
Induction of Heme Oxygenase-1 by 15d-Prostaglandin J2 Mediated via a ROS-Dependent Sp1 and AP-1 Cascade Suppresses Lipopolysaccharide-Triggered Interleukin-6 Expression in Mouse Brain Microvascular Endothelial Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Cell Cultures and Treatment
2.3. Protein Preparation and Western Blot Analysis
2.4. Transient Transfection with siRNAs in bEnd.3 Cells
2.5. Quantitative Real-Time PCR Analysis
2.6. Measurement of Intracellular ROS Accumulation
2.7. NOX Activity Assay
2.8. Cytoplasmic and Nuclear Protein Extraction
2.9. Chromatin Immunoprecipitation Assay (ChIP)
2.10. Preparation of Recombinant Adenovirus
2.11. Measurement of IL-6 Generation
2.12. Statistical Analysis of Data
3. Results
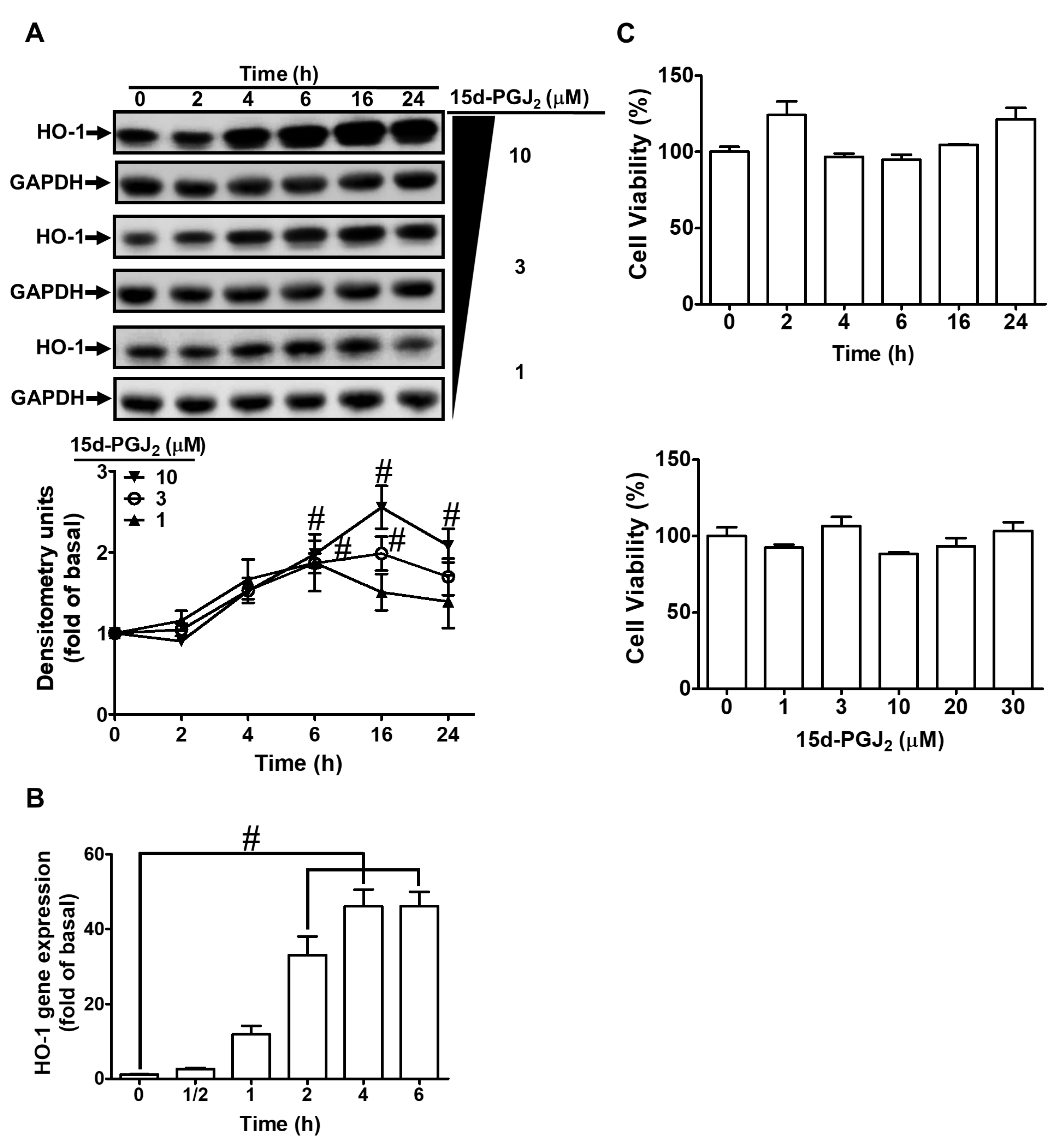
3.1. 15d-PGJ2 Induces Expression of HO-1 mRNA and Protein in bEnd.3 Cells
3.2. 15d-PGJ2 Induces HO-1 Expression via Mitochondria- and NOX-Generated ROS
3.3. Involvement of PKCδ in 15d-PGJ2-Induced Expression of HO-1
3.4. 15d-PGJ2 Enhances Expression of HO-1 via JNK1/2
3.5. Sp1 Is Involved in 15d-PGJ2 Induced HO-1 Upregulation
3.6. c-Jun Is Involved in 15d-PGJ2 Stimulated Expression of HO-1
3.7. 15d-PGJ2-Induced HO-1 Upregulation Attenuates LPS-Stimulated IL-6 Secretion
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Erickson, M.A.; Banks, W.A. Blood-brain barrier dysfunction as a cause and consequence of Alzheimer’s disease. J. Cereb. Blood Flow Metab. 2013, 33, 1500–1513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.; Pienaar, I.S. Disruption of the blood-brain barrier in Parkinson’s disease: Curse or route to a cure? Front. Biosci. 2014, 19, 272–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortiz, G.G.; Pacheco-Moises, F.P.; Macias-Islas, M.A.; Flores-Alvarado, L.J.; Mireles-Ramirez, M.A.; Gonzalez-Renovato, E.D.; Hernandez-Navarro, V.E.; Sanchez-Lopez, A.L.; Alatorre-Jimenez, M.A. Role of the blood-brain barrier in multiple sclerosis. Arch. Med. Res. 2014, 45, 687–697. [Google Scholar] [CrossRef] [PubMed]
- Thal, S.C.; Neuhaus, W. The blood-brain barrier as a target in traumatic brain injury treatment. Arch. Med. Res. 2014, 45, 698–710. [Google Scholar] [CrossRef] [PubMed]
- Corsini, E.; Dufour, A.; Ciusani, E.; Gelati, M.; Frigerio, S.; Gritti, A.; Cajola, L.; Mancardi, G.L.; Massa, G.; Salmaggi, A. Human brain endothelial cells and astrocytes produce IL-1β but not IL-10. Scand. J. Immunol. 1996, 44, 506–511. [Google Scholar] [CrossRef] [PubMed]
- Fabry, Z.; Fitzsimmons, K.M.; Herlein, J.A.; Moninger, T.O.; Dobbs, M.B.; Hart, M.N. Production of the cytokines interleukin 1 and 6 by murine brain microvessel endothelium and smooth muscle pericytes. J. Neuroimmunol. 1993, 47, 23–34. [Google Scholar] [CrossRef]
