sulfasalazine | Inhibit system Xc | Induce | [149] |
| Glutamate | Inhibit system Xc | Induce | [150] |
| RSL3 | Block GPx4 | Induce | [135] |
| ML162 | Block GPx4 | Induce | [136] |
| ML210 | Block GPx4 | Induce | [137] |
| NSC144988 | Block GPx4 | Induce | [138] |
| Ferrostatin1 | Radical trapping of lipid peroxide | Inhibit | [151] |
| Liprostatin1 | Radical trapping of lipid peroxide | Inhibit | [151] |
| α-Tocophenol | Radical trapping of lipid peroxide | Inhibit | [23] |
| Idebenone | CoQ10 anolog- Radical trapping of lipid peroxide | Inhibit | [152] |
| 4-Chlorobenzoic acid | Block CoQ10 pathway | Induce | [140] |
| iFSP1 | Block CoQ10 pathway | Induce | [139] |
| Deferoxamine | Iron chelator | Inhibit | [23] |
9. Ferroptosis, Mitochondrial Dysfunction, and Alzheimer’s Disease
10. Future Perspectives in Alzheimer’s Disease
11. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Wiley, J. Alzheimer’s disease facts and figures. Alzheimers Dement. 2021, 17, 327–406. [Google Scholar]
- Coyle, J.T.; Price, D.L.; DeLong, M.R. Alzheimer’s disease: A disorder of cortical cholinergic innervation. Science 1983, 219, 1184–1190. [Google Scholar] [PubMed]
- Collaborators, G.B.D.N. Global, regional, and national burden of neurological disorders 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 459–480. [Google Scholar]
- Wang, W.; Zhao, F.; Ma, X.; Perry, G.; Zhu, X. Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: Recent advances. Mol. Neurodegener. 2020, 15, 30. [Google Scholar] [CrossRef] [PubMed]
- Plascencia-Villa, G.; Perry, G. Iron.ic facts about dementia. J. Neurol. Neurosurg. Psychiatry 2018, 89, 446–447. [Google Scholar] [PubMed]
- Khachaturian, Z.S. Diagnosis of Alzheimer’s disease. Arch. Neurol. 1985, 42, 1097–1105. [Google Scholar]
- McKean, N.E.; Handley, R.R.; Snell, R.G. A Review of the Current Mammalian Models of Alzheimer’s Disease and Challenges That Need to Be Overcome. Int. J. Mol. Sci. 2021, 22, 13168. [Google Scholar]
- Rossmann, M.P.; Dubois, S.M.; Agarwal, S.; Zon, L.I. Mitochondrial function in development and disease. Dis. Models Mech. 2021, 14, dmm048912. [Google Scholar]
- Martinez-Reyes, I.; Chandel, N.S. Mitochondrial TCA cycle metabolites control physiology and disease. Nat. Commun. 2020, 11, 102. [Google Scholar]
- Pagliarini, D.J.; Rutter, J. Hallmarks of a new era in mitochondrial biochemistry. Genes Dev. 2013, 27, 2615–2627. [Google Scholar]
- Harris, D.A.; Das, A.M. Control of mitochondrial ATP synthesis in the heart. Biochem. J. 1991, 280 Pt 3, 561–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crichton, R.R.; Dexter, D.T.; Ward, R.J. Brain iron metabolism and its perturbation in neurological diseases. J. Neural. Transm. 2011, 118, 301–314. [Google Scholar] [CrossRef] [PubMed]
- Belaidi, A.A.; Bush, A.I. Iron neurochemistry in Alzheimer’s disease and Parkinson’s disease: Targets for therapeutics. J. Neurochem. 2016, 139 (Suppl. S1), 179–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, R.J.; Zucca, F.A.; Duyn, J.H.; Crichton, R.R.; Zecca, L. The role of iron in brain ageing and neurodegenerative disorders. Lancet Neurol. 2014, 13, 1045–1060. [Google Scholar] [CrossRef] [Green Version]
- Weinreb, O.; Mandel, S.; Youdim, M.B.H.; Amit, T. Targeting dysregulation of brain iron homeostasis in Parkinson’s disease by iron chelators. Free Radic. Biol. Med. 2013, 62, 52–64. [Google Scholar] [CrossRef]
- Singh, N.; Haldar, S.; Tripathi, A.K.; Horback, K.; Wong, J.; Sharma, D.; Beserra, A.; Suda, S.; Anbalagan, C.; Dev, S.; et al. Brain iron homeostasis: From molecular mechanisms to clinical significance and therapeutic opportunities. Antioxid. Redox Signal. 2014, 20, 1324–1363. [Google Scholar] [CrossRef] [Green Version]
- Hentze, M.W.; Kuhn, L.C. Molecular control of vertebrate iron metabolism: MRNA-based regulatory circuits operated by iron, nitric oxide, and oxidative stress. Proc. Natl. Acad. Sci. USA 1996, 93, 8175–8182. [Google Scholar] [CrossRef] [Green Version]
- Kaplan, J.; Ward, D.M.; de Domenico, I. The molecular basis of iron overload disorders and iron-linked anemias. Int. J. Hematol. 2011, 93, 14–20. [Google Scholar] [CrossRef]
- Muckenthaler, M.U.; Galy, B.; Hentze, M.W. Systemic iron homeostasis and the iron-responsive element/iron-regulatory protein (IRE/IRP) regulatory network. Annu. Rev. Nutr. 2008, 28, 197–213. [Google Scholar] [CrossRef]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Mailloux, R.J.; McBride, S.L.; Harper, M.E. Unearthing the secrets of mitochondrial ROS and glutathione in bioenergetics. Trends Biochem. Sci. 2013, 38, 592–602. [Google Scholar] [CrossRef] [PubMed]
- Mailloux, R.J. Teaching the fundamentals of electron transfer reactions in mitochondria and the production and detection of reactive oxygen species. Redox Biol. 2015, 4, 381–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [Green Version]
