
Altered Mitochondrial Opa1-Related Fusion in Mouse Promotes Endothelial Cell Dysfunction and Atherosclerosis

 , , , , and
, , , , and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Mice
- − Ldlr−/−Opa1+/− and their littermate control Ldlr−/−Opa1+/+
- − Ldlr−/−EC-Opa1 mice and their littermate control Ldlr−/−EC-WT.
2.2. Flow-Mediated Dilation in Mesenteric Arteries In Vitro
2.3. Perfused Isolated Mouse Kidney
2.4. Determination of ATP and H2O2 Levels in the Kidney Perfusate
2.5. Cell Culture
2.6. RNA Interference
2.7. Cell Migration Assay
2.8. In Vitro Exposure of Endothelial Cells to Shear Stress
2.9. Measurement of Endothelial Cells Alignment and Elongation
2.10. Circular Flow Assay
2.11. Analysis of mRNA Levels by RT-qPCR
2.12. Immunofluorescence Analyses
2.13. Mitochondrial Shape Measurement in Endothelial Cells
2.14. Analysis of Protein Expression Levels by Western Blot
2.15. Endothelial Cell Isolation from Mouse Mesenteric Arteries
2.16. Analyses of Atherosclerosis Lesions
2.17. Lipids and Glucose Blood Level in Mice
2.18. Statistical Analyses
3. Results
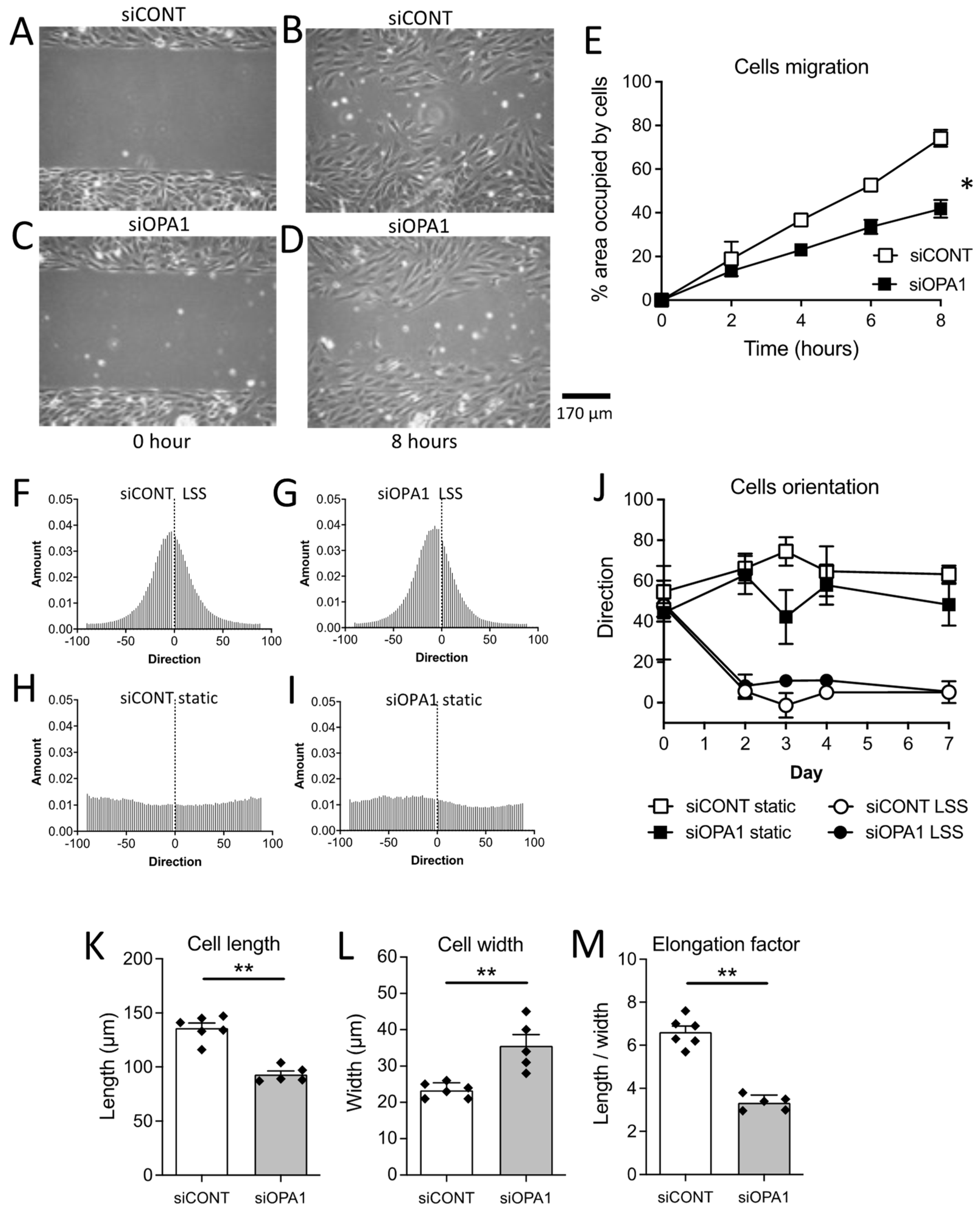
3.1. In Vitro Endothelial Cell Migration
3.2. In Vitro Flow-Mediated Endothelial Cell Alignment and Elongation
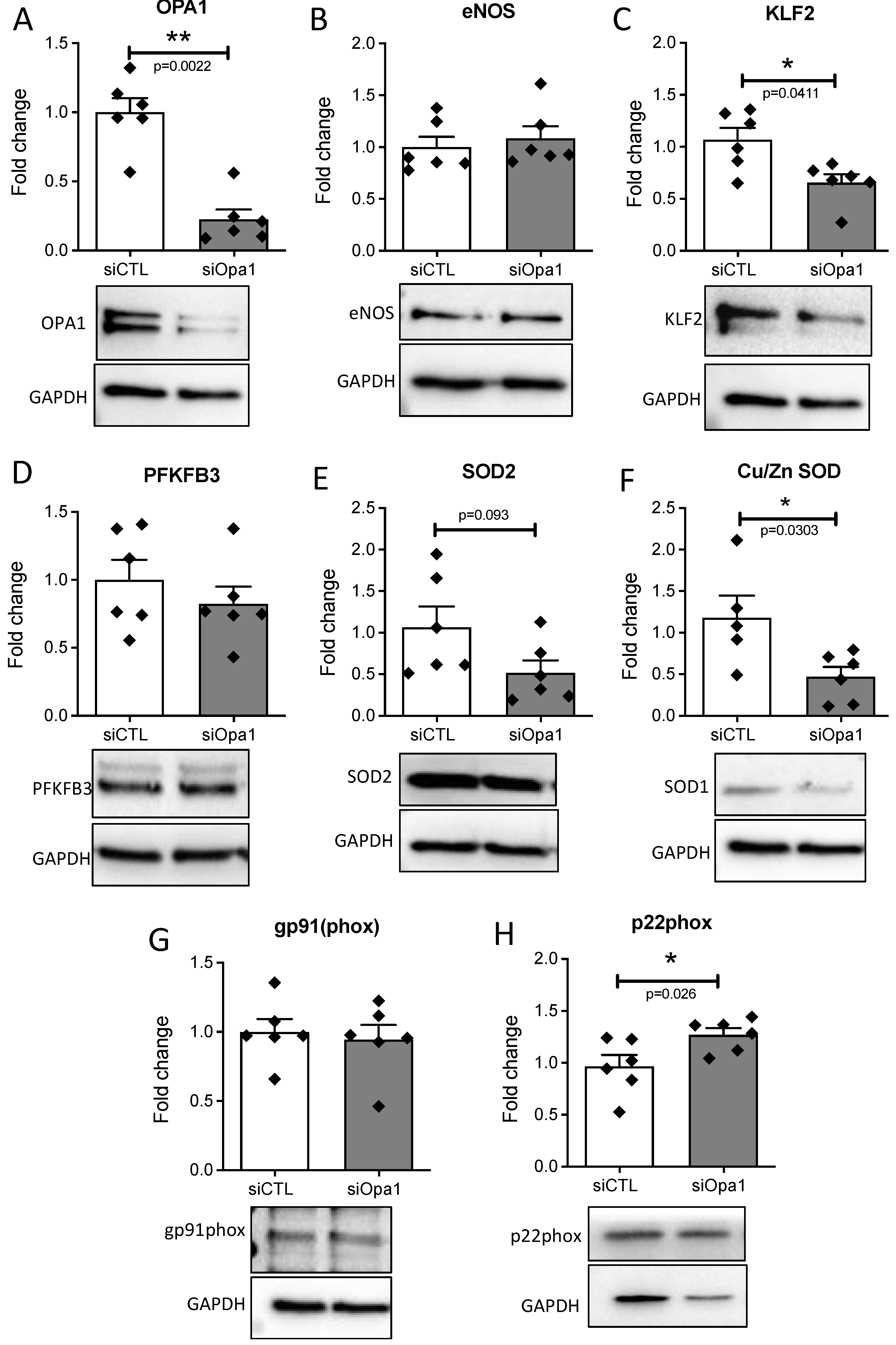
3.3. OPA1 Silencing Altered the Response of Endothelial Cells to Laminar Flow
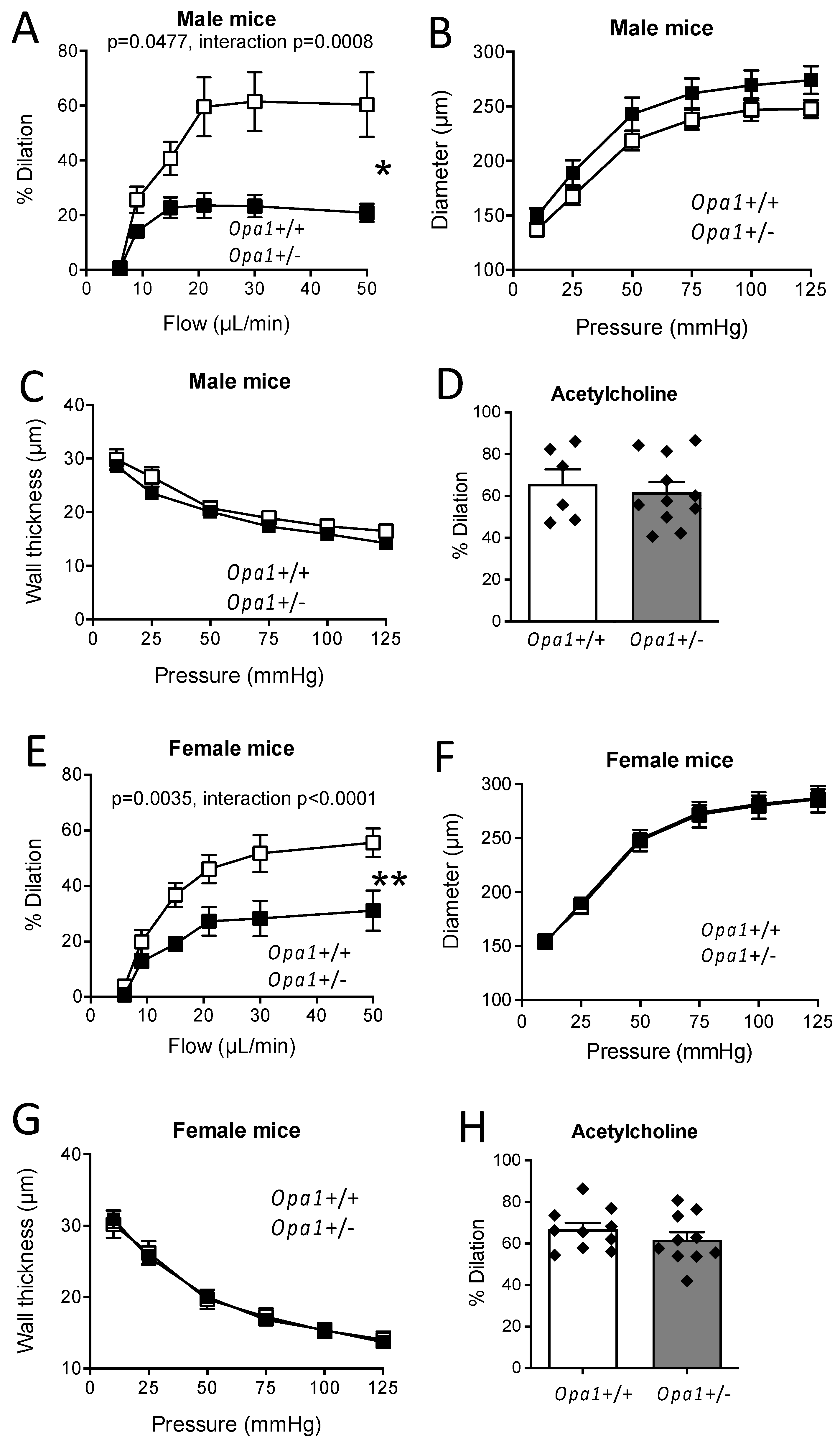
3.4. In Vitro Flow-Mediated Dilation in Opa1+/− Mouse Resistance Arteries
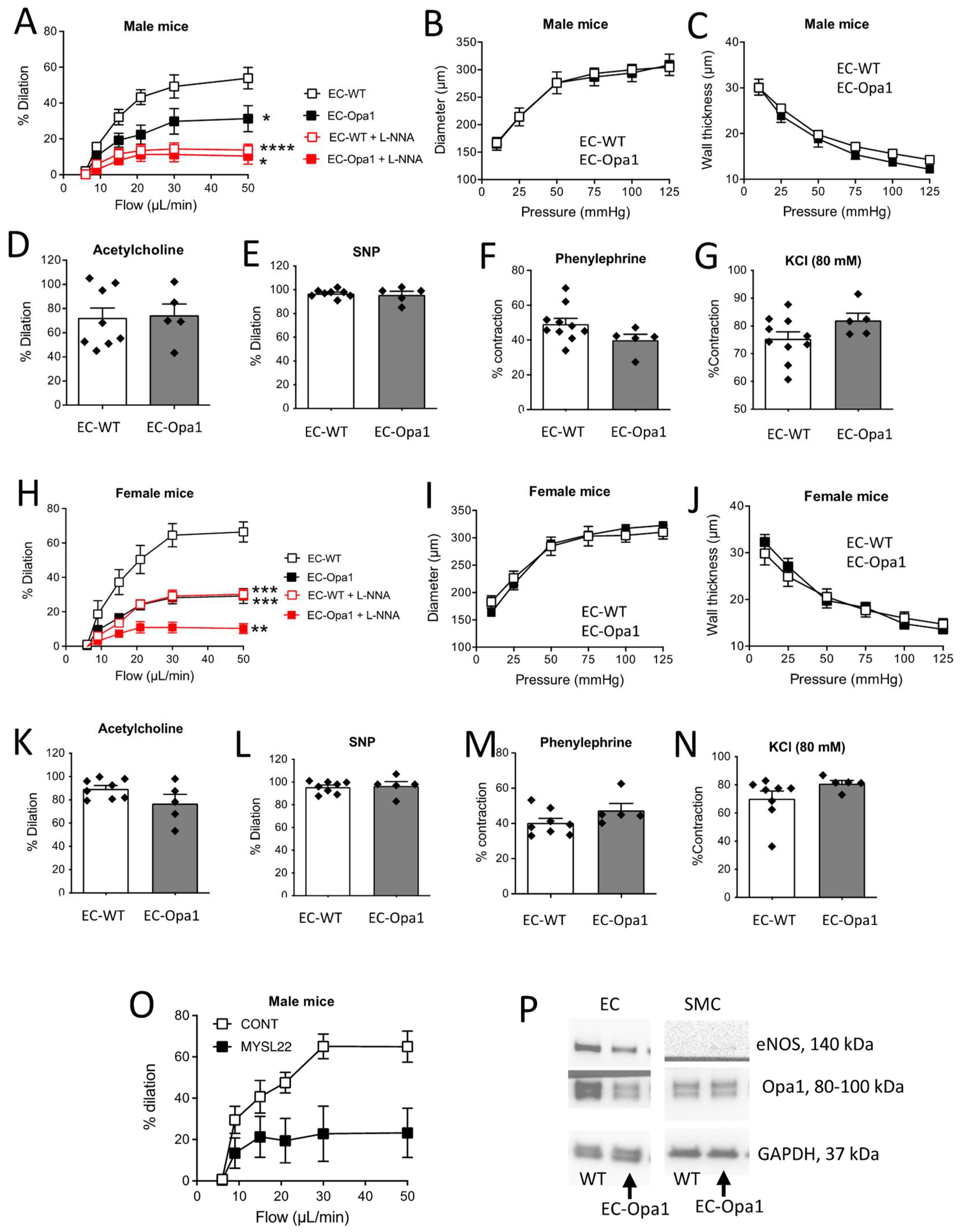
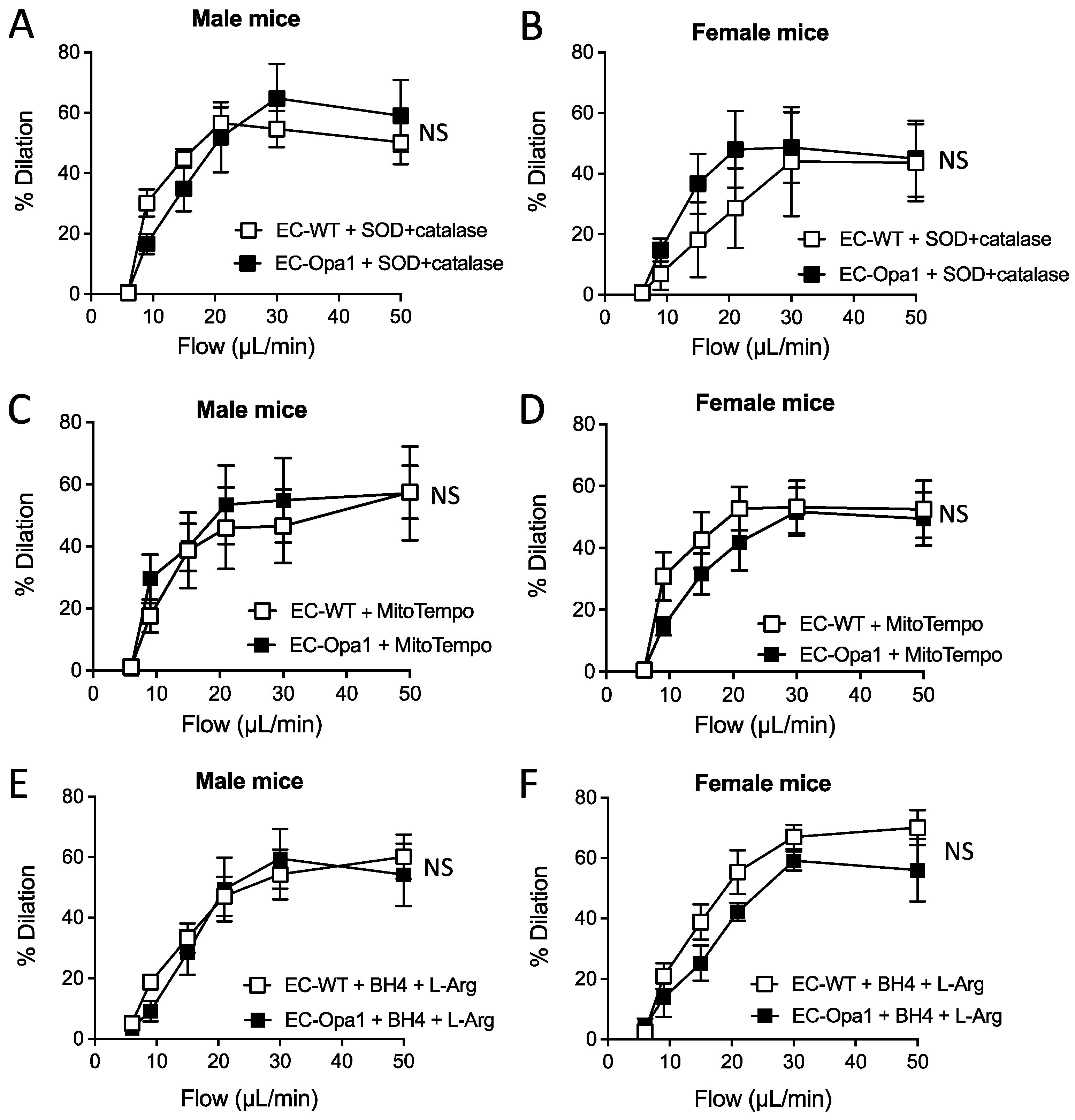
3.5. Flow-Mediated Dilation in Mouse Lacking Opa1 in Endothelial Cells
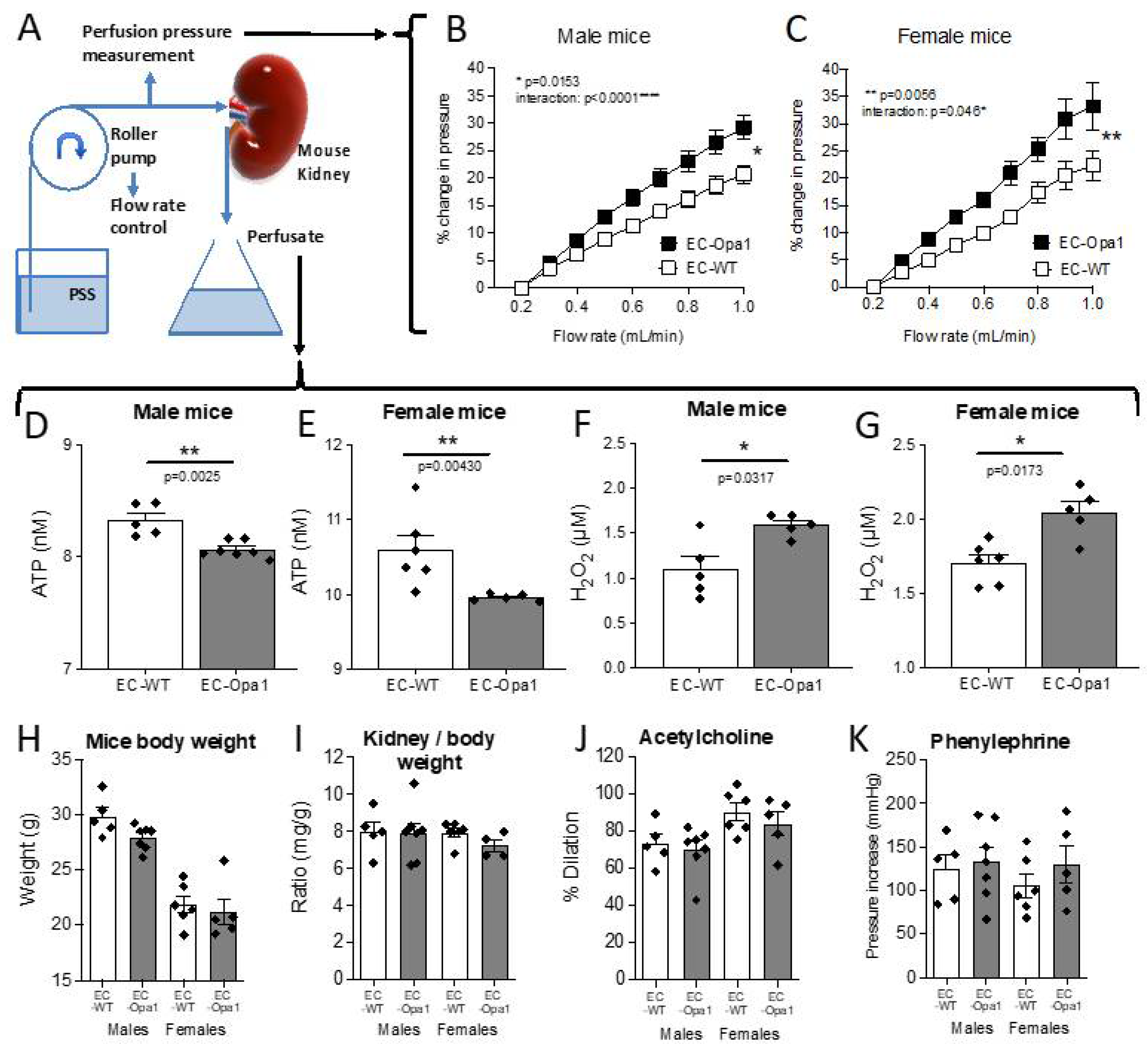
3.6. Flow-Pressure Relationship, ATP and H2O2 Production in Ex-Vivo Perfused Kidney
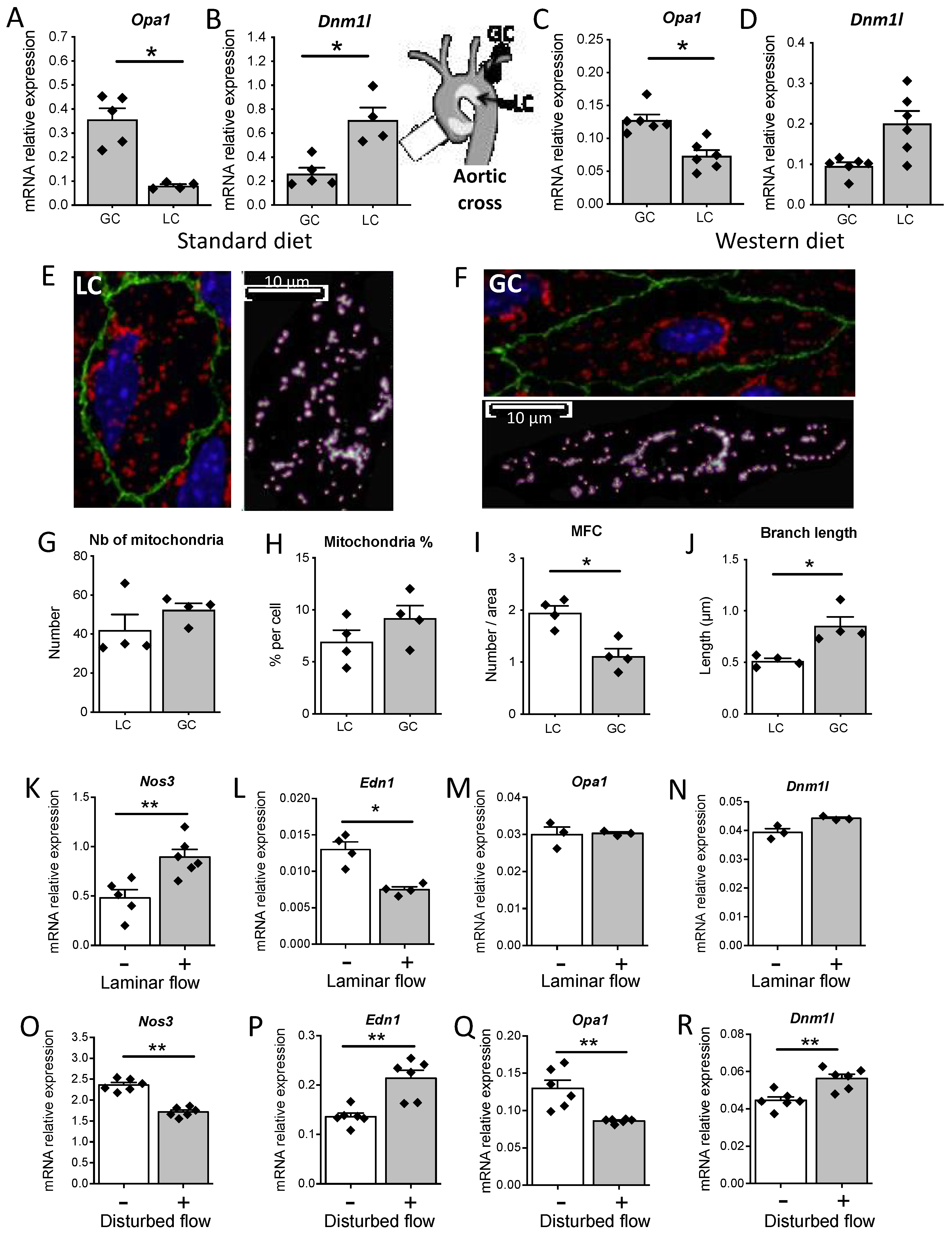
3.7. Disturbed Flow In Vivo and In Vitro Reduced Opa1 Level and Mitochondrial Length
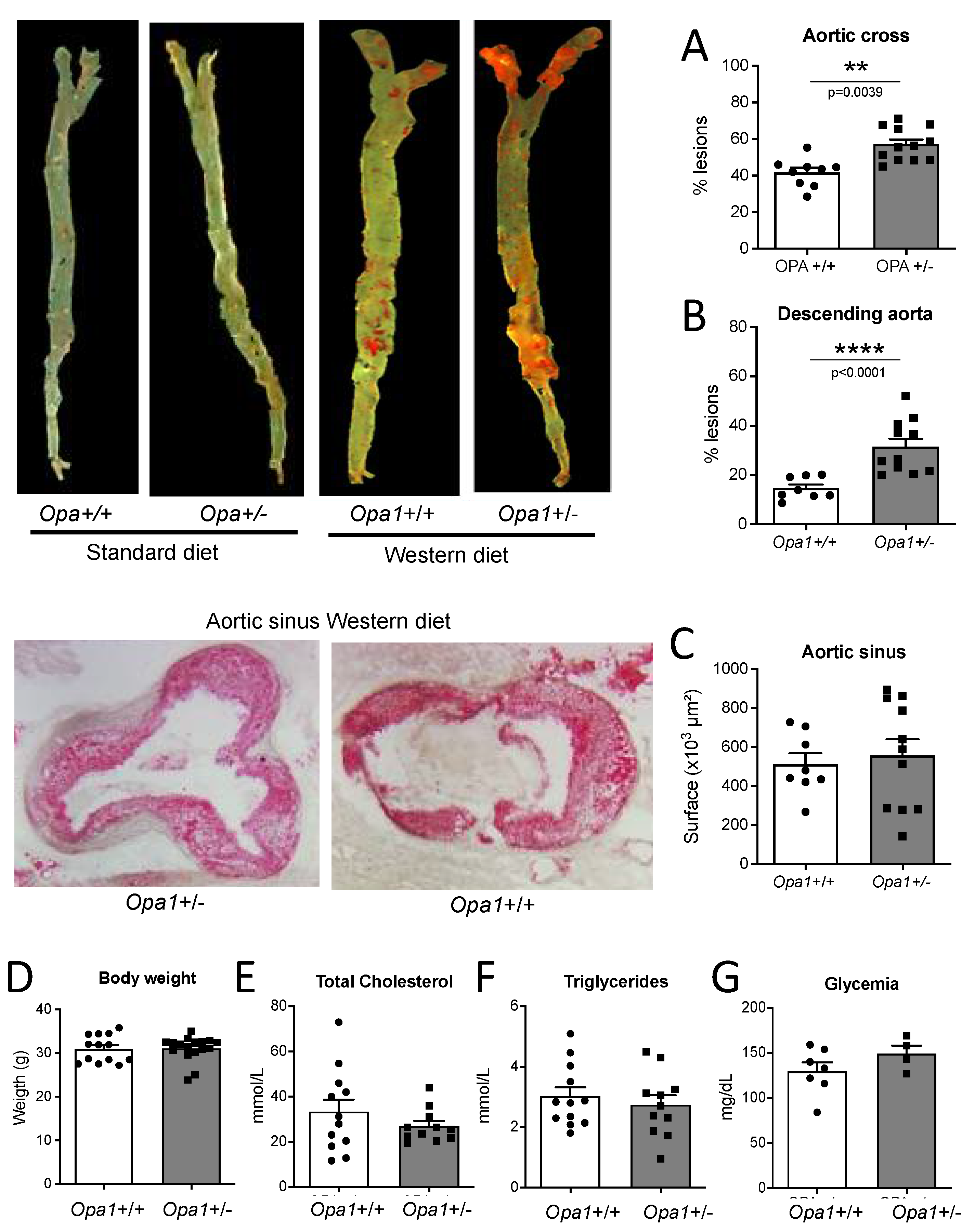
3.8. Lipid Deposits in the Aorta of Opa1+/− Mice
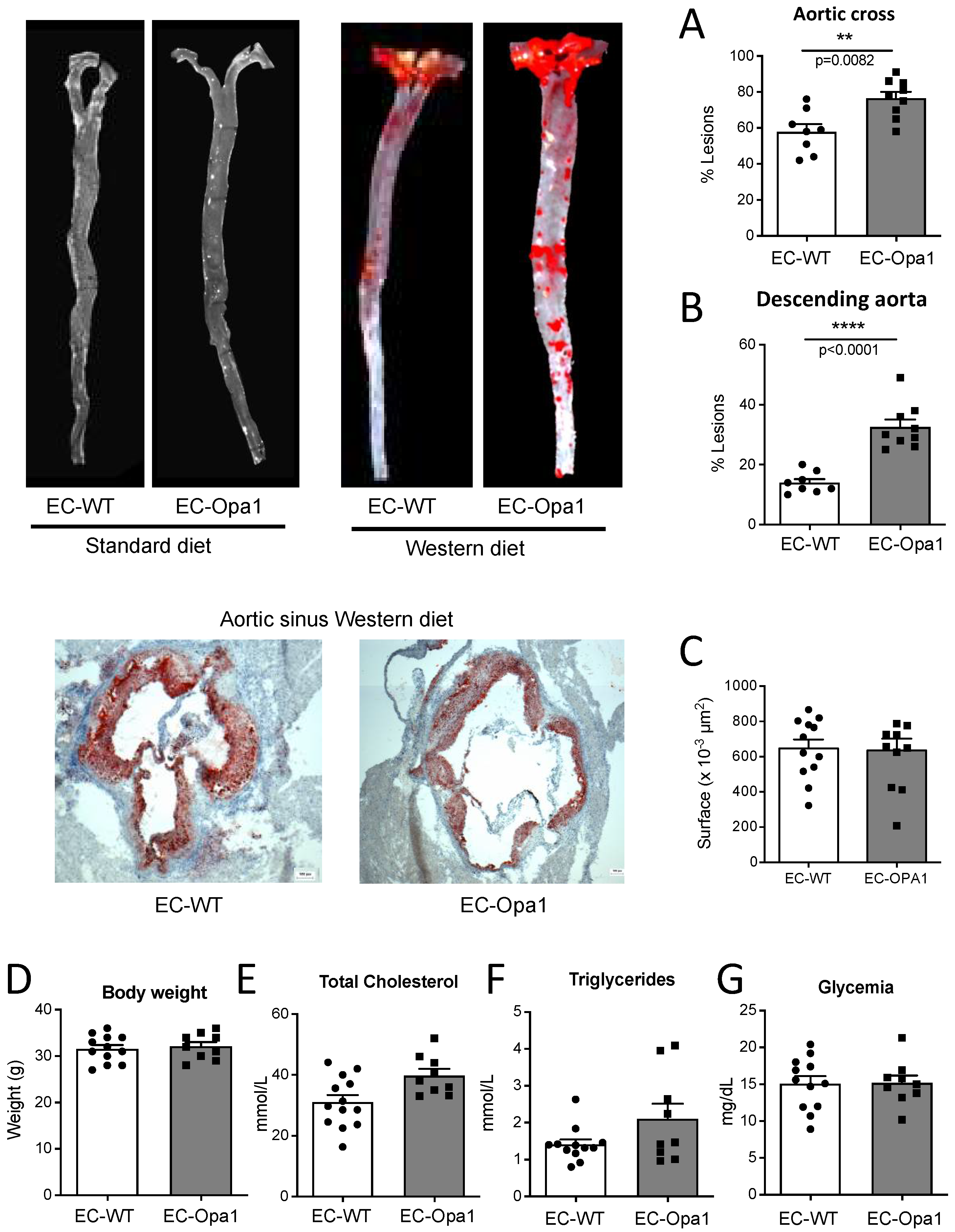
3.9. Lipid Deposits in the Aorta of Mice Lacking Opa1 in Endothelial Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Struijker-Boudier, H.A.; Rosei, A.E.; Bruneval, P.; Camici, P.G.; Christ, F.; Henrion, D.; Lévy, B.I.; Pries, A.; Vanoverschelde, J.-L. Evaluation of the microcirculation in hypertension and cardiovascular disease. Eur. Heart J. 2007, 28, 2834–2840. [Google Scholar] [CrossRef]
- Segal, S.S. Regulation of blood flow in the microcirculation. Microcirculation 2005, 12, 33–45. [Google Scholar] [CrossRef]