- Gotsch, U.; Jager, U.; Dominis, M.; Vestweber, D. Expression of P-selectin on endothelial cells is upregulated by LPS and TNF-α in vivo. Cell Adhes. Commun. 1994, 2, 7–14. [Google Scholar] [CrossRef]
- Wong, D.; Dorovini-Zis, K. Upregulation of intercellular adhesion molecule-1 (ICAM-1) expression in primary cultures of human brain microvessel endothelial cells by cytokines and lipopolysaccharide. J. Neuroimmunol. 1992, 39, 11–21. [Google Scholar] [CrossRef]
- Gonzalez-Velasquez, F.; Reed, J.W.; Fuseler, J.W.; Matherly, E.E.; Kotarek, J.A.; Soto-Ortega, D.D.; Moss, M.A. Activation of brain endothelium by soluble aggregates of the amyloid-β protein involves nuclear factor-κB. Curr. Alzheimer Res. 2011, 8, 81–94. [Google Scholar] [CrossRef]
- Yang, Y.M.; Shang, D.S.; Zhao, W.D.; Fang, W.G.; Chen, Y.H. Microglial TNF-α-dependent elevation of MHC class I expression on brain endothelium induced by amyloid-β promotes T cell transendothelial migration. Neurochem. Res. 2013, 38, 2295–2304. [Google Scholar] [CrossRef]
- Ricciotti, E.; FitzGerald, G.A. Prostaglandins and inflammation. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 986–1000. [Google Scholar] [CrossRef] [PubMed]
- Haskew-Layton, R.E.; Payappilly, J.B.; Xu, H.; Bennett, S.A.; Ratan, R.R. 15-Deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2) protects neurons from oxidative death via an Nrf2 astrocyte-specific mechanism independent of PPARγ. J. Neurochem. 2013, 124, 536–547. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Song, N.Y.; Kim, E.H.; Na, H.K.; Joe, Y.; Chung, H.T.; Surh, Y.J. 15-Deoxy-Δ12,14-prostaglandin J2 induces p53 expression through Nrf2-mediated upregulation of heme oxygenase-1 in human breast cancer cells. Free Radic. Res. 2014, 48, 1018–1027. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Bueno, B.; Madrigal, J.L.; Lizasoain, I.; Moro, M.A.; Lorenzo, P.; Leza, J.C. The anti-inflammatory prostaglandin 15d-PGJ2 decreases oxidative/nitrosative mediators in brain after acute stress in rats. Psychopharmacology 2005, 180, 513–522. [Google Scholar] [CrossRef]
- Scher, J.U.; Pillinger, M.H. 15d-PGJ2: The anti-inflammatory prostaglandin? Clin. Immunol. 2005, 114, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Schipper, H.M. Heme oxygenase expression in human central nervous system disorders. Free Radic. Biol. Med. 2004, 37, 1995–2011. [Google Scholar] [CrossRef]
- Ding, Y.; Chen, M.; Wang, M.; Li, Y.; Wen, A. Posttreatment with 11-Keto-β-boswellic acid ameliorates cerebral ischemia-reperfusion injury: Nrf2/HO-1 pathway as a potential mechanism. Mol. Neurobiol. 2015, 52, 1430–1439. [Google Scholar] [CrossRef]
- Jiao, C.; Gao, F.; Ou, L.; Yu, J.; Li, M.; Wei, P.; Miao, F. Tetrahydroxy stilbene glycoside (TSG) antagonizes Aβ-induced hippocampal neuron injury by suppressing mitochondrial dysfunction via Nrf2-dependent HO-1 pathway. Biomed. Pharmacother. 2017, 96, 222–228. [Google Scholar] [CrossRef]