- Cao, J.Y.; Dixon, S.J. Mechanisms of ferroptosis. Cell Mol. Life Sci. 2016, 73, 2195–2209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Comish, P.B.; Tang, D.; Kang, R. Characteristics and Biomarkers of Ferroptosis. Front. Cell Dev. Biol. 2021, 9, 637162. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Hou, W.; Song, X.; Yu, Y.; Huang, J.; Sun, X.; Kang, R.; Tang, D. Ferroptosis: Process and function. Cell Death Differ. 2016, 23, 369–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doll, S.; Conrad, M. Iron and ferroptosis: A still ill-defined liaison. IUBMB Life 2017, 69, 423–434. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.S.; Stockwell, B.R. Ferroptosis: Death by Lipid Peroxidation. Trends Cell Biol. 2016, 26, 165–176. [Google Scholar] [CrossRef] [Green Version]
- Conrad, M.; Kagan, V.E.; Bayir, H.; Pagnussat, G.C.; Head, B.; Traber, M.G.; Stockwell, B.R. Regulation of lipid peroxidation and ferroptosis in diverse species. Genes Dev. 2018, 32, 602–619. [Google Scholar] [CrossRef] [Green Version]
- Goldstein, S.; Meyerstein, D.; Czapski, G. The Fenton reagents. Free Radic. Biol. Med. 1993, 15, 435–445. [Google Scholar] [CrossRef]
- Winterbourn, C.C. Toxicity of iron and hydrogen peroxide: The Fenton reaction. Toxicol Lett. 1995, 82, 969–974. [Google Scholar] [CrossRef]
- Minotti, G.; Aust, S.D. The role of iron in oxygen radical mediated lipid peroxidation. Chem. Biol. Interact. 1989, 71, 1–19. [Google Scholar] [CrossRef]
- Leitao, E.F.V.; Ventura, E.; de Souza, M.A.F.; Riveros, J.M.; do Monte, S.A. Spin-Forbidden Branching in the Mechanism of the Intrinsic Haber-Weiss Reaction. Chem. Open 2017, 6, 360–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patron, M.; Raffaello, A.; Granatiero, V.; Tosatto, A.; Merli, G.; De Stefani, D.; Wright, L.; Pallafacchina, G.; Terrin, A.; Mammucari, C.; et al. The mitochondrial calcium uniporter (MCU): Molecular identity and physiological roles. J. Biol. Chem. 2013, 288, 10750–10758. [Google Scholar] [CrossRef] [Green Version]
- Lill, R.; Hoffmann, B.; Molik, S.; Pierik, A.J.; Rietzschel, N.; Stehling, O.; Uzarska, M.A.; Webert, H.; Wilbrech, C.; Mühlenhoff, U. The role of mitochondria in cellular iron-sulfur protein biogenesis and iron metabolism. Biochim. Biophys. Acta 2012, 1823, 1491–1508. [Google Scholar] [CrossRef] [Green Version]
- Stehling, O.; Wilbrecht, C.; Lill, R. Mitochondrial iron-sulfur protein biogenesis and human disease. Biochimie 2014, 100, 61–77. [Google Scholar] [CrossRef]
- Paul, B.T.; Manz, D.H.; Torti, F.M.; Torti, S.V. Mitochondria and Iron: Current questions. Expert Rev. Hematol. 2017, 10, 65–79. [Google Scholar] [CrossRef] [Green Version]
- Schultz, I.J.; Chen, C.; Paw, B.H.; Hamza, I. Iron and porphyrin trafficking in heme biogenesis. J. Biol. Chem. 2010, 285, 26753–26759. [Google Scholar] [CrossRef] [Green Version]
- Chung, J.; Chen, C.; Paw, B.H. Heme metabolism and erythropoiesis. Curr. Opin. Hematol. 2012, 19, 156–162. [Google Scholar] [CrossRef]
- Poulos, T.L. Heme enzyme structure and function. Chem. Rev. 2014, 114, 3919–3962. [Google Scholar] [CrossRef] [Green Version]
- Hanna, D.A.; Martinez-Guzman, O.; Reddi, A.R. Heme Gazing: Illuminating Eukaryotic Heme Trafficking, Dynamics, and Signaling with Fluorescent Heme Sensors. Biochemistry 2017, 56, 1815–1823. [Google Scholar] [CrossRef] [PubMed]
- Reddi, A.R.; Hamza, I. Heme Mobilization in Animals: A Metallolipid’s Journey. Acc. Chem. Res. 2016, 49, 1104–1110. [Google Scholar] [CrossRef] [Green Version]
- Raghuram, S.; Stayrook, K.R.; Huang, P.; Rogers, P.M.; Nosie, A.K.; McClure, D.B.; Burris, L.L.; Khorasanizadeh, S.; Burris, T.P.; Rastinejad, F. Identification of heme as the ligand for the orphan nuclear receptors REV-ERBalpha and REV-ERBbeta. Nat. Struct. Mol. Biol. 2007, 14, 1207–1213. [Google Scholar] [CrossRef] [Green Version]
- Hou, S.; Reynolds, M.F.; Horrigan, F.T.; Heinemann, S.H.; Hoshi, T. Reversible binding of heme to proteins in cellular signal transduction. Acc. Chem. Res. 2006, 39, 918–924. [Google Scholar] [CrossRef] [PubMed]
- Chiabrando, D.; Vinchi, F.; Fiorito, V.; Mercurio, S.; Tolosano, E. Heme in pathophysiology: A matter of scavenging, metabolism and trafficking across cell membranes. Front. Pharmacol. 2014, 5, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Severance, S.; Hamza, I. Trafficking of heme and porphyrins in metazoa. Chem. Rev. 2009, 109, 4596–4616. [Google Scholar] [CrossRef] [Green Version]
- Brzoska, K.; Meczynska, S.; Kruszewski, M. Iron-sulfur cluster proteins: Electron transfer and beyond. Acta Biochim. Pol. 2006, 53, 685–691. [Google Scholar] [CrossRef] [Green Version]
- Imlay, J.A. Iron-sulphur clusters and the problem with oxygen. Mol. Microbiol. 2006, 59, 1073–1082. [Google Scholar] [CrossRef]
- Maio, N.; Rouault, T.A. Iron-sulfur cluster biogenesis in mammalian cells: New insights into the molecular mechanisms of cluster delivery. Biochim. Biophys. Acta 2015, 1853, 1493–1512. [Google Scholar] [CrossRef] [Green Version]
- Rouault, T.A. The role of iron regulatory proteins in mammalian iron homeostasis and disease. Nat. Chem. Biol. 2006, 2, 406–414. [Google Scholar] [CrossRef]