- Zhou, J.; Li, Y.-S.; Chien, S. Shear Stress–Initiated Signaling and Its Regulation of Endothelial Function. Arter. Thromb. Vasc. Biol. 2014, 34, 2191–2198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deanfield, J.E.; Halcox, J.P.; Rabelink, T.J. Endothelial function and dysfunction: Testing and clinical relevance. Circulation 2007, 115, 1285–1295. [Google Scholar] [CrossRef] [PubMed]
- Naiberg, M.R.; Newton, D.F.; Goldstein, B.I. Flow-mediated dilation and neurocognition: Systematic review and future directions. Psychosom. Med. 2016, 78, 192–207. [Google Scholar] [CrossRef]
- Chehaitly, A.; Vessieres, E.; Guihot, A.L.; Henrion, D. Flow-mediated outward arterial remodeling in aging. Mech. Ageing Dev. 2021, 194, 111416. [Google Scholar] [CrossRef] [PubMed]
- Freed, J.K.; Beyer, A.M.; LoGiudice, J.A.; Hockenberry, J.C.; Gutterman, D.D. Ceramide changes the mediator of flow-induced vasodilation from nitric oxide to hydrogen peroxide in the human microcirculation. Circ. Res. 2014, 115, 525–532. [Google Scholar] [CrossRef] [Green Version]
- Marín-García, J.; Akhmedov, A.T. Mitochondrial dynamics and cell death in heart failure. Heart Fail. Rev. 2016, 21, 123–136. [Google Scholar] [CrossRef]
- Culic, O.; Gruwel, M.L.; Schrader, J. Energy turnover of vascular endothelial cells. Am. J. Physiol. Physiol. 1997, 273, C205–C213. [Google Scholar] [CrossRef]
- Kadlec, A.O.; Beyer, A.M.; Ait-Aissa, K.; Gutterman, D.D. Mitochondrial signaling in the vascular endothelium: Beyond reactive oxygen species. Basic Res. Cardiol. 2016, 111, 1–12. [Google Scholar] [CrossRef]
- Vásquez-Trincado, C.; García-Carvajal, I.; Pennanen, C.; Parra, V.; Hill, J.A.; Rothermel, B.A.; Lavandero, S. Mitochondrial dynamics, mitophagy and cardiovascular disease. J. Physiol. 2016, 594, 509–525. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.-B.; Subrayan, S.; Lim, S.Y.; Yellon, D.M.; Davidson, S.M.; Hausenloy, D.J. Inhibiting mitochondrial fission protects the heart against ischemia/reperfusion injury. Circulation 2010, 121, 2012–2022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marsboom, G.; Toth, P.; Ryan, J.J.; Hong, Z.; Wu, X.; Fang, Y.-H.; Thenappan, T.; Piao, L.; Zhang, H.J.; Pogoriler, J.; et al. Dynamin-Related Protein 1–Mediated Mitochondrial Mitotic Fission Permits Hyperproliferation of Vascular Smooth Muscle Cells and Offers a Novel Therapeutic Target in Pulmonary Hypertension. Clin. Trans. Res. 2012, 110, 1484–1497. [Google Scholar] [CrossRef]
- Salabei, J.K.; Hill, B.G. Mitochondrial fission induced by platelet-derived growth factor regulates vascular smooth muscle cell bioenergetics and cell proliferation. Redox Biol. 2013, 1, 542–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potente, M.; Carmeliet, P. The Link Between Angiogenesis and Endothelial Metabolism. Annu. Rev. Physiol. 2017, 79, 43–66. [Google Scholar] [CrossRef]