- Zhang, G.; Zhang, F.; Zhang, T.; Gu, J.; Li, C.; Sun, Y.; Yu, P.; Zhang, Z.; Wang, Y. Tetramethylpyrazine nitrone improves neurobehavioral functions and confers neuroprotection on rats with traumatic brain injury. Neurochem. Res. 2016, 41, 2948–2957. [Google Scholar] [CrossRef]
- Loboda, A.; Damulewicz, M.; Pyza, E.; Jozkowicz, A.; Dulak, J. Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: An evolutionarily conserved mechanism. Cell Mol. Life Sci. 2016, 73, 3221–3247. [Google Scholar] [CrossRef] [Green Version]
- Ryter, S.W.; Alam, J.; Choi, A.M. Heme oxygenase-1/carbon monoxide: From basic science to therapeutic applications. Physiol. Rev. 2006, 86, 583–650. [Google Scholar] [CrossRef] [PubMed]
- Yabluchanskiy, A.; Sawle, P.; Homer-Vanniasinkam, S.; Green, C.J.; Foresti, R.; Motterlini, R. CORM-3, a carbon monoxide-releasing molecule, alters the inflammatory response and reduces brain damage in a rat model of hemorrhagic stroke. Crit. Care Med. 2012, 40, 544–552. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, D.; Fu, X.; Yu, L.; Lu, Z.; Gao, Y.; Liu, X.; Man, J.; Li, S.; Li, N.; et al. Carbon monoxide-releasing molecule-3 protects against ischemic stroke by suppressing neuroinflammation and alleviating blood-brain barrier disruption. J. Neuroinflamm. 2018, 15, 188. [Google Scholar] [CrossRef] [PubMed]
- Cairo, G.; Tacchini, L.; Pogliaghi, G.; Anzon, E.; Tomasi, A.; Bernelli-Zazzera, A. Induction of ferritin synthesis by oxidative stress. Transcriptional and post-transcriptional regulation by expansion of the “free” iron pool. J. Biol. Chem. 1995, 270, 700–703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berberat, P.O.; Katori, M.; Kaczmarek, E.; Anselmo, D.; Lassman, C.; Ke, B.; Shen, X.; Busuttil, R.W.; Yamashita, K.; Csizmadia, E.; et al. Heavy chain ferritin acts as an antiapoptotic gene that protects livers from ischemia reperfusion injury. FASEB J. 2003, 17, 1724–1726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, T.N.; Cheung, W.M.; Wu, J.S.; Chen, J.J.; Lin, H.; Chen, J.J.; Liou, J.Y.; Shyue, S.K.; Wu, K.K. 15d-prostaglandin J2 protects brain from ischemia-reperfusion injury. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 481–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koyani, C.N.; Kitz, K.; Rossmann, C.; Bernhart, E.; Huber, E.; Trummer, C.; Windischhofer, W.; Sattler, W.; Malle, E. Activation of the MAPK/Akt/Nrf2-Egr1/HO-1-GCLc axis protects MG-63 osteosarcoma cells against 15d-PGJ2-mediated cell death. Biochem. Pharmacol. 2016, 104, 29–41. [Google Scholar] [CrossRef] [Green Version]
- Lim, H.J.; Lee, K.S.; Lee, S.; Park, J.H.; Choi, H.E.; Go, S.H.; Kwak, H.J.; Park, H.Y. 15d-PGJ2 stimulates HO-1 expression through p38 MAP kinase and Nrf-2 pathway in rat vascular smooth muscle cells. Toxicol. Appl. Pharmacol. 2007, 223, 20–27. [Google Scholar] [CrossRef]