- White, M.F.; Dillingham, M.S. Iron-sulphur clusters in nucleic acid processing enzymes. Curr. Opin. Struct. Biol. 2012, 22, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Rich, P.R.; Marechal, A. The mitochondrial respiratory chain. Essays Biochem. 2010, 47, 1–23. [Google Scholar] [PubMed] [Green Version]
- Nicholls, D.G. Mitochondrial ion circuits. Essays Biochem. 2010, 47, 25–35. [Google Scholar] [PubMed]
- Rees, D.M.; Leslie, A.G.; Walker, J.E. The structure of the membrane extrinsic region of bovine ATP synthase. Proc. Natl. Acad. Sci. USA 2009, 106, 21597–21601. [Google Scholar] [CrossRef] [Green Version]
- Walker, J.E. The ATP synthase: The understood, the uncertain and the unknown. Bioche Soc. Trans. 2013, 41, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Read, A.D.; Bentley, R.E.T.; Archer, S.L.; Dunham-Snary, K.J. Mitochondrial iron-sulfur clusters: Structure, function, and an emerging role in vascular biology. Redox Biol. 2021, 47, 102164. [Google Scholar] [CrossRef]
- Hagerhall, C. Succinate: Quinone oxidoreductases: Variations on a conserved theme. Biochim. Biophys. Acta 1997, 1320, 107–141. [Google Scholar] [CrossRef] [Green Version]
- Page, C.C.; Moser, C.C.; Chen, X.; Dutton, P.L. Natural engineering principles of electron tunnelling in biological oxidation-reduction. Nature 1999, 402, 47–52. [Google Scholar] [CrossRef]
- Crofts, A.R. The cytochrome bc1 complex: Function in the context of structure. Annu. Rev. Physiol. 2004, 66, 689–733. [Google Scholar] [CrossRef] [Green Version]
- Beinert, H.; Kennedy, M.C. Aconitase, a two-faced protein: Enzyme and iron regulatory factor. FASEB J. 1993, 7, 1442–1449. [Google Scholar] [CrossRef]
- Gardner, P.R. Superoxide-driven aconitase FE-S center cycling. Biosci. Rep. 1997, 17, 33–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, W.H.; Rouault, T.A. Metabolic regulation of citrate and iron by aconitases: Role of iron-sulfur cluster biogenesis. Biometals 2007, 20, 549–564. [Google Scholar] [CrossRef] [PubMed]
- Onukwufor, J.O.; Berry, B.J.; Wojtovich, A.P. Physiologic Implications of Reactive Oxygen Species Production by Mitochondrial Complex I Reverse Electron Transport. Antioxidants 2019, 8, 285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adam-Vizi, V.; Chinopoulos, C. Bioenergetics and the formation of mitochondrial reactive oxygen species. Trends Pharmacol. Sci. 2006, 27, 639–645. [Google Scholar] [CrossRef]
- Onukwufor, J.O.; Stevens, D.; Kamunde, C. Combined effects of cadmium, temperature and hypoxia-reoxygenation on mitochondrial function in rainbow trout (Oncorhynchus mykiss). Aquat. Toxicol. 2017, 182, 129–141. [Google Scholar] [CrossRef]
- Onukwufor, J.O.; Kibenge, F.; Stevens, D.; Kamunde, C. Hypoxia-reoxygenation differentially alters the thermal sensitivity of complex I basal and maximal mitochondrial oxidative capacity. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 2016, 201, 87–94. [Google Scholar] [CrossRef]
- Harley, A.; Cooper, J.M.; Schapira, A.H. Iron induced oxidative stress and mitochondrial dysfunction: Relevance to Parkinson’s disease. Brain Res. 1993, 627, 349–353. [Google Scholar] [CrossRef]
- Walter, P.B.; Knutson, M.D.; Paler-Martinez, A.; Lee, S.; Xu, Y.; Viteri, F.E.; Ames, B.N. Iron deficiency and iron excess damage mitochondria and mitochondrial DNA in rats. Proc. Natl. Acad. Sci. USA 2002, 99, 2264–2269. [Google Scholar] [CrossRef] [Green Version]
- Loschen, G.; Azzi, A.; Richter, C.; Flohé, L. Superoxide radicals as precursors of mitochondrial hydrogen peroxide. FEBS Lett. 1974, 42, 68–72. [Google Scholar] [CrossRef] [Green Version]
- Butterfield, D.A.; Lauderback, C.M. Lipid peroxidation and protein oxidation in Alzheimer’s disease brain: Potential causes and consequences involving amyloid beta-peptide-associated free radical oxidative stress. Free Radic. Biol. Med. 2002, 32, 1050–1060. [Google Scholar] [CrossRef]
- Bradley, M.A.; Markesbery, W.R.; Lovell, M.A. Increased levels of 4-hydroxynonenal and acrolein in the brain in preclinical Alzheimer disease. Free Radic. Biol. Med. 2010, 48, 1570–1576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradley, M.A.; Xiong-Fister, S.; Markesbery, W.R.; Lovell, M.A. Elevated 4-hydroxyhexenal in Alzheimer’s disease (AD) progression. Neurobiol. Aging 2012, 33, 1034–1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Blanco, A.; Baquero, M.; Vento, M.; Gil, E.; Bataller, L.; Chafer-Pericas, C. Potential oxidative stress biomarkers of mild cognitive impairment due to Alzheimer disease. J. Neurol. Sci. 2017, 373, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Bradley-Whitman, M.A.; Lovell, M.A. Biomarkers of lipid peroxidation in Alzheimer disease (AD): An update. Arch. Toxicol. 2015, 89, 1035–1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mills, E.; Dong, X.P.; Wang, F.; Xu, H. Mechanisms of brain iron transport: Insight into neurodegeneration and CNS disorders. Future Med. Chem. 2010, 2, 51–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hentze, M.W.; Muckenthaler, M.U.; Galy, B.; Camaschella, C. Two to tango: Regulation of Mammalian iron metabolism. Cell 2010, 142, 24–38. [Google Scholar] [CrossRef] [Green Version]