- Bretón-Romero, R.; Acín-Perez, R.; Rodríguez-Pascual, F.; Martínez-Molledo, M.; Brandes, R.P.; Rial, E.; Enríquez, J.A.; Lamas, S. Laminar shear stress regulates mitochondrial dynamics, bioenergetics responses and PRX3 activation in endothelial cells. Biochim. et Biophys. Acta 2014, 1843, 2403–2413. [Google Scholar] [CrossRef] [Green Version]
- Scheitlin, C.G.; Julian, J.A.; Shanmughapriya, S.; Madesh, M.; Tsoukias, N.M.; Alevriadou, B.R. Endothelial mitochondria regulate the intracellular Ca2+ response to fluid shear stress. Am. J. Physiol.-Cell Physiol. 2016, 310, C479–C490. [Google Scholar] [CrossRef] [Green Version]
- Herkenne, S.; Ek, O.; Zamberlan, M.; Pellattiero, A.; Chergova, M.; Chivite, I.; Novotná, E.; Rigoni, G.; Fonseca, T.B.; Samardzic, D.; et al. Developmental and Tumor Angiogenesis Requires the Mitochondria-Shaping Protein Opa1. Cell Metab. 2020, 31, 987–1003. [Google Scholar] [CrossRef]
- Gutterman, D.D.; Chabowski, D.S.; Kadlec, A.O.; Durand, M.J.; Freed, J.K.; Ait-Aissa, K.; Beyer, A.M. The Human Microcirculation: Regulation of Flow and Beyond. Circ. Res. 2016, 118, 157–172. [Google Scholar] [CrossRef] [Green Version]
- Sarzi, E.; Angebault, C.; Seveno, M.; Gueguen, N.; Chaix, B.; Bielicki, G.; Boddaert, N.; Bonnefont, A.-L.; Cazevieille, C.; Rigau, V.; et al. The human OPA1delTTAG mutation induces premature age-related systemic neurodegeneration in mouse. Brain 2012, 135, 3599–3613. [Google Scholar] [CrossRef]
- Lenaers, G.; Hamel, C.; Delettre, C.; Amati-Bonneau, P.; Procaccio, V.; Bonneau, D.; Reynier, P.; Milea, D. Dominant optic atrophy. Orphanet J. Rare Dis. 2012, 7, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Votruba, M.; Aijaz, S.; Moore, A.T. A review of primary hereditary optic neuropathies. J. Inherit. Metab. Dis. 2003, 26, 209–227. [Google Scholar] [CrossRef] [PubMed]
- Cesareo, M.; Giannini, C.; Di Marino, M.; Aloe, G.; Martucci, A.; Aiello, F.; Cusumano, A.; Mancino, R.; Ricci, F.; Sorge, R.P.; et al. Optical coherence tomography angiography in the multimodal assessment of the retinal posterior pole in autosomal dominant optic atrophy. Acta Ophthalmol. 2021, 100, e798–e806. [Google Scholar] [CrossRef] [PubMed]
- Ciurică, S.; Lopez-Sublet, M.; Loeys, B.L.; Radhouani, I.; Natarajan, N.; Vikkula, M.; Maas, A.H.E.M.; Adlam, D.; Persu, A. Arterial Tortuosity. Hypertension 2019, 73, 951–960. [Google Scholar] [CrossRef]
- Kim, D.; Votruba, M.; Roy, S. Opa1 Deficiency Promotes Development of Retinal Vascular Lesions in Diabetic Retinopathy. Int J. Mol. Sci. 2021, 22, 5928. [Google Scholar] [CrossRef]