- Lin, C.C.; Hsieh, H.L.; Chi, P.L.; Yang, C.C.; Hsiao, L.D.; Yang, C.M. Upregulation of COX-2/PGE2 by ET-1 mediated through Ca2+-dependent signals in mouse brain microvascular endothelial cells. Mol. Neurobiol. 2014, 49, 1256–1269. [Google Scholar] [CrossRef]
- Yang, C.-C.; Hsiao, L.-D.; Shih, Y.-F.; Yu, Z.-Y.; Yang, C.-M. Anti-inflammatory effects of rhamnetin on bradykinin-induced matrix metalloproteinase-9 expression and cell migration in rat brain astrocytes. Int. J. Mol. Sci. 2022, 23, 609. [Google Scholar] [CrossRef]
- Cho, R.L.; Yang, C.C.; Tseng, H.C.; Hsiao, L.D.; Lin, C.C.; Yang, C.M. Haem oxygenase-1 up-regulation by rosiglitazone via ROS-dependent Nrf2-antioxidant response elements axis or PPARγ attenuates LPS-mediated lung inflammation. Br. J. Pharmacol. 2018, 175, 3928–3946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, I.T.; Luo, S.F.; Lee, C.W.; Wang, S.W.; Lin, C.C.; Chang, C.C.; Chen, Y.L.; Chau, L.Y.; Yang, C.M. Overexpression of HO-1 protects against TNF-α-mediated airway inflammation by down-regulation of TNFR1-dependent oxidative stress. Am. J. Pathol. 2009, 175, 519–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, K.; Li, J.; Li, S.; Feng, J.; Wu, L.; Liu, T.; Zhang, R.; Xu, S.; Cheng, K.; Zhou, Y.; et al. 15d-PGJ2 alleviates ConA-induced acute liver injury in mice by up-regulating HO-1 and reducing hepatic cell autophagy. Biomed. Pharmacother. 2016, 80, 183–192. [Google Scholar] [CrossRef]
- Shih, R.H.; Cheng, S.E.; Hsiao, L.D.; Kou, Y.R.; Yang, C.M. Cigarette smoke extract upregulates heme oxygenase-1 via PKC/NADPH oxidase/ROS/PDGFR/PI3K/Akt pathway in mouse brain endothelial cells. J. Neuroinflamm. 2011, 8, 104. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Kim, S.R.; Je, J.; Jeong, K.; Kim, S.; Kim, H.J.; Chang, K.C.; Park, S.W. The proximal tubular α7 nicotinic acetylcholine receptor attenuates ischemic acute kidney injury through Akt/PKC signaling-mediated HO-1 induction. Exp. Mol. Med. 2018, 50, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castrillo, A.; Traves, P.G.; Martin-Sanz, P.; Parkinson, S.; Parker, P.J.; Bosca, L. Potentiation of protein kinase C ζ activity by 15-deoxy-Δ12,14-prostaglandin J2 induces an imbalance between mitogen-activated protein kinases and NF-κB that promotes apoptosis in macrophages. Mol. Cell Biol. 2003, 23, 1196–1208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.J.; Kim, M.S.; Park, J.Y.; Woo, J.S.; Kim, Y.K. 15-Deoxy-Δ12,14-prostaglandin J2 induces apoptosis via JNK-mediated mitochondrial pathway in osteoblastic cells. Toxicology 2008, 248, 121–129. [Google Scholar] [CrossRef]