- Moos, T. Brain iron homeostasis. Dan. Med. Bull. 2002, 49, 279–301. [Google Scholar]
- Andrews, N.C.; Schmidt, P.J. Iron homeostasis. Annu. Rev. Physiol. 2007, 69, 69–85. [Google Scholar] [CrossRef]
- Duce, J.A.; Tsatsanis, A.; Cater, M.A.; James, S.A.; Robb, E.; Wikhe, K.; Leong, S.L.; Perez, K.; Johanssen, T.; Greenough, M.A.; et al. Iron-export ferroxidase activity of beta-amyloid precursor protein is inhibited by zinc in Alzheimer’s disease. Cell 2010, 142, 857–867. [Google Scholar] [CrossRef] [Green Version]
- Bush, A.I. The metal theory of Alzheimer’s disease. J. Alzheimers Dis. 2013, 33 (Suppl. S1), S277–S281. [Google Scholar] [CrossRef]
- Das, N.; Raymick, J.; Sarkar, S. Role of metals in Alzheimer’s disease. Metab. Brain Dis. 2021, 36, 1627–1639. [Google Scholar] [CrossRef] [PubMed]
- Haacke, E.M.; Cheng, N.Y.; House, M.J.; Liu, Q.; Neelavalli, J.; Ogg, R.J.; Khan, A.; Ayaz, M.; Kirsch, W.; Obenaus, A. Imaging iron stores in the brain using magnetic resonance imaging. Magn. Reson. Imaging 2005, 23, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Haacke, E.M.; Miao, Y.; Liu, M.; Habib, C.A.; Katkuri, Y.; Liu, T.; Yang, Z.; Lang, Z.; Hu, J.; Wu, J. Correlation of putative iron content as represented by changes in R2* and phase with age in deep gray matter of healthy adults. J. Magn. Reson. Imaging 2010, 32, 561–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raven, E.P.; Lu, P.H.; Tishler, T.A.; Heydari, P.; Bartzokis, G. Increased iron levels and decreased tissue integrity in hippocampus of Alzheimer’s disease detected in vivo with magnetic resonance imaging. J. Alzheimers Dis. 2013, 37, 127–136. [Google Scholar] [CrossRef]
- Langkammer, C.; Schweser, F.; Krebs, N.; Deistung, A.; Goessler, W.; Scheurer, E.; Sommer, K.; Reishofer, G.; Yen, K.; Fazekas, F.; et al. Quantitative susceptibility mapping (QSM) as a means to measure brain iron? A post mortem validation study. Neuroimage 2012, 62, 1593–1599. [Google Scholar] [CrossRef] [Green Version]
- Jara, H.; Sakai, O.; Mankal, P.; Irving, R.P.; Norbash, A.M. Multispectral quantitative magnetic resonance imaging of brain iron stores: A theoretical perspective. Top. Magn. Reson. Imaging 2006, 17, 19–30. [Google Scholar] [CrossRef]
- Du, L.; Zhao, Z.; Cui, A.; Zhu, Y.; Zhang, L.; Liu, J.; Shi, S.; Fu, C.; Han, X.; Gao, W.; et al. Increased Iron Deposition on Brain Quantitative Susceptibility Mapping Correlates with Decreased Cognitive Function in Alzheimer’s Disease. ACS Chem. Neurosci. 2018, 9, 1849–1857. [Google Scholar] [CrossRef]
- Sun, Y.; Ge, X.; Han, X.; Cao, W.; Wang, Y.; Ding, W.; Cao, M.; Zhang, Y.; Xu, Q.; Zhou, Y.; et al. Characterizing Brain Iron Deposition in Patients with Subcortical Vascular Mild Cognitive Impairment Using Quantitative Susceptibility Mapping: A Potential Biomarker. Front. Aging Neurosci. 2017, 9, 81. [Google Scholar] [CrossRef]
- Qin, Z.; Wu, W.; Liu, D.; Zheng, C.; Kang, J.; Zhou, H.; Meng, X.; Haacke, E.M.; Wang, L. Quantitative Susceptibility Mapping of Brain Iron Relating to Cognitive Impairment in Hypertension. J. Magn. Reson. Imaging 2022, 28043. [Google Scholar] [CrossRef]
- Tambasco, N.; Nigro, P.; Chiappiniello, A.; Paoletti, F.P.; Scialpi, S.; Simoni, S.; Chiarini, P.; Parnetti, L. An Updated Overview of the Magnetic Resonance Imaging of Brain Iron in Movement Disorders. Behav. Neurol. 2022, 2022, 3972173. [Google Scholar] [CrossRef]
- Ruetten, P.P.R.; Gillard, J.H.; Graves, M.J. Introduction to Quantitative Susceptibility Mapping and Susceptibility Weighted Imaging. Br. J. Radiol. 2019, 92, 20181016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Bergen, J.M.G.; Li, X.; Quevenco, F.C.; Gietl, A.F.; Treyer, V.; Meyer, R.; Buck, A.; Kaufmann, P.A.; Nitsch, R.M.; van Zijl, P.C.M.; et al. Simultaneous quantitative susceptibility mapping and Flutemetamol-PET suggests local correlation of iron and beta-amyloid as an indicator of cognitive performance at high age. Neuroimage 2018, 174, 308–316. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.A.; Zhu, X.; Tabaton, M.; Liu, G.; McKeel, D.W.; Cohen, M.L.; Wang, X.; Siedlak, S.L.; Dwyer, B.E.; Hayashi, T.; et al. Increased iron and free radical generation in preclinical Alzheimer disease and mild cognitive impairment. J. Alzheimers Dis. 2010, 19, 363–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Everett, J.; Céspedes, E.; Shelford, L.R.; Exley, C.; Collingwood, J.F.; Dobson, J.; van der Laan, G.; Jenkins, C.A.; Arenholz, E.; Telling, N.D. Ferrous iron formation following the co-aggregation of ferric iron and the Alzheimer’s disease peptide beta-amyloid (1–42). J. R. Soc. Interface 2014, 11, 20140165. [Google Scholar] [CrossRef]