- Zerem, A.; Yosovich, K.; Rappaport, Y.C.; Libzon, S.; Blumkin, L.; Ben-Sira, L.; Lev, D.; Lerman-Sagie, T. Metabolic stroke in a patient with bi-allelic OPA1 mutations. Metab. Brain Dis. 2019, 34, 1043–1048. [Google Scholar] [CrossRef]
- Robert, P.; Nguyen, P.M.C.; Richard, A.; Grenier, C.; Chevrollier, A.; Munier, M.; Grimaud, L.; Proux, C.; Champin, T.; Lelièvre, E.; et al. Protective role of the mitochondrial fusion protein OPA1 in hypertension. FASEB J. 2021, 35, e21678. [Google Scholar] [CrossRef]
- Rodríguez-Nuevo, A.; Díaz-Ramos, A.; Noguera, E.; Díaz-Sáez, F.; Duran, X.; Muñoz, J.P.; Romero, M.; Plana, N.; Sebastián, D.; Tezze, C.; et al. Mitochondrial DNA and TLR9 drive muscle inflammation upon Opa1 deficiency. EMBO J. 2018, 37, e96553. [Google Scholar] [CrossRef]
- Billon-Galés, A.; Fontaine, C.; Douin-Echinard, V.; Delpy, L.; Berges, H.; Calippe, B.; Lenfant, F.; Laurell, H.; Guery, J.-C.; Gourdy, P.; et al. Endothelial Estrogen Receptor-α Plays a Crucial Role in the Atheroprotective Action of 17β-Estradiol in Low-Density Lipoprotein Receptor–Deficient Mice. Circulation 2009, 120, 2567–2576. [Google Scholar] [CrossRef] [Green Version]
- Iglarz, M.; Matrougui, K.; Lévy, B.I.; Henrion, D. Chronic blockade of endothelin ETA receptors improves flow dependent dilation in resistance arteries of hypertensive rats. Cardiovasc. Res. 1998, 39, 657–664. [Google Scholar] [CrossRef] [Green Version]
- Henrion, D.; Terzi, F.; Matrougui, K.; Duriez, M.; Boulanger, C.M.; Colucci-Guyon, E.; Babinet, C.; Briand, P.; Friedlander, G.; Poitevin, P.; et al. Impaired flow-induced dilation in mesenteric resistance arteries from mice lacking vimentin. J. Clin. Investig. 1997, 100, 2909–2914. [Google Scholar] [CrossRef] [PubMed]
- Bouvet, C.; de Chantemele, E.B.; Guihot, A.-L.; Vessieres, E.; Bocquet, A.; Dumont, O.; Jardel, A.; Loufrani, L.; Moreau, P.; Henrion, D. Flow-Induced Remodeling in Resistance Arteries From Obese Zucker Rats Is Associated With Endothelial Dysfunction. Hypertension 2007, 50, 248–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Favre, J.; Vessieres, E.; Guihot, A.-L.; Grimaud, L.; Proux, C.; Loufrani, L.; Lenfant, F.; Fontaine, C.; Arnal, J.-F.; Henrion, D. Early Inactivation of Membrane Estrogen Receptor Alpha (ERα) Recapitulates the Endothelial Dysfunction of Aged Mouse Resistance Arteries. Int. J. Mol. Sci. 2022, 23, 2862. [Google Scholar] [CrossRef] [PubMed]
- Favre, J.; Vessieres, E.; Guihot, A.-L.; Proux, C.; Grimaud, L.; Rivron, J.; Garcia, M.C.; Réthoré, L.; Zahreddine, R.; Davezac, M.; et al. Membrane estrogen receptor alpha (ERα) participates in flow-mediated dilation in a ligand-independent manner. eLife 2021, 10, e68695. [Google Scholar] [CrossRef]