- Ho, T.C.; Chen, S.L.; Yang, Y.C.; Chen, C.Y.; Feng, F.P.; Hsieh, J.W.; Cheng, H.C.; Tsao, Y.P. 15-deoxy-Δ12,14-prostaglandin J2 induces vascular endothelial cell apoptosis through the sequential activation of MAPKS and p53. J. Biol. Chem. 2008, 283, 30273–30288. [Google Scholar] [CrossRef] [Green Version]
- Sanada, Y.; Tan, S.J.O.; Adachi, N.; Miyaki, S. Pharmacological targeting of heme oxygenase-1 in osteoarthritis. Antioxidants 2021, 10, 419. [Google Scholar] [CrossRef]
- Lee, T.S.; Tsai, H.L.; Chau, L.Y. Induction of heme oxygenase-1 expression in murine macrophages is essential for the anti-inflammatory effect of low dose 15-deoxy-Δ12,14-prostaglandin J2. J. Biol. Chem. 2003, 278, 19325–19330. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.D.; Tsai, S.H.; Lin, S.Y.; Ho, Y.S.; Hung, L.F.; Pan, S.; Ho, F.M.; Lin, C.M.; Liang, Y.C. Thiol antioxidant and thiol-reducing agents attenuate 15-deoxy-Δ12,14-prostaglandin J2-induced heme oxygenase-1 expression. Life Sci. 2004, 74, 2451–2463. [Google Scholar] [CrossRef] [PubMed]
- Haddad, J.J.; Safieh-Garabedian, B.; Saadé, N.E.; Kanaan, S.A.; Land, S.C. Chemioxyexcitation (ΔpO2/ROS)-dependent release of IL-1β, IL-6 and TNF-α: Evidence of cytokines as oxygen-sensitive mediators in the alveolar epithelium. Cytokine 2001, 13, 138–147. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.C.; Hsiao, L.D.; Lin, H.H.; Tseng, H.C.; Situmorang, J.H.; Leu, Y.L.; Yang, C.M. Induction of HO-1 by 5, 8-dihydroxy-4’,7-dimethoxyflavone via activation of ROS/p38 MAPK/Nrf2 attenuates thrombin-induced connective tissue growth factor expression in human cardiac fibroblasts. Oxid. Med. Cell. Longev. 2020, 2020, 1080168. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.C.; Yang, C.C.; Chen, Y.W.; Hsiao, L.D.; Yang, C.M. Arachidonic acid induces ARE/Nrf2-dependent heme oxygenase-1 transcription in rat brain astrocytes. Mol. Neurobiol. 2018, 55, 3328–3343. [Google Scholar] [CrossRef]
- Yang, C.M.; Lin, C.C.; Yang, C.C.; Cho, R.L.; Hsiao, L.D. Mevastatin-induced AP-1-dependent HO-1 expression suppresses vascular cell adhesion molecule-1 expression and monocyte adhesion on human pulmonary alveolar epithelial cells challenged with TNF-α. Biomolecules 2020, 10, 381. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.L.; Lin, H.H.; Kim, K.J.; Lin, A.; Ou, J.H.; Ann, D.K. PKCδ signaling: A dual role in regulating hypoxic stress-induced autophagy and apoptosis. Autophagy 2009, 5, 244–246. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.G.; Yang, J.H. PKC-δ mediates TCDD-induced apoptosis of chondrocyte in ROS-dependent manner. Chemosphere 2010, 81, 1039–1044. [Google Scholar] [CrossRef]
- Lee, S.E.; Jeong, S.I.; Yang, H.; Park, C.S.; Jin, Y.H.; Park, Y.S. Fisetin induces Nrf2-mediated HO-1 expression through PKC-δ and p38 in human umbilical vein endothelial cells. J. Cell. Biochem. 2011, 112, 2352–2360. [Google Scholar] [CrossRef]