- Mastroeni, D.; Khdour, O.M.; Delvaux, E.; Nolz, J.; Olsen, G.; Berchtold, N.; Cotman, C.; Hecht, S.M.; Coleman, P.D. Nuclear but not mitochondrial-encoded oxidative phosphorylation genes are altered in aging, mild cognitive impairment, and Alzheimer’s disease. Alzheimers Dement. 2017, 13, 510–519. [Google Scholar] [CrossRef] [Green Version]
- Casley, C.S.; Canevari, L.; Land, J.M.; Clark, J.B.; Sharpe, M.A. Beta-amyloid inhibits integrated mitochondrial respiration and key enzyme activities. J. Neurochem. 2002, 80, 91–100. [Google Scholar] [CrossRef]
- Huang, X.T.; Liu, X.; Ye, C.Y.; Tao, L.X.; Zhou, H.; Zhang, H.Y. Iron-induced energy supply deficiency and mitochondrial fragmentation in neurons. J. Neurochem. 2018, 147, 816–830. [Google Scholar] [CrossRef] [Green Version]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Sumimoto, H. Structure, regulation and evolution of Nox-family NADPH oxidases that produce reactive oxygen species. FEBS J. 2008, 275, 3249–3277. [Google Scholar] [CrossRef]
- Suh, Y.A.; Arnold, R.S.; Lassegue, B.; Shi, J.; Xu, X.; Sorescu, D.; Chung, A.B.; Griendling, K.K.; Lambeth, J.D. Cell transformation by the superoxide-generating oxidase Mox1. Nature 1999, 401, 79–82. [Google Scholar] [CrossRef]
- Tarafdar, A.; Pula, G. The Role of NADPH Oxidases and Oxidative Stress in Neurodegenerative Disorders. Int. J. Mol.Sci. 2018, 19, 3824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panday, A.; Sahoo, M.K.; Osorio, D.; Batra, S. NADPH oxidases: An overview from structure to innate immunity-associated pathologies. Cell. Mol. Immunol. 2015, 12, 5–23. [Google Scholar] [CrossRef] [Green Version]
- Sorce, S.; Krause, K.H. NOX enzymes in the central nervous system: From signaling to disease. Antioxid. Redox Signal. 2009, 11, 2481–2504. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.M.; Zhou, H.; Hong, J.S. NADPH oxidases: Novel therapeutic targets for neurodegenerative diseases. Trends Pharmacol. Sci. 2012, 33, 295–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leto, T.L.; Geiszt, M. Role of Nox family NADPH oxidases in host defense. Antioxid. Redox Signal. 2006, 8, 1549–1561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johri, A. Disentangling Mitochondria in Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 11520. [Google Scholar] [CrossRef]
- Huang, Z.; Yan, Q.; Wang, Y.; Zou, Q.; Li, J.; Liu, Z.; Cai, Z. Role of Mitochondrial Dysfunction in the Pathology of Amyloid-beta. J. Alzheimers Dis. 2020, 78, 505–514. [Google Scholar] [CrossRef]
- Liang, W.S.; Reiman, E.M.; Valla, J.; Dunckley, T.; Beach, T.G.; Grover, A.; Niedzielko, T.L.; Schneider, L.E.; Mastroeni, D.; Caselli, R.; et al. Alzheimer’s disease is associated with reduced expression of energy metabolism genes in posterior cingulate neurons. Proc. Natl. Acad. Sci. USA 2008, 105, 4441–4446. [Google Scholar] [CrossRef] [Green Version]
- Brooks, W.M.; Lynch, P.J.; Ingle, C.C.; Hatton, A.; Emson, P.C.; Faull, R.L.M.; Starkey, M.P. Gene expression profiles of metabolic enzyme transcripts in Alzheimer’s disease. Brain Res. 2007, 1127, 127–135. [Google Scholar] [CrossRef]
- Saito, E.R.; Miller, J.B.; Harari, O.; Cruchaga, C.; Mihindukulasuriya, K.A.; Kauwe, J.S.K.; Bikman, B.T. Alzheimer’s disease alters oligodendrocytic glycolytic and ketolytic gene expression. Alzheimers Dement. 2021, 17, 1474–1486. [Google Scholar] [CrossRef]
- Malkov, A.; Popova, I.; Ivanov, A.; Jang, S.S.; Yoon, S.Y.; Osypov, A.; Huang, Y.; Zilberter, Y.; Zilberter, M. Abeta initiates brain hypometabolism, network dysfunction and behavioral abnormalities via NOX2-induced oxidative stress in mice. Commun. Biol. 2021, 4, 1054. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Perry, G.; Smith, M.A.; Wang, X. Abnormal mitochondrial dynamics in the pathogenesis of Alzheimer’s disease. J. Alzheimers Dis. 2013, 33 (Suppl. S1), S253–S262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Detmer, S.A.; Chan, D.C. Functions and dysfunctions of mitochondrial dynamics. Nat. Rev. Mol. Cell Biol. 2007, 8, 870–879. [Google Scholar] [CrossRef] [PubMed]
- Trewin, A.J.; Berry, B.J.; Wojtovich, A.P. Exercise and Mitochondrial Dynamics: Keeping in Shape with ROS and AMPK. Antioxidants 2018, 7, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, D.C. Mitochondrial fusion and fission in mammals. Annu. Rev. Cell Dev. Biol. 2006, 22, 79–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.; Lee, D.G.; Kim, B.; Park, S.J.; Kim, J.H.; Lee, S.R.; Chang, K.T.; Lee, H.S.; Lee, D.S. Iron overload triggers mitochondrial fragmentation via calcineurin-sensitive signals in HT-22 hippocampal neuron cells. Toxicology 2015, 337, 39–46. [Google Scholar] [CrossRef]
- Slupe, A.M.; Merrill, R.A.; Flippo, K.H.; Lobas, M.A.; Houtman, J.C.; Strack, S. A calcineurin docking motif (LXVP) in dynamin-related protein 1 contributes to mitochondrial fragmentation and ischemic neuronal injury. J. Biol. Chem. 2013, 288, 12353–12365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munoz, P.; Humeres, A.; Elgueta, C.; Kirkwood, A.; Hidalgo, C.; Núñez, M.T. Iron mediates N-methyl-D-aspartate receptor-dependent stimulation of calcium-induced pathways and hippocampal synaptic plasticity. J. Biol. Chem. 2011, 286, 13382–13392. [Google Scholar] [CrossRef] [Green Version]