- Mack, J.J.; Mosqueiro, T.S.; Archer, B.J.; Jones, W.M.; Sunshine, H.; Faas, G.C.; Briot, A.; Aragón, R.L.; Su, T.; Romay, M.C.; et al. NOTCH1 is a mechanosensor in adult arteries. Nat. Commun. 2017, 8, 1620. [Google Scholar] [CrossRef]
- Salek, M.M.; Sattari, P.; Martinuzzi, R.J. Analysis of fluid flow and wall shear stress patterns inside partially filled agitated culture well plates. Ann. Biomed. Eng. 2012, 40, 707–728. [Google Scholar] [CrossRef]
- Malek, A.; Izumo, S. Physiological fluid shear stress causes downregulation of endothelin-1 mRNA in bovine aortic endothelium. Am. J. Physiol. Physiol. 1992, 263, C389–C396. [Google Scholar] [CrossRef]
- Dancu, M.B.; Berardi, D.E.; Heuvel, J.P.V.; Tarbell, J.M. Asynchronous Shear Stress and Circumferential Strain Reduces Endothelial NO Synthase and Cyclooxygenase-2 but Induces Endothelin-1 Gene Expression in Endothelial Cells. Arter. Thromb. Vasc. Biol. 2004, 24, 2088–2094. [Google Scholar] [CrossRef]
- Morawietz, H.; Talanow, R.; Szibor, M.; Rueckschloss, U.; Schubert, A.; Bartling, B.; Darmer, D.; Holtz, J. Regulation of the endothelin system by shear stress in human endothelial cells. J. Physiol. 2000, 525, 761–770. [Google Scholar] [CrossRef]
- Lavallée, M.; Takamura, M.; Parent, R.; Thorin, E. Crosstalk between endothelin and nitric oxide in the control of vascular tone. Heart Fail. Rev. 2001, 6, 265–276. [Google Scholar] [CrossRef]
- Ishibazawa, A.; Nagaoka, T.; Takahashi, T.; Yamamoto, K.; Kamiya, A.; Ando, J.; Yoshida, A. Effects of Shear Stress on the Gene Expressions of Endothelial Nitric Oxide Synthase, Endothelin-1, and Thrombomodulin in Human Retinal Microvascular Endothelial Cells. Investig. Opthalmol. Vis. Sci. 2011, 52, 8496–8504. [Google Scholar] [CrossRef]
- The dMIQE Group; Huggett, J.F. The Digital MIQE Guidelines Update: Minimum Information for Publication of Quantitative Digital PCR Experiments for 2020. Clin. Chem. 2020, 66, 1012–1029. [Google Scholar] [CrossRef] [PubMed]
- Parra, V.; Altamirano, F.; Hernández-Fuentes, C.P.; Tong, D.; Kyrychenko, V.; Rotter, D.; Pedrozo, Z.; Hill, J.A.; Eisner, V.; Lavandero, S.; et al. Down Syndrome Critical Region 1 Gene, Rcan1, Helps Maintain a More Fused Mitochondrial Network. Circ. Res. 2018, 122, e20–e33. [Google Scholar] [CrossRef] [PubMed]
- Forrester, S.J.; Preston, K.J.; Cooper, H.A.; Boyer, M.J.; Escoto, K.M.; Poltronetti, A.J.; Elliott, K.J.; Kuroda, R.; Miyao, M.; Sesaki, H.; et al. Mitochondrial Fission Mediates Endothelial Inflammation. Hypertension 2020, 76, 267–276. [Google Scholar] [CrossRef]