- Zhang, H.; Forman, H.J. Acrolein induces heme oxygenase-1 through PKC-δ and PI3K in human bronchial epithelial cells. Am. J. Respir. Cell Mol. Biol. 2008, 38, 483–490. [Google Scholar] [CrossRef]
- Cho, R.L.; Lin, W.N.; Wang, C.Y.; Yang, C.C.; Hsiao, L.D.; Lin, C.C.; Yang, C.M. Heme oxygenase-1 induction by rosiglitazone via PKCα/AMPKα/p38 MAPKα/SIRT1/PPARγ pathway suppresses lipopolysaccharide-mediated pulmonary inflammation. Biochem. Pharmacol. 2018, 148, 222–237. [Google Scholar] [CrossRef]
- Chien, P.T.; Lin, C.C.; Hsiao, L.D.; Yang, C.M. Induction of HO-1 by carbon monoxide releasing molecule-2 attenuates thrombin-induced COX-2 expression and hypertrophy in primary human cardiomyocytes. Toxicol. Appl. Pharmacol. 2015, 289, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Maqueda, M.; El Bekay, R.; Alba, G.; Monteseirín, J.; Chacón, P.; Vega, A.; Martín-Nieto, J.; Bedoya, F.J.; Pintado, E.; Sobrino, F. 15-deoxy-Δ12,14-prostaglandin J2 induces heme oxygenase-1 gene expression in a reactive oxygen species-dependent manner in human lymphocytes. J. Biol. Chem. 2004, 279, 21929–21937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, S.W.; Seo, C.Y.; Han, H.; Han, J.Y.; Jeong, J.S.; Kwak, J.Y.; Park, J.I. 15d-PGJ2 induces apoptosis by reactive oxygen species-mediated inactivation of Akt in leukemia and colorectal cancer cells and shows in vivo antitumor activity. Clin. Cancer Res. 2009, 15, 5414–5425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deshane, J.; Kim, J.; Bolisetty, S.; Hock, T.D.; Hill-Kapturczak, N.; Agarwal, A. Sp1 regulates chromatin looping between an intronic enhancer and distal promoter of the human heme oxygenase-1 gene in renal cells. J. Biol. Chem. 2010, 285, 16476–16486. [Google Scholar] [CrossRef] [Green Version]
- Hock, T.D.; Liby, K.; Wright, M.M.; McConnell, S.; Schorpp-Kistner, M.; Ryan, T.M.; Agarwal, A. JunB and JunD regulate human heme oxygenase-1 gene expression in renal epithelial cells. J. Biol. Chem. 2007, 282, 6875–6886. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Zarjou, A.; Traylor, A.M.; Bolisetty, S.; Jaimes, E.A.; Hull, T.D.; George, J.F.; Mikhail, F.M.; Agarwal, A. In vivo regulation of the heme oxygenase-1 gene in humanized transgenic mice. Kidney Int. 2012, 82, 278–291. [Google Scholar] [CrossRef] [Green Version]
- Rojo, A.I.; Salina, M.; Salazar, M.; Takahashi, S.; Suske, G.; Calvo, V.; de Sagarra, M.R.; Cuadrado, A. Regulation of heme oxygenase-1 gene expression through the phosphatidylinositol 3-kinase/PKC-ζ pathway and Sp1. Free Radic. Biol. Med. 2006, 41, 247–261. [Google Scholar] [CrossRef]