- Guan, W.; Xia, M.; Ji, M.; Chen, B.; Li, S.; Zhang, M.; Liang, S.; Chen, B.; Gong, W.; Dong, C.; et al. Iron induces two distinct Ca2+ signalling cascades in astrocytes. Commun. Biol. 2021, 4, 525. [Google Scholar] [CrossRef]
- Lee, D.G.; Park, J.; Lee, H.S.; Lee, S.R.; Lee, D.S. Iron overload-induced calcium signals modulate mitochondrial fragmentation in HT-22 hippocampal neuron cells. Toxicology 2016, 365, 17–24. [Google Scholar] [CrossRef]
- Wiemerslage, L.; Lee, D. Quantification of mitochondrial morphology in neurites of dopaminergic neurons using multiple parameters. J. Neurosci. Methods 2016, 262, 56–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manczak, M.; Reddy, P.H. Abnormal interaction between the mitochondrial fission protein Drp1 and hyperphosphorylated tau in Alzheimer’s disease neurons: Implications for mitochondrial dysfunction and neuronal damage. Hum. Mol. Genet. 2012, 21, 2538–2547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manczak, M.; Kandimalla, R.; Fry, D.; Sesaki, H.; Reddy, P.H. Protective effects of reduced dynamin-related protein 1 against amyloid beta-induced mitochondrial dysfunction and synaptic damage in Alzheimer’s disease. Hum. Mol. Genet. 2016, 25, 5148–5166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, P.H.; Yin, X.; Manczak, M.; Kumar, S.; Pradeepkiran, J.A.; Vijayan, M.; Reddy, A.P. Mutant APP and amyloid beta-induced defective autophagy, mitophagy, mitochondrial structural and functional changes and synaptic damage in hippocampal neurons from Alzheimer’s disease. Hum. Mol. Genet. 2018, 27, 2502–2516. [Google Scholar] [CrossRef] [PubMed]
- Manczak, M.; Kandimalla, R.; Yin, X.; Reddy, P.H. Hippocampal mutant APP and amyloid beta-induced cognitive decline, dendritic spine loss, defective autophagy, mitophagy and mitochondrial abnormalities in a mouse model of Alzheimer’s disease. Hum. Mol. Genet. 2018, 27, 1332–1342. [Google Scholar] [CrossRef] [Green Version]
- Sanderson, T.H.; Raghunayakula, S.; Kumar, R. Release of mitochondrial Opa1 following oxidative stress in HT22 cells. Mol. Cell Neurosci. 2015, 64, 116–122. [Google Scholar] [CrossRef]
- Lobas, M.A.; Tao, R.; Nagai, J.; Kronschläger, M.T.; Borden, P.M.; Marvin, J.S.; Looger, L.L.; Khakh, B.S. A genetically encoded single-wavelength sensor for imaging cytosolic and cell surface ATP. Nat. Commun. 2019, 10, 711. [Google Scholar] [CrossRef] [Green Version]
- Tao, R.; Zhao, Y.; Chu, H.; Wang, A.; Zhu, J.; Chen, X.; Zou, Y.; Shi, M.; Liu, R.; Su, N.; et al. Genetically encoded fluorescent sensors reveal dynamic regulation of NADPH metabolism. Nat. Methods 2017, 14, 720–728. [Google Scholar] [CrossRef]
- Kagan, V.E.; Mao, G.; Qu, F.; Angeli, J.P.; Doll, S.; Croix, C.S.; Dar, H.H.; Liu, B.; Tyurin, V.A.; Ritov, V.B. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol. 2017, 13, 81–90. [Google Scholar] [CrossRef]
- Mikulska-Ruminska, K.; Anthonymuthu, T.S.; Levkina, A.; Shrivastava, I.H.; Kapralov, A.A.; Bayır, H.; Kagan, V.E.; Bahar, I. NO• Represses the Oxygenation of Arachidonoyl PE by 15LOX/PEBP1: Mechanism and Role in Ferroptosis. Int. J. Mol. Sci. 2021, 22, 5253. [Google Scholar] [CrossRef]
- Doll, S.; Proneth, B.; Tyurina, Y.Y.; Panzilius, E.; Kobayashi, S.; Ingold, I.; Irmler, M.; Beckers, J.; Aichler, M.; Walch, A.; et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 2017, 13, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Yang, L.; Geng, L.; He, J.; Chen, L.; Sun, Q.; Zhao, J.; Wang, X. Inhibition of Acyl-CoA Synthetase Long-Chain Family Member 4 Facilitates Neurological Recovery After Stroke by Regulation Ferroptosis. Front. Cell Neurosci. 2021, 15, 632354. [Google Scholar] [CrossRef] [PubMed]
- Kagan, V.E.; Tyurina, Y.Y.; Vlasova, I.I.; Kapralov, A.A.; Amoscato, A.A.; Anthonymuthu, T.S.; Tyurin, V.A.; Shrivastava, I.H.; Cinemre, F.B.; Lamade, A.; et al. Redox Epiphospholipidome in Programmed Cell Death Signaling: Catalytic Mechanisms and Regulation. Front. Endocrinol. 2020, 11, 628079. [Google Scholar] [CrossRef] [PubMed]
- Hambright, W.S.; Fonseca, R.S.; Chen, L.; Na, R.; Ran, Q. Ablation of ferroptosis regulator glutathione peroxidase 4 in forebrain neurons promotes cognitive impairment and neurodegeneration. Redox Biol. 2017, 12, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; Stockwell, B.R. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem. Biol. 2008, 15, 234–245. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [Green Version]
- Weïwer, M.; Bittker, J.A.; Lewis, T.A.; Shimada, K.; Yang, W.S.; MacPherson, L.; Dandapani, S.; Palmer, M.; Stockwell, B.R.; Schreiber, S.L.; et al. Development of small-molecule probes that selectively kill cells induced to express mutant RAS. Bioorg. Med. Chem. Lett. 2012, 22, 1822–1826. [Google Scholar] [CrossRef] [Green Version]