- Hemel, I.M.; Engelen, B.P.; Luber, N.; Gerards, M. A hitchhiker’s guide to mitochondrial quantification. Mitochondrion 2021, 59, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Guivarc’h, E.; Buscato, M.; Guihot, A.L.; Favre, J.; Vessieres, E.; Grimaud, L.; Wakim, J.; Melhem, N.J.; Zahreddine, R.; Adlanmerini, M.; et al. Predominant Role of Nuclear Versus Membrane Estrogen Receptor α in Arterial Protection: Implications for Estrogen Receptor α Modulation in Cardiovascular Prevention/Safety. J. Am. Heart Assoc. 2018, 7, e008950. [Google Scholar] [CrossRef] [Green Version]
- Davies, P.F. Hemodynamic shear stress and the endothelium in cardiovascular pathophysiology. Nat. Clin. Pract. Cardiovasc. Med. 2009, 6, 16–26. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, S.; Fisher, A.B. Mechanotransduction in the endothelium: Role of membrane proteins and reactive oxygen species in sensing, transduction, and transmission of the signal with altered blood flow. Antioxid. Redox Signal 2014, 20, 899–913. [Google Scholar] [CrossRef]
- Varin, R.; Mulder, P.; Richard, V.; Tamion, F.; Devaux, C.; Henry, J.P.; Lallemand, F.; Lerebours, G.; Thuillez, C. Exercise improves flow-mediated vasodilatation of skeletal muscle arteries in rats with chronic heart failure: Role of nitric oxide, prostanoids, and oxidant stress. Circulation 1999, 99, 2951–2957. [Google Scholar] [CrossRef] [Green Version]
- Alexander, Y.; Osto, E.; Schmidt-Trucksäss, A.; Shechter, M.; Trifunovic, D.; Duncker, D.J.; Aboyans, V.; Bäck, M.; Badimon, L.; Cosentino, F.; et al. Endothelial function in cardiovascular medicine: A consensus paper of the European Society of Cardiology Working Groups on Atherosclerosis and Vascular Biology, Aorta and Peripheral Vascular Diseases, Coronary Pathophysiology and Microcirculation, and Thrombosis. Cardiovasc. Res. 2020, 117, 29–42. [Google Scholar] [CrossRef] [Green Version]
- Korkmaz, H.; Onalan, O. Evaluation of endothelial dysfunction: Flow-mediated dilation. Endothelium 2008, 15, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Blouin, A.; Bolender, R.P.; Weibel, E.R. Distribution of organelles and membranes between hepatocytes and nonhepatocytes in the rat liver parenchyma. A stereological study. J. Cell Biol. 1977, 72, 441–455. [Google Scholar] [CrossRef] [PubMed]
- Cruys, B.; Wong, B.W.; Kuchnio, A.; Verdegem, D.; Cantelmo, A.R.; Conradi, L.C.; Vandekeere, S.; Bouché, A.; Cornelissen, I.; Vinckier, S.; et al. Glycolytic regulation of cell rearrangement in angiogenesis. Nat. Commun. 2016, 7, 12240. [Google Scholar] [CrossRef] [Green Version]