- Liang, J.Q.; Xu, H.B.; Wu, Y.L.; Sun, S.R.; Jia, Z.H.; Wei, C.; You, J.H. Effect of serum from overfatigue rats on JNK/c-Jun/HO-1 pathway in human umbilical vein endothelial cells and the intervening effect of Tongxinluo superfine powder. Chin. J. Integr. Med. 2009, 15, 121–127. [Google Scholar] [CrossRef]
- Chi, P.L.; Lin, C.C.; Chen, Y.W.; Hsiao, L.D.; Yang, C.M. CO induces Nrf2-dependent heme oxygenase-1 transcription by cooperating with Sp1 and c-Jun in rat brain astrocytes. Mol. Neurobiol. 2015, 52, 277–292. [Google Scholar] [CrossRef]
- De Vries, H.E.; Blom-Roosemalen, M.C.; van Oosten, M.; de Boer, A.G.; van Berkel, T.J.; Breimer, D.D.; Kuiper, J. The influence of cytokines on the integrity of the blood-brain barrier in vitro. J. Neuroimmunol. 1996, 64, 37–43. [Google Scholar] [CrossRef]
- Mogi, M.; Harada, M.; Narabayashi, H.; Inagaki, H.; Minami, M.; Nagatsu, T. Interleukin (IL)-1β, IL-2, IL-4, IL-6 and transforming growth factor-α levels are elevated in ventricular cerebrospinal fluid in juvenile parkinsonism and Parkinson’s disease. Neurosci. Lett. 1996, 211, 13–16. [Google Scholar] [CrossRef]
- Biswas, C.; Shah, N.; Muthu, M.; La, P.; Fernando, A.P.; Sengupta, S.; Yang, G.; Dennery, P.A. Nuclear heme oxygenase-1 (HO-1) modulates subcellular distribution and activation of Nrf2, impacting metabolic and anti-oxidant defenses. J. Biol. Chem. 2014, 289, 26882–26894. [Google Scholar] [CrossRef] [PubMed] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, C.-C.; Hsiao, L.-D.; Shih, Y.-F.; Chang, C.-I.; Yang, C.-M. Induction of Heme Oxygenase-1 by 15d-Prostaglandin J2 Mediated via a ROS-Dependent Sp1 and AP-1 Cascade Suppresses Lipopolysaccharide-Triggered Interleukin-6 Expression in Mouse Brain Microvascular Endothelial Cells. Antioxidants 2022, 11, 719. https://doi.org/10.3390/antiox11040719
Yang C-C, Hsiao L-D, Shih Y-F, Chang C-I, Yang C-M. Induction of Heme Oxygenase-1 by 15d-Prostaglandin J2 Mediated via a ROS-Dependent Sp1 and AP-1 Cascade Suppresses Lipopolysaccharide-Triggered Interleukin-6 Expression in Mouse Brain Microvascular Endothelial Cells. Antioxidants. 2022; 11(4):719. https://doi.org/10.3390/antiox11040719
Chicago/Turabian StyleYang, Chien-Chung, Li-Der Hsiao, Ya-Fang Shih, Ching-I Chang, and Chuen-Mao Yang. 2022. "Induction of Heme Oxygenase-1 by 15d-Prostaglandin J2 Mediated via a ROS-Dependent Sp1 and AP-1 Cascade Suppresses Lipopolysaccharide-Triggered Interleukin-6 Expression in Mouse Brain Microvascular Endothelial Cells" Antioxidants 11, no. 4: 719. https://doi.org/10.3390/antiox11040719
APA StyleYang, C.-C., Hsiao, L.-D., Shih, Y.-F., Chang, C.-I., & Yang, C.-M. (2022). Induction of Heme Oxygenase-1 by 15d-Prostaglandin J2 Mediated via a ROS-Dependent Sp1 and AP-1 Cascade Suppresses Lipopolysaccharide-Triggered Interleukin-6 Expression in Mouse Brain Microvascular Endothelial Cells. Antioxidants, 11(4), 719. https://doi.org/10.3390/antiox11040719