- Eaton, J.K.; Ruberto, R.A.; Kramm, A.; Viswanathan, V.S.; Schreiber, S.L. Diacylfuroxans Are Masked Nitrile Oxides That Inhibit GPX4 Covlently. J. Am. Chem. Soc. 2019, 141, 20407–20415. [Google Scholar] [CrossRef]
- Doll, S.; Freitas, F.P.; Shah, R.; Aldrovandi, M.; da Silva, M.C.; Ingold, I.; Goya Grocin, A.; Xavier da Silva, T.N.; Panzilius, E.; Scheel, C.H.; et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 2019, 575, 693–698. [Google Scholar] [CrossRef]
- Bersuker, K.; Hendricks, J.M.; Li, Z.; Magtanong, L.; Ford, B.; Tang, P.H.; Roberts, M.A.; Tong, B.; Maimone, T.J.; Zoncu, R.; et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 2019, 575, 688–692. [Google Scholar] [CrossRef]
- Wu, M.; Xu, L.G.; Li, X.; Zhai, Z.; Shu, H.B. AMID, an apoptosis-inducing factor-homologous mitochondrion-associated protein, induces caspase-independent apoptosis. J. Biol. Chem. 2002, 277, 25617–25623. [Google Scholar] [CrossRef] [PubMed]
- Ohiro, Y.; Garkavtsev, I.; Kobayashi, S.; Sreekumar, K.R.; Nantz, R.; Higashikubo, B.T.; Duffy, S.L.; Higashikubo, R.; Usheva, A.; Gius, D.; et al. A novel p53-inducible apoptogenic gene, PRG3, encodes a homologue of the apoptosis-inducing factor (AIF). FEBS Lett. 2002, 524, 163–171. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Hambright, W.S.; Na, R.; Ran, Q. Ablation of the Ferroptosis Inhibitor Glutathione Peroxidase 4 in Neurons Results in Rapid Motor Neuron Degeneration and P.Paralysis. J. Biol. Chem. 2015, 290, 28097–28106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farazi, T.A.; Waksman, G.; Gordon, J.I. The biology and enzymology of protein N-myristoylation. J. Biol. Chem. 2001, 276, 39501–39504. [Google Scholar] [CrossRef] [Green Version]
- Udenwobele, D.I.; Su, R.C.; Good, S.V.; Ball, T.B.; Varma Shrivastav, S.; Shrivastav, A. Myristoylation: An Important Protein Modification in the Immune Response. Front. Immunol. 2017, 8, 751. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Dai, T.; Sun, W.; Wei, Y.; Ren, J.; Zhang, L.; Zhang, M.; Zhou, F. Protein N-myristoylation: Functions and mechanisms in control of innate immunity. Cell. Mol. Immunol. 2021, 18, 878–888. [Google Scholar] [CrossRef]
- Dolma, S.; Lessnick, S.L.; Hahn, W.C.; Stockwell, B.R. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell 2003, 3, 285–296. [Google Scholar] [CrossRef] [Green Version]
- Dixon, S.J.; Patel, D.N.; Welsch, M.; Skouta, R.; Lee, E.D.; Hayano, M.; Thomas, A.G.; Gleason, C.E.; Tatonetti, N.P.; Slusher, B.S.; et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife 2014, 3, e02523. [Google Scholar] [CrossRef]
- Abbott, M.A.; Prater, S.N.; Banugaria, S.G.; Richards, S.M.; Young, S.P.; Rosenberg, A.S.; Kishnani, P.S. Atypical immunologic response in a patient with CRIM-negative Pompe disease. Mol. Genet. Metab. 2011, 104, 583–586. [Google Scholar] [CrossRef] [Green Version]
- Murphy, T.H.; Miyamoto, M.; Sastre, A.; Schnaar, R.L.; Coyle, J.T. Glutamate toxicity in a neuronal cell line involves inhibition of cystine transport leading to oxidative stress. Neuron 1989, 2, 1547–1558. [Google Scholar] [CrossRef]
- Zilka, O.; Shah, R.; Li, B.; Friedmann Angeli, J.P.; Griesser, M.; Conrad, M.; Pratt, D.A. On the Mechanism of Cytoprotection by Ferrostatin-1 and Liproxstatin-1 and the Role of Lipid Peroxidation in Ferroptotic Cell Death. ACS Central Sci. 2017, 3, 232–243. [Google Scholar] [CrossRef] [PubMed]
- Shimada, K.; Skouta, R.; Kaplan, A.; Yang, W.S.; Hayano, M.; Dixon, S.J.; Brown, L.M.; Valenzuela, C.A.; Wolpaw, A.J.; Stockwell, B.R.; et al. Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat. Chem. Biol. 2016, 12, 497–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.M.; Cho, S.S.; Ki, S.H. Emerging roles of ferroptosis in liver pathophysiology. Arch. Pharmacal Res. 2020, 43, 985–996. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Zhang, H.; Yang, S.K.; Wu, X.; He, D.; Cao, K.; Zhang, W. Emerging Role of Ferroptosis in Acute Kidney Injury. Oxid. Med. Cell. Longev. 2019, 2019, 8010614. [Google Scholar] [CrossRef]
- Reichert, C.O.; de Freitas, F.A.; Sampaio-Silva, J.; Rokita-Rosa, L.; Barros, P.L.; Levy, D.; Bydlowski, S.P. Ferroptosis Mechanisms Involved in Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 8765. [Google Scholar] [CrossRef]
- Ye, Z.; Liu, W.; Zhuo, Q.; Hu, Q.; Liu, M.; Sun, Q.; Zhang, Z.; Fan, G.; Xu, W.; Ji, S.; et al. Ferroptosis: Final destination for cancer? Cell Prolif. 2020, 53, e12761. [Google Scholar] [CrossRef]
- Magtanong, L.; Dixon, S.J. Ferroptosis and Brain Injury. Dev. Neurosci. 2018, 40, 382–395. [Google Scholar] [CrossRef]
- Wu, X.; Li, Y.; Zhang, S.; Zhou, X. Ferroptosis as a novel therapeutic target for cardiovascular disease. Theranostics 2021, 11, 3052–3059. [Google Scholar] [CrossRef]
- Bao, W.D.; Pang, P.; Zhou, X.T.; Hu, F.; Xiong, W.; Chen, K.; Wang, J.; Wang, F.; Xie, D.; Hu, Y.Z.; et al. Loss of ferroportin induces memory impairment by promoting ferroptosis in Alzheimer’s disease. Cell Death Differ. 2021, 28, 1548–1562. [Google Scholar] [CrossRef]