- Feletou, M.; Vanhoutte, P.M. The third pathway: Endothelium-dependent hyperpolarization. J. Physiol. Pharmacol. 1999, 50, 525–534. [Google Scholar] [PubMed]
- Vercauteren, M.; Remy, E.; Devaux, C.; Dautreaux, B.; Henry, J.-P.; Bauer, F.; Mulder, P.; van Huijsduijnen, R.H.; Bombrun, A.; Thuillez, C.; et al. Improvement of Peripheral Endothelial Dysfunction by Protein Tyrosine Phosphatase Inhibitors in Heart Failure. Circulation 2006, 114, 2498–2507. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Mathur, J.; Vessières, E.; Hammack, S.; Nonomura, K.; Favre, J.; Grimaud, L.; Petrus, M.; Francisco, A.; Li, J.; et al. GPR68 Senses Flow and Is Essential for Vascular Physiology. Cell 2018, 173, 762–775.e16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Förstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Iring, A.; Strilic, B.; Juárez, J.A.; Kaur, H.; Troidl, K.; Tonack, S.; Burbiel, J.C.; Müller, C.E.; Fleming, I.; et al. P2Y2 and Gq/G11 control blood pressure by mediating endothelial mechanotransduction. J. Clin. Investig. 2015, 125, 3077–3086. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Chennupati, R.; Kaur, H.; Iring, A.; Wettschureck, N.; Offermanns, S. Endothelial cation channel PIEZO1 controls blood pressure by mediating flow-induced ATP release. J. Clin. Investig. 2016, 126, 4527–4536. [Google Scholar] [CrossRef]
- Makino, A.; Scott, B.T.; Dillmann, W.H. Mitochondrial fragmentation and superoxide anion production in coronary endothelial cells from a mouse model of type 1 diabetes. Diabetologia 2010, 53, 1783–1794. [Google Scholar] [CrossRef] [Green Version]
- Paltauf-Doburzynska, J.; Malli, R.; Graier, W.F. Hyperglycemic conditions affect shape and Ca2+ homeostasis of mitochondria in endothelial cells. J. Cardiovasc. Pharmacol. 2004, 44, 423–436. [Google Scholar] [CrossRef] [PubMed]
- Shenouda, S.M.; Widlansky, M.E.; Chen, K.; Xu, G.; Holbrook, M.; Tabit, C.E.; Hamburg, N.M.; Frame, A.A.; Caiano, T.L.; Kluge, M.A.; et al. Altered Mitochondrial Dynamics Contributes to Endothelial Dysfunction in Diabetes Mellitus. Circulation 2011, 124, 444–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baratchi, S.; Khoshmanesh, K.; Woodman, O.L.; Potocnik, S.; Peter, K.; McIntyre, P. Molecular Sensors of Blood Flow in Endothelial Cells. Trends Mol. Med. 2017, 23, 850–868. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Accession number | Forward | Reverse |
|---|---|---|---|
| Nos3 | NM_008713.4 | CCAGTGCCCTGCTTCATC | GCAGGGCAAGTTAGGATCAG |
| Edn1 | NM_010104.4 | TGCTGTTCGTGACTTTCCAA | GGGCTCTGCACTCCATTCT |
| Opa1 | NM_001199177.1 | ACCAGGAGAAGTAGACTGTGTCAA | TCTTCAAATAAACGCAGAGGTG |
| Dnml1 | NM_001276340.1 | AGATCGTCGTAGTGGGAACG | CCACTAGGCTTTCCAGCACT |
| Hprt | NM_013556.2 | AAGACATTCTTTCCAGTTAAAGTTGAG | AAGACATTCTTTCCAGTTAAAGTTGAG |
| Gapdh | NM_008084.2 | CCGGGGCTGGCATTGCTCTC | GGGGTGGGTGGTCCAGGGTT |
| Gusb | NM_010368.1 | CTCTGGTGGCCTTACCTGAT | CAGTTGTTGTCACCTTCACCTC |
| Target Antigen | Vendor or Source | Catalog # | Working Concentration | Lot # | Persistent ID/URL |
|---|---|---|---|---|---|
| eNOS | BD Biosciences | #610297 | 1/1000 | 8199630 | AB_397691 |
| OPA1 | BD Biosciences | #612606 | 1/1000 | 1025917 | AB_399888 |
| Cyp1B1 | Santa Cruz biotechnologies | #sc-374228 | 1/500 | K0620 | AB_10990317 |
| PFKFB3 | Cell Signaling Technology | #13123 | 1/1000 | 2 | AB_2617178 |
| Klf2 | Atlas antibodies | #HPA055964 | 0.5 µg/mL | AB_2682989 | |
| SOD2 | Cell Signaling Technology | #13141 | 1/1000 | 2 | AB_2636921 |
| SOD3 | Enzo | #ADI-SOD-101-E | 0.5 µg/mL | 01101319 | AB_2039584 |
| gp91phox | BD Biosciences | #611414 | 1/500 | 6226646 | AB_398936 |
| gp91phox | AssayGenie | CAB11966 | 1/500 | 1 | AB_2915942 |
| p22phox | Cell Signaling Technology | #27297 | 1/1000 | 1 | |
| GAPDH | Cell Signaling Technology | #2118 | 1/2000 | 14 | AB_561053 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chehaitly, A.; Guihot, A.-L.; Proux, C.; Grimaud, L.; Aurrière, J.; Legouriellec, B.; Rivron, J.; Vessieres, E.; Tétaud, C.; Zorzano, A.; et al. Altered Mitochondrial Opa1-Related Fusion in Mouse Promotes Endothelial Cell Dysfunction and Atherosclerosis. Antioxidants 2022, 11, 1078. https://doi.org/10.3390/antiox11061078
Chehaitly A, Guihot A-L, Proux C, Grimaud L, Aurrière J, Legouriellec B, Rivron J, Vessieres E, Tétaud C, Zorzano A, et al. Altered Mitochondrial Opa1-Related Fusion in Mouse Promotes Endothelial Cell Dysfunction and Atherosclerosis. Antioxidants. 2022; 11(6):1078. https://doi.org/10.3390/antiox11061078
Chicago/Turabian StyleChehaitly, Ahmad, Anne-Laure Guihot, Coralyne Proux, Linda Grimaud, Jade Aurrière, Benoit Legouriellec, Jordan Rivron, Emilie Vessieres, Clément Tétaud, Antonio Zorzano, and et al. 2022. "Altered Mitochondrial Opa1-Related Fusion in Mouse Promotes Endothelial Cell Dysfunction and Atherosclerosis" Antioxidants 11, no. 6: 1078. https://doi.org/10.3390/antiox11061078
APA StyleChehaitly, A., Guihot, A.-L., Proux, C., Grimaud, L., Aurrière, J., Legouriellec, B., Rivron, J., Vessieres, E., Tétaud, C., Zorzano, A., Procaccio, V., Joubaud, F., Reynier, P., Lenaers, G., Loufrani, L., & Henrion, D. (2022). Altered Mitochondrial Opa1-Related Fusion in Mouse Promotes Endothelial Cell Dysfunction and Atherosclerosis. Antioxidants, 11(6), 1078. https://doi.org/10.3390/antiox11061078