- Chen, L.; Dar, N.J.; Na, R.; McLane, K.D.; Yoo, K.; Han, X.; Ran, Q. Enhanced defense against ferroptosis ameliorates cognitive impairment and reduces neurodegeneration in 5xFAD mice. Free Radic. Biol. Med. 2022, 180, 1–12. [Google Scholar] [CrossRef]
- Ates, G.; Goldberg, J.; Currais, A.; Maher, P. CMS121, a fatty acid synthase inhibitor, protects against excess lipid peroxidation and inflammation and alleviates cognitive loss in a transgenic mouse model of Alzheimer’s disease. Redox Biol. 2020, 36, 101648. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.H.; Wang, D.W.; Xu, S.F.; Zhang, S.; Fan, Y.G.; Yang, Y.Y.; Guo, S.Q.; Wang, S.; Guo, T.; Wang, Z.Y.; et al. alpha-Lipoic acid improves abnormal behavior by mitigation of oxidative stress, inflammation, ferroptosis, and tauopathy in P301S Tau transgenic mice. Redox Biol. 2018, 14, 535–548. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Li, W.J.; Zheng, X.R.; Liu, Q.L.; Du, Q.; Lai, Y.J.; Liu, S.Q. Eriodictyol ameliorates cognitive dysfunction in APP/PS1 mice by inhibiting ferroptosis via vitamin D receptor-mediated Nrf2 activation. Mol. Med. 2022, 28, 11. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.J.; Huang, L.F.; Deng, J.L.; Wang, Y.M.; Guo, C.; Peng, X.N.; Liu, Z.; Gao, J.M. Cognitive enhancement and neuroprotective effects of OABL, a sesquiterpene lactone in 5xFAD Alzheimer’s disease mice model. Redox Biol. 2022, 50, 102229. [Google Scholar] [CrossRef] [PubMed]
- Tong, L.; Harwood, H.J., Jr. Acetyl-coenzyme A carboxylases: Versatile targets for drug discovery. J. Cell. Biochem. 2006, 99, 1476–1488. [Google Scholar] [CrossRef] [Green Version]
- Abu-Elheiga, L.; Brinkley, W.R.; Zhong, L.; Chirala, S.S.; Woldegiorgis, G.; Wakil, S.J. The subcellular localization of acetyl-CoA carboxylase 2. Proc. Natl. Acad. Sci. USA 2000, 97, 1444–1449. [Google Scholar] [CrossRef] [Green Version]
- Hopwood, D.A.; Sherman, D.H. Molecular genetics of polyketides and its comparison to fatty acid biosynthesis. Annu. Rev. Genet. 1990, 24, 37–66. [Google Scholar] [CrossRef]
- Long, J.M.; Holtzman, D.M. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 2019, 179, 312–339. [Google Scholar] [CrossRef]
- Sevigny, J.; Chiao, P.; Bussière, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The antibody aducanumab reduces Abeta plaques in Alzheimer’s disease. Nature 2016, 537, 50–56. [Google Scholar] [CrossRef]
- Bastrup, J.; Hansen, K.H.; Poulsen, T.B.G.; Kastaniegaard, K.; Asuni, A.A.; Christensen, S.; Belling, D.; Helboe, L.; Stensballe, A.; Volbracht, C. Anti-Abeta Antibody Aducanumab Regulates the Proteome of Senile Plaques and Closely Surrounding Tissue in a Transgenic Mouse Model of Alzheimer’s Disease. J. Alzheimers Dis. 2021, 79, 249–265. [Google Scholar] [CrossRef]
- Tolar, M.; Abushakra, S.; Hey, J.A.; Porsteinsson, A.; Sabbagh, M. Aducanumab, gantenerumab, BAN2401, and ALZ-801-the first wave of amyloid-targeting drugs for Alzheimer’s disease with potential for near term approval. Alzheimers Res. Ther. 2020, 12, 95. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. Alzheimer disease and aducanumab: Adjusting our approach. Nat. Rev. Neurol. 2019, 15, 365–366. [Google Scholar] [CrossRef] [PubMed]
- Ostrowitzki, S.; Lasser, R.A.; Dorflinger, E.; Scheltens, P.; Barkhof, F.; Nikolcheva, T.; Ashford, E.; Retout, S.; Hofmann, C.; Delmar, P.; et al. A phase III randomized trial of gantenerumab in prodromal Alzheimer’s disease. Alzheimers Res. Ther. 2017, 9, 95. [Google Scholar] [CrossRef] [PubMed]
- Klein, G.; Delmar, P.; Voyle, N.; Rehal, S.; Hofmann, C.; Abi-Saab, D.; Andjelkovic, M.; Ristic, S.; Wang, G.; Bateman, R.; et al. Gantenerumab reduces amyloid-beta plaques in patients with prodromal to moderate Alzheimer’s disease: A PET substudy interim analysis. Alzheimers Res. Ther. 2019, 11, 101. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, K.; Moein, A.; Bittner, T.; Ostrowitzki, S.; Lin, H.; Honigberg, L.; Jin, J.Y.; Quartino, A. Pharmacokinetics and pharmacodynamic effect of crenezumab on plasma and cerebrospinal fluid beta-amyloid in patients with mild-to-moderate Alzheimer’s disease. Alzheimers Res. Ther. 2020, 12, 16. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Onukwufor, J.O.; Dirksen, R.T.; Wojtovich, A.P. Iron Dysregulation in Mitochondrial Dysfunction and Alzheimer’s Disease. Antioxidants 2022, 11, 692. https://doi.org/10.3390/antiox11040692
Onukwufor JO, Dirksen RT, Wojtovich AP. Iron Dysregulation in Mitochondrial Dysfunction and Alzheimer’s Disease. Antioxidants. 2022; 11(4):692. https://doi.org/10.3390/antiox11040692
Chicago/Turabian StyleOnukwufor, John O., Robert T. Dirksen, and Andrew P. Wojtovich. 2022. "Iron Dysregulation in Mitochondrial Dysfunction and Alzheimer’s Disease" Antioxidants 11, no. 4: 692. https://doi.org/10.3390/antiox11040692
APA StyleOnukwufor, J. O., Dirksen, R. T., & Wojtovich, A. P. (2022). Iron Dysregulation in Mitochondrial Dysfunction and Alzheimer’s Disease. Antioxidants, 11(4), 692. https://doi.org/10.3390/antiox11